

Abstract
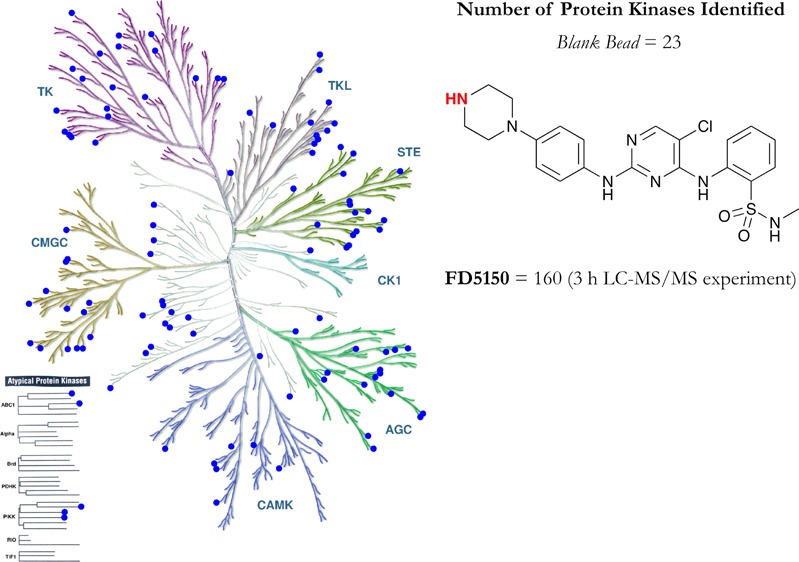
The effects of compound loading on the identification of protein kinases (PKs) was examined using two previously reported sepharose-supported PK inhibitors (PKIs): bisindolylmaleimide X (S1) and CZC8004 (S2). Compound loadings of 0.1, 0.5, 2.5, 5, 10, 25, and 50% content and an ethanolamine-blocked control bead (no compound) were investigated. A 50% bead loading gave the highest level of PK identification for both S1 and S2, extracting 34 and 55 PKs, respectively, from a single cell lysate. Control beads allowed overall identification of 23 PKs, which we term the kinase beadome, whereas sepharose-supported sunitinib (S7; 50% loading) identified 20, 11 of which were common to the control beads. The reliability of bead pull-downs was examined in duplicate experiments using two independently synthesized batches each of S1 and S2. Bead S1 showed high similarity in the absolute numbers of PKs identified across two experiments, at 40 and 35 PKs, of which 26 were common across the two batches of beads, with 14 and 9 unique PKs identified in each experiment. The S2 beads extracted 61 and 64 PKs with 55 PKs common across the two bead batches examined. We also report on the development and use of a novel promiscuous PKI analogue, 2-[(5-chloro-2{[4-(piperazin-1-yl)phenyl]amino}pyrimidin-4-yl)amino]-N-methylbenzene-sulfonamide (S15), which extracted 12 additional unique PKs over the two parent compounds from which it was designed, the combination of which identifies 160 unique PKs. S15 was based on the common pyrimidine core scaffold of S9 and S10. Thus, S15 expands the utility of kinobeads by broadening the kinome coverage for bead-based pull-down. Combining the data for all beads across 90 and 180 min liquid chromatography–mass spectrometry (LC–MS)/MS analysis identified a total of 160 unique PKs.
Introduction
From studying their molecular mechanisms, it is apparent that small molecules and drugs, even targeted drugs, are typically promiscuous. With most small molecule compounds, the mantra “one drug, one target” should be replaced with “one drug, multiple targets”.1 Polypharmacology drugs may be advantageous when compared with highly selective pharmacological agents.2−4 Multitarget activities may lead to increased efficacy with reduced susceptibility to drug resistance. Examples of effective multitarget drugs include aspirin (inflammation) and various selective serotonin reuptake inhibitors (SSRIs; depression) and imatinib (cancer).5−10
Traditionally, drug discovery efforts have been focused on in vitro assays comprising a subset of the target protein family used as purified recombinant proteins. Although this strategy performs well in identifying early leads, it offers only a limited possibility of revealing off-targets. This can partially explain some of the common discrepancies that arise when correlating in vitro assay results with efficacy in cellular or animal models.11,12
Mass spectrometry (MS)-based chemical proteomics has recently emerged as a powerful tool for identification of both the polypharmacology and off-target actions of drug candidates.12−14 Advances in quantitative MS enable relatively routine largescale experimentation. It is possible to quantitate thousands of proteins in a single experiment using both labeled and label-free approaches.15 This can be done on a system-wide basis or using various methodologies to selectively study specific cellular components.16,17
The human genome contains 538 protein kinases (PKs) and more than 2000 other ATP and purine-binding proteins, thus examination of the polypharmacological activities and “off-targets” associated with kinase inhibitors is essential.13,18 A multitude of PK inhibitors (PKIs) target the ATP-binding site of kinases, which have conserved structural elements across the family, and thus is unlikely to be selective toward a single kinase. A chemical proteomics approach based on Kinobead technology has proved to be important in the development of a kinase inhibitor target and off-target profile, confirming the polypharmacology of many clinical drugs.5−7,10,19,20
The development of kinobeads and related chemical proteomics approaches for affinity-based protein profiling requires the immobilization of the small molecule inhibitor to a solid support.21−24 The immobilization step itself presents several challenges, as it may not be synthetically possible with the parent small molecule compound, requiring potentially complex synthetic modifications, which may lead to reduced on-target potency or the introduction of additional off-target effects. The existing functional groups suitable for coupling may be critical binding residues, and thus coupling potentially removes target affinity. These issues have begun to be addressed through the development of multiple generations of kinobeads.15 After bead immobilization, the probe must be still capable of accessing the target ATP-binding site, and this can be affected by the nature of the solid support. Probe-target accessibility may also be affected by the nature of the surface packing and the effect of reactive moieties on the surface of the solid support, which in turn may act synergistically, antagonistically, or present multiple issues through nonspecific interactions with proteins within the cell lysate. To date, there has been no systematic evaluation of the amount of affinity probe loading onto the solid support or the use of unloaded control beads to determine probe specificity, nor has the effect of surface capping to reduce nonspecific binding been evaluated.25 Herein, we report our findings in these areas.
Results and Discussion
Most of the current PKIs target the highly conserved ATP-binding pocket of PKs; thus, an expedient approach to probe development has been based on known promiscuous PKIs, such as bisindolylmaleimide X (BIMX; 1) and CZC8004 (2). These two PKIs are commercially available, and have been the subject of previous studies profiling their binding affinity for PK and purine-binding proteins, and they both can be directly coupled to NHS-sepharose (Figure 1).26,27
Figure 1.
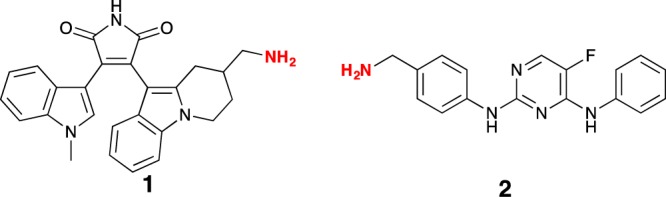
Chemical structures of BIMX (1) and CZC8004 (2), the highlighted NH2 moieties represent the point of attachment to NHS-sepharose beads.
Effect of Bead Loading on PK Capture
To determine the optimal inhibitor loading for capturing PKs on beads, we investigated the effects of 0.1, 0.5, 1, 2, 5, 10, 25, and 50% loadings of BIMX (1) and CZC8004 (2) in a series of pull-down experiments from the lysates of HeLa cells (S1 and S2, respectively, where “S” indicates that the PKI is sepharose-bound). The pull-down data revealed moderate to high levels of PK binding, based on 90 min liquid chromatography (LC)–MS/MS elution and MS evaluation (Figure 2). Efforts to examine higher bead loadings (75 and 100%) were prevented due to lack of compound solubility and poor reproducibility of the synthesis outcomes. Therefore, 50% loading was the highest feasible in this study, while ensuring appropriate quality control over synthesized beads.
Figure 2.

(A) Number of PKs identified by S1 as a function of percentage of the PKI loading of 1 to sepharose beads (S1). (B) As for (A) using 2. This data includes only PKs identified using a 90 min LC–MS/MS analysis. Analyses were conducted in duplicates (see Experimental Procedures).
Upon decreasing the S1 bead loading from 50 to 10%, there was a decrease in the number of PKs identified, and further decreases to 5–0.1% produced variable data. The number of bound PKs with S1 at 50 and 0.1% loading was identical, suggestive of high levels of nonspecific binding in the 0.1% loading S1. With the S2 beads, there was a clearer dose–response from 50 to 2% bead loading, with a decreasing number of PKs observed. These data show that the highest number of PKs is observed with 50% bead loading with both inhibitors. Despite both 1 and 2 being classified as promiscuous PKIs, relatively low numbers of PKs were identified in HeLa cell lysates, with S1 binding 34 and S2 55 PKs, respectively.24,28,29 The relatively lower affinity with S1 exacerbated the nonspecific binding observed (Figure 2). Interestingly, the use of a blank, ethanolamine-blocked control bead (identical to S1 and S2 beads, with 0% PKI loading) reproducibly bound 14 PKs (based on a 90 min LC–MS/MS analysis). Around 1100 proteins were identified in all pull-downs, regardless of the bead loading. There was no clear trend with lower bead loading causing more “off-target” binding, or vice versa (Table S1). The only notable shift was in the number of PKs bound as the bead loading increased (data not shown).
