

Abstract
The stability of antibody–drug conjugates (ADCs) in circulation is critical for maximum efficacy and minimal toxicity. An ADC reaching the intended target intact can deliver the highest possible drug load to the tumor and reduce off-target toxicity from free drug in the blood. As such, assessment of ADC stability is a vital piece of data during development. However, traditional ADC stability assays can be manually intensive, low-throughput, and require large quantities of ADC material. Here, we introduce an automated, high-throughput plasma stability assay for screening drug release and aggregation over 144 h for up to 40 ADCs across five matrices simultaneously. The amount of ADC material during early drug development is often limited, so this assay was implemented in 384-well format to minimize material requirements to <100 μg of each ADC and 100 μL of plasma per species type. Drug release and aggregation output were modeled using nonlinear regression equations to calculate formation rates for each data type. A set of 15 ADCs with different antibodies and identical valine–citrulline–p-aminobenzylcarbamate–monomethylauristatin E linker-drug payloads was tested and formation rates were compared across ADCs and between species, revealing several noteworthy trends. In particular, a wide range in aggregation was found when altering only the antibody, suggesting a key role for plasma stability screening early in the development process to find and remove antibody candidates with the potential to create unstable ADCs. The assay presented here can be leveraged to provide stability data on new chemistry and antibody screening initiatives, select the best candidate for in vivo studies, and provide results that highlight stability issues inherent to particular ADC designs throughout all stages of ADC development.
Introduction
Antibody–drug conjugates (ADCs) have steadily gained attention over the last two decades for their potential as targeted oncology therapeutics. Two ADCs are currently approved by the Food and Drug Administration for the treatment of cancer and more than 50 are undergoing clinical trials for a variety of different indications. In combining the targeting capabilities of an antibody with a highly potent small molecule, ADCs aim to increase the therapeutic index through improved efficacy and decreased systemic toxicity by selective delivery of the toxin only to the target tumor cells.1 The cytotoxic agents are chemically attached to an antibody, generally through either lysine amines or free thiols.2 A chemical linker tethers the antibody and drug together and is usually designed to be enzymatically cleaved in such a way as to release the toxin in an active form.3 Throughout this article, we refer to the cytotoxic agent interchangeably as drug, toxin, or warhead. Payload is used to signify the linker-drug portion of the ADC. The release of the drug is carried out by intracellular proteases in the endolysosomal compartments of the cell and occurs after the receptor–ADC complex is internalized and trafficked to the proper intracellular location.4 Noncleavable linkers are also commonly used; however, these constructs require catabolism of the antibody to release the active warhead.5,6
For exclusive and selective delivery of drug to the target cells, the construct needs to arrive at its destination with a full drug load. If active drug is released before the ADC can reach the target, off-target toxicity may result, and less drug will be delivered to the site of action.7 Factors that negatively impact safe delivery of intact ADC include plasma-labile linkers,8 blood-proteases,9 and aggregation-prone molecules, which can lead to immunogenicity or nonspecific uptake.10 First-generation ADC programs were hampered by unstable drug linkage, such as hydrazone linkers.11 The knowledge base of linker technologies yielding highly stable circulating ADCs has grown, and studies over the last few years have highlighted the wide effect different chemistries can have on the stability of an ADC.12 Linker stability questions become more complex when considering species differences. Plasma stability assessments performed on a single species may not be representative and could potentially mislead data interpretation of efficacy and tolerability studies during ADC development.13,14
Drug development is a time-consuming and costly endeavor. Biologics can further lengthen the process, with extended in vivo t1/2 necessitating longer duration studies compared to those of most small molecules. To interrogate how chemical modifications (i.e., changes to the linker or drug) affect the pharmacokinetic properties of an ADC, multiweek animal studies are often needed. These studies can take months to complete from initial study design to final data analysis, resulting in long cycle times for rounds of structure–activity relationship investigation. In vitro assays able to triage ADCs with poor properties prior to the initiation of animal studies could significantly speed the developmental process and lessen animal usage. Key roadblocks to the utilization of in vitro assays in the past have been the limited amounts of ADC material during early developmental phases as well as time-intensive assays often performed in a low-throughput manner. Here, we have outlined an automated and multiplexed ADC plasma stability assay that can provide thorough and rapid characterization of ADC drug release and aggregation for up to 40 ADCs simultaneously. By testing ADC stability in multiple species, this assay can serve as a front line evaluation for ADCs prior to in vivo studies. Additionally, we deployed this methodology on a set of ADCs and modeled formation rates of released drug and aggregations for comparison of ADC plasma stability across the set and between species. This assay can serve as a critical first-tier, in vitro screening tool for new immunoconjugates, ultimately leading to better ADC candidates and decreased cycle times for successive generations of ADC molecules.
Results and Discussion
Protein Stability
The stability of ADCs in buffer and plasma from four species was monitored over 144 h at 37 °C. Stability was assessed in human plasma as well as plasma from relevant preclinical species: cynomolgus monkey, Sprague Dawley rat, and CD1 mouse. Additionally, a solution consisting of phosphate-buffered saline (PBS) at physiological pH, referred to as “buffer” throughout this work, was included to monitor the behavior of ADCs in a typical storage solution. Species were chosen from ones commonly used during various stages in the ADC development process. Mice are the primary efficacy model for oncology, with xenograft studies among the most frequently utilized studies. Rats are useful as preclinical pharmacology and toxicology models, especially if the antibody is cross-reactive to the target in rat. Cynomolgus monkeys are often the nonhuman primate of choice for investigational new drug (IND)-enabling toxicology studies. Incubations in this study were run for 144 h to reach equilibrium for the majority of tested molecules. A 96 h incubation has been recommended by the IQ Consortium and is the common incubation duration for IND submissions.7,15
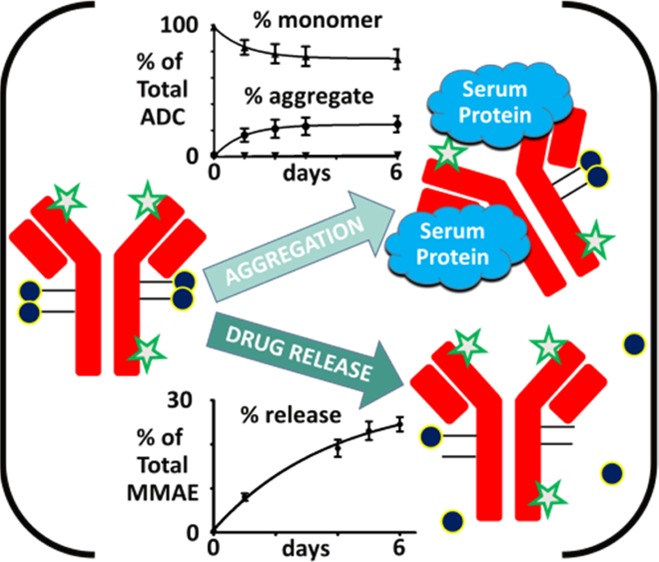
Samples were analyzed by size exclusion chromatography (SEC) to obtain the percentage of antibody fragments and high molecular weight species (HMWS) formed in plasma relative to the amount of ADC monomer present. The terms aggregates and HMWS are used interchangeably throughout this work. Fluorophore was added to the antibodies of the ADCs (Figure 1A) for differentiation of ADCs from endogenous plasma protein by fluorescence detection.
