

Abstract

The current work focuses on the fabrication of high-molecular-weight stereocomplex poly(lactic acid)/nanohydroxyapatite (sPLA/n-HAP)-based bionanocomposite for three-dimensional (3D)-printed orthopedic implants and high-temperature engineering applications. To achieve the same, n-HAP is grafted with poly(d-lactic acid) (PDLA) via in situ ring-opening polymerization of d-lactide, followed by blending with poly(l-lactic acid) (PLLA), which yields sPLA/n-HAP biocomposite with improved storage modulus even at temperatures higher than 140 °C. X-ray diffraction and calorimetric analysis ensure the presence of 100% stereocomplex crystallites of biocomposite along with significant improvement in the melting temperature (∼227 °C). Noteworthy improvements in the mechanical and gas-barrier properties of the developed biocomposites are achieved due to the uniform dispersion of n-HAP (∼60 nm) confirmed by morphological studies. An unusual improvement in elongation at break (∼130% at 1 wt % HAP loading) makes this composite a toughened material. However, the tensile strength is improved by ∼16%, whereas oxygen permeability and water vapor transmission rate are found to reduce by ∼48 and ∼34%, respectively. Interestingly, the developed material is processed as monofilament, followed to 3D printing to yield a middle phalanx bone as a representative example of orthopedic implants. In vitro studies reveal that cell adhesion and proliferation on the surface of the developed biocomposite support its biocompatible nature. This signifies the possible future aspects of the material in commercial biomedical and high-temperature engineering applications.
Introduction
In the recent past, poly(lactic acid) (PLA) has been considered as a potential candidate to replace the traditional petroleum-based thermoplastics for several applications, such as textile, agriculture, biomedicine, packaging, and other engineering disciplines.1 PLA can be produced using lactic acid monomer, which is a chiral molecule and has been derived from renewable agricultural resources.2 Because of the chiral properties of lactic acid, PLA has two semicrystalline stereoisomers: poly(l-lactic acid) (PLLA) and poly(d-lactic acid) (PDLA). It is known that PLLA and PDLA can be crystallized in several polymorphs, such as α, β, and γ, formed under different processing conditions.3 In 1987, Ikada et al. reported the formation of a special type of polymorph in PLA by mixing PLLA and PDLA, called stereocomplex, made by combining right- and left-handed helical polymer chains, and found its melting temperature to be 50 °C higher than that of the normal enantiomeric pure PLA.4 A similar phenomenon was also seen by Miyamoto et al. in stereospecific poly(methyl methacrylate).5 Because of the intermolecular hydrogen bonding between PLLA and PDLA molecules, stereocomplex crystallites undergo compact polymer chain packing than being homocrystals.6 Stereocomplexation in PLA promises superior thermal,7,8 mechanical,9,10 thermomechanical,11 and barrier properties12 to enantiomeric pure PLA, which makes it an interesting polymorph to study.13
The formation of stereocomplex crystallites is highly dependent on the specific arrangement of PLLA and PDLA chains in the blend, and it becomes complicated due to the similar temperature range for crystallization of homocrystals and stereocomplex crystallites. It has been confirmed that stereocomplex crystallites with a trace amount of homocrystals can be formed by mixing PLLA and PDLA in 1:1 ratio.9 Furthermore, it is limited to polymers with low molecular weight, such as less than 100 kDa, and the amount of homocrystals has been found to be excessively high in case of high-molecular-weight polymers (higher than 100 kDa).14 Therefore, development of PLA with a higher content of stereocomplex crystallites is the prevailing issue among polymer scientists to obtain PLA with enhanced mechanical, barrier, and thermal properties.
In this context, several research groups are trying to develop different techniques, such as solid-state polymerization,15 development of stereo diblock copolymer,16 supercritical fluid technology,17,18 layer-by-layer assembly,19 and so forth, to prepare relatively high-molecular-weight PLLA/PDLA blends with high content of stereocomplex crystallites. Some of the researchers have used modified or unmodified fillers, such as nanocrystalline cellulose,20 nanographite,21 graphene oxide,22,23 carbon nanotube,24−26 lignin,27 and other polymers.28−30 The present work demonstrates the use of hydroxyapatite as a filler into the PLA matrix. Nanohydroxyapatite (n-HAP) is a bioactive nontoxic complex form of calcium phosphate, which constitutes 60–70% of mammalian bones. It can be produced by several biological or synthetic methods, such as precipitation, hydrothermal and sol–gel method, hydrolysis, and solid-state synthesis31 from bioresources, such as eggshells, seashells, plants, animal bones, and so forth.32 Because of its similarity to mammalian hard tissues, n-HAP is one of the most investigated synthetic biomaterials. Substantial research has been carried out by a number of researchers for the fabrication of PLA/n-HAP biocomposite for different applications.33−36 It is essential to modify n-HAP due to its poor interfacial adhesion with PLA and poor mechanical properties.37 To avoid the agglomeration of n-HAP particles, researchers have grafted n-HAP with PLA via in situ ring-opening polymerization (ROP). Du and his colleague developed the poly(d,l-lactic acid) (PDLLA)-grafted n-HAP via solution ROP in toluene and explored it for the shape memory application.38 Qiu and his group modified the n-HAP surface with lactic acid in toluene before grafting with PLLA at 130 °C for 80 h and blended with PLLA at 170 °C. The prepared composite displayed good mechanical properties and uniform microstructure.39 In the same direction, Hong et al. found good dispersion of n-HAP in polymer matrix after grafting with PLA in xylene solution, which led to the improvement in the mechanical properties.40 Similarly, Wang and his group modified the surface of n-HAP with lactic acid oligomer and found the improved dispersion, which led to the enhancement in the properties of the end product.41 A number of researchers have grafted n-HAP, which involves solvents, and these methods are not industrially viable. To our knowledge, no study addressing the modification of n-HAP in bulk and its application in the development of stereocomplex poly(lactic acid) (sPLA) has been conducted so far.
Therefore, the current work was dedicated to develop a facile process to graft n-HAP via in situ bulk ROP of lactide. sPLA/n-HAP biocomposite was developed with different n-HAP content. The molecular structure and the mechanical, thermal, thermomechanical, and barrier properties of the prepared biocomposite were investigated, which evidently demonstrated the usability of grafted n-HAP in the enhancement of stereocomplexation in PLA and its ultimate high-temperature engineering application. It promises better compatibility with PLA molecules, which was determined by morphological studies. sPLA and the developed composites were further tested for cell adhesion and proliferation to explore their potential as a biomaterial. The developed material was melt-processed to form filaments for three-dimensional (3D) printing to explore its possible application as orthopedic implant.
Results and Discussion
Characterization of Fabricated n-HAP
n-HAP is characterized by electron microscopy to identify the morphology and size of the particles, as shown in Figure 1. At 20k× magnification, it was difficult to clearly identify separate n-HAP particles (Figure 1a). The images at higher magnification (50k×) show the particles of n-HAP. The particle size is found to be in the range of 30–60 nm. However, some fraction of particles are found as agglomerates as big as ∼3 μm. The contents of phosphate (P) and calcium (Ca) are measured by energy-dispersive X-ray (EDX) analysis, as shown in Figure 2. The average ratio Ca/P is found to be 1.68, which confirms the fabrication of n-HAP.42
Figure 1.

Field emission scanning electron microscopy (FESEM) images of n-HAP particles at 20k× (a) and 50k× (b).
Figure 2.
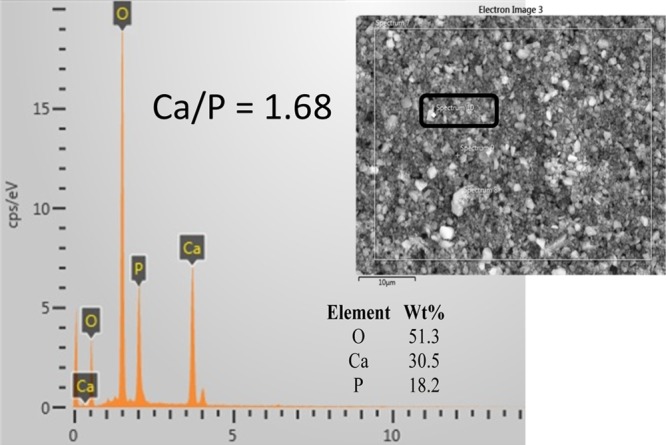
EDX analysis of n-HAP nanoparticles (inset: SEM image of bulk n-HAP, 10 μm scale).
Raman spectroscopy is a powerful tool for the analysis of minerals.43 Raman spectra of the prepared n-HAP are investigated and presented in Figure 3 against wavenumber. The observed Raman bands are found in agreement with the literature.44 The characteristic peak of phosphate ion associated with fully symmetric P–O–P stretching mode in n-HAP is found at 962 cm–1.45 The band at 157 cm–1 belongs to Ca-PO4 in double-bending mode, whereas the bands at 430 and 589 cm–1 are assigned to PO43– in a triply degenerate bending mode. The 1051 cm–1 peak corresponds to triply degenerate antisymmetric stretching mode of PO43–.46
Figure 3.
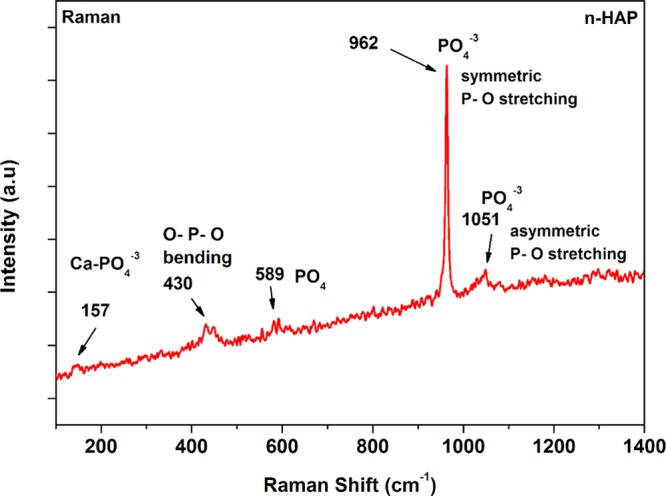
Raman spectra of n-HAP and peak assignments.