Reproducibly of PK Identification
We investigated the variability between individual measurements with bound inhibitors from two independently synthesized batches of beads through a duplicate extraction and LC–MS/MS profiling of S1 and S2 in HeLa cell lysates. Bead S1 showed high similarity in the absolute numbers of PKs identified at 40 and 35; of these, 26 were common across the pull-down experiments with each bead batch, thus there were 14 and 9 additional PKs identified in each experiment. The combined evaluation of S1, in this manner, afforded identification of a total of 49 PKs (Figure 3). The S2 beads afforded a more robust interrogation of the HeLa cell lysates with the identification of 61 and 64 PKs, with 55 in common across both batches of S2 beads, for a combined total of 70 PKs. Each S2 based experiment identified six and nine unique PKs (Figure 3). For the S2 beads, PKs that bound to only one batch of beads were typically bound at low levels as revealed by a low peptide identification count (data not shown). Given the stochastic nature of peptide selection for MS/MS analysis and the complex nature of the samples, this is not surprising. A similar outcome was evident for the S1 beads, excepting four PKs that were identified by >10 peptides in one batch and absent in the other (data not shown). The reason for this is unclear at this time.
Figure 3.
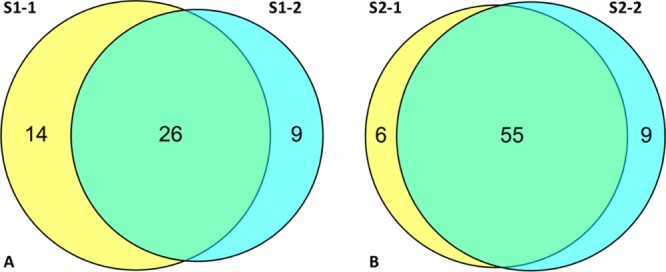
Identification of PKs from HeLa cell lysates using two separately synthesized batches of beads, S1 and S2. (A) S1 bead variation highlighted in the Venn diagram of the S1 batch 1 (S1-1) vs that of the batch 2 (S1-2), showing PK binding overlap; (B) as for A using the S2 batch 1 (S2-1) versus batch 2 (S2-2) showing PK binding overlap. Analyses were conducted in duplicate (Experimental Procedures).
Expanded Bead Catalog
Having established that 50% loading was optimal, we then synthesized a range of other beads based on purvalanol B (3), sunitinib (4), dasatinib (5) (the modified compounds for NHS-sepharose affixing are 6, 7, and 8, respectively, and the synthesis methods are described in the Supporting Information), CTx-0294885 (9), and the Kuster compound 19 (KC19; 10) (Figure 4). PKIs, 9 and 10, were synthesized inhouse (Supporting Information and below). At 50% loading, the residual active NHS ester sites were blocked through treatment with ethanolamine. An ethanolamine-blocked control bead was also prepared, to allow in situ correction for nonspecific binding effects.
Figure 4.

Chemical structures of purvalanol B (3), sunitinib (4), dasatinib (5) (the modified compounds for NHS-sepharose affixing are 6, 7, and 8, respectively) CTx-0294885 (9), and the Kuster compound 19 (KC19; 10). The highlighted NH2 moieties represent the point of NHS-sepharose bead attachment.
Exposure of the ethanolamine-blocked control beads (no attached PKI) to a HeLa cell lysate allowed identification of 23 PKs (Table S1), higher than that observed with the sunitinib S7 probe (23 vs 20 PKs, with 9 PKs being unique to the S7 probe and 11 matching those in the blank probe); Table 1). We call the 23 kinases that bound nonspecifically the “kinase beadome”. In all instances, exposure of the corresponding sepharose-supported analogues (S6, S7, S8, S9, and S10) to a HeLa cell lysate allowed identification of moderate to high numbers of PKs, from 20 (sunitinib (S7)) to 105 (CTx-0294885 (S9)) PKs (Table 1). Although S9 and S10 resulted in 105 and 100 PKs being identified, there was approximately an 80% overlap in the identified PKs. This was not surprising, given their structural similarity, but suggests that additional structural modification might afford either a more diverse or broader identification of the kinome in HeLa cells.
Table 1. Bead Probe Small Molecule and the Number of PKs Identified on Data Combined from 90 and 180 min LC–MS/MS Analyses of the Bead Pull-Down of a HeLa Lysate.
| bead | number of PKs identifieda | number of PKs overlapping with blank beads |
|---|---|---|
| blocked beads (control) | 23 | NA |
| BIMX (S1) | 40 | 20 |
| CZC8004 (S2) | 61 | 19 |
| purvalanol B (S6) | 65 | 17 |
| sunitinib (S7) | 20 | 11 |
| dasatinib (S8) | 64 | 19 |
| CTx-0294885 (S9) | 105 | 21 |
| KC19 (S10) | 100 | 22 |
Analyses were conducted as duplicates, using 90 and 180 min runs.
Of the PKIs examined in Table 1, S9 (Ctx-0298845) and S10 (KC19) were significantly more effective than S1–S8. The former two compounds comprised a common pyrimidine core, differing only in the pendent moieties, with the S9-amide represented by the isosteric sulfonamide in S10. We therefore further explored the development of an S9/S10 hybrid through the synthesis of a novel compound FD5180 (15). We merged the key structural features of 9 and 10 while retaining the common pyrimidine core (Figure 5), leveraging our prior synthesis of 9 and our in-house synthesis of 10.30,31
Figure 5.

Merging of the common pharmacophoric elements of 9 (CTx-0298845) and 10 (KC19) to afford the hybrid analogue 15 (FD5180) for subsequent attachment to sepharose beads.
Synthesis commenced with the generation of sulfonamide 12 from 2-nitrobenzenesulfonyl chloride 11 and methylamine, followed by a facile Fe/AcOH-mediated reduction of 12’s nitro moiety, affording 13.32 Several bases and reaction conditions were trialed in relation to the nucleophilic aromatic substitution of 13 to afford 14, and employing cesium carbonate as base with microwave irradiation led to the highest yield, albeit still modest at 29%, of pyrimidine 14. The final step toward the synthesis of both KC19 (10) and FD5180 (15) involved a second SNAr reaction with the requisite amine (Scheme 1). In the generation of 10, tert-butyl 4-aminobenzylcarbamate was employed to prevent competing addition via the benzylamine moiety. The nucleophilic substitution proceeded smoothly with simultaneous boc-deprotection giving 10. With hybrid 15, tert-butyl 4-(4-aminophenyl)piperazine-1-yl)aniline was employed and, in a similar manner, led to the generation of boc-deprotected 15 in a moderate yield (41%).
Scheme 1. Reagents and Conditions.

(i) CH3NH2, (CH3CH2)3N, CH2Cl2, rt, 1 h; (ii) Fe powder, glacial AcOH, ethanol, sonication, rt; 1 h; (iii) 2,4,5-trichloropyrimidine, Cs2CO3, CH3CN, MW 150 °C, 1 h; (iv) tert-butyl 4-aminobenzylcarbamate, MW 150 °C, 30 min; (v) tert-butyl 4-(4-aminophenyl)piperazine-1-carboxylate, n-butanol, MW 150 °C, 1 h.
Characterization of PKs Identified by Hybrid S15 (FD5180)
The use of S15 (FD5180) in a series of pull-down experiments from HeLa cell lysates with an LC–MS/MS elution of 90 and 180 min resulted in the identification of 111 PKs (with an overlap of 22 PKs with the control bead), a slightly larger number than that observed with CTx-0298845 (S9) and KC19 (S10) (Table 1 and Figure 6). Importantly, each of the three beads identified a number of distinct kinases (Figure 6). Combining the data acquired for all beads across 90 and 180 min LC–MS/MS analysis identified a total of 160 unique PKs.
Figure 6.
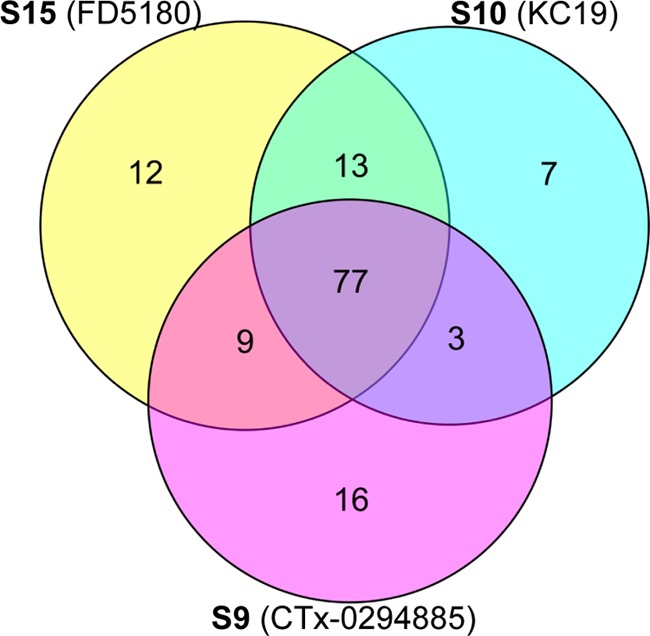
Venn diagram showing the overlap of unique PKs arising from pull down of a HeLa lysate with S9, S10, or S15, after analysis using a 90 and 180 min LC–MS/MS elution. Analyses were conducted in duplicate from 90 and 180 min high-performance liquid chromatography (HPLC) runs.