Figure 1.

Measurement of ADC aggregate formation in plasma over time. (A) Schematic of ADCs with broad payload distribution (blue circles) and stochastic labeling with Alexa Fluor 488 (green stars) for differentiation of conjugates from endogenous plasma proteins during SEC analysis to determine the degree of protein aggregation. (B) Monomer, fragments, and aggregates were measured over a 6 day time course in human plasma for Ab095–maleimide–caproyl−valine–citrulline–p-aminobenzylcarbamate–monomethylauristatin E (Ab095–vc–MMAE) conjugates, with an average drug-to-antibody ratio (DAR) of 3.35. The aggregation data was fit with eq 1, and the best-fit result is shown above the aggregation curve. Each data point is the mean of two biological replicates with two technical replicates of each, and the error bars are the standard deviation. (C–E) The effect of DAR change on the total aggregation was assessed for DAR species of 2.4, 3.4, and 4.6 in monkey, mouse, and rat plasma. The data shown is the mean of two replicates for each time point and sample.
Here, we used a human IgG antibody, Ab095. A valine–citrulline linker to monomethylauristatin E (MMAE) with a p-aminobenzylcarbamate (PABC) spacer was attached to Ab095 through maleimide–caproyl on the free cysteines of the antibody (Ab095–mc–vc–PABC–MMAE, referred to as Ab095–vc–MMAE for succinctness).16 Aggregation was measured for two conjugation lots of Ab095–vc–MMAE, with an average DAR of 3.35. The preparations were composed of a broad distribution of drug to antibody, with 0, 2, 4, 6, and 8 species present.17 In Figure 1B, Ab095–vc–MMAE monomer decreased in human plasma over the incubation time course. Total monomer levels declined from 98 ± 0.5% monomer of the total fluorescence levels at day 0 to 70 ± 2.3% by the sixth day of incubation. The formation of antibody fragments containing fluorophore contributed little to the monomer decrease, as only 1.7% of the total fluorescence signal was attributed to antibody fragments by the conclusion of the incubation. In comparison, HMWS were at 1.2% in PBS, HMWS levels increased to 2 ± 0.4% upon human plasma addition, and levels were at 28.4 ± 0.04% after 6 days of incubation. The aggregation of therapeutic antibodies and ADCs can result from a variety of complex mechanisms,18 and intricate mathematical models have accordingly been put forth in the past to describe such phenomena.19 Here, we elected to use a simple equation to model aggregation data and capture HMWS formation over multiday time periods. With the goal of enabling differentiation of aggregation between ADCs in a high-throughput screening assay, a mechanistic description of the underlying cause of aggregation was deemed to be beyond the scope of this work. The aggregation data points were fit by eq 1, a nonlinear regression, defined as follows
| 1 |
where A is the level of aggregation at time t, kagg is the rate of aggregate formation, A0 is the amount of aggregation at time 0, and Aequilibrium is the projected steady state level of aggregation. Eq 1 is well suited to model the formation kinetics of HMWS from these data, where the overall aggregation levels increase over time until a plateau is reached, either at a steady state value or at the maximum aggregation level (i.e., 100% of the total signal is composed of HMWS). Additionally, as a nonlinear regression, eq 1 can fit both linear and nonlinear growth patterns. Using eq 1 on the aggregation data in Figure 1B, a fit was determined with an R2 value of 0.998, an Aequilibrium of 23.1%, and a kagg of 1.02 day–1. The A0 aggregation levels of 1.4% were added to Aequilibrium to give an adjusted Aequilibrium of 24.5% aggregation, which we termed Amax. As Amax is nearly equal to the measured day six aggregation value (Aday6), both the numerical data and the graphical data agree with the aggregation plateaus at 28%.
To study the impact of higher DAR on aggregation, SEC profiles over 6 days were acquired for three DAR variants (DAR 2.4, 3.4, and 4.6) of Ab095–vc–MMAE (Figures 1C–E and 2A). The different DAR variants were found to have greatly differing levels of HMWS in each of the plasmas used. The DAR 4.6 material demonstrated significant initial aggregation that increased over time. For each of the four plasmas from different species, at least 17% aggregation was observed at day 0 for the ADC with the highest DAR. The other two DAR variants had lower initial aggregation (<5% total HMWS) that rapidly increased, with day 6 levels >16% HMWS for all species with the DAR 3.4 lot and >14% HMWS for the DAR 2.4 lot. The degree of aggregation was fairly consistent across the DAR variants within each species, as the highest DAR Ab095–vc–MMAE yielded the most aggregation and the lowest DAR ADC yielded the least aggregation.
Figure 2.

Rate of aggregation formation for Ab095–vc–MMAE in human plasma. (A) Aggregation values over 6 days are plotted for three lots of Ab095–vc–MMAE containing different DARs. The data was fit with eq 1. (B) Parameters from the curve fitting, Aequilibrium, kagg, Aday6, Aday0, and R2 values are displayed.
These results are consistent with previous observations of increasing aggregation as the DAR is increased.20 Other studies have found the CH2 domain of IgG to become destabilized upon higher DAR conjugation, leading to formation of HMWS in buffer and likely more susceptibility toward aggregation in plasma as well.21 Higher DAR variants of ADCs with vc–MMAE have also been shown to lead to altered pharmacokinetics due to increased clearance.22 The mechanism of accelerated clearance of high DAR species may be attributed to higher hydrophobicity producing aggregation,15 which leads to ADC uptake by liver cells; Kupffer and sinusoidal endothelial cells uptake has been demonstrated to be minimized by decreasing overall hydrophobicity of the ADC molecule.23 These data show that one can increase the drug load without sacrificing ADC plasma stability and pharmacokinetics through design of the ADC constructs with lower hydrophobicity and aggregation.