Figure 4 presents the X-ray diffraction (XRD) spectra of fabricated n-HAP nanoparticles, showing intense well-resolved peaks. The crystalline peaks are observed at 25.8, 28.9, 31.9, 32.8, 42.1, 46.7, 49.5, and 53.3° corresponding to (002), (210), (211), (300)(202), (310), (311), (222), (213), and (004) planes.47 The obtained results are in agreement with the hydroxyapatite fingerprint reported in the literature.48
Figure 4.
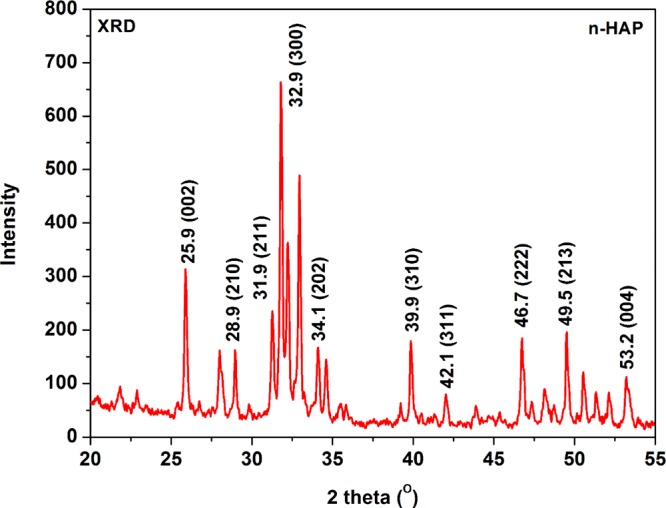
XRD spectra of n-HAP.
Chemical Modification of n-HAP
To improve the dispersion and homogeneity into PLA matrix, n-HAP is modified by grafting with PDLA via bulk ROP of d-lactide. It was assumed that the hydroxyl group present in n-HAP molecule would contribute as a coinitiator for the ROP of lactide by coordination insertion mechanism along with catalyst tin octoate.49 It is clearly known that n-HAP is not soluble in solvents like chloroform; however, it partially dissolved and formed a stable suspension when grafted with PDLA (Figure 5). The 13C NMR study of PDLA- and PDLA-grafted n-HAP is done, and the results are presented in Figure 6. It is found that the peaks present at chemical shifts 16.6, 69.1, and 169.4 ppm correspond to the methyl, methine, and carbonyl carbons of the PLA chain. In comparison to the spectra of PLA/n-HAP, one additional chemical shift was found at 175.3 ppm, which corresponds to the terminal carbonyl carbon connected with the oxygen–calcium element of n-HAP. A similar mechanism of grafting of PLA with starch has been described elsewhere.50 The schematic representation of the grafting of n-HAP with PDLA is shown in Figure 7.
Figure 5.

Dispersion of grafted n-HAP in comparison with pristine n-HAP in chloroform.
Figure 6.
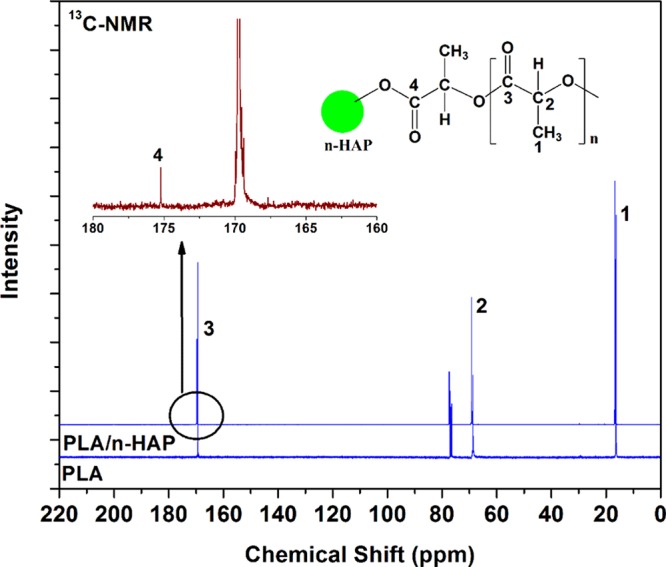
Comparison of 13C NMR spectra of PLA and PLA/n-HAP.
Figure 7.

Schematic representation of grafting of n-HAP with PDLA.
Further, it is known that n-HAP is an inorganic material that is thermally stable below 900 °C and does not lose its molecular structure at higher temperature.51 In this case, thermogravimetric analysis (TGA) can be an effective method to calculate the amount of PDLA chain connected with n-HAP. A comparison of TGA graphs of n-HAP and PDLA/n-HAP is shown in Figure 8. Approximately 2.2% reduction in weight loss is found in case of PDLA/n-HAP compared to pristine n-HAP, which suggests that ∼2.2% of PDLA is chemically bound to n-HAP molecules, which degrade against increase of temperature.
Figure 8.
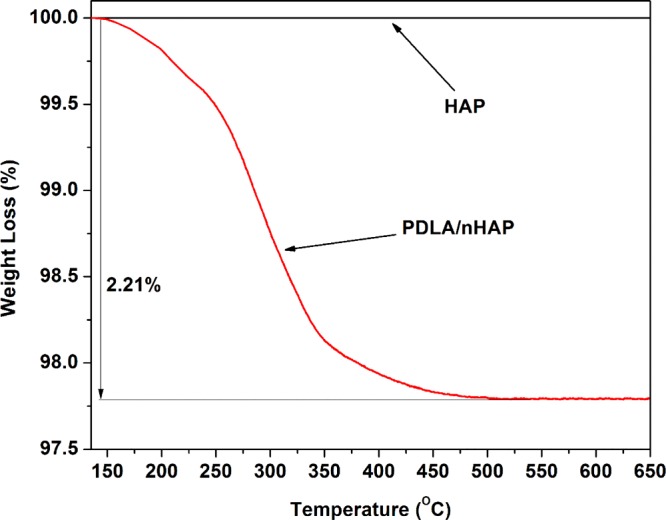
Comparison of weight loss of PLA/n-HAP and n-HAP against temperature.
sPLA/n-HAP Biocomposite
Fourier transform infrared (FTIR) spectra of sPLA and sPLA/n-HAP biocomposites are presented in Figure 9a. All of the samples display similar spectra to sPLA. The peaks at 2923 and 2847 cm–1 are assigned to the stretching vibration of −C–H of PLA. The band at 1747 cm–1 corresponds to the carbonyl (C=O) stretching of PLA. The band at 1452 cm–1 corresponds to the bending of methyl group, and the peak at 863 cm–1 is assigned to −C–C– stretching of the backbone of PLA molecule. FTIR spectra are highly sensitive to the arrangement of the chains in crystalline segment. The peak at 908 cm–1 in Figure 9b is attributed to the 31 β helical structure of sPLA chains.7 The intensity of the peak is increased as the content of n-HAP is enhanced, which qualitatively suggests the increase in the content of stereocomplex crystallites in the matrix. The interaction between carbonyl carbon of one enantiomeric PLA and methyl group of the other enantiomeric PLA, which form the hydrogen bond, leads to the formation of stereocomplex crystallites. This hydrogen bonding affects the vibration peak at 1038 cm–1 assigned to the C–CH3 group of PLA,52 which red-shifted to 1035 cm–1 corresponding to the C=O···CH3 stretching mode (Figure 9c), confirming the formation of stereocomplex crystallites.53
Figure 9.
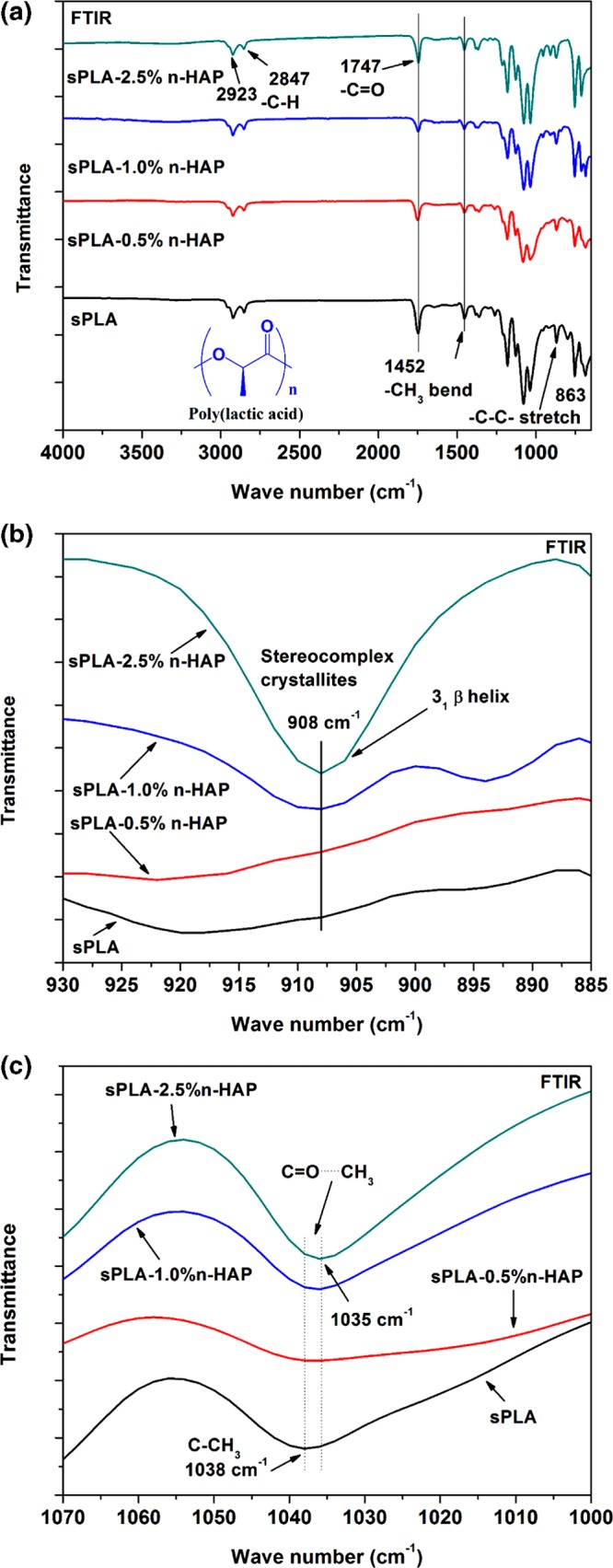
(a) Complete FTIR spectra of sPLA and sPLA/n-HAP biocomposites. (b) Magnified FTIR spectra (930–885 cm–1) of sPLA and sPLA/n-HAP biocomposites. (c) Magnified FTIR spectra (1070–1000 cm–1) of sPLA and sPLA/n-HAP biocomposites.