Despite the similar structural chemical motif spanning S9, S10, and S15, only 77 PKs were common to all three beads. Individually, S9, S10, and S15 yielded 16, 7, and 12 unique PKs, respectively. Examination of the kinase classes identified highlighted that S15 showed good coverage of all major classes (TK, TKL, STE, CMGC, CAMK, and AGC), only lacking in coverage of the CK1 class, as does S10, with only S9 showing a low coverage (Figure 7). Probe S15 showed more pronounced (than S9 and S10) coverage, in particular, of the AGC and CMGC kinase classes. The kinase coverage by these beads is shown in Figures S25–S34. These data suggest that the simple modifications made herein impart additional promiscuity to this class of already broad spectrum PKIs.
Figure 7.

Comparison of PKs pulled down using CTx-0294885 (S9), KC19 (S10), and hybrid FD5180 (S15) kinase affinity beads. (A) PKs common to all three beads (blue). (B) PKs common between hybrid FD5180 (S15) and KC19 (S10) (red); PKs common between hybrid FD5180 (S15) and CTx-0294885 (S9) (green); PKs common between CTx-0294885 (S9) and KC19 (S10) (magenta). (C) PKs unique to CTx-0294885 (S9, black), KC19 (S10, green), and hybrid FD5180 (S15, red). Illustration was drawn courtesy of Cell Signaling Technology, Inc., www.cellsignal.com, and the kinome render database comprising 523 PKs.
Impact of Control Beads on Kinome Analysis
This is the first evaluation of ethanolamine-blocked blank beads, which bind 23 PKs nonspecifically (Table 2). Relative quantitation shows that of the 23 PKs bound, 14 bind to all beads (including inhibitor-loaded S6–S10 and S15) at a similar level (Figures S1–S14), consistent with the proposed nonspecific nature of this binding. Of the remaining nine PKs, most bind at a similar level across S6–S10 and S15, again suggesting a high level of relatively nonspecific binding (Figures S15–S22).
Table 2. Kinase Beadomea.
| PK uniprot code | PK identity | supp figure | PK uniprot code | PK identity | supp figure |
|---|---|---|---|---|---|
| P51812 | ribosomal protein S6 kinase α-3 | S12 | Q9H2G2 | STE20-like serine/threonine-PK | S24 |
| P78527 | DNA-dependent PK catalytic subunit | S13 | P46734 | dual specificity mitogen-activated PK 3 | S25 |
| Q13882 | protein-tyrosine kinase 6 | S14 | P06213 | insulin receptor | S26 |
| P08581 | hepatocyte growth factor receptor | S15 | P16591 | tyrosine-PK Fer | S27 |
| P42345 | serine/threonine-PK mTOR | S16 | Q9UQM7 | calcium/calmodulin-dependent PK type II subunit α | S28 |
| P23458 | tyrosine-PK JAK1 | S17 | P07948 | tyrosine-PK Lyn | S29 |
| P06241 | tyrosine-PK Fyn | S18 | O60674 | tyrosine-PK JAK2 | S30 |
| P49841 | glycogen synthase kinase-3 β | S19 | Q86TW2 | uncharacterized aarF domain-containing PK 1 | S31 |
| O94804 | serine/threonine-PK 10 | S20 | P10398 | serine/threonine-PK A-Raf | S32 |
| P52564 | dual specificity mitogen-activated PK 6 | S21 | P54760 | ephrin type-B receptor 4 | S33 |
| P29317 | ephrin type-A receptor 2 | S22 | P53355 | death-associated PK 1 | S34 |
| Q99683 | mitogen-activated PK 5 | S23 |
List of the PKs detected with ethanolamine-blocked sepharose (agarose) blank control beads, based on a 90 and 180 min LC–MS/MS analysis of a HeLa cell lysate.
Conclusions
Our data demonstrates that the level of chemical biology probe (inhibitor) affixed to a solid support impacts the number of PKs identified in a HeLa cell lysate. Examination of S1 and S2 beads loaded with 0.1, 0.5, 2.5, 5, 10, 25, and 50% inhibitor revealed the optimal bead loading to be 50%. The use of control beads, for the first time in a kinobead experiment, revealed significant levels of nonspecific binding (what we term the “kinase beadome”), which in one case surpassed the number of PKIs identified by inhibitor bound beads, for example, sunitinib (S7) at 20 versus 23 PKs identified. These observations highlight the key role of including appropriate control bead validation studies as an essential part of the identification of a lead target compound.
Our study is the first we are aware of that compares two separate synthetic batches of the same bead used for pull-downs, demonstrating reasonable reliability, robustness, and quality control. Using HeLa cell lysate S1 afforded identification of 40 and 35 PKs, respectively, with 14 and 9 unique PKs across S1-1 and S1-2. The S2 beads afforded a more robust interrogation of the HeLa cell lysate with the identification of 61 and 64 PKs with 55 PKs in common across S2-1 and S2-2. Although potentially adversely impacting the quantitation, this supports an expansion of the kinome for a particular target compound, on the basis of sequential analysis and pooling of the data attained. The data demonstrates the need for better quality control in bead validation studies.
Recent reports have positioned KC19 (S10) and CTx-0294885 (S9) as highly promiscuous PKIs, with a significant level of structural similarity, which suggested further potential to enhance the promiscuity of these compounds. Our data validates this hypothesis through the synthesis and evaluation of 2-[(5-chloro-2{[4-(piperazin-1-yl)phenyl]amino}pyrimidin-4-yl)amino]-N-methylbenzene-sulfonamide (FD5180; S15) in a HeLa cell lysate with the identification of 111 PKs in this single experiment from a single cell type. S15 performs at a similar overall level to S10 and S9 at 100 and 105 PKs, respectively. However, these three compounds were each isolated from 7 to 16 kinases that were unique to each ligand. The addition of S15 allowed a maximum of 160 or 147 PKs to be identified over a 180 min or a 90 min LC–MS/MS experiment, respectively, across these three beads. On the basis of a single evaluation comparison of S15, S10, and S9, there were 77 common PKs identified, whereas S9, S10, and S15 further yielded 16, 7, and 12 unique PKs, respectively. Therefore, 2-[(5-chloro-2{[4-(piperazin-1-yl)phenyl]amino}pyrimidin-4-yl)amino]-N-methylbenzene-sulfonamide (FD5180; S15) expands the utility of kinobeads by broadening the kinome covered.
Using a range of beads, a user can select broadly reactive (S15), or more specific (Sunitinib, S7) beads, depending on the experimental requirements and the kinases being targeted by each compound. The beads can be combined for greater overall kinome coverage. Our study shows that for target validation, control beads are an imperative.
Experimental Procedures
Chemistry General Procedures
All reactions were performed using standard laboratory equipment and standard laboratory glassware. Solvents and reagents were purchased from Sigma-Aldrich, Matrix Scientific, or Lancaster Synthesis and used as received. Organic solvents were bulk quality and were redistilled from glass prior to use. Flash chromatography was carried out using silica gel 200–400 mesh (60 Å). Organic solvent extracts were dried with magnesium sulfate (MgSO4) and dried under reduced pressure with either Büchi or Heidolph rotary evaporators. Melting points were recorded in open capillaries on a Brüker 565 Melting Point Apparatus and are expressed in degrees Celsius (°C). Electrospray mass spectra were recorded using 10% dimethyl sulfoxide (DMSO)/H2O or HPLC-grade methanol or acetonitrile as carrier solvents on a Shimadzu LC–MS 2010 EV spectrometer or an Agilent 1260 LCMS system. All compounds were of ≥95% purity as determined by HPLC and LC–MS analysis.
NMR spectroscopy was performed on a Brüker Avance III 400 MHz (or 600 MHz) spectrometer, where 1H NMR and 13C NMR were acquired at 400 and 101 MHz (or 600 and 150 MHz), respectively. All spectra were recorded in deuterated dimethyl sulfoxide (DMSO-d6) or deuterated chloroform (CDCl3), obtained from Sigma-Aldrich or Cambridge Isotope Laboratories Inc., unless otherwise stated, with the residual solvent peaks used as the internal reference (δ 2.49 (quintet) and δ 39.7 (septet) for 1H NMR and 13C NMR, respectively). Chemical shifts (δ) were measured in parts per million (ppm) and referenced against the internal reference peaks. Coupling constants (J) were measured in Hertz (Hz). Multiplicities are denoted as singlet (s), broad singlet (br s), doublet (d), doublet of doublets (dd), doublet of doublet of doublets (ddd), triplet (t), triplet of doublets (td), doublet of triplets (dt), quartet (q), quintet (quin), and multiplet (m). Peaks are listed in increasing chemical shift in the following format: chemical shift (integration (1H), multiplicity (1H), coupling constant (1H)).