Although the presence of a fluorophore could impact HMWS formation, the average number of fluorophores added is less than one, representing a small molecular weight change (∼600 Da per fluorophore) to the much larger ADC (>150 kDa). Furthermore, different ADCs conjugated with fluorophores have been shown to have a wide range of HMWS levels (data not shown), demonstrating that HMWS arise from ADC properties and interactions with plasma components and not as a result of fluorophore addition. Accordingly, this approach provided the ability to rank order ADCs on the basis of relative aggregation levels.
A numerical approach was used to compare the data for the three DAR variants from incubations with human plasma, and eq 1 was utilized to determine the rate of aggregate formation (Figure 2A). Four values from the regression fitting were most useful for the purpose of comparison. The metrics of kagg, Aequilibrium, Aday0, and Aday6 displayed in Figure 2B are enough to summarize the data. The rate of each DAR species shows that the middle DAR has the fastest aggregation, with a 1.07 day–1kagg, whereas the high DAR species has a kagg of 0.15 day–1, a slow rate compared to that of the other two. The Aday6 values of the DAR 3.4 and 4.6 species are similar, with DAR 3.4 having slightly higher Aday6 aggregation levels. However, the initial aggregation value for DAR 4.6 starts much higher at 22.3%, and slower, more linear growth kinetics leads to an Amax of 31.5%, whereas the Amax of the DAR 3.4 lot remains about the same as that of the Aday6 value because the steady state had already been achieved.
The extension of these numerical analyses to larger data sets can aid in comprehension of the data. The amount of data can quickly scale due to an analysis matrix of five different incubation conditions, with an array of changing antibodies, attachments, linker chemistries, and drugs. As such, numerical analyses could become a necessity for a given series of ADCs. Accordingly, the focus here is to provide a simplistic way to compare large data sets to differentiate the most well-behaved ADCs and inform future iterations of ADCs, especially if a performance benchmark has been determined for a particular target. Because many facets dictate the general aggregation levels of ADCs, it may be unadvisable to draw a hard line of acceptable aggregation to be used for all ADC series. However, a benchmark for each target can advise on whether changes to ADC chemistry have positive or negative outcomes on plasma stability.
Drug Release
In addition to protein stability, the levels of released warhead in plasma were measured over time to assess the stability of the linker-drug. Limiting free circulating warhead is a critical consideration when designing an ADC molecule. The separation of toxin from the antibody before its arrival at the target cells can lead to off-target toxicity. Additionally, antibodies with no or reduced drug load can compete against antibodies containing drug for binding to target receptors, which may lead to a loss of efficacy. Knowledge of ADC behavior in plasma of preclinical species can not only help interpret outcomes from toxicity and efficacy studies but also inform researchers of potential liabilities prior to in vivo studies. For example, a poorly behaved ADC in a mouse efficacy study may show poor efficacy than a similar but more stable ADC. Another example would be if rat was initially chosen as a toxicity model but was found to have much greater release kinetics than that predicted for human. Plasma stability data in such an instance could help guide selection of the best preclinical species in which to perform toxicity studies to best predict human outcomes.
The measurement of the released drug was performed in parallel with the aggregation analysis to gain insight on the stability of the linker-drug in buffer and plasmas over time. The amount of drug release from the ADC was quantitated by liquid chromatography–mass spectrometry (LC–MS) and expressed as a percentage of the theoretical starting drug concentration in the system. To enable the LC–MS analysis, plasma proteins were precipitated with organic solvent and removed by centrifugation, an efficient sample cleanup procedure previously shown to remove >90% protein while retaining small molecules in solution.24 Under these conditions, both free and previously noncovalently protein-bound drug will be present in the supernatant for measurement as unconjugated drug.25 Accurate quantitation of the drug was performed using six-point standard curves prepared in the appropriate matrix and processed alongside of the samples. For an ADC with DAR 4, the standard curve was prepared to be able to quantify release amounts from 0.02 to 112%. This wide range enables quantitation of drug release for both stable (e.g., <0.1% release) and unstable (e.g., >10% release) ADCs in the same experiment.
Levels of unconjugated MMAE from Ab095–vc–MMAE ADC were <1% of the theoretical starting amount after 6 days of incubation in PBS buffer, human plasma, and cynomolgus plasma. Conversely, rodent plasma produced significantly higher concentrations of free MMAE (Figure 3A). In rat plasma, free MMAE levels were 2.5% of the theoretical total MMAE present after 6 days. Levels in CD1 mouse plasma were approximately 10-fold higher, at nearly 25% after 6 days. The higher degree of linker proteolysis observed in mouse plasma compared to that of the other species can be attributed to the enzyme carboxylesterase 1C, of which the valine–citrulline linker on our ADC is a substrate.9,13 Rat plasma may also contain carboxylesterases with activity toward valine–citrulline, albeit not to the same extent as mouse plasma.9
Figure 3.

MMAE release over time from Ab095–vc–MMAE in different matrices. The percent of released MMAE over 6 days at 37 °C was measured by LC−MS in mouse, rat, monkey, and human plasma, as well as in PBS. (A) Data is from the mean of four biological replicates, each with averaged data from two technical replicates, and error bars are standard error. (B) The stability of MMAE was measured in the matrices, with the amount remaining at each time point normalized to the initial amount at day 0. (C) The data was fit using the nonlinear regression outlined in eq 2. The inset on the right is a zoomed-in section of the data.