It is well known that the stereocomplex crystallites in PLA have a higher melting temperature than normal PLA homocrystals.4 The differential scanning calorimetry (DSC) analysis of sPLA and other sPLA/n-HAP biocomposites is shown in Figure 10a. The thermogram indicates that sPLA is found to have the crystals of homopolymers with trace amount of stereocomplex crystallites. The endotherm at 152 and ∼178 °C is associated to the melting of homocrystals, which may be the result of α′ and α forms of crystallites,54 and melting peak at ∼210 °C corresponds to the stereocomplex crystallites. An increase in the content of grafted n-HAP in polymer matrix leads to increase in the melting enthalpy (ΔHm,sc) of the stereocomplex crystallites from ∼17 J g–1 for sPLA to ∼55 J g–1 for sPLA-2.5% n-HAP and helps in the reduction of melting enthalpy (ΔHm,hc) of homocrystals from ∼28 J g–1 (sPLA) to 0 J g–1 (sPLA-5% n-HAP) (Table 1). Increase in melting temperature from ∼210 °C for sPLA to ∼227 °C for sPLA-2.5% n-HAP suggests the perfectness and increased amount of stereocomplex crystallites. Reduction in the degree of crystallization (Xc,hc) for homocrystals is found from ∼30% for sPLA to 0% for sPLA-2.5% n-HAP. The degree of crystallization (Xc,sc) for stereocomplex crystals was improved from ∼12% for sPLA to ∼39% for sPLA-2.5% n-HAP with no trace of melting enthalpy for homocrystals, which reaches to 100% fraction of stereocomplex crystallites, as shown in Figure 10b.
Figure 10.
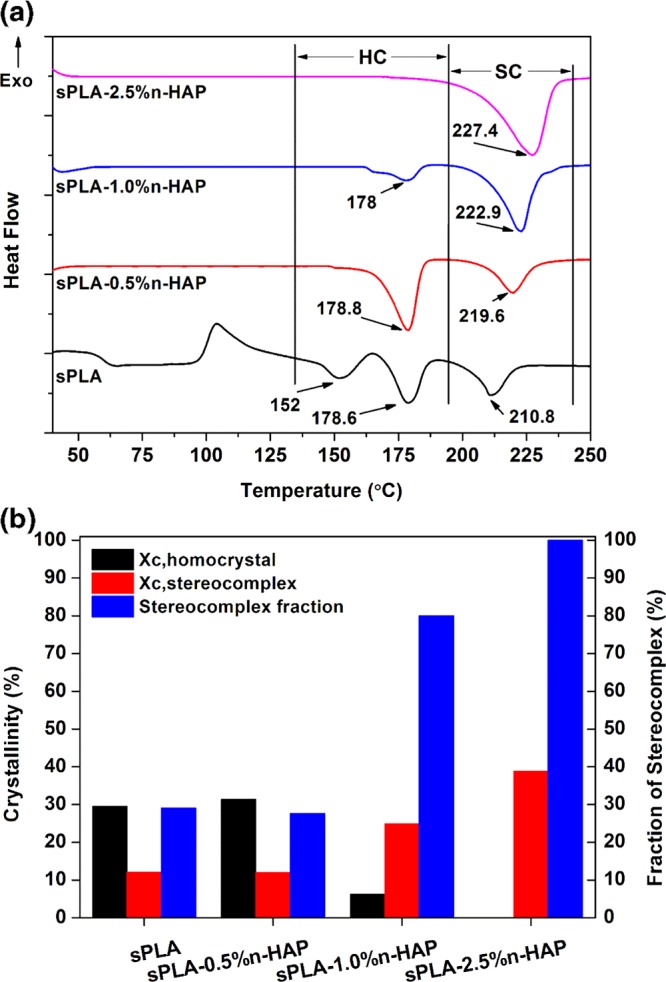
(a) DSC thermogram of sPLA and sPLA/n-HAP biocomposites. (b) Crystallinity (homocrystal, stereocomplex) and fraction of stereocomplexation of sPLA and sPLA/n-HAP biocomposites.
Table 1. DSC Results for sPLA and sPLA/n-HAP Biocomposites.
| sample name | ΔHm,hc (J g–1) | ΔHm,sc (J g–1) | Tm,hc (°C) | Tm,sc (°C) |
|---|---|---|---|---|
| sPLA | 27.6 | 17.1 | 152, 178.6 | 210.8 |
| sPLA-0.5% n-HAP | 29.5 | 17 | 178.8 | 219.6 |
| sPLA-1.0% n-HAP | 5.8 | 35.4 | 178 | 222.9 |
| sPLA-2.5% n-HAP | 0 | 55.1 | 227.4 |
Further, the effect of grafted n-HAP on stereocomplexation in PLA matrix is analyzed using XRD. The XRD patterns of sPLA and sPLA/n-HAP biocomposites are shown in Figure 11. It is known that the sPLA chains arrange themselves in a triclinic unit cell with dimensions of a = 0.916 nm, b = 0.916 nm, c = 0.870 nm, α = β = 109.2°, and γ = 109.8°, which is having a 31 helical structure (3 Å increase per unit of monomer).55 The XRD patterns at peaks 11.8, 20.6, and 23.9° are related to stereocomplex crystallites and correspond to the (110), (300)/(030), and (220) lattice planes, whereas the homopolymer PLA crystallizes (α form) in a pseudo-orthorhombic unit cell of dimensions a = 1.07 nm, b = 0.595, and c = 2.78 nm, holding a 103 helical structure (10 Å increase per 3 units of monomer). Peaks for PLA at 14.7, 16.6, and 19° in XRD correspond to (010), (200/110), and (203) crystal planes. It is clear from the pattern that the peaks related to the stereocomplex evolved and the peaks related to the homocrystals are diminished with increase in the grafted n-HAP content.
Figure 11.
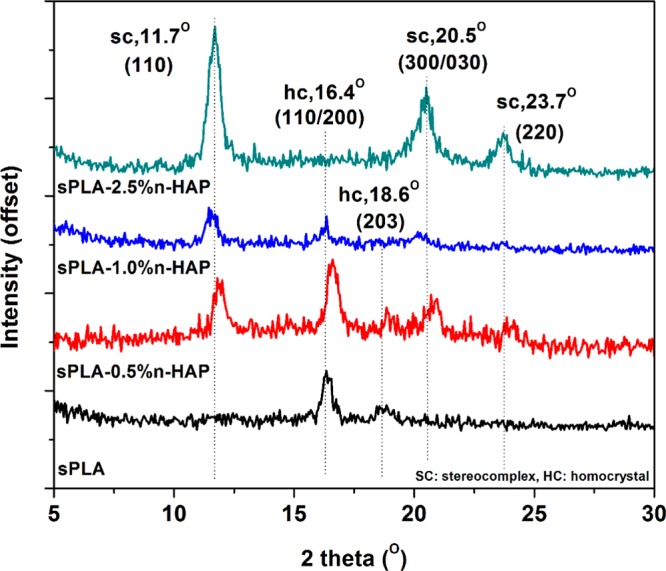
XRD of sPLA and sPLA/n-HAP biocomposites.
The above discussion suggests that the presence of grafted n-HAP in the polymer matrix helps in the development of stereocomplex crystallites, thus limiting the evolution of the homocrystals. This improvement leads to the enhancement of other properties of biocomposites.
Effect of Grafted n-HAP on the Mechanical and Thermomechanical Properties of sPLA
The presence of a filler in the polymer matrix enhances the mechanical and thermomechanical properties; however, homogenous dispersion of fillers is the most critical parameter. Effective ways to improve the dispersion is the modification of fillers with diluting matrix element. In this work, the filler, that is, n-HAP, is grafted with PDLA, which was miscible in the PLLA matrix, which led to the uniform dispersion of n-HAP. The FESEM analysis of the fractured surface of sPLA and sPLA/n-HAP biocomposites is shown in Figure 12. The fractured surface of sPLA (Figure 12a) is smooth and free of any foreign particles, whereas in the sPLA/n-HAP (Figure 12b) biocomposite, the n-HAP particles of ∼60 nm are found to be uniformly dispersed.
Figure 12.

FESEM images of fractured surface of sPLA (a) and sPLA/n-HAP (b) biocomposites.
The presence of grafted n-HAP in PLA matrix is playing a dual role: as homogeneously reinforced filler into the matrix and providing the extended molecular surface area, in the form of PDLA chains, which efficiently interacts with PLLA chains forming the stereocomplex crystallites. Load–elongation curve for sPLA and sPLA/n-HAP biocomposites is presented in Figure 13a. The ultimate tensile strength (UTS) is found to improve by ∼16%, that is, 40.2 MPa, for sPLA-2.5% n-HAP compared to that of sPLA, that is, 33.8 MPa. Improvement in the degree of crystallinity suggests significantly more intermolecular bonding and cross-linking in the form of stereocomplexation, which may be responsible for the increment in the tensile strength. It also suggests the strong interfacial bonding between n-HAP and polymer chains. The elongation at break is found to be 131.6% for sPLA-1% n-HAP compared to 6.3% for sPLA (Figure 13b), which implies that the ductility of sPLA is significantly improved with incorporation of modified n-HAP. It is known that the improvement in ductility ensures the delay in fracture, which reduces the abrupt failure of the biocomposite. The integrated molecular surface area in the form of PDLA on modified n-HAP acts as a bridge between the sliding and elongating polymeric chains and prolongs the breakage process. It also suggests a strong interaction between matrix and filler and may be a result of chain uncoiling or chain sliding in the biocomposite.56 However, the elongation at break is reduced to ∼55% in case of sPLA-2.5% n-HAP. Increase in the content of n-HAP in the matrix ensures the upsurge in the tensile strength due to the confinement of polymer chains and the formation of stereocomplex crystallites. However, n-HAP particles present in the matrix at higher amount may generate voids during the elongation process, which ultimately induce breakage and rupture of bonds, resulting in the reduction in elongation at break in sPLA-2.5% n-HAP compared to sPLA-1% n-HAP.