Synthesis
N-Methyl-2-nitrobenzenesulfonamide (12)
2-Nitrobenzenesulfonyl chloride 11 (1.001 g, 4.51 mmol) was added to a stirring solution of methylamine (0.5 mL, 5.42 mmol) and triethylamine (1.0 mL, 6.77 mmol) in CH2Cl2 (40 mL) and stirred at room temperature (rt) for 1 h. After this time, 1 M HCl (40 mL) was added, and the organic layer was extracted with CH2Cl2 (2 × 40 mL). The aqueous phase was washed again with CH2Cl2 (2 × 40 mL). The combined organic extracts were washed with saturated NaHCO3 (50 mL), dried over MgSO4, and evaporated in vacuo to afford 12 (0.887 g, 91%) as an off-white solid. M.p. 112–114 °C. 1H NMR (400 MHz, acetone-d6) δ 8.12–8.10 (m, 1H), 7.98–7.88 (m, 3H), 6.49 (s, 1H), 2.75 (s, 3H); 13C NMR (101 MHz, acetone-d6) δ 148.5, 134.1, 132.5, 132.2, 130.8, 124.9, 28.9; Mass spectrum (ESI, −ve) m/z 216 [(M – H)−, 100%], IR (cm–1) 3335, 3096, 1533, 1381, 1352, 1339, 1167, 1115, 731.
2-Amino-N-methylbenzenesulfonamide (13)
To a suspension of 12 (0.150 g, 0.69 mmol) in glacial acetic acid (2 mL), ethanol (2 mL), and water (1 mL) was added iron powder (0.200 mg, 3.45 mmol).32 The suspension was sonicated for 1 h at room temperature. After this time, the reaction mixture was filtered through a pad of Celite, washed with EtOAc (50 mL), and evaporated in vacuo. The resulting residue was partitioned between 1 M NaOH (40 mL) and EtOAc (40 mL). The separated aqueous layer was extracted with EtOAc (3 × 40 mL), the combined organic layers were washed with water (40 mL) and brine (40 mL), dried over MgSO4, and evaporated in vacuo to afford the desired 13 as a yellow cloudy oil (0.103 g, 80%). 1H NMR (400 MHz, acetone-d6) δ 7.69 (dd, J = 8.0, 1.4 Hz, 1H), 7.32–7.30 (m, 1H), 6.82–6.76 (m, 2H), 4.88 (s, 2H), 4.81 (s, 1H), 2.56 (d, J = 5.4 Hz, 3H); 13C NMR (151 MHz, acetone-d6) δ 146.6, 133.6, 129.6, 119.8, 117.2, 115.9, 28.4; mass spectrum (ESI, +ve) m/z 187 [(M + H)+, 100%], IR (cm–1) 3480, 3377, 2939, 1619, 1600, 1483, 1454, 1315, 1143, 1064, 749.
2-[(2,5-Dichloropyrimidin-4-yl)amino]-N-methylbenzenesulfonamide (14)
A mixture comprising 13 (0.250 g, 1.34 mmol), 2,4,5-trichloropyrimidine (0.271 g, 1.48 mmol), and cesium carbonate (0.875 g, 2.68 mmol) in acetonitrile (5 mL) was heated under microwave irradiation for 1 h at 150 °C. The reaction was cooled, evaporated under reduced pressure, and dissolved in EtOAc (40 mL) and water (40 mL). The aqueous layer was washed with EtOAc (2 × 40 mL), and the organic extracts were combined, washed with brine (40 mL), dried over MgSO4, and evaporated in vacuo. The residue obtained was purified by flash chromatography (hexane: ethyl acetate 100:0 → 50:50) to furnish 14 (0.290 g, 29%) as a white solid. M.p. 162 °C (dec.); 1H NMR (400 MHz, CDCl3) δ 9.62 (s, 1H), 8.54 (dd, J = 8.4, 1.1 Hz, 1H), 8.28 (s, 1H), 7.95 (dd, J = 8.0, 1.6 Hz, 1H), 7.67 (ddd, J = 8.7, 7.4, 1.7 Hz, 1H), 7.29 (ddd, J = 8.5, 7.5, 1.2 Hz, 1H), 4.65 (d, J = 5.6 Hz, 1H), 2.66 (d, J = 5.4 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 158.0, 156.5, 155.6, 135.6, 134.3, 130.1, 127.1, 124.4, 123.1, 115.3, 29.5; mass spectrum (ESI, +ve) m/z 333 [(M)+ 100%], 335 [(M + 2)+, 65%], 337 [(M + 4)+, 12%], IR (cm–1) 3325, 3105, 2853, 1595, 1568, 1559, 1481, 1447, 1358, 1309, 1148, 1052, 976, 759, 541.
2-[(2-{[4-(Aminomethyl)phenyl]amino}-5-chloropyrimidin-4-yl)amino]-N-methylbenzene-sulfonamide (10)
A mixture comprising 14 (0.104 g, 0.31 mmol) and tert-butyl 4-aminobenzylcarbamate (0.058 mg, 0.26 mmol) in n-butanol (5 mL) was heated under microwave irradiation at 150 °C for 30 min. The mixture was cooled to room temperature, concentrated in vacuo, and taken up in CH2Cl2 (20 mL), and the recovered starting material 14 was collected by filtration. The filtrate was evaporated in vacuo to afford the titled compound 10 as a pale brown solid (0.02 g, 16%). M.p. 142 °C (dec.); 1H NMR (400 MHz, CD3OD) δ 8.57 (d, J = 8.2 Hz, 1H), 8.10 (s, 1H), 7.89 (dd, J = 8.0, 1.4 Hz, 1H), 7.61–7.55 (m, 3H), 7.54–7.49 (m, 2H), 7.28–7.20 (m, 2H), 7.15 (d, J = 8.5 Hz, 2H), 4.43 (s, 2H), 3.58 (s, 1H), 2.51 (s, 3H); 13C NMR (101 MHz, CD3OD) δ 159.4, 156.9, 155.9, 140.1, 138.0, 134.9, 130.5, 129.3 (2C), 128.8, 128.7, 125.1, 124.3, 121.4 (2C), 106.8, 55.5, 29.1; mass spectrum (ESI, −ve) m/z 417 [(M(35Cl) – H)−, 100%], 419 [(M(37Cl) – H)−, 35%]; IR νmax (cm–1) 3340, 3296, 2922, 2852, 1601, 1575, 1566, 1520, 1415, 1321, 773, 582.
2-[(5-Chloro-2{[4-(piperazin-1-yl)phenyl]amino}pyrimidin-4-yl)amino]-N-methylbenzene-sulfonamide (15)
A mixture comprising 14 (0.200 g, 0.60 mmol) and tert-butyl 4-(4-aminophenyl)piperazine-1-carboxylate (0.45 mmol, 6.2 mL of 0.072 M stock solution in n-BuOH) was heated under microwave irradiation at 150 °C for 1 h. After this time, the reaction mixture was concentrated in vacuo, and the residue was purified by flash chromatography (hexane/ethyl acetate 100:0 → 50:50) to afford the titled compound 15 as a pale brown solid (0.102 g, 41%); 1H NMR (400 MHz, CD3OD) δ 8.59 (d, J = 8.3 Hz, 1H), 8.07 (s, 1H), 7.88 (dd, J = 8.0, 1.5 Hz, 1H), 7.64–7.46 (m, 1H), 7.42 (d, J = 9.0 Hz, 2H), 7.32–7.18 (m, 1H), 6.93 (d, J = 9.0 Hz, 2H), 3.11 (dd, J = 6.3, 3.6 Hz, 4H), 3.01 (dd, J = 6.2, 3.7 Hz, 4H), 2.51 (s, 3H); 13C NMR (101 MHz, CD3OD) δ 159.8, 157.0, 155.8, 149.1, 138.1, 134.4, 134.3, 130.5, 128.4, 124.9, 124.1, 123.4 (2C), 118.3 (2C), 106.3, 51.8 (2C), 46.5 (2C), 29.0; mass spectrum (ESI, −ve) m/z 472 [(M(35Cl) – H)−, 100%], 474 [(M(37Cl) – H)−, 35%]; IR (cm–1) 3340, 3290, 3084, 2823, 1606, 1576, 1516, 1419, 1321, 773, 586.
Modification of Sunitnib
tert-Butyl (6-(5-Formyl-2,4-dimethyl-1H-pyrrole-3-carboxamido)hexyl)carbamate
A solution of 5-formyl-2,4-dimethyl-1H-pyrrole-3-carboxylic acid (150 mg, 0.90 mmol) in acetonitrile (10 mL) was stirred at room temperature and HBTU (510 mg, 1.35 mmol), iPr2NEt (480 μL, 2.70 mmol), and then tert-butyl (6-aminohexyl)carbamate (242 μL, 1.08 mmol) added. The ensuing mixture was heated to 60 °C overnight. After this time, the mixture was cooled to room temperature and concentrated in vacuo to afford a dark solid that was immediately dissolved in EtOAc (50 mL). The solution was washed with water (2 × 20 mL), brine (50 mL), dried over MgSO4, filtered, and the filtrate concentrated in vacuo to give a brown solid that was purified by flash chromatography (CH3OH/CH2Cl2 0:1 → 1:9 v/v), and concentration of the relevant fractions (Rf = 0.2 in 1:9 v/v CH3OH/CH2Cl2) afforded the titled compound as a white solid (194 mg, 59%). M.p. 178–179 °C; 1H NMR (600 MHz, CDCl3 + CD3OD) δ 9.57 (s, 1H), 9.39 (s, 1H), 5.65 (s, 1H), 4.54 (s, 1H), 3.11 (s, 2H), 2.50 (s, 3H), 2.49 (s, 3H), 1.61–1.59 (m, 2H), 1.50–1.36 (m, 17H); 13C NMR (151 MHz, CDCl3 + CD3OD) δ 177.0, 165.1, 156.1, 139.0, 130.7, 128.1, 119.3, 79.1, 40.2, 39.2, 30.04, 29.8, 28.4 (3 overlapping signals), 26.4, 26.1, 13.5, 10.2; mass spectrum (ESI, +ve) m/z 366 [(M + H)+ 20%], 388 [(M + Na)+, 13%], 310 [(M – Boc + 2Na – H)+, 100%], 266 [(M – Boc + H)+, 65%]; IR (cm–1) 3374, 3214, 2981, 2936, 1688, 1665, 1623, 1522, 1442, 1363, 1342, 1279, 1248, 1175, 1039, 1021, 976, 810, 780, 713, 621.