Because this assay used a triple quadrupole mass spectrometer, our analysis was focused on a specific molecular weight, defined prior to analysis of the samples. Metabolism of drug could change the released product mass and lead to lower detectable levels of the targeted molecular weight species measured. Any metabolism or instability (e.g., precipitation) of the drug in the matrices could produce misleading results on the degree of drug release by underrepresenting the actual amounts of drug release from the ADC. To better understand the measured warhead levels from this assay, the stability of the warhead in the different matrices must be measured to properly interpret the ADC released drug data. In Figure S1A, the amount of release of a different warhead, specified here as Drug #1, from an ADC, referred to as ADC #1, was nearly 2% in mouse plasma, which was significantly higher than the negligible amount of released drug observed for monkey plasma and buffer. When the stability of Drug #1 was assessed in the matrices, Drug #1 was shown to be unstable in mouse and monkey plasma (Figure S1B). These data highlight the possibility of release in monkey plasma with drug instability possibly limiting overall drug accumulation from ADC in plasma over time. Furthermore, the 2% of release in mouse plasma was likely an underestimate of the actual released drug amounts because instability decreases the amount of drug measured. Simulations were run to visualize the impact drug instability (Figure S1C) has on overall measured release levels (Figure S1D). Even with a warhead half-life of 4–5 days, the overall effect on measured levels of free warhead can be greater than 50%. Poor warhead stability can quickly diminish the levels of detectable warhead, which could make an unstable warhead with high release appear stable if only ADC plasma stability is used, as observed in monkey plasma for ADC #1. With the effect of drug stability in mind, the MMAE drug was incubated in the different matrices to capture the stability of MMAE on its own. Our observations show MMAE to be relatively stable in the five matrices tested, with at least 92% MMAE remaining for all matrices after 6 days at 37 °C (Figure 3B). When ADC and drug stability data are considered together, greater confidence can be placed in the Ab095–vc–MMAE stability data for buffer, human, and cynomolgus plasma.
As with the aggregation data, the ADC plasma release data can be captured with a similar nonlinear equation. The regression equation follows the same format as that of the equation described above, with different naming conventions
| 2 |
where R is the percentage of release of the theoretical total amount of the conjugated drug at time t, Requilibrium is the release as a percentage of the total warhead at steady state, krelease is the rate of unconjugated drug formation, and R0 is the amount of unconjugated drug at the start of the incubation. Ab095–vc–MMAE release data was evaluated using eq 2 (Figure 3C). The best-fit equation for mouse plasma generated a nearly 10-fold higher krelease value than that of the other plasma matrices and had an Requilibrium of 30.2%, which was similar to the measured day six level. Because of the close proximity between the two values, the release in mouse appears to be near completion after 6 days. In contrast, the projected Requilibrium values extend much past their Rday6 values for the other matrices tested, indicating that release would continue after day 6. All projections have visual alignment with the graphical data, as mouse seems to have reached equilibrium by day 6, whereas the other plasma matrices produced more linear data.
The release profile of an ADC containing a noncleavable linker was also investigated. The construct contained Ab095 conjugated directly to another auristatin molecule, monomethylauristatin F, through a maleimide–caproyl attachment (Ab095–mc–MMAF). In Ab095–mc–MMAF, 0.02–0.03% of the total MMAF was released in the various plasmas after the full incubation time course (Figure S2A). MMAF was also shown to be stable in all matrices (Figure S2B). This low level of release indicates the mc attachment portion of the molecule was not susceptible to cleavage in plasma. However, cysteine switching of the maleimide onto the free cysteine-34 of serum albumin has been shown to occur and can lead to significant DAR loss over time.26 Because we are only monitoring unconjugated warhead, our method would not detect this payload transfer. In addition to payload transfer, alterations to the attachment chemistry can have a substantial change on the release profile of an ADC containing a dipeptide linker. In Figure S2C, two ADCs with the same antibody backbone, linker, and warhead but different attachment chemistries had the levels of released drug measured. One attachment, a maleimide–caproyl attachment, yielded >24.6% release in mouse plasma, and the other attachment, a stable diaminopropionic acid attachment,27 had 2.4% release on average over three replicates. The stabilization, which promotes hydrolysis of the maleimide ring to limit retro-Michael reaction, appears to have also altered the susceptibility of the valine–citrulline to be cleaved in plasma. These results demonstrate that all portions of an ADC can contribute to plasma stability of the linker-drug.
Screening and Characterization of ADCs
A set of 15 ADCs was analyzed to determine the formation of aggregation and drug release levels in plasma over 6 days. The ADCs were comprised of different antibodies conjugated with the same vc–MMAE payload. The immunoconjugates were incubated in the five matrices detailed above, and aliquots were taken for parallel analysis by MS and SEC. Aggregation levels of the ADCs after 6 days in different plasma matrices are shown in Figure 4A. The median levels of HMWS were 25.3% for rat, 26.0% for mouse, 20.3% for monkey, and 24.2% for human plasma. Aggregation was much lower in buffer, with a median level of 2.3%. Levels of aggregation were varied among the set of ADCs investigated. For example, in rat plasma, the highest percentage of aggregation after the 6 day incubation was 33.3% of the total amount of ADC signal and the lowest aggregation was 17.0%. The data was fit with eq 1 (rat plasma aggregation in Figure 4B and the other four matrices in Figure S3A–C). All fitted data had equations with R2 values >0.98, except one ADC in the monkey plasma set, which had an R2 value of 0.95. Interestingly, although monkey plasma produced the lowest overall aggregation levels of the plasma matrices, the monkey plasma had the highest kagg with >2-fold higher median kagg values compared to the rodent species (Figure S4A). The aggregation levels increased quickly in the monkey plasma but reached steady state in less time and at a lower value than in the other plasma matrices. More generally, the difference in kagg values between rodent plasma and the primate plasmas distinguished the shapes of the aggregation formation, as faster rates for primate plasma lead to steady state being achieved sooner than in rodent. The data fits were used to calculate predicted levels of aggregation after 28 days, which represents approximately two t1/2 of a long-lasting ADC. The difference between the measured Aday6 (Figure 4A) and the predicted Aday28 (Figure 4C) levels are small, at a mean change of 0.38% aggregation across all species. These small changes between Aday6 and Aday28 correspond well with the visual observation that by day 6 the data has either already reached or would soon reach the steady state. Although these data are mostly at equilibrium by the end of our experiment, this may not always be the case and the differences between Aday6 and Amax would be informative in such a case.
Figure 4.

Stability screening of 15 vc–MMAE containing ADCs in different plasma matrices. ADCs containing the same vc–MMAE payload were conjugated to different antibodies and assessed for both (top) aggregation and (bottom) release levels in parallel over 6 days. (A) A box and whisker plot shows the aggregation levels at day 6 for the ADCs in the four plasma matrices. The box covers the 25th through 75th percentile of data and the whiskers extend to the 10th and 90th percentiles. The line through the box is the median value. (B) Aggregation data from rat plasma was fit with eq 1. (C) The predicted level of aggregation at day 28 is represented with box and whisker plots. (D) Release data at day six is represented by a box and whisker plot. (E) The fit released drug data from rat plasma using eq 2 is graphed. (F) The predicted amount of release at day 28 is represented with box and whisker plots.