Figure 13.
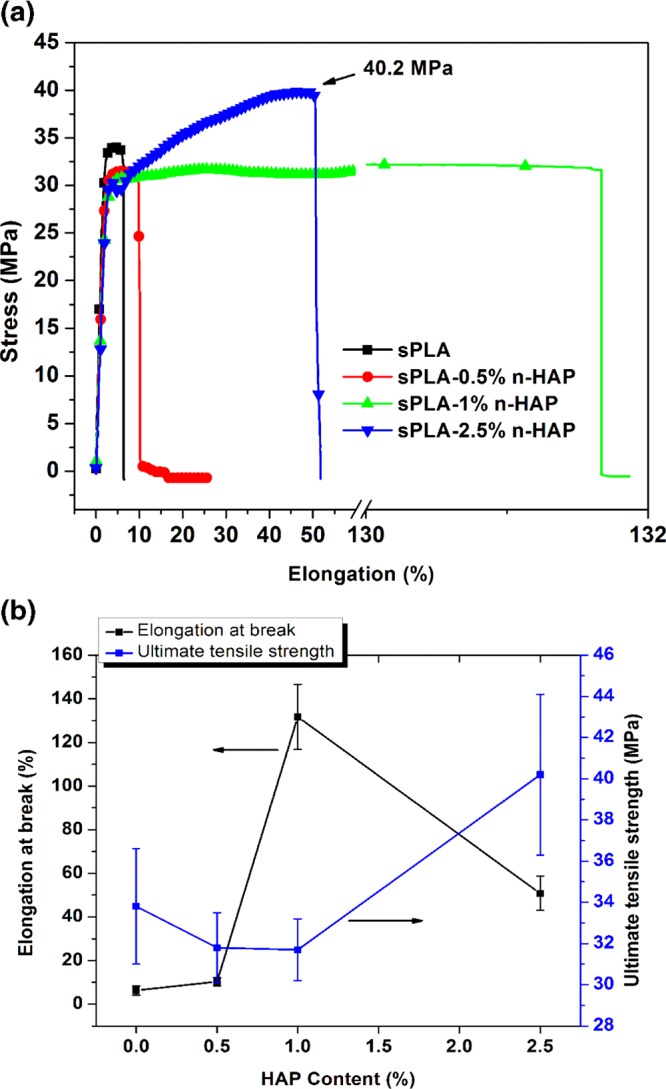
(a) Representative data for stress of sPLA and sPLA/n-HAP biocomposites against elongation percentage at break. (b) UTS and elongation at break of sPLA and sPLA/n-HAP biocomposites.
The effect of grafted n-HAP on the thermal stability of the sPLA/n-HAP biocomposite is analyzed by measuring the storage modulus and tan δ in the temperature range of 30–180 °C on applied dynamic force using dynamic mechanical analysis (DMA) in tensile mode. The storage modulus of sPLA and sPLA/n-HAP biocomposites is shown in Figure 14a. The storage modulus at 30 °C is found to be ∼1243 MPa in case of sPLA, which is enhanced to ∼2350 MPa for sPLA-2.5% n-HAP and remained higher than that of sPLA or other sPLA/n-HAP biocomposites throughout the temperature range. This improvement in the storage modulus of biocomposites indicates the stiffness of viscoelastic polymer, which resists the deformation on the action of applied force and can also be attributed to the increased polymer chain rigidity. The loss tangent curve is shown in Figure 14b as a function of temperature. The peaks are related to the glass transition of the polymers, indicating the movement of amorphous polymeric chains, which is the transition of material from hard glassy state to soft rubbery state. Changes in the peak shape with increase in n-HAP content indicate the relationship between polymer structure and internal molecular motion. The fillers present in the polymeric system affect the glass transition by developing hindrance to the movement of polymer chains. The peak becomes flattened in case of sPLA-2.5% n-HAP, which may be the result of hindrance to polymer chains developed by n-HAP nanoparticles as well as increase in the stereocomplex crystallites content. This increase in the stereocomplexation suppresses the intensity of tan δ peak for sPLA-2.5% n-HAP compared to sPLA, as discussed in the previous section that the stereocomplex crystallites are more tightly packed than the homocrystals, which induces chain rigidity.57 This analysis suggests that the prepared biocomposite was thermally more stable than pristine sPLA at elevated temperature.
Figure 14.
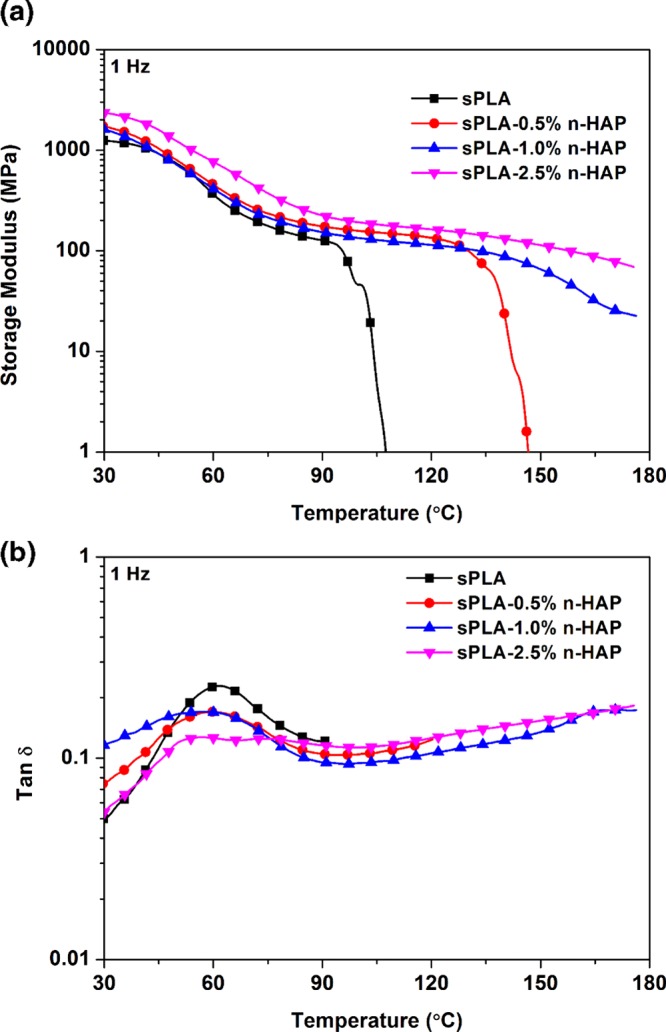
(a) Storage modulus of sPLA and sPLA/n-HAP biocomposites against temperature; (b) tan δ of sPLA and sPLA/n-HAP biocomposites against temperature.
From the above discussion, it can be concluded that the grafted n-HAP plays a dual role, that is, to develop the homogeneously dispersed biocomposite and supporting the formation of stereocomplex crystallites in sPLA/n-HAP biocomposite via providing the extended molecular surface area in the form of PDLA, which interacts with PLLA and form the stereocomplex crystallites. The formation of stereocomplex crystallites in the presence of grafted n-HAP was delineated in the schematic shown in Figure 15.
Figure 15.

Schematic for the formation of stereocomplex crystallites in sPLA/n-HAP biocomposite.
Barrier Properties
The barrier properties of the polymers are the crucial parameters to decide their targeted applications. The prepared biocomposite films are analyzed for the oxygen permeability and water vapor permeability. oxygen transmission rate (OTR) results are shown in Figure 16 at different temperatures, which show a significant drop of nearly 48% in oxygen permeability at 23 °C. The molecular compactness and degree of crystallinity are the parameters that highly affect the gas permeability. The formation of stereocomplex crystallites due to the presence of grafted n-HAP increases the compactness of the polymer chains. Increase in the crystalline density directly affects the oxygen permeability as the diffusion of oxygen molecules is hindered in crystalline domain.58 The FESEM analysis of fractured surface, as discussed in the previous section, showed the homogeneous dispersion of n-HAP indicating the interaction of polymer chains with n-HAP particles, which are responsible for the formation of stereocomplex crystallites. n-HAP may also be contributing to the reduction in the oxygen permeability by developing a tortuous path for the oxygen molecules.
Figure 16.
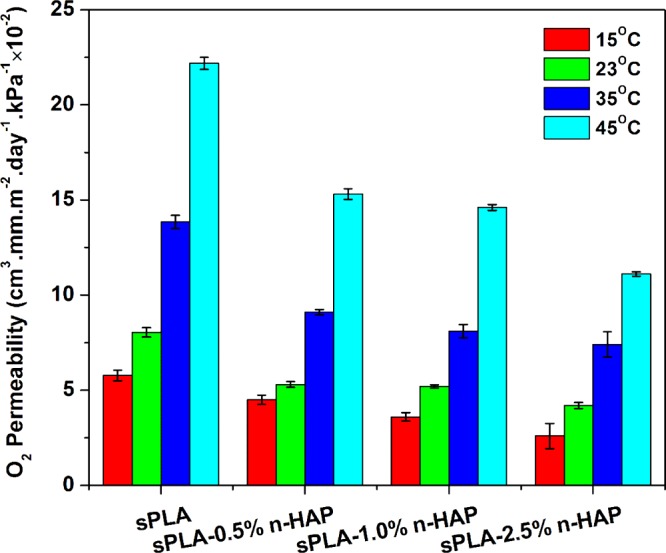
Oxygen permeability of sPLA and sPLA/n-HAP biocomposites at different temperatures.
Water vapor permeability of sPLA and sPLA/n-HAP biocomposites is shown in Figure 17, in which a 34% reduction was observed for sPLA-2.5% n-HAP compared to sPLA. The grafting of the n-HAP with PDLA chains enhances the interaction of n-HAP with polymeric chains, which increase the dispersion, resulting in the enhancement of stereocomplexation. Improvement in the degree of crystallinity affects the bulk surface property of sPLA and makes it resistant to moisture. The development of stereocomplex crystallites may be responsible for the hindrance to the diffusion of water molecules.