(Z)-tert-Butyl (6-(5-((5-Fluoro-2-oxoindolin-3-ylidene)methyl)-2,4-dimethyl-1H-pyrrole-3-carboxamido)hexyl)carbamate
A solution of tert-butyl (6-(5-formyl-2,4-dimethyl-1H-pyrrole-3-carboxamido)hexyl)carbamate (124 mg, 0.34 mmol) in ethanol (10 mL) was stirred at room temperature, and 5-fluoroindolin-2-one (57 mg, 0.37 mmol) and piperidine (1 drop) were added. The ensuing mixture was heated at reflux overnight and then cooled to room temperature. The resulting slurry was filtered, and the precipitate was washed with cold ethanol (10 mL), cold diethyl ether (10 mL), and air dried to afford the titled compound as a yellow solid (127 mg, 75%). M.p. 243–244 °C; 1H NMR (600 MHz, DMSO-d6) δ 13.67 (s, 1H), 10.88 (s, 1H), 7.75 (d, J = 9.0 Hz, 1H), 7.70 (s, 1H), 7.62 (t, J = 5.2 Hz, 1H), 6.92 (t, J = 9.0 Hz, 1H), 6.85–6.83 (m, 1H), 6.77 (s, 1H), 3.20 (dd, J = 12.3, 6.2 Hz, 2H), 2.90 (dd, J = 12.3, 6.2 Hz, 2H), 2.42 (s, 3H), 2.40 (s, 3H), 1.53–1.45 (m, 2H), 1.37 (s, 9H), 1.31– 1.27 (m, 4H) (signals due to amide group not observed); 13C NMR (151 MHz, DMSO-d6) δ 170.0, 165.0, 158.7 (d, 1JCF = 234.1 Hz), 156.1, 136.7, 135.0, 130.7, 127.7 (d, 3JCF = 9.5 Hz), 126.2, 125.4, 121.7, 114.9 (d, 4JCF = 2.9 Hz), 112.8 (d, 2JCF = 24.2 Hz), 110.5 (d, 3JCF = 8.7 Hz), 106.5, 106.3, 77.8, 39.1, 30.0, 29.8, 28.7 (3 overlapping signals), 26.7, 26.5, 13.7, 10.9; mass spectrum (ESI, +ve) m/z 499 [(M + H)+ 100%], 443 [(M – Boc + 2Na – H)+, 55%], 399 [(M – Boc + H)+, 25%]; IR (cm–1) 3363, 3149, 2936, 1686, 1674, 1567, 1524, 1478, 1327, 1276, 1241, 1171, 1144, 1043, 997, 926, 793, 782, 669, 606.
(Z)-N-(6-Aminohexyl)-5-((5-fluoro-2-oxoindolin-3-ylidene)methyl)-2,4-dimethyl-1H-pyrrole-3-carboxamide
A slurry of (Z)-tert-butyl (6-(5-((5-fluoro-2-oxoindolin-3-ylidene)methyl)-2,4-dimethyl-1H-pyrrole-3-carboxamido)hexyl)carbamate (116 mg, 2.33 mmol) in CH2Cl2 (10 mL) was stirred at 0 °C and treated dropwise with HCl (233 μL, 0.93 mmol of a 4 M dioxane solution). The ensuing mixture was stirred and allowed to warm to room temperature over 4 h. The resulting slurry was filtered, and the precipitate washed with cold ether (10 mL) and air dried to afford the titled compound (70 mg, 75%) as a yellow solid. This material was used directly in the bead coupling due to degradation (product turned a tan color) on sitting. The product and purity were confirmed by LC–MS analysis. Mass spectrum (ESI, +ve) m/z 443 [(M + H)+ 100%].
Modification of Dasatinib
N-(2-Chloro-6-methylphenyl)-2-((6-(4-(2-chloroethyl)piperazin-1-yl)-2-methylpyrimidin-4-yl)amino)thiazole-5-carboxamide
A slurry of dasatinib (346 mg; 0.71 mmol) in anhydrous dimethylformamide (DMF) (1 mL) was stirred at 0 °C and treated dropwise with thionyl chloride (210 μL, 2.90 mmol). The ensuing mixture was stirred and allowed to warm to room temperature over 2 h; it was then poured onto iced water (50 mL), and the pH adjusted to 8 by the addition of saturated NaHCO3. The resulting slurry was filtered, washed with water (2 × 25 mL), and air dried to afford the titled compound as a white solid (230 mg, 64%); m.p. 241–244 °C); 1H NMR (600 MHz, DMSO-d6) δ 11.46 (s, 1H), 9.87 (s, 1H), 8.22 (s, 1H), 7.40 (d, J = 7.6 Hz, 1H), 7.29–7.25 (m, 2H), 6.05 (s, 1H), 3.72 (t, J = 5.7 Hz, 2H), 3.52 (s, 4H), 2.69 (t, J = 5.7 Hz, 2H), 2.52 (s, 4H, overlapped by residual DMSO signal), 2.41 (s, 3H), 2.24 (s, 3H); 13C NMR (151 MHz, DMSO-d6) δ 165.2, 162.6, 162.3, 159.9, 157.0, 138.8, 133.5, 132.4, 129.0, 128.2, 127.0, 125.7, 82.7, 59.1, 52.1 (2 overlapping signals), 43.5 (2 overlapping signals), 41.4, 25.6, 18.3; Mass spectrum (ESI, +ve) m/z 507 [(M + H)+ 80%]; IR (cm–1) 3195, 2943, 2821, 1623, 1583, 1512, 1411, 1394, 1295, 1194, 1131, 1003, 983, 862, 812, 862, 812, 770, 734, 686, 637.
2-((6-(4-(2-Azidoethyl)piperazin-1-yl)-2-methylpyrimidin-4-yl)amino)-N-(2-chloro-6-methylphenyl)thiazole-5-carboxamide
A solution of chlorodasatinib (481 mg, 0.95 mmol) in DMF (5 mL) was stirred at room temperature and sodium azide (150 mg, 2.34 mmol) added. The mixture was heated to 85 °C overnight, cooled to room temperature, and poured into water (50 mL). The resulting slurry was filtered; the precipitate washed with water (2 × 25 mL) and air dried to afford the titled compound as a white solid (437 mg; 90%). M.p. 233 °C (dec.); 1H NMR (600 MHz, DMSO magnetically stirred) δ 9.87 (s, 1H), 8.22 (s, 1H), 7.40 (d, J = 7.7 Hz, 1H), 7.30–7.24 (m, 2H), 6.06 (s, 1H), 3.52 (s, 4H), 3.41 (t, J = 5.7 Hz, 2H), 2.58 (t, J = 5.7 Hz, 2H), 2.52 (s, 4H, obscured by residual DMSO signal), 2.41 (s, 3H), 2.24 (s, 3H) (signals due to secondary amine not observed); 13C NMR (151 MHz, DMSO-d6) δ 165.7, 163.0, 162.84, 160.4, 157.4, 141.3, 139.3, 134.0, 132.9, 129.5, 128.6, 127.5, 126.2, 83.2, 57.0, 52.5 (2 overlapping signals), 47.6, 44.1 (2 overlapping signals), 26.1, 18.8; mass spectrum (ESI, −ve) m/z 511 [(M – H)−, 80%]; IR (cm–1) 3199, 2856, 2104, 1619, 1575, 1506, 1413, 1392, 1293, 1186, 1104, 1009, 976, 860, 770, 633.
2-((6-(4-(2-Aminoethyl)piperazin-1-yl)-2-methylpyrimidin-4-yl)amino)-N-(2-chloro-6-methylphenyl)thiazole-5-carboxamide
A slurry of azidodasatinib (437 mg, 0.85 mmol) in 1:1:1 v/v/v – CH3OH/CH3CH2OH/EtOAc (50 mL) was stirred at room temperature, and 10% palladium on carbon (44 mg) was added. This was bubbled with hydrogen gas, and this mixture stirred at room temperature under a hydrogen atmosphere (1 atm). The dark mixture was filtered (Celite); the filter cake washed with CH3OH (2 × 10 mL); and the filtrate was concentrated in vacuo to afford an off-white solid, which was purified by flash chromatography (Grace Amino column, 0:1 → 1:9 v/v CH3OH/CH2Cl2), and the concentration of the relevant fractions afforded the titled compound as a white solid (261 mg, 55%) m.p. 213 °C (dec.); 1H NMR (600 MHz, DMSO-d6) δ 9.87 (s, 1H), 8.22 (s, 1H), 7.40 (d, J = 7.2 Hz, 1H), 7.37–7.18 (m, 2H), 6.05 (s, 1H), 3.52 (s, 4H), 2.70 (2H), 2.45–2.41 (m, 9H), 2.24 (s, 3H) (signals due to secondary amine not observed); 13C NMR (151 MHz, DMSO-d6) δ 165.2, 162.6, 162.4, 159.9, 157.0, 140.8, 138.8, 133.5, 132.4, 129.0, 128.2, 127.0, 125.7, 82.6, 59.7, 52.4 (2 overlapping signals), 43.6 (2 overlapping signals), 38.1, 25.6, 18.3; mass spectrum (ESI, +ve) m/z 487 [(M + H)+, 40%]; IR (cm–1) 3999, 2886, 2817, 1621, 1577, 1507, 1413, 1293, 1194, 1001, 986, 770.