The release data for the set of ADCs was similar to the release levels seen with Ab095–vc–MMAE in that there was, on average, >20% release in mouse plasma, >4% release in rat plasma, and <1% release in human and monkey plasma after 6 days of incubation (Figure 4D). As above, data was fit with eq 2 (Figures 4E and S3D–F). The majority of the fit data had R2 values >0.99, but there were five instances in the human and monkey data sets where a fit <0.9 was achieved. In contrast to the small differences between Aday6 and Aday28, Rday6 and Rday28 were significantly different. Several projected Rmax values reached the maximum 100% release due to linearity in free drug formation. In the case of these linear data, steady state conditions were not approached by the end of the incubation. To make the values more pharmacologically relevant, day 28 was used as above, which resulted in higher release levels than those seen at day 6, but significantly less than 100% release due to the slow release rates. In contrast to the other species, equilibriums were achieved in mouse plasma, as the small difference between Rmax and Rday6 indicates. The change in release between Rmax and Rday6 of 2.2% in mouse shows much less of a difference than that in the other species, wherein the mean differences were between 13.6 and 40.2%. Looking at the krelease values, mouse plasma produces a release rate almost three-fold higher than any of the other species (Figure S4B). Taking these data together, the mouse has rapid initial drug release until reaching the steady state, whereas the other ADCs have slower but steadier free drug formation kinetics.
We next were interested in the correlation between aggregation and drug release. The data sets were linearly fit, and a poor correlation was found between the data sets, as the highest R2 value was 0.03 (Figure S5). Therefore, in these data, no connection exists between the release of drug and formation of HMWS. In Figure S6, we graphed the species demonstrating the most aggregation and toxin release. The mouse plasma produced the highest release values for all but one ADC, which had slightly more release in the rat plasma. The higher toxin levels were expected due to the presence of carboxylesterase 1C in mouse plasma, as mentioned above. The aggregation data had less of a rodent bias, with human and mouse producing the most aggregation in seven ADCs each.
Overall, each ADC displayed fairly consistent behavior. If high aggregation was found for an ADC in one species, it could be expected the other ADCs would display similar HMWS levels. When the aggregation of one species was plotted against the aggregation of another species, the linear fits of the graphs produced positive slopes (i.e., as aggregation increased in one species, it likely increased in other species too) and R2 values between 0.60 and 0.85 (Figure S7A). Similarly, the release levels of the ADCs had positive slopes, although with less correlation (R2 values between 0.16 and 0.74) than that of the aggregation data (Figure S7B). However, these data also highlight the wide range in aggregation, with the only variation in the ADCs coming from a change in the antibody backbone. In addition, a 6 day incubation appears to be excessive, especially for assessing aggregation, as most of the “action” has taken place after 2 or 3 days; the equations presented here could be used to model predicted results at extended time points, further streamlining the assay in the future. These results suggest potentially testing a panel of antibodies during antibody selection, as the specific structure and conformation may influence ADC stability.
Conclusions
Protein aggregation is important to monitor, as aggregation can negatively impact ADC pharmacokinetics and potentially lead to immunogenicity.28,29 Many of the ADCs shown in this work had high levels of aggregation and toxin release and would likely not have the physiochemical properties necessary to progress further into development. By utilizing the data from this assay correctly, these molecules could be prescreened with only the top ADCs moving forward, saving valuable time and effort in the pursuit of the best candidates. In addition, the plasma stability of one species may not be reflective of others, so correct interpretation of in vivo studies warrants testing in the appropriate matrix. As described above, many factors can influence the aggregation of ADCs, including the DAR, attachment chemistry, payload, and antibody backbone. Additionally, knowledge of payload stability for an ADC is crucial, as any significant stability issues can greatly diminish efficacy and lead to off-target toxicity driven by the unconjugated drug present in the bloodstream. By designing ADCs with their plasma stability in mind, monomeric ADCs with fully intact payloads can arrive at their target destination. Furthermore, through miniaturization, automation, and parallel SEC and LC−MS measurements, a rapid and timely assessment of ADC stability at the earliest stages of ADC development efforts can be performed. Using these data earlier in drug discovery will allow for accelerated decision making, reduction in animal and material costs, and improvement in the probability of success.
Future methodological additions, with an eye toward further understanding of ADC plasma stability and increased usability for uncharacterized ADCs, could include metabolite profiling and toxin accounting. Qualitative survey scans or neutral loss could be implemented for metabolite identification by finding precursor ions with characteristic fragment ions corresponding to the drug of interest; such a workflow would be beneficial for new linker-drug combinations in which the released product is unknown. These workflows could help identify various toxin containing components (e.g., linker + drug or attachment + linker + drug) that could additionally be liberated in plasma. Further, determination of the warhead fate after plasma incubation could serve to highlight important characteristics of the ADC, including cysteine switching to albumin proteins.24 A possible procedure would be to measure ADC-associated warhead, the warhead in the soluble fraction, and the warhead in the protein fraction. These data together could be used to determine a “mass balance” of drug in the system. One could also add in a hydrophobic interaction chromatography measurement to determine the composition of DAR for each ADC,30 although the method does not always sufficiently separate different DAR species. In summary, these focused efforts on understanding the stability of ADCs in plasma through in vitro assessments will enable more efficient drug development processes and furthered learnings about the complex interactions between biologics and biomatrices.
Experimental Procedures
ADC Preparation
A solution of 10 mM tris(2-carboxyethyl)phosphine was added to a 10 mg/mL solution of antibody, and the reaction mixture was incubated at 37 °C for 1 h. A solution of 3.3 mM linker-warhead payload in dimethyl sulfoxide (DMSO) was added to the reduced antibody and gently mixed for 30 min. The reaction solution was washed and purified on a PD10 desalting column and then filtered through a 0.2 μm, low protein-binding 13 mm syringe-filter and stored at 4 °C.