Figure 17.
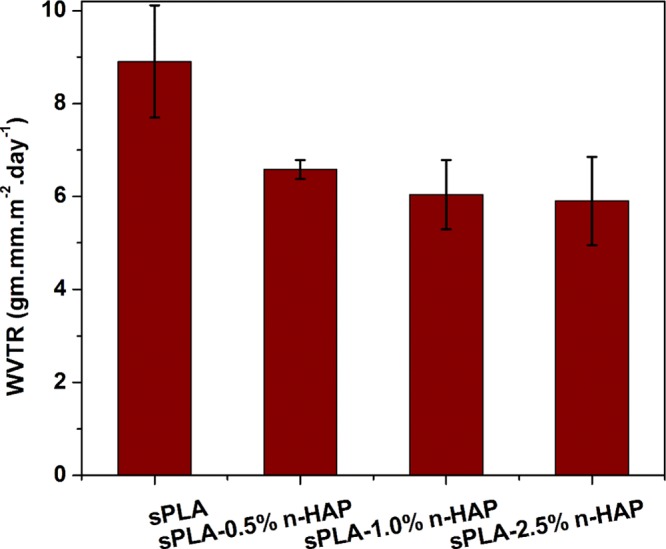
WVTR of sPLA and sPLA/n-HAP biocomposites.
Cell Adhesion and Proliferation
BHK-21 cells are used for the determination of the biocompatible nature of the developed material. The cell viability assay indicates that the cell survivabilities on sPLA, sPLA-1.0% n-HAP and sPLA-2.5% n-HAP (Figure 18) are almost similar, which suggests no adverse effect of the addition of filler (hydroxyapatite) in the sPLA matrix. The nuclei of the cells stained with 4’,6-diamidino-2-phenylindole (DAPI) showed its intact nature, indicating the cell adhesion and proliferation on the surfaces of sPLA, sPLA-1.0% n-HAP, and sPLA- 2.5% n-HAP (Figures 19 and 20). These results suggest the potential biomedical application of the fabricated n-HAP-based biocomposite.
Figure 18.
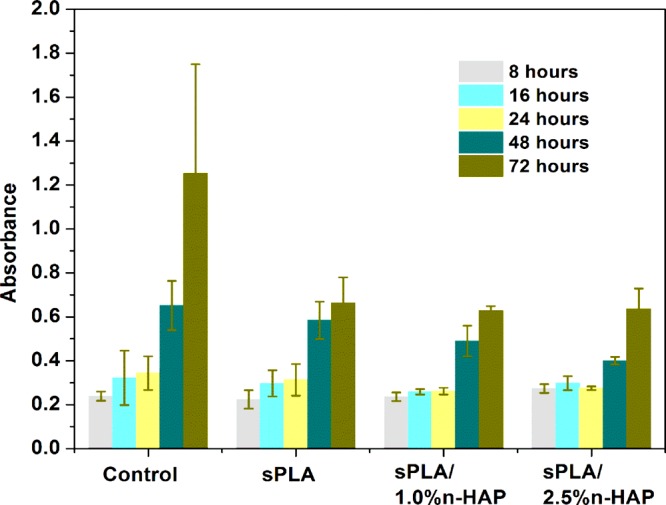
BHK-21 cell viability determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay for control, sPLA, sPLA-1.0% n-HAP, and sPLA-2.5% n-HAP.
Figure 19.
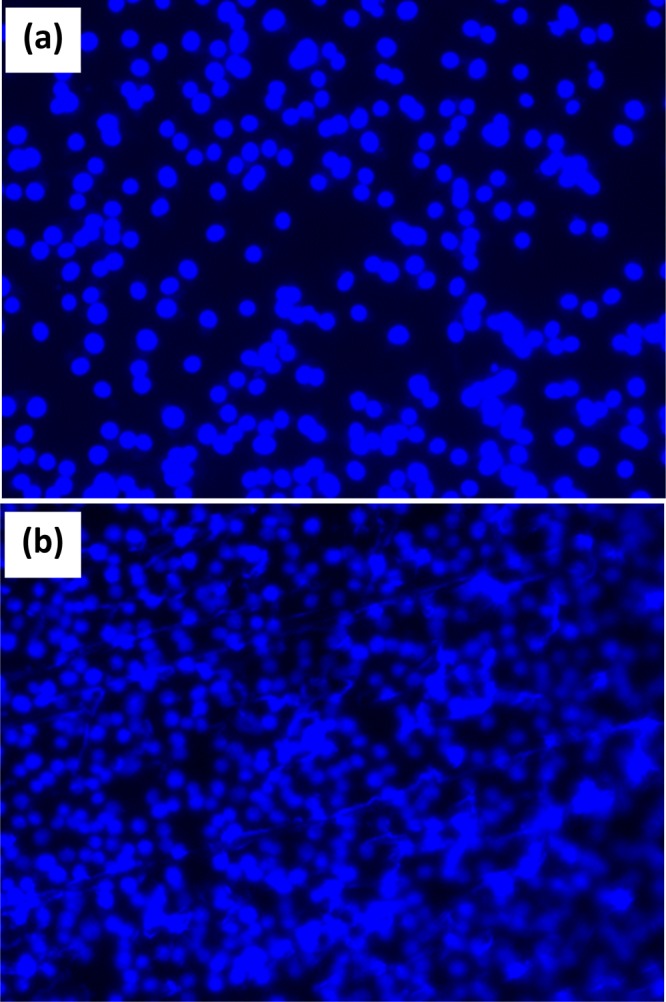
BHK-21 cell proliferation on control (a) sPLA after incubation for 48 h and staining with DAPI (b).
Figure 20.

BHK-21 cell proliferation on sPLA-1.0% n-HAP (c) and sPLA-2.5% n-HAP after incubation for 48 h and staining with DAPI (d).
The fabricated biocomposite is melt-processed with a twin-screw mini-extruder to produce the filament with ∼1.6 mm diameter, which was used for 3D printing of the representative middle phalanx bone successfully, as shown in Figure 21. This ensures that the developed material had the ability to be processed using an industrially viable process, that is, melt processing, which is further utilized for designing the desired product using 3D-printing technique.
Figure 21.

Synthesis, processing, and application of sPLA/n-HAP biocomposite.
Conclusions
Successful fabrication and grafting of n-HAP (∼60 nm) with PDLA via in situ bulk ROP of d-lactide is carried out, which is confirmed by 13C NMR and TGA, leading to the production of high-molecular-weight PDLA/n-HAP biocomposite. sPLA/n-HAP biocomposite is produced with 100% stereocomplex crystallite content with a melting temperature of ∼227 °C. It is concluded that the grafted n-HAP plays a dual role, that is, to develop homogeneous dispersion in the polymer and the formation of stereocomplex crystallites by providing the extended molecular surface area in the form of PDLA chains. Improved stereocomplexation ultimately affects the elongation at break (132%), tensile strength (40.2 MPa), and storage modulus (∼47%). The enhancement in the stereocomplexation results in 48 and 34% reduction in oxygen permeability and water vapor transmission rate (WVTR), respectively. The concise summary can be portrayed by displaying the application of the lab-synthesized material in high-temperature engineering as well as biomedical applications. The synthesized material was tested for cell viability, melt-processed, and further utilized for 3D printing of the finger (middle phalanx) bone. This highlights the importance of the developed material for designing commercial-value products. The finger bone made using 3D-printing technique can further be tested for osteoblast cell proliferation and differentiation to determine the rate of formation of natural bone synthesized by bone cells. This could further give new directions to the scientific approach.
Experimental Section
Materials
l-Lactide and d-lactide were produced from l-lactic acid (Purac, India) and d-lactic acid (Musashino, Japan), respectively, by a two-step polymerization and depolymerization procedure. l-Lactic acid or d-lactic acid was dehydrated to obtain oligomeric PLLA or PDLA, which was further depolymerized in the presence of tin oxide to obtain l-lactide or d-lactide, respectively, and used after purification. Tin oxide (SnO) and stannous octoate (tin octoate) were procured from Sigma-Aldrich. Catalyst solution was prepared by dissolving 1 g of tin octoate in 10 mL of toluene (Merck, India). high-performance liquid chromatography (HPLC)-grade chloroform and hydrochloric acid (HCl) were purchased from Merck, India. Sodium hydroxide (NaOH) was obtained from HiMedia Laboratories, India. All of the chemicals were used as received.
Preparation of n-HAP from Fish Scale
Hydroxyapatite nanopowder (n-HAP) was fabricated in the laboratory using fish scales (Labeorohita), which are considered as biowaste. Raw fish scales were collected from a nearby local fish market in Guwahati, Assam, India. Initially, the fish scales were washed several times in running tap water to remove dirt and other contaminants. The washed fish scales were deprotonated by stirring with a 1 M HCl solution for 24 h at room temperature. Thereafter, the fish scales were neutralized with a 1 M NaOH solution, followed by dilution with deionized water with pH 7. Excess water was removed by filtration through a filtration unit. Cleaned deprotonated fish scales were dried in a hot-air oven at 50 °C for 4–5 h to remove the excess amount of moisture. The dried scales were then calcinated in a programmable box furnace (B/F 14-15, VB Ceramic Consultants, India) at 1150 °C for 3 h to evaporate or remove the organic content. The nanosized HAP was obtained by wet-milling the calcinated flakes of fish scales in the mono ball milling machine (Planetary Mono Mill PULVERISETTE 6 classic line, Fritsch, Germany) for 3 h at 290 revolutions per minute (rpm) at room temperature.59,60 Before wet milling, the calcinated scales were mixed with deionized water to make the slurry, and zirconia balls (0.8–1.0 mm diameter) were added in 10:1 weight ratio. After removing the zirconia balls by filtration, homogenization was carried out by a homogenizer (Digital Homogenizer, IKA) with 6000 rpm at 5 °C for 15 min and the obtained slurry was lyophilized (LaboGene, SCANVAC) for 72 h at −95 °C to obtain a nanoparticulate of HAP. The obtained n-HAP was stored for further experiments.