Modification of Purvalanol B
(R)-4-((2-((1-((tert-Butyldimethylsilyl)oxy)-3-methylbutan-2-yl)amino)-9-isopropyl-9H-purin-6-yl)amino)-2-chlorobenzoic Acid [O-TBDMS Purvalanol B]
A solution of purvalanol B (50 mg, 0.49 mmol) in DMF (1 mL) was stirred at room temperature, and imidazole (20 mg; 0.98 mmol) and TBDMS chloride (80 mg, 0.74 mmol), added. The ensuing mixture was stirred at room temperature overnight, diluted with water (20 mL), and then extracted with ethyl acetate (4 × 20 mL). The organic extracts were washed with water (2 × 50 mL), brine (1 × 30 mL), dried over MgSO4, and concentrated in vacuo to give a yellow oil. Purification by flash chromatography (Grace Amino column, 0:1 → 1:9 v/v CH3OH/CH2Cl2) and concentration of the relevant fractions afforded the titled compound as a clear oil (48 mg, 55%); 1H NMR (600 MHz, CDCl3) δ 10.46 (s, 1H), 8.59 (d, J = 8.2 Hz, 1H), 8.32 (s, 1H), 8.15 (d, J = 8.7 Hz, 1H), 7.73 (s, 1H), 5.10 (d, J = 9.4 Hz, 1H), 4.71–4.69 (m, 1H), 3.97 (s, 2H), 3.82–3.79 (m, 1H), 3.69–3.67 (m, 1H), 2.07–2.03 (m, 1H), 1.55 (d, J = 6.8 Hz, 3H), 1.54 (d, J = 6.8 Hz, 3H), 1.01 (d, J = 7.0 Hz, 3H), 1.00 (d, J = 7.0 Hz, 3H), 0.99 (s, 9H), 0.06 (s, 6H); 13C NMR (151 MHz, CDCl3) δ 169.2, 159.8, 151.9, 151.7, 145.2, 136.5, 134.8, 133.4, 122.5, 121.4, 117.2, 113.4, 63.1 (2 overlapping signals), 57.87 (very slow relaxation, determined by heteronuclear single quantum coherence (HSQC) experiments), 46.6 (very slow relaxation, determined by HSQC experiments), 29.2, 25.9 (3 overlapping signals), 22.6, 22.5, 19.9, 18.3, −5.38 (2 overlapping signals); IR (cm–1) 3294, 3195, 3103, 2955, 2932, 2859, 1686, 1634, 1587, 1567, 1499, 1463, 1396, 1293, 1253, 1236, 1106, 1045, 1007, 837, 787, 738.
(R)-tert-Butyl (6-(4-((2-((1-((tert-Butyldimethylsilyl)oxy)-3-methylbutan-2-yl)amino)-9-isopropyl-9H-purin-6-yl)amino)-2-chlorobenzamido)hexyl)carbamate
A solution of O-TBDMS purvalanol B (237 mg, 0.43 mmol) in DMF (1 mL) was stirred at room temperature, and HBTU (427 mg, 0.65 mmol), HOBt (64 mg, 0.47 mmol), iPr2NEt2 (230 μL, 1.29 mmol), and tert-butyl (6-aminohexyl)carbamate (105 μL, 0.47 mmol) were added. The ensuing mixture was stirred at room temperature overnight, diluted with water (50 mL), and extracted with ethyl acetate (4 × 20 mL). The organic extracts were washed with 1 M NaOH (1 × 20 mL), water (2 × 20 mL), brine (40 mL), dried over MgSO4 and concentrated in vacuo to afford a tan-colored residue. Purification by flash chromatography (0:1 → 1:19 v/v CH3OH/CH2Cl2) and concentration of the relevant fraction (Rf = 0.4 in 1:9 v/v CH3OH/CH2Cl2) afforded the titled compound as a gold-colored resin (306 mg, 96%); 1H NMR (600 MHz, CDCl3 + CD3OD) δ 8.22 (s, 1H), 7.86 (s, 1H), 7.71 (d, J = 8.5 Hz, 1H), 7.57 (s, 1H), 7.49 (d, J = 7.3 Hz, 1H), 6.47 (s, 1H), 5.13 (d, J = 9.5 Hz, 1H), 4.74–4.62 (m, 1H), 4.57 (s, 1H), 3.97 (dt, J = 10.8, 4.3 Hz, 1H), 3.83 (dd, J = 10.2, 3.6 Hz, 1H), 3.71 (dd, J = 10.2, 4.6 Hz, 1H), 3.45 (dd, J = 13.0, 6.9 Hz, 2H), 3.11–3.09 (m, 2H), 2.07–2.04 (m, 1H), 1.64–1.60 (m, 2H), 1.56 (d, J = 6.8 Hz, 3H), 1.55 (d, J = 6.8 Hz, 3H), 1.51–1.35 {m, 15H (obscured by H2O (2H)}, 1.03 (d, J = 6.9 Hz, 3H), 1.02 (d, J = 6.9 Hz, 3H), 0.88 (s, 9H), 0.03 (s, 6H); 13C NMR (151 MHz, CDCl3 + CD3OD) δ 166.1, 161.4, 159.5, 156.2, 151.6, 151.5, 142.5, 135.8, 131.4, 131.1, 127.9, 120.3, 117.5, 115.1, 79.2, 63.2, 58.1, 46.6, 40.5, 40.1, 30.1, 29.5, 29.4, 28.6 (3 overlapping signals), 26.7, 26.0, 25.9 (3 overlapping signals), 22.6, 22.6, 19.8, 18.4, −5.41, −5.43; IR (cm–1) 3321, 2932, 2656, 1693, 1628, 1587, 1493, 1460, 1401, 1365, 1302, 1251, 1169, 1109, 837, 776, 640.
(R)-N-(6-Aminohexyl)-2-chloro-4-((2-((1-hydroxy-3-methylbutan-2-yl)amino)-9-isopropyl-9H-purin-6-yl)amino)benzamide
A solution of (R)-tert-butyl (6-(4-((2-((1-((tert-butyldimethylsilyl)oxy)-3-methylbutan-2-yl)amino)-9-isopropyl-9H-purin-6-yl)amino)-2-chlorobenzamido)hexyl)carbamate (308 mg, 0.41 mmol) in THF (5 mL) was stirred at 0 °C. HCl (1.65 mL of a 4 M dioxane solution) was added dropwise, and the mixture was stirred and allowed to warm to room temperature overnight. The mixture was diluted with water (20 mL), and the pH adjusted to 8 with the addition of saturated NaHCO3 and extracted with ethyl acetate (4 × 20 mL). The combined organic extracts were washed with water (2 × 20 mL), brine (20 mL), dried over MgSO4, and concentrated in vacuo to give a white sticky solid. Purification by flash chromatography (Grace Amino, 2.5:97.5 → 1:9 v/v CH3OH/CH2Cl2) and concentration of the relevant fractions (Rf = 0.15 in 1:9 v/v CH3OH/CH2Cl2) afforded the titled compound as an orange resin (130 mg, 60%). 1H NMR (600 MHz, CD3OD) δ 8.24 (s, 1H), 7.89 (s, 1H), 7.71 (s, 1H), 7.41 (d, J = 8.4 Hz, 1H), 4.70–4.65 (m, 1H), 4.01–4.00 (m, 1H), 3.80–3.69 (m, 2H), 3.45–3.45 (m, 1H), 2.99 (s, 1H), 2.77–2.52 (m, 2H), 2.05 (dq, J = 13.6, 6.8 Hz, 1H), 1.64–1.43 (m, 14H), 1.05 (d, J = 9.2 Hz, 3H), 1.04 (d, J = 9.2 Hz, 3H) (signals due to amide, primary, and secondary amine protons not observed); 13C NMR (151 MHz, CD3OD) δ 169.9, 161.2, 152.9, 152.9, 143.8, 137.8, 132.3, 130.8, 130.4, 121.1, 118.5, 115.2, 63.4, 59.9, 48.1, 42.4, 40.8, 33.6, 30.6, 30.3, 27.9, 27.6, 22.6, 22.5, 20.1, 19.4; IR (cm–1) 3176, 2924, 1617, 1577, 1506, 1413, 1293, 1194, 1003, 986, 864, 814, 768, 747, 634, 532.
Preparation of Sepharose Beads
Solutions and Buffers
All buffers and solutions were degassed before use and stored at 4 °C. Wash solution: 1 mM HCl; organic coupling buffer (pH 8.3): 1:1 v/v DMSO/H2O, 100 mM NaHCO3; blocking agent (pH 8.3): 0.5 M ethanolamine, 0.5 M NaCl; acetate buffer (pH 4.4): 0.1 M acetic acid and 0.1 M sodium acetate; sodium bicarbonate buffer (pH 8.3): 0.2 M NaHCO3.