Antibody Labeling with Fluorophore
ADCs were diluted to a target concentration of 0.5 mg/mL with pH 7.4 PBS. Aliquots of 50 μL were transferred to a 96-well plate. The ADCs were then fluorescently labeled with Alexa Fluor 488 TFP (Thermo Scientific, Waltham, MA), which reacts with the free amines present on the antibody. To enable antibody labeling, 6 μL of 1 M sodium bicarbonate was first added to the plate, followed by 3 μL of Alexa Fluor 488 TFP reconstituted to 1 mg/mL in DMSO. Samples were incubated in the dark at room temperature with shaking for 2 h. The labeling reaction was quenched with 60 μL of 0.2 M Tris–HCl pH 7.4. On average, less than one fluorophore was added to each ADC.
Sample Processing
Following labeling, 10 μL was transferred into a 384-well plate and diluted with 90 μL of PBS or plasma containing 1.8 mg/mL ethylenediaminetetraacetic acid and 10 mM sodium azide. The different plasmas (BioreclamationIVT, Baltimore, MD) used were from CD1 mice, Sprague Dawley rats, cynomolgus monkeys, and humans.
The ADC-containing buffer/plasma samples were incubated at 37 °C with 5% CO2. Samples were collected after incubation times of 0, 4, 24, 48, 72, and 144 h with a Biomek FXP Laboratory Automation Workstation (Beckman Coulter, Indianapolis, IN). The plasma plates were retrieved from the Cytomat Plate Hotel in the incubator. Five microliters from each well was transferred to two 384-well plates, with one containing 70 μL of 95:5 acetonitrile/methanol, with 50 nM carbutamide as the internal standard. Using a Multi-drop Combi Reagent Dispenser (Thermo Scientific), 20 μL of water and 5 μL of DMSO were added to the plate containing the organic solvent. Plates were spun at 3000 rpm for 5 min at 4 °C prior to further analysis.
Aggregate, Fragment, and Monomer Determination
SEC was performed on an Infinity II Bio-Inert 1260 HPLC (Agilent Technologies, Santa Clara, CA) comprised of a quaternary pump (G5611A), temperature-controlled well plate autosampler (G5667A, G1330B), column compartment (G1316C), and fluorescent detector (G1321B). A Waters SEC column (Waters Corp., Milford, MA) with a guard column was used as the stationary phase (Acquity BEH200 SEC; 30 × 4.6 mm2 and 150 × 4.6 mm2, 1.7 μm particle size, 200 Å pore size). The mobile phase was comprised of 100 mM sodium phosphate and 150 mM sodium chloride in water with the pH adjusted to 7.0. Typical peak elution was for 3.5–3.6 min at a flow rate of 0.4 mL/min, with a column temperature of 30 °C. All samples were chilled to 8 °C during analysis. Data peaks were processed with Atlas Chromatography Data System version 9.0 (Thermo Scientific) to determine the percentages of aggregate, fragment, and monomer.
Standard Curve Preparation and Method Optimization
Standard curves for each unconjugated drug were prepared in 384-well plates. Six concentration points were delivered per compound in 5 μL of DMSO. These plates were then stamped with 5 μL of matrix, 70 μL of organic solution with internal standard (described previously), and 20 μL of water. Plates were then sealed and spun at 3000 rpm for 5 min prior to MS injection.
Small molecules were diluted in 50:50 acetonitrile/water with 0.1% formic acid at a final concentration between 0.2 and 0.5 μM and tuned through DiscoveryQuant Optimize version 2.1.3. The QuickTune workflow is used to generate the tune parameters. The precursor ion scan type is set to Q3 MS, with a spectrum window width of ±0.5 amu and a high collision gas setting. The MS2 product ion will display five fragments, with a minimum fragment loss of 20 amu and a starting mass of 55 amu. Compounds were tuned in positive mode to start and then negative mode if required. The settings consisted of a precursor threshold of 1 × 105 counts/s and fragment threshold of 1 × 104 counts/s, with a mass range between 50 and 1200 amu.
The MRM methods were made using DiscoveryQuant Analyze version 2.1.3 (Sciex). Each method consisted of one transition for the analyte of interest and a transition for the carbutamide internal standard. The MMAE transition had a Q1 mass of 718.7, a Q3 mass of 686.5, and a collision energy setting of 42 V. The Q1 mass for carbutamide was 272.2 Da, the Q3 mass was 108.1 Da, and collision energy was 35 V.
LC–MS Analysis
Measurements of unconjugated drug concentrations were performed with a 5500 Qtrap (Sciex) mass spectrometer coupled to an Agilent 1290 LC system with a CTC PAL autosampler. A Kinetex 5 μm 100 Å C18, 30 × 2.1 mm2 column was used with a 0.9 min LC gradient. The mobile phase A was composed of high-performance liquid chromatography (HPLC)-grade water with 0.1% formic acid, and mobile phase B consisted of 0.1% formic acid in HPLC-grade acetonitrile. The starting gradient conditions were 95% solvent A and 5% solvent B, with a steep ramp to 98% solvent B by 0.3 min. The high percent solvent B was held for 0.4 min, then quickly returned to starting conditions for column equilibration prior to the next injection.
Data was processed in Sound Review Analytics software version 2.2.0.8688 (Sound Analytics), which allows for autointegration of the peaks. Standard curves were fit with a quadratic regression, with 1/x weighting. The integration algorithm uses “IntelliQuan” with a baseline subtraction window of 1 min, a noise percentage of 10%, a peak splitting factor equal to 10, a minimum width of 0 s, and a minimum height of 1 × 104 counts/s.
Integrated peak data was imported into IDBS E-Workbook Suite 9.1.0. A Biobook template automatically calculated the concentration of warhead at each time point, and then the corresponding percentage of release compared to the total amount of warhead in the system was calculated.
Postprocessing Data Analysis
Aggregation and drug release data were fit using GraphPad Prism 5.0 for Windows. Nonlinear regressions, defined in eqs 1 and 2, were used after making custom analysis equations in Prism. Aequilibrium and Requilibrium were constrained with maximum values of 100, and curves were fit using a maximum of 1 × 103 iterations. Simulations of drug stability and measured warhead release were performed in Matlab SimBiology 5.2.
Acknowledgments
We would like to thank the members of the AbbVie DMPK department for their support. In particular, we would like to thank Trish Stuart for early work on the development of the high-throughput assay, the AbbVie Oncology Chemistry department for preparation of the ADCs, Lance Heinle and Mary Larsen for MS assistance, Donna Montavon and Shelley Kaliszewski for aggregation work, and Cory Kalvass for helpful discussions. All authors are employees of AbbVie. The design, study conduct, and financial support for this research were provided by AbbVie. AbbVie participated in the interpretation of data, review, and approval of the publication.