Preparation of PDLA-Grafted n-HAP
n-HAP was grafted with PDLA via bulk ROP of d-lactide. A dried ampoule equipped with a magnetic stirrer was filled with required amount of purified d-lactide, n-HAP, and tin octoate. Lactide to catalyst molar ratio ([L]/[C]) was maintained at 2000:1. The ampoule was purged with argon gas for 1 h to remove moisture and oxygen. Vacuum was applied to the ampoule for 2 h to eliminate the excess amount of toluene. The ampoule was sealed using a heating torch under vacuum condition, and oil temperature was increased to 105 °C. Mixing of monomer, n-HAP (1, 2, and 5 wt %), and catalyst was done for 2 h; then, the temperature was increased to 160 °C and the ampoule was left for further 2 h for ROP. The obtained PDLA-grafted n-HAP was recovered by breaking the ampoule and named as PDLA/n-HAP; some amount of ungrafted PDLA was also formed, which remained with the PDLA/n-HAP composite. In the same fashion, PLLA and PDLA were also synthesized. The granulated forms of PLLA, PDLA, and PDLA/n-HAP were kept in a vacuum oven at 40 °C for 12 h to remove the residual monomer.
Preparation of sPLA/n-HAP Biocomposite
To prepare the sPLA/n-HAP biocomposite, PDLA/n-HAP and PLLA in 1:1 weight ratio were dissolved in 70 mL of chloroform (5% hexafluoroisopropanol solution) in a single-necked round-bottom flask at room temperature for 48 h. The obtained solution was cast on a poly(tetrafluoroethylene) (PTFE, 150 mm diameter) Petri dish kept at room temperature. The Petri dish was covered with perforated aluminum foil to reduce the rate of evaporation of solvent. After drying for 24 h, the obtained film was kept in a vacuum oven at 60 °C for 48 h to remove the residual content of solvent. The sPLA biocomposites with final content of 0.5, 1, and 2.5% n-HAP were denoted as sPLA-0.5% n-HAP, sPLA-1% n-HAP, and sPLA-2.5% n-HAP, respectively, whereas the blend of pristine PLLA and PDLA was indicated as sPLA. The prepared biocomposite films were used for further characterization.
Characterization
Raman spectra of n-HAP was recorded by Laser Micro Raman System (model: LabRam HR, make: Horiba Jobin Yvon) at room temperature. The spectra were recorded from wavenumber 2000 to 100 cm–1. Plasma-filtered laser light (514 nm) was focused onto the sample with 50× objective exposed for 15 s. The prepared n-HAP powder was directly analyzed, and the spectra were recorded by 10 accumulation.
Gel permeation chromatography (Shimazdu, Japan) was used to determine the molecular weight of the prepared biocomposite at 40 °C. The eluent used was HPLC-grade chloroform with a flow rate of 1.0 mL min–1. The system was calibrated using the monodispersed polystyrene standard. The prepared biocomposite was dissolved in chloroform and filtered through 0.45 μm PTFE syringe filters before analysis. The molecular weight and specific rotation of the PLLA, PDLA, and PDLA/n-HAP biocomposites are tabulated in Table 2.
Table 2. Molecular Weight of Fabricated Biocomposite.
| sample name | weight average molecular weight (Mw) | polydispersity index (PDI) | specific rotation [α]58925 |
|---|---|---|---|
| PLLA | 197 | 1.9 | –157 |
| PDLA | 255 | 1.8 | 156 |
| PDLA-1.0% n-HAP | 299 | 1.7 | |
| PDLA-2.0% n-HAP | 299 | 1.6 | |
| PDLA-5.0% n-HAP | 275 | 1.7 |
The chemical structures of PDLA and PDLA-grafted n-HAP were determined by a 600 MHz nuclear magnetic resonance (NMR) spectroscope (Bruker, Germany). 13C NMR spectra of samples were recorded after dissolving the samples in deuterated chloroform (CDCl3) for 48 h.
Calculation of the amount of PDLA grafted on n-HAP was made by TGA (PerkinElmer). Weight loss of the sample (6–8 mg) was recorded by heating under the temperature range of 30–700 °C at a heating rate of 10 °C min–1 under an inert N2 environment. PDLA-grafted n-HAP was purified by removing the unreacted PDLA chains from the system via dissolution and centrifugation, followed by a drying process before analysis.
The FTIR spectra of the biocomposites were obtained by attenuated total reflection (ATR) mode in Frontier FT-IR spectrometer (PerkinElmer) at room temperature. The spectra were recorded after 16 scans from wavenumber 4000 to 650 cm–1. The prepared biocomposite was directly analyzed, and the spectra were recorded.
Field emission scanning electron microscope (FESEM) connected with an EDX spectrometer was used to determine the topography of the fractured surface of different samples placed on carbon tape. Gold sputtering was carried out for 30 s, followed by characterization using FESEM (Sigma, Zeiss, GmbH) at an accelerating voltage of 2–4 kV.
The thermal behavior of the biocomposite was measured using a differential scanning calorimeter (DSC) (Phoenix DSC 204 F1NETZSCH, GmbH) under nitrogen atmosphere. The thermograph was recorded by heating the sample from 20 to 250 °C at a 10 °C min–1 heating rate. The percentage crystallinity of sPLA and biocomposite was calculated as per the following equations
| 1 |
| 2 |
| 3 |
where Xc,sc and Xc,hc are the degrees of crystallinity of stereocomplex crystallites and homocrystals, respectively; ΔHm,sc and ΔHm,hc are the enthalpies of fusion at melting for stereocomplex crystallites and homocrystals, respectively; ΔHmp,sc and ΔHmp,hc are the heats of fusion of a perfect crystal (93.6 J g–1 for homocrystals and 142 J g–1 for stereocomplex crystallites);55 ΔHc is the enthalpy of cold crystallization; and fsc (%) is the percentage of fraction of stereocomplex crystallites into the system.
Model-D8 Advance system diffractometer (Bruker, Germany) was used for the XRD analysis. The instrument was equipped with Cu Kα radiation (λ = 0.1541 nm) as X-ray source (40 kV, 40 mA) operating at a scan rate of 3° per min in the 2θ ranges of 5–40° (for composites films) and 20–55° (for n-HAP power) to generate the spectra.
The tensile strength and percentage elongation of prepared samples (7 mm width, ∼0.2 mm thickness and 15 mm gauge length) were measured using Tensile Stress Testing Stage (TST 350, Linkam) equipped with a 20 N load cell at a constant cross speed of 1 mm min–1 in tensile mode. The stage was controlled by system controller PE95/T95 with the system software Linksys 32. Each sample was analyzed with five replicates, and the average results were reported along with standard deviation.
The thermomechanical stability of the prepared biocomposites at higher temperatures with dynamic force application was measured using DMA (DMA 242 E model, NETZSCH, GmbH) in the temperature range of 25–190 °C at a 2 °C min–1 heating rate, 1 Hz frequency, and 10 μm displacement amplitude.
OX2/231 oxygen permeability tester (Labthink, China) was used to determine the OTR for the biocomposites with varying amount of n-HAP. The measurements were performed as per ASTM D3985 standard at 15, 23, 35, and 45 °C using high-purity oxygen gas (99.999%) on a film having an area of 50 cm2. Pure oxygen (99.9%) at a pressure of 0.5 bar and a flow rate of 20 mL min–1 was maintained in the upper half of the sample chamber during analysis, whereas nitrogen gas was maintained in the lower half of the chamber. The chambers were purged for 6 h before measurement. The test was carried out for at least 6 h so as to reach the steady state.
PERMATRAN-W model 1/50 (Mocon) was used to determine the WVTR as per ASTM standard E398-03. The relative humidity (RH) was fixed as 100 and 10% in the dry and wet chambers, respectively, yielding a driving force of 90% RH. The film of 50 cm2 area was analyzed at 37.8 ± 0.1 °C under atmospheric pressure.
In Vitro Studies
Cell Seeding and Cultivation
Baby Hamster Kidney fibroblast cells (BHK-21) were obtained from National Centre for Cell Science (Pune, India). The BHK-21 cells were cultured in T25 culture flasks containing Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen) supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, 1% l-glutamine, and 1% pyruvate (Invitrogen). The cells were incubated at 37 °C in a humidified atmosphere containing 5% CO2. The cells were recultured and maintained further for cell adhesion and proliferation assay. The cells were stained with trypan blue (SRL) and counted using Countess II FL Automated Cell counter (Thermo Fisher Scientific). The circular films of sPLA, sPLA-1.0% n-HAP, and sPLA-2.5% n-HAP were placed in the wells of a 96-well plate (Nunc) after sterilization by autoclave. To examine the cell viability, 2 × 104 cells/well were seeded onto the films using 100 μL of DMEM per well with polystyrene microplate as a control. All of the studies were carried out in triplicates, and the average data of the obtained results were reported with standard deviation.
MTT Assay
Mitochondrial activity of BHK-21 cells seeded onto the developed biocomposite films was assessed by enzymatic conversion of tetrazolium dye MTT (Sigma-Aldrich) after the time intervals of 8, 16, 24, 48, and 72 h. MTT reagent was prepared by dissolving 5 mg of MTT in 1 mL of phosphate-buffered saline (PBS). After each interval, 10 μL of MTT reagent was added to every well and incubated for 3 h at 37 °C in a humidified atmosphere. Later, the MTT reagent was removed from the wells and 100 μL of dimethyl sulfoxide (Merck) was added to dissolve the formosan crystals. Absorbance were measured using plate reader (Thermo Fischer Scientific) at 570 nm with reference wavelength of 650 nm.
Cell Staining
To determine the biocompatibility of the developed films, BHK-21 cells were seeded on a 12-well plate coated with circular films of sPLA, sPLA-1.0% n-HAP, and sPLA-2.5% n-HAP and maintained. The cell nuclei were stained with DAPI (dilactate) following 36 h of incubation (Sigma-Aldrich). DAPI stock (5 mg mL–1) was prepared in water and diluted to 1:2500 in 1% PBS solution. The cell medium was then aspirated completely, followed by rinsing with PBS thrice. The cells were fixed with 4% formaldehyde, incubated for 10 min, and further rinsed with PBS thrice. The cells were then permeabilized in 0.2% Triton X-100 after 5 min, allowed to aspirate, and rinsed thrice with PBS. DAPI (300 μL) was added to the wells and incubated for 15 min in dark. This was followed by aspiration and rinsing the cells thrice with PBS. Fluorescence was captured by exciting DAPI with ultraviolet light and was detected through a blue/cyan filter. The images were captured using FLoid Cell Imaging Station (Thermo Fischer Scientific) at 20× resolution.