Coupling Inhibitors to Sepharose Beads
NHS-activated Sepharose 4 fast flow (1 mL, 50% slurry, ex. GE Healthcare Life Sciences) was added to a mini Bio-Spin column and treated with wash solution (10 × 0.5 mL) followed by organic coupling buffer (2 × 0.5 mL). To the washed beads were added a stock solution of the inhibitor (of desired concentration) and a 5:1 v/v DMSO/coupling buffer solution (0.5 mL), and the reaction mixture was rotated end-over-end for 24 h. After this time, the matrices were washed with coupling buffer (4 × 0.5 mL) (filtrate was retained for coverage analysis, via LC–MS), treated with blocking agent, and then rotated end-over-end for 24 h in the dark. After this time, the matrices were drained and washed with acetate buffer (3 × 1 mL) and bicarbonate buffer (3 × 1 mL); this sequence was repeated four times before storing over 20% aqueous ethanol at 4 °C.
Using LC–MS analysis, a quantitative study was carried out to determine the exact amount of the inhibitor bound to sepharose. With the exception of CZC8004 (2), which returned a bead loading of 29–34%, all other compounds returned bead loading values of 10–20%.
Preparation of Control Beads
NHS-activated Sepharose 4 fast flow (1 mL, 50% slurry, ex. GE Healthcare Life Sciences) was added to a mini Bio-Spin column and washed with wash solution (10 × 0.5 mL) followed by organic coupling buffer (2 × 0.5 mL). Blocking agent was added, and the matrices were rotated end-over-end for 24 h. After this time, the resin was washed with acetate buffer (3 × 1 mL) and bicarbonate buffer (3 × 1 mL); this sequence was repeated four times before storing over 20% aqueous ethanol at 4 °C.
Kinomics: Pull-Down
A HeLa S3 cell pellet was resuspended in lysis buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 20 mM NaF, 1 mM DTT, 1.5 mM MgCl2·6H2O, 5% (v/v) glycerol, 0.8% (v/v) Nonidet P-40, 0.1 mM PMSF, 1 μg/mL leupeptin, 1 Roche 11873 580001, EDTA-free protease inhibitor cocktail tablet per 50 mL lysate) and sonicated for 60 s with a probe sonicator. A lipid removal agent (Sigma) was added at 0.01 g/mL of lysate and incubated, with mixing for 30 min at 4 °C. The lysate was centrifuged for 20 min at 45 000g, 4 °C. The supernatant was collected and protein concentration, determined using a Millipore Direct Detect. For pull down, 30 μL of a 50% slurry of each bead was used in a Bio-Rad mini bio-spin column. Beads were washed with lysis buffer prior to use. To each washed bead, 10.9 mg of lysate was added and incubated for 60 min with mixing at 4 °C. All reactions were conducted in duplicate. Following incubation, the beads were washed three times (800 μL) with lysis buffer, then twice (800 μL) with 50 mM NH4HCO3, pH 7.8. An on-bead digest was performed overnight with trypsin (Promega) at 37 °C. Peptides were collected via centrifugation, and the beads were washed first with 100 μL of 50 mM NH4HCO3, pH 7.8, and subsequently with 80 μL of 70% (v/v) acetonitrile and 0.5% (v/v) formic acid. The appropriate washes were pooled and vacuum-concentrated prior to resuspension in 0.5% (v/v) formic acid and desalting using Empore 1 mL SPE cartridges (Sigma). Following cleanup, the eluate was vacuum concentrated and resuspended in 0.5% (v/v) formic acid prior to MS analysis. Each bead was used individually for pull-down.
LC–MS/MS
Peptides were separated on a Eksigent NanoLC Ultra fitted with an SGE C18P trap column (10 mm × 300 μm) and a self-packed C18 (Dr. Maisch, Reprosil-C18-AQ, 3 μm) 26 cm × 75 μm column. Peptides were separated over a 90 or 180 min run at 350 nL/min and fed directly into an AB SCIEX 5600 MS. The top 30 peptides were subjected to MS/MS, and the data searched against human proteins in Uniprot (301 202 sequences) using Mascot 2.5.1, X!Tandem Sledgehammer (2013.09.01.1), MSGF + (v10282), and Comet (2015.02 rev 4), the latter three utilizing SearchGUI 2.4.1.33 Data were postprocessed using PeptideShaker 1.7.1, and the peptide and protein FDR rates were set to 1%.34 The list of identified proteins from each LC–MS/MS run was compared to a list of human PKs to determine which PKs bound to each bead.
MS1 Quantitation
Skyline 3.5.0.9319 (64-bit) was used for MS1 quantitation of samples analyzed using the 180 min run.35 A total of 30 light standard peptides were spiked into each sample to allow for loading normalization. These peptides were appended to the database used for searching. The sequence for each of the identified PKs was added to Skyline, the peptide search for each sample was loaded into Skyline, and the MS1 data were used for quantitation of peaks within ±2 min of MS/MS identifications. Peak integration was scored using the mProphet algorithm, and only peaks with q-values <0.05 were used. Peak areas are corrected for loading by dividing each peak by the sum total peak area of added light standard peptides. As such, all peak areas are expressed relative to the amount of the standard peptides. Data was exported in the CSV format and filtered using Excel; peptide data was combined into proteins using the InfernoRDN R-rollup function.36 Relative quantitation is only included for proteins in which there were five or more peptides. Venn diagrams were produced using the program VennDIS 1.0.1.37
Acknowledgments
The authors thank the Australian Cancer Research Foundation, the Ramaciotti Foundation, and the Australian Research Council for project support. P.J.R. is the recipient of grants and Fellowships from the NHMRC (Australia).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b00020.
1H and 13C NMR spectra for the compounds synthesized herein; plots detailing the kinase identification from beads based pull-down experiments (PDF)
Author Contributions
§ F.M.D., A.J.S.L., and P.G.H. contributed equally to this project.
The authors declare no competing financial interest.
Supplementary Material
References
- Lee J.; Bogyo M. Target Deconvolution Techniques in Modern Phenotypic Profiling. Curr. Opin. Chem. Biol. 2013, 17, 118–126. 10.1016/j.cbpa.2012.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins A. L. Network Pharmacology: the Next Paradigm in Drug Discovery. Nat. Chem. Biol. 2008, 4, 682–690. 10.1038/nchembio.118. [DOI] [PubMed] [Google Scholar]
- Hu Y.; Bajorath J. Compound Promiscuity: What Can We Learn From Current Data?. Drug Discovery Today 2013, 18, 644–650. 10.1016/j.drudis.2013.03.002. [DOI] [PubMed] [Google Scholar]
- Peters J.-U. Polypharmacology - Foe or Friend?. J. Med. Chem. 2013, 56, 8955–8971. 10.1021/jm400856t. [DOI] [PubMed] [Google Scholar]
- Kroeze W. K.; Roth B. L.. Polypharmacological Drugs: “Magic Shotguns” for Psychiatric Diseases; Peters/Polypharmacology; John Wiley & Sons, Inc.: Hoboken, NJ, 2012; pp 133–148. [Google Scholar]
- Roth B. L.; Sheffler D. J.; Kroeze W. K. Magic Shotguns Versus Magic Bullets: Selectively Non-Selective Drugs for Mood Disorders and Schizophrenia. Nat. Rev. Drug Discovery 2004, 3, 353–359. 10.1038/nrd1346. [DOI] [PubMed] [Google Scholar]
- Petrelli A.Polypharmacological Kinase Inhibitors: New Hopes for Cancer Therapy; Peters/Polypharmacology; John Wiley & Sons, Inc.: Hoboken, NJ, 2012; pp 149–165. [Google Scholar]
- Reddy A. S.; Zhang S. Polypharmacology: Drug Discovery for the Future. Expert Rev. Clin. Pharmacol. 2013, 6, 41–47. 10.1586/ecp.12.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boran A. D. W.; Iyengar R. Systems Approaches to Polypharmacology and Drug Discovery. Curr. Opin. Drug Discovery Dev. 2010, 13, 297–309. [PMC free article] [PubMed] [Google Scholar]
- Knight Z. A.; Lin H.; Shokat K. M. Targeting the Cancer Kinome Through Polypharmacology. Nat. Rev. Cancer 2010, 10, 130–137. 10.1038/nrc2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fliri A. F.; Loging W. T.; Thadeio P. F.; et al. Analysis of Drug-Induced Effect Patterns to Link Structure and Side Effects of Medicines. Nat. Chem. Biol. 2005, 1, 389–397. 10.1038/nchembio747. [DOI] [PubMed] [Google Scholar]