Glossary
Abbreviations
- ADC
antibody–drug conjugate
- DAR
drug-to-antibody ratio
- SEC
size exclusion chromatography
- MS
mass spectrometry
- LC–MS/MS
liquid chromatography–tandem mass spectrometry
- MMAE
monomethylauristatin E
- vc–MMAE
maleimide–caproyl–valine–citrulline–PABC–MMAE
- HMWS
high-molecular-weight species
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b00452.
Supplemental figures as described in the text included (PDF)
The authors declare the following competing financial interest(s): All authors are employees of AbbVie.
Supplementary Material
References
- Schwartz R. S. Paul Ehrlich’s magic bullets. N. Engl. J. Med. 2004, 350, 1079–1080. 10.1056/NEJMp048021. [DOI] [PubMed] [Google Scholar]
- Wu A. M.; Senter P. D. Arming antibodies: prospects and challenges for immunoconjugates. Nat. Biotechnol. 2005, 23, 1137–1146. 10.1038/nbt1141. [DOI] [PubMed] [Google Scholar]
- Alley S. C.; Okeley N. M.; Senter P. D. Antibody-drug conjugates: targeted drug delivery for cancer. Curr. Opin. Chem. Biol. 2010, 14, 529–537. 10.1016/j.cbpa.2010.06.170. [DOI] [PubMed] [Google Scholar]
- Ritchie M.; Tchistiakova L.; Scott N. Implications of receptor-mediated endocytosis and intracellular trafficking dynamics in the development of antibody drug conjugates. mAbs 2013, 5, 13–21. 10.4161/mabs.22854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doronina S. O.; Mendelsohn B. A.; Bovee T. D.; Cerveny C. G.; Alley S. C.; Meyer D. L.; Oflazoglu E.; Toki B. E.; Sanderson R. J.; Zabinski R. F.; Wahl A. F.; Senter P. D. Enhanced activity of monomethylauristatin F through monoclonal antibody delivery: effects of linker technology on efficacy and toxicity. Bioconjugate Chem. 2006, 17, 114–124. 10.1021/bc0502917. [DOI] [PubMed] [Google Scholar]
- Adair J. R.; Howard P. W.; Hartley J. A.; Williams D. G.; Chester K. A. Antibody-drug conjugates - a perfect synergy. Expert Opin. Biol. Ther. 2012, 12, 1191–1206. 10.1517/14712598.2012.693473. [DOI] [PubMed] [Google Scholar]
- Saber H.; Leighton J. K. An FDA oncology analysis of antibody-drug conjugates. Regul. Toxicol. Pharmacol. 2015, 71, 444–452. 10.1016/j.yrtph.2015.01.014. [DOI] [PubMed] [Google Scholar]
- Doronina S. O.; Toki B. E.; Torgov M. Y.; Mendelsohn B. A.; Cerveny C. G.; Chace D. F.; DeBlanc R. L.; Gearing R. P.; Bovee T. D.; Siegall C. B.; Francisco J. A.; Wahl A. F.; Meyer D. L.; Senter P. D. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat. Biotechnol. 2003, 21, 778–784. 10.1038/nbt832. [DOI] [PubMed] [Google Scholar]
- Dorywalska M.; Dushin R.; Moine L.; Farias S. E.; Zhou D.; Navaratnam T.; Lui V.; Hasa-Moreno A.; Casas M. G.; Tran T. T.; Delaria K.; Liu S. H.; Foletti D.; O’Donnell C. J.; Pons J.; Shelton D. L.; Rajpal A.; Strop P. Molecular Basis of Valine-Citrulline-PABC Linker Instability in Site-Specific ADCs and Its Mitigation by Linker Design. Mol. Cancer Ther. 2016, 15, 958–970. 10.1158/1535-7163.MCT-15-1004. [DOI] [PubMed] [Google Scholar]
- Jeffrey S. C.; Burke P. J.; Lyon R. P.; Meyer D. W.; Sussman D.; Anderson M.; Hunter J. H.; Leiske C. I.; Miyamoto J. B.; Nicholas N. D.; Okeley N. M.; Sanderson R. J.; Stone I. J.; Zeng W.; Gregson S. J.; Masterson L.; Tiberghien A. C.; Howard P. W.; Thurston D. E.; Law C. L.; Senter P. D. A potent anti-CD70 antibody-drug conjugate combining a dimeric pyrrolobenzodiazepine drug with site-specific conjugation technology. Bioconjugate Chem. 2013, 24, 1256–1263. 10.1021/bc400217g. [DOI] [PubMed] [Google Scholar]
- Ducry L.; Stump B. Antibody-drug conjugates: linking cytotoxic payloads to monoclonal antibodies. Bioconjugate Chem. 2010, 21, 5–13. 10.1021/bc9002019. [DOI] [PubMed] [Google Scholar]
- Jain N.; Smith S. W.; Ghone S.; Tomczuk B. Current ADC Linker Chemistry. Pharm. Res. 2015, 32, 3526–3540. 10.1007/s11095-015-1657-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokter W.; Ubink R.; van der Lee M.; van der Vleuten M.; van Achterberg T.; Jacobs D.; Loosveld E.; van den Dobbelsteen D.; Egging D.; Mattaar E.; Groothuis P.; Beusker P.; Coumans R.; Elgersma R.; Menge W.; Joosten J.; Spijker H.; Huijbregts T.; de Groot V.; Eppink M.; de Roo G.; Verheijden G.; Timmers M. Preclinical profile of the HER2-targeting ADC SYD983/SYD985: introduction of a new duocarmycin-based linker-drug platform. Mol. Cancer Ther. 2014, 13, 2618–2629. 10.1158/1535-7163.MCT-14-0040-T. [DOI] [PubMed] [Google Scholar]
- Wang H.; Rangan V. S.; Sung M. C.; Passmore D.; Kempe T.; Wang X.; Thevanayagam L.; Pan C.; Rao C.; Srinivasan M.; Zhang Q.; Gangwar S.; Deshpande S.; Cardarelli P.; Marathe P.; Yang Z. Pharmacokinetic characterization of BMS-936561, an anti-CD70 antibody-drug conjugate, in preclinical animal species and prediction of its pharmacokinetics in humans. Biopharm. Drug Dispos. 2016, 37, 93–106. 10.1002/bdd.1953. [DOI] [PubMed] [Google Scholar]