Three-Dimensional Printing of Middle Phalanx Bone
The middle phalanx bone as a representative orthopedic implant was printed using 3D printer (model: Hydra 250, make: REDD Robotics, India) at 220 °C with a bed temperature of 110 °C. The layer resolution was kept constant at 0.2 mm. The print speed was 50 mm s–1, fill density was 40%, and bottom and top thicknesses were kept to be constant (0.6 mm). The filament used for the 3D printer was fabricated via a twin-screw mini-extruder (HAAKE MiniLab II from Thermo Scientific) with a 1.5 mm diameter circular die. The filament diameter was found to be 1.6 ± 0.2 mm.
Acknowledgments
The authors acknowledge the Centre of Excellence for Sustainable Polymers (CoE-SusPol) funded by Department of Chemicals and Petrochemicals (DCPC) and Central Instruments Facility (CIF) at Indian Institute of Technology Guwahati (IIT Guwahati), India, for providing research and analytical facilities.
The authors declare no competing financial interest.
References
- Nagarajan V.; Mohanty A. K.; Misra M. Perspective on Polylactic Acid (PLA) based Sustainable Materials for Durable Applications: Focus on Toughness and Heat Resistance. ACS Sustainable Chem. Eng. 2016, 4, 2899–2916. 10.1021/acssuschemeng.6b00321. [DOI] [Google Scholar]
- Garlotta D. A Literature Review of Poly(Lactic Acid). J. Polym. Environ. 2001, 9, 63–84. 10.1023/A:1020200822435. [DOI] [Google Scholar]
- Lin T. T.; Liu X. Y.; He C. A DFT study on poly(lactic acid) polymorphs. Polymer 2010, 51, 2779–2785. 10.1016/j.polymer.2010.03.062. [DOI] [Google Scholar]
- Ikada Y.; Jamshidi K.; Tsuji H.; Hyon S. H. Stereocomplex formation between enantiomeric poly(lactides). Macromolecules 1987, 20, 904–906. 10.1021/ma00170a034. [DOI] [Google Scholar]
- Miyamoto T.; Inagaki H. The Stereocomplex Formation in Poly(methyl methacrylate) and the Stereospecific Polymerization of Its Monomer. Polym. J. 1970, 1, 46–54. 10.1295/polymj.1.46. [DOI] [Google Scholar]
- Sawai D.; Tsugane Y.; Tamada M.; Kanamoto T.; Sungil M.; Hyon S.-H. Crystal density and heat of fusion for a stereo-complex of poly(L-lactic acid) and poly(D-lactic acid). J. Polym. Sci., Part B: Polym. Phys. 2007, 45, 2632–2639. 10.1002/polb.21270. [DOI] [Google Scholar]
- Tran H. T.; Ajiro H.; Hsiao Y.-J.; Akashi M. Thermally resistant polylactide layer-by-layer film prepared using an inkjet approach. Polym. J. 2017, 49, 327–334. 10.1038/pj.2016.119. [DOI] [Google Scholar]
- Tsuji H.; Fukui I. Enhanced thermal stability of poly(lactide)s in the melt by enantiomeric polymer blending. Polymer 2003, 44, 2891–2896. 10.1016/S0032-3861(03)00175-7. [DOI] [Google Scholar]
- Tsuji H.; Ikada Y. Stereocomplex formation between enantiomeric poly(lactic acid)s. XI. Mechanical properties and morphology of solution-cast films. Polymer 1999, 40, 6699–6708. 10.1016/S0032-3861(99)00004-X. [DOI] [Google Scholar]
- Sawai D.; Tamada M.; Kanamoto T. Development of Oriented Morphology and Mechanical Properties upon Drawing of Stereo-Complex of Poly(L-lactic acid) and Poly(D-lactic acid) by Solid-State Coextrusion. Polym. J. 2007, 39, 953–960. 10.1295/polymj.PJ2007038. [DOI] [Google Scholar]
- Bai H.; Liu H.; Bai D.; Zhang Q.; Wang K.; Deng H.; Chen F.; Fu Q. Enhancing the melt stability of polylactide stereocomplexes using a solid-state cross-linking strategy during a melt-blending process. Polym. Chem. 2014, 5, 5985–5993. 10.1039/C4PY00700J. [DOI] [Google Scholar]
- Tsuji H.; Tsuruno T. Water Vapor Permeability of Poly(L-lactide)/Poly(D-lactide) Stereocomplexes. Macromol. Mater. Eng. 2010, 295, 709–715. 10.1002/mame.201000071. [DOI] [Google Scholar]
- Tsuji H. Poly(lactic acid) stereocomplexes: A decade of progress. Adv. Drug Delivery Rev. 2016, 107, 97–135. 10.1016/j.addr.2016.04.017. [DOI] [PubMed] [Google Scholar]
- Pan P.; Bao J.; Han L.; Xie Q.; Shan G.; Bao Y. Stereocomplexation of high-molecular-weight enantiomeric poly(lactic acid)s enhanced by miscible polymer blending with hydrogen bond interactions. Polymer 2016, 98, 80–87. 10.1016/j.polymer.2016.06.014. [DOI] [Google Scholar]
- Fukushima K.; Furuhashi Y.; Sogo K.; Miura S.; Kimura Y. Stereoblock Poly(lactic acid): Synthesis via Solid-State Polycondensation of a Stereocomplexed Mixture of Poly(L-lactic acid) and Poly(D-lactic acid). Macromol. Biosci. 2005, 5, 21–29. 10.1002/mabi.200400121. [DOI] [PubMed] [Google Scholar]
- Han L.; Xie Q.; Bao J.; Shan G.; Bao Y.; Pan P. Click chemistry synthesis, stereocomplex formation, and enhanced thermal properties of well-defined poly(l-lactic acid)-b-poly(d-lactic acid) stereo diblock copolymers. Polym. Chem. 2017, 8, 1006–1016. 10.1039/C6PY01989G. [DOI] [Google Scholar]
- Bibi G.; Jung Y.; Lim J.-C.; Kim S. H. Novel Strategy of Lactide Polymerization Leading to Stereocomplex Polylactide Nanoparticles Using Supercritical Fluid Technology. ACS Sustainable Chem. Eng. 2016, 4, 4521–4528. 10.1021/acssuschemeng.6b00446. [DOI] [Google Scholar]
- Purnama P.; Kim S. H. Stereocomplex Formation of High-Molecular-Weight Polylactide Using Supercritical Fluid. Macromolecules 2010, 43, 1137–1142. 10.1021/ma902536p. [DOI] [Google Scholar]
- Akagi T.; Fujiwara T.; Akashi M. Inkjet Printing of Layer-by-Layer Assembled Poly(lactide) Stereocomplex with Encapsulated Proteins. Langmuir 2014, 30, 1669–1676. 10.1021/la404162h. [DOI] [PubMed] [Google Scholar]
- Jiang L.; Lv P.; Ma P.; Bai H.; Dong W.; Chen M. Stereocomplexation kinetics of enantiomeric poly(l-lactide)/poly(d-lactide) blends seeded by nanocrystalline cellulose. RSC Adv. 2015, 5, 71115–71119. 10.1039/C5RA12586C. [DOI] [Google Scholar]
- Gardella L.; Furfaro D.; Galimberti M.; Monticelli O. On the development of a facile approach based on the use of ionic liquids: preparation of PLLA (sc-PLA)/high surface area nano-graphite systems. Green Chem. 2015, 17, 4082–4088. 10.1039/C5GC00964B. [DOI] [Google Scholar]
- Sun Y.; He C. Synthesis and Stereocomplex Crystallization of Poly(lactide)–Graphene Oxide Nanocomposites. ACS Macro Lett. 2012, 1, 709–713. 10.1021/mz300131u. [DOI] [PubMed] [Google Scholar]
- Xu H.; Wu D.; Yang X.; Xie L.; Hakkarainen M. Thermostable and Impermeable “Nano-Barrier Walls” Constructed by Poly(lactic acid) Stereocomplex Crystal Decorated Graphene Oxide Nanosheets. Macromolecules 2015, 48, 2127–2137. 10.1021/ma502603j. [DOI] [Google Scholar]
- Sun Y.; He C. Synthesis, stereocomplex crystallization, morphology and mechanical property of poly(lactide)-carbon nanotube nanocomposites. RSC Adv. 2013, 3, 2219–2226. 10.1039/c2ra23179d. [DOI] [Google Scholar]
- Yang S.; Zhong G.-J.; Xu J.-Z.; Li Z.-M. Preferential formation of stereocomplex in high-molecular-weight polylactic acid racemic blend induced by carbon nanotubes. Polymer 2016, 105, 167–171. 10.1016/j.polymer.2016.10.034. [DOI] [Google Scholar]
- Tsuji H.; Kawashima Y.; Takikawa H.; Tanaka S. Poly(l-lactide)/nano-structured carbon composites: Conductivity, thermal properties, crystallization, and biodegradation. Polymer 2007, 48, 4213–4225. 10.1016/j.polymer.2007.05.040. [DOI] [Google Scholar]
- Sun Y.; Yang L.; Lu X.; He C. Biodegradable and renewable poly(lactide)-lignin composites: synthesis, interface and toughening mechanism. J. Mater. Chem. A 2015, 3, 3699–3709. 10.1039/C4TA05991C. [DOI] [Google Scholar]
- Nagahama K.; Aoki R.; Saito T.; Ouchi T.; Ohya Y.; Yui N. Enhanced stereocomplex formation of enantiomeric polylactides grafted on a polyrotaxane platform. Polym. Chem. 2013, 4, 1769–1773. 10.1039/c3py21055c. [DOI] [Google Scholar]