- Bantscheff M.; Scholten A.; Heck A. J. R. Revealing Promiscuous Drug-Target Interactions by Chemical Proteomics. Drug Discovery Today 2009, 14, 1021–1029. 10.1016/j.drudis.2009.07.001. [DOI] [PubMed] [Google Scholar]
- Manning G.; Whyte D. B.; Martinez R.; Hunter T.; Sudarsanam S. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- Klaeger S.; Gohlke B.; Perrin J.; Gupta V.; Heinzlmeir S.; Helm D.; Qiao H.; Bergamini G.; Handa H.; Savitski M. M.; Bantscheff M.; Médard G.; Preissner R.; Kuster B. Chemical Proteomics Reveals Ferrochelatase as a Common Off-Target of Kinase Inhibitors. ACS Chem. Biol. 2016, 11, 1245–1254. 10.1021/acschembio.5b01063. [DOI] [PubMed] [Google Scholar]
- Médard G.; Pachl F.; Ruprecht B.; Klaeger S.; Heinzlmeir S.; Helm D.; Qiao H.; Ku X.; Wilhelm M.; Kuehne T.; Wu Z.; Dittmann A.; Hopf C.; Kramer K.; Kuster B. Optimized Chemical Proteomics Assay for Kinase Inhibitor Profiling. J. Proteome Res. 2015, 14, 1574–1586. 10.1021/pr5012608. [DOI] [PubMed] [Google Scholar]
- Moghaddas Gholami A.; Hahne H.; Wu Z.; Auer F. J.; Meng C.; Wilhelm M.; Kuster B. Global Proteome Analysis of the NCI-60 Cell Line Panel. Cell Rep. 2013, 4, 609–620. 10.1016/j.celrep.2013.07.018. [DOI] [PubMed] [Google Scholar]
- Wu Z.; Doondeea J. B.; Gholami A. M.; Janning M. C.; Lemeer S.; Kramer K.; Eccles S. A.; Gollin S. M.; Grenman R.; Walch A.; Feller S. M.; Kuster B. Quantitative Chemical Proteomics Reveals New Potential Drug Targets in Head and Neck Cancer. Mol. Cell. Proteomics 2011, 10, M111-011635 10.1074/mcp.M111.011635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng F.; Jia P.; Wang Q.; Zhao Z. Quantitative Network Mapping of the Human Kinome Interactome Reveals New Clues for Rational Kinase Inhibitor Discovery and Individualized Cancer Therapy. Oncotarget 2014, 5, 3697–3710. 10.18632/oncotarget.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaman M. W.; Herrgard S.; Treiber D. K.; Gallant P.; Atteridge C. E.; Campbell B. T.; Chan K. W.; Ciceri P.; Davis M. I.; Edeen P. T.; Faraoni R.; Floyd M.; Hunt J. P.; Lockhart D. J.; Milanov Z. V.; Morrison M. J.; Pallares G.; Patel H. K.; Pritchard S.; Wodicka L. M.; Zarrinkar P. P. A Quantitative Analysis of Kinase Inhibitor Selectivity. Nat. Biotechnol. 2008, 26, 127–132. 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- Reddy A. S.; Zhang S. Polypharmacology: Drug Discovery for the Future. Expert Rev. Clin. Pharmacol. 2013, 6, 41–47. 10.1586/ecp.12.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becher I.; Savitski M. M.; Savitski M. F.; Hopf C.; Bantscheff M.; Drewes G. Affinity Profiling of the Cellular Kinome for the Nucleotide Cofactors ATP, ADP, and GTP. ACS Chem. Biol. 2013, 8, 599–607. 10.1021/cb3005879. [DOI] [PubMed] [Google Scholar]
- Lemeer S.; Zörgiebel C.; Ruprecht B.; Kohl K.; Kuster B. Comparing Immobilized Kinase Inhibitors and Covalent ATP Probes for Proteomic Profiling of Kinase Expression and Drug Selectivity. J. Proteome Res. 2013, 12, 1723–1731. 10.1021/pr301073j. [DOI] [PubMed] [Google Scholar]
- Willems L. I.; Overkleeft H. S.; van Kasteren S. I. Current Developments in Activity-Based Protein Profiling. Bioconjugate Chem. 2014, 25, 1181–1191. 10.1021/bc500208y. [DOI] [PubMed] [Google Scholar]
- Médard G.; Pachl F.; Ruprecht B.; Klaeger S.; Heinzlmeir S.; Helm D.; Qiao H.; Ku X.; Wilhelm M.; Kuehne T.; Wu Z.; Dittmann A.; Hopf C.; Kramer K.; Kuster B. Optimized Chemical Proteomics Assay for Kinase Inhibitor Profiling. J. Proteome Res. 2015, 14, 1574–1586. 10.1021/pr5012608. [DOI] [PubMed] [Google Scholar]
- Ong S.-E.; Schenone M.; Margolin A. A.; Li X.; Do K.; Doud M. K.; Mani D. R.; Kuai L.; Wang X.; Wood J. L.; Tolliday N. J.; Koehler A. N.; Marcaurelle L. A.; Golub T. R.; Gould R. J.; Schreiber S. L.; Carr S. A. Identifying the Proteins to Which Small-Molecule Probes and Drugs Bind in Cells. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 4617–4622. 10.1073/pnas.0900191106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bantscheff M.; Eberhard D.; Abraham Y.; Bastuck S.; Boesche M.; Hobson S.; Mathieson T.; Perrin J.; Raida M.; Rau C.; Reader V.; Sweetman G.; Bauer A.; Bouwmeester T.; Hopf C.; Kruse U.; Neubauer G.; Ramsden N.; Rick J.; Kuster B.; Drewes G. Quantitative Chemical Proteomics Reveals Mechanisms of Action of Clinical ABL Kinase Inhibitors. Nat. Biotechnol. 2007, 25, 1035–1044. 10.1038/nbt1328. [DOI] [PubMed] [Google Scholar]
- Brehmer D.; Godl K.; Zech B.; Wissing J.; et al. Proteome-Wide Identification of Cellular Targets Affected by Bisindolylmaleimide-Type Protein Kinase C Inhibitors. Mol. Cell. Proteomics 2004, 490. 10.1074/mcp.M300139-MCP200. [DOI] [PubMed] [Google Scholar]
- Daub H.; Olsen J. V.; Bairlein M.; Gnad F.; Oppermann F. S.; Körner R.; Greff Z.; Kéri G.; Stemmann O.; Mann M. Kinase-Selective Enrichment Enables Quantitative Phosphoproteomics of the Kinome Across the Cell Cycle. Mol. Cell 2008, 31, 438–448. 10.1016/j.molcel.2008.07.007. [DOI] [PubMed] [Google Scholar]
- Fang B.; Haura E. B.; Smalley K. S.; Eschrich S. A.; Koomen J. M. Methods for Investigation of Targeted Kinase Inhibitor Therapy Using Chemical Proteomics and Phosphorylation Profiling. Biochem. Pharmacol. 2010, 80, 739–747. 10.1016/j.bcp.2010.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Holmes I. P.; Hochgräfe F.; Walker S. R.; Ali N. A.; Humphrey E. S.; Wu J.; de Silva M.; Kersten W. J. A.; Connor T.; Falk H.; Allan L.; Street I. P.; Bentley J. D.; Pilling P. A.; Monahan B. J.; Peat T. S.; Daly R. J. Characterization of the Novel Broad-Spectrum Kinase Inhibitor CTx-0294885 as an Affinity Reagent for Mass Spectrometry-Based Kinome Profiling. J. Prot. Res. 2013, 12, 3104–3116. 10.1021/pr3008495. [DOI] [PubMed] [Google Scholar]
- Russell C.; Lin A. J. S.; Hains P.; Simone M. I.; Robinson P. J.; McCluskey A. An Integrated Flow and Microwave Approach to a Broad Spectrum Protein Kinase Inhibitor. RSC Adv. 2015, 5, 93433–93437. 10.1039/C5RA09426G. [DOI] [Google Scholar]
- Gamble A. B.; Garner J.; Gordon C. P. Aryl Nitro Reduction with Iron Powder or Stannous Chloride Under Ultrasonic Irradiation. Syn. Commun. 2007, 37, 2777–2786. 10.1080/00397910701481195. [DOI] [Google Scholar]
- Vaudel M.; Barsnes H.; Berven F. S.; Sickmann A.; Martens L. SearchGUI: an Open-Source Graphical User Interface for Simultaneous OMSSA and X!Tandem Searches. Proteomics 2011, 11, 996–999. 10.1002/pmic.201000595. [DOI] [PubMed] [Google Scholar]
- Vaudel M.; Burkhart J. M.; Zahedi R. P.; Oveland E.; Berven F. S.; Sickmann A.; Martens L.; Barsnes H. PeptideShaker Enables Reanalysis of MS-Derived Proteomics Data Sets. Nat. Biotechnol. 2015, 33, 22–24. 10.1038/nbt.3109. [DOI] [PubMed] [Google Scholar]
- MacLean B.; Tomazela D. M.; Shulman N.; Chambers M.; Finney G. L.; Frewen B.; Kern R.; Tabb D. L.; Liebler D. C.; MacCoss M. J. Skyline: an Open Source Document Editor for Creating and Analyzing Targeted Proteomics Experiments. Bioinformatics 2010, 26, 966–968. 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polpitiya A. D.; Qian W.-J.; Jaitly N.; Petyuk V. A.; Adkins J. N.; Camp D. G.; Anderson G. A.; Smith R. D. DAnTE: a Statistical Tool for Quantitative Analysis of -Omics Data. Bioinformatics 2008, 24, 1556–1558. 10.1093/bioinformatics/btn217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatchenko V.; Ignatchenko A.; Sinha A.; Boutros P. C.; Kislinger T. VennDIS: a JavaFX-Based Venn and Euler Diagram Software to Generate Publication Quality Figures. Proteomics 2015, 15, 1239–1244. 10.1002/pmic.201400320. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.