- Kraynov E.; Kamath A. V.; Walles M.; Tarcsa E.; Deslandes A.; Iyer R. A.; Datta-Mannan A.; Sriraman P.; Bairlein M.; Yang J. J.; Barfield M.; Xiao G.; Escandon E.; Wang W.; Rock D. A.; Chemuturi N. V.; Moore D. J. Current Approaches for Absorption, Distribution, Metabolism, and Excretion Characterization of Antibody-Drug Conjugates: An Industry White Paper. Drug Metab. Dispos. 2016, 44, 617–623. 10.1124/dmd.115.068049. [DOI] [PubMed] [Google Scholar]
- Francisco J. A.; Cerveny C. G.; Meyer D. L.; Mixan B. J.; Klussman K.; Chace D. F.; Rejniak S. X.; Gordon K. A.; DeBlanc R.; Toki B. E.; Law C. L.; Doronina S. O.; Siegall C. B.; Senter P. D.; Wahl A. F. cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 2003, 102, 1458–1465. 10.1182/blood-2003-01-0039. [DOI] [PubMed] [Google Scholar]
- Boswell C. A.; Mundo E. E.; Zhang C.; Bumbaca D.; Valle N. R.; Kozak K. R.; Fourie A.; Chuh J.; Koppada N.; Saad O.; Gill H.; Shen B. Q.; Rubinfeld B.; Tibbitts J.; Kaur S.; Theil F. P.; Fielder P. J.; Khawli L. A.; Lin K. Impact of drug conjugation on pharmacokinetics and tissue distribution of anti-STEAP1 antibody-drug conjugates in rats. Bioconjugate Chem. 2011, 22, 1994–2004. 10.1021/bc200212a. [DOI] [PubMed] [Google Scholar]
- Arvinte T.; Palais C.; Green-Trexler E.; Gregory S.; Mach H.; Narasimhan C.; Shameem M. Aggregation of biopharmaceuticals in human plasma and human serum: implications for drug research and development. mAbs 2013, 5, 491–500. 10.4161/mabs.24245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singla A.; Bansal R.; Joshi V.; Rathore A. S. Aggregation Kinetics for IgG1-Based Monoclonal Antibody Therapeutics. AAPS J. 2016, 18, 689–702. 10.1208/s12248-016-9887-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckley N. S.; Lazzareschi K. P.; Chih H. W.; Sharma V. K.; Flores H. L. Investigation into temperature-induced aggregation of an antibody drug conjugate. Bioconjugate Chem. 2013, 24, 1674–1683. 10.1021/bc400182x. [DOI] [PubMed] [Google Scholar]
- Guo J.; Kumar S.; Chipley M.; Marcq O.; Gupta D.; Jin Z.; Tomar D. S.; Swabowski C.; Smith J.; Starkey J. A.; Singh S. K. Characterization and Higher-Order Structure Assessment of an Interchain Cysteine-Based ADC: Impact of Drug Loading and Distribution on the Mechanism of Aggregation. Bioconjugate Chem. 2016, 27, 604–615. 10.1021/acs.bioconjchem.5b00603. [DOI] [PubMed] [Google Scholar]
- Hamblett K. J.; Senter P. D.; Chace D. F.; Sun M. M.; Lenox J.; Cerveny C. G.; Kissler K. M.; Bernhardt S. X.; Kopcha A. K.; Zabinski R. F.; Meyer D. L.; Francisco J. A. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. 10.1158/1078-0432.CCR-04-0789. [DOI] [PubMed] [Google Scholar]
- Lyon R. P.; Bovee T. D.; Doronina S. O.; Burke P. J.; Hunter J. H.; Neff-LaFord H. D.; Jonas M.; Anderson M. E.; Setter J. R.; Senter P. D. Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat. Biotechnol. 2015, 33, 733–735. 10.1038/nbt.3212. [DOI] [PubMed] [Google Scholar]
- Kay R.; Barton C.; Ratcliffe L.; Matharoo-Ball B.; Brown P.; Roberts J.; Teale P.; Creaser C. Enrichment of low molecular weight serum proteins using acetonitrile precipitation for mass spectrometry based proteomic analysis. Rapid Commun. Mass Spectrom. 2008, 22, 3255–3260. 10.1002/rcm.3729. [DOI] [PubMed] [Google Scholar]
- Polson C.; Sarkar P.; Incledon B.; Raguvaran V.; Grant R. Optimization of protein precipitation based upon effectiveness of protein removal and ionization effect in liquid chromatography-tandem mass spectrometry. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2003, 785, 263–275. 10.1016/S1570-0232(02)00914-5. [DOI] [PubMed] [Google Scholar]
- Alley S. C.; Benjamin D. R.; Jeffrey S. C.; Okeley N. M.; Meyer D. L.; Sanderson R. J.; Senter P. D. Contribution of linker stability to the activities of anticancer immunoconjugates. Bioconjugate Chem. 2008, 19, 759–765. 10.1021/bc7004329. [DOI] [PubMed] [Google Scholar]
- Lyon R. P.; Setter J. R.; Bovee T. D.; Doronina S. O.; Hunter J. H.; Anderson M. E.; Balasubramanian C. L.; Duniho S. M.; Leiske C. I.; Li F.; Senter P. D. Self-hydrolyzing maleimides improve the stability and pharmacological properties of antibody-drug conjugates. Nat. Biotechnol. 2014, 32, 1059–1062. 10.1038/nbt.2968. [DOI] [PubMed] [Google Scholar]
- Hermeling S.; Crommelin D. J.; Schellekens H.; Jiskoot W. Structure-immunogenicity relationships of therapeutic proteins. Pharm. Res. 2004, 21, 897–903. 10.1023/B:PHAM.0000029275.41323.a6. [DOI] [PubMed] [Google Scholar]
- Rosenberg A. S. Effects of protein aggregates: an immunologic perspective. AAPS J. 2006, 8, E501–E507. 10.1208/aapsj080359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakankar A.; Chen Y.; Gokarn Y.; Jacobson F. S. Analytical methods for physicochemical characterization of antibody drug conjugates. mAbs 2011, 3, 161–172. 10.4161/mabs.3.2.14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.