- Zhang W.; Zhang D.; Fan X.; Bai G.; Jiang Y.; Hu Z. Preparation and characterization of stereocomplex aggregates based on PLA-P188-PLA. RSC Adv. 2016, 6, 50543–50552. 10.1039/C6RA04537E. [DOI] [Google Scholar]
- Watanabe J.; Eriguchi T.; Ishihara K. Stereocomplex Formation by Enantiomeric Poly(lactic acid) Graft-Type Phospholipid Polymers for Tissue Engineering. Biomacromolecules 2002, 3, 1109–1114. 10.1021/bm025586r. [DOI] [PubMed] [Google Scholar]
- Kweh S. W. K.; Khor K. A.; Cheang P. The production and characterization of hydroxyapatite (HA) powders. J. Mater. Process. Technol. 1999, 89–90, 373–377. 10.1016/S0924-0136(99)00061-8. [DOI] [Google Scholar]
- Akram M.; Ahmed R.; Shakir I.; Ibrahim W. A. W.; Hussain R. Extracting hydroxyapatite and its precursors from natural resources. J. Mater. Sci. 2014, 49, 1461–1475. 10.1007/s10853-013-7864-x. [DOI] [Google Scholar]
- Salerno A.; Fernandez-Gutierrez M.; San Roman del Barrio J.; Pascual C. D. Macroporous and nanometre scale fibrous PLA and PLA-HA composite scaffolds fabricated by a bio safe strategy. RSC Adv. 2014, 4, 61491–61502. 10.1039/C4RA07732F. [DOI] [Google Scholar]
- Dadbin S.; Kheirkhah Y. Gamma irradiation of melt processed biomedical PDLLA/HAP nanocomposites. Radiat. Phys. Chem. 2014, 97, 270–274. 10.1016/j.radphyschem.2013.12.001. [DOI] [Google Scholar]
- Lin P.-L.; Fang H.-W.; Tseng T.; Lee W.-H. Effects of hydroxyapatite dosage on mechanical and biological behaviors of polylactic acid composite materials. Mater. Lett. 2007, 61, 3009–3013. 10.1016/j.matlet.2006.10.064. [DOI] [Google Scholar]
- Ishii S.; Tamura J.; Furukawa T.; Nakamura T.; Matsusue Y.; Shikinami Y.; Okuno M. Long-term study of high-strength hydroxyapatite/poly(L-lactide) composite rods for the internal fixation of bone fractures: A 2–4-year follow-up study in rabbits. J. Biomed. Mater. Res., Part B 2003, 66B, 539–547. 10.1002/jbm.b.10027. [DOI] [PubMed] [Google Scholar]
- Sun H.; Ai M.; Zhu S.; Jia X.; Cai Q.; Yang X. Polylactide-hydroxyapatite nanocomposites with highly improved interfacial adhesion via mussel-inspired polydopamine surface modification. RSC Adv. 2015, 5, 95631–95642. 10.1039/C5RA21010K. [DOI] [Google Scholar]
- Du K.; Gan Z. Shape memory behaviour of HA-g-PDLLA nanocomposites prepared via in situ polymerization. J. Mater. Chem. B 2014, 2, 3340–3348. 10.1039/C3TB21861A. [DOI] [PubMed] [Google Scholar]
- Qiu X.; Hong Z.; Hu J.; Chen L.; Chen X.; Jing X. Hydroxyapatite Surface Modified by l-Lactic Acid and Its Subsequent Grafting Polymerization of l-Lactide. Biomacromolecules 2005, 6, 1193–1199. 10.1021/bm049502l. [DOI] [PubMed] [Google Scholar]
- Hong Z.; Qiu X.; Sun J.; Deng M.; Chen X.; Jing X. Grafting polymerization of l-lactide on the surface of hydroxyapatite nano-crystals. Polymer 2004, 45, 6699–6706. 10.1016/j.polymer.2004.07.036. [DOI] [Google Scholar]
- Wang Z.; Xu Y.; Wang Y.; Ito Y.; Zhang P.; Chen X. Enhanced in Vitro Mineralization and in Vivo Osteogenesis of Composite Scaffolds through Controlled Surface Grafting of l-Lactic Acid Oligomer on Nanohydroxyapatite. Biomacromolecules 2016, 17, 818–829. 10.1021/acs.biomac.5b01543. [DOI] [PubMed] [Google Scholar]
- Tlotleng M.; Akinlabi E.; Shukla M.; Pityana S. Microstructures, hardness and bioactivity of hydroxyapatite coatings deposited by direct laser melting process. Mater. Sci. Eng., C 2014, 43, 189–198. 10.1016/j.msec.2014.06.032. [DOI] [PubMed] [Google Scholar]
- Kontoyannis C. G.; Bouropoulos N. C.; Koutsoukos P. G. Raman spectroscopy: A tool for the quantitative analysis of mineral components of solid mixtures. The case of calcium oxalate monohydrate and hydroxyapatite. Vib. Spectrosc. 1997, 15, 53–60. 10.1016/S0924-2031(97)00025-8. [DOI] [Google Scholar]
- Leitune V. C. B.; Collares F. M.; Trommer R. M.; Andrioli D. G.; Bergmann C. P.; Samuel S. M. W. The addition of nanostructured hydroxyapatite to an experimental adhesive resin. J. Dent. 2013, 41, 321–327. 10.1016/j.jdent.2013.01.001. [DOI] [PubMed] [Google Scholar]
- Yilmaz B.; Evis Z. Raman Spectroscopy Investigation of Nano Hydroxyapatite Doped with Yttrium and Fluoride Ions. Spectrosc. Lett. 2014, 47, 24–29. 10.1080/00387010.2013.778296. [DOI] [Google Scholar]
- Ciobanu C. S.; Iconaru S. L.; Le Coustumer P.; Predoi D. Vibrational Investigations of Silver-Doped Hydroxyapatite with Antibacterial Properties. J. Spectrosc. 2013, 2013, 1. 10.1155/2013/471061. [DOI] [Google Scholar]
- Mobasherpour I.; Heshajin M. S.; Kazemzadeh A.; Zakeri M. Synthesis of nanocrystalline hydroxyapatite by using precipitation method. J. Alloys Compd. 2007, 430, 330–333. 10.1016/j.jallcom.2006.05.018. [DOI] [Google Scholar]
- Ślósarczyk A.; Paszkiewicz Z.; Paluszkiewicz C. FTIR and XRD evaluation of carbonated hydroxyapatite powders synthesized by wet methods. J. Mol. Struct. 2005, 744–747, 657–661. 10.1016/j.molstruc.2004.11.078. [DOI] [Google Scholar]
- Masutani K.; Kimura Y.. PLA Synthesis. From the Monomer to the Polymer. In Poly(lactic acid) Science and Technology: Processing, Properties, Additives and Applications; The Royal Society of Chemistry, 2015; Chapter 1, pp 1–36. [Google Scholar]
- Wu C.-S. Improving Polylactide/Starch Biocomposites by Grafting Polylactide with Acrylic Acid – Characterization and Biodegradability Assessment. Macromol. Biosci. 2005, 5, 352–361. 10.1002/mabi.200400159. [DOI] [PubMed] [Google Scholar]
- Liao C.-J.; Lin F.-H.; Chen K.-S.; Sun J.-S. Thermal decomposition and reconstitution of hydroxyapatite in air atmosphere. Biomaterials 1999, 20, 1807–1813. 10.1016/S0142-9612(99)00076-9. [DOI] [PubMed] [Google Scholar]
- Kister G.; Cassanas G.; Vert M. Effects of morphology, conformation and configuration on the IR and Raman spectra of various poly(lactic acid)s. Polymer 1998, 39, 267–273. 10.1016/S0032-3861(97)00229-2. [DOI] [Google Scholar]
- Zhang J.; Sato H.; Tsuji H.; Noda I.; Ozaki Y. Infrared Spectroscopic Study of CH3···OC Interaction during Poly(l-lactide)/Poly(d-lactide) Stereocomplex Formation. Macromolecules 2005, 38, 1822–1828. 10.1021/ma047872w. [DOI] [Google Scholar]
- Su Z.; Li Q.; Liu Y.; Hu G.-H.; Wu C. Multiple melting behavior of poly(lactic acid) filled with modified carbon black. J. Polym. Sci., Part B: Polym. Phys. 2009, 47, 1971–1980. 10.1002/polb.21790. [DOI] [Google Scholar]
- Tsuji H. Poly(lactide) Stereocomplexes: Formation, Structure, Properties, Degradation, and Applications. Macromol. Biosci. 2007, 7, 1299. 10.1002/mabi.200700275. [DOI] [PubMed] [Google Scholar]
- Karak N.Fundamentals Of Polymers: Raw Materials To Finish Products; Prentice-Hall Of India Pvt. Limited, 2009. [Google Scholar]
- Oyama H. T.; Abe S. Stereocomplex Poly(lactic acid) Alloys with Superb Heat Resistance and Toughness. ACS Sustainable Chem. Eng. 2015, 3, 3245–3252. 10.1021/acssuschemeng.5b00832. [DOI] [Google Scholar]
- Bai H.; Huang C.; Xiu H.; Zhang Q.; Deng H.; Wang K.; Chen F.; Fu Q. Significantly Improving Oxygen Barrier Properties of Polylactide via Constructing Parallel-Aligned Shish-Kebab-Like Crystals with Well-Interlocked Boundaries. Biomacromolecules 2014, 15, 1507–1514. 10.1021/bm500167u. [DOI] [PubMed] [Google Scholar]
- Prasad A.; Devendar B.; Sankar M. R.; Robi P. S. Micro-Scratch Based Tribological Characterization of Hydroxyapatite (HAp) Fabricated through Fish Scales. Mater. Today: Proc. 2015, 2, 1216–1224. 10.1016/j.matpr.2015.07.034. [DOI] [Google Scholar]
- Prasad A.; Mohan Bhasney S.; Sankar M. R.; Katiyar V. Fish Scale Derived Hydroxyapatite reinforced Poly (Lactic acid) Polymeric Bio-films: Possibilities for Sealing/locking the Internal Fixation Devices. Mater. Today: Proc. 2017, 4, 1340–1349. 10.1016/j.matpr.2017.01.155. [DOI] [Google Scholar]