

Abstract
Infections caused by multidrug-resistant (MDR) bacteria are a rapidly growing threat to human health, in many cases exacerbated by their presence in biofilms. We report here a biocompatible oil-in-water crosslinked polymeric nanocomposite that degrades in the presence of physiologically relevant biomolecules. These degradable nanocomposites demonstrated broad-spectrum penetration and elimination of MDR bacteria, eliminating biofilms with no toxicity to co-cultured mammalian fibroblast cells. Notably, serial passaging revealed that bacteria were unable to develop resistance towards these nanocomposites, highlighting the therapeutic promise of this platform.
Keywords: Nanocomposite, Crosslinked, Nanoemulsion, Multidrug-Resistance, Biodegradable
Table of Contents
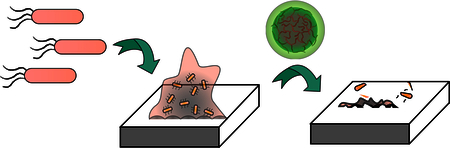
Introduction
Multidrug-resistant (MDR) bacteria infect more than two million people annually in the U.S., resulting in significant loss of life and limb, with treatment requiring prolonged and costly therapeutic regimens. 1,2,3 These dangerous pathogens, including P. aeruginosa and S. aureus, further frustrate treatment due to their innate ability to micro-colonize into biofilms. 4,5,6 Bacterial biofilm infections are challenging to treat on wounds and indwelling medical devices, as the extracellular polymeric substances (EPS) in these biofilms both inhibit antibiotic penetration and aid bacteria to evade host immune responses. 7,8,9 Furthermore, the slow growth rate of bacteria and the biofilm microenvironment act together to facilitate the development of antibiotic resistance. 10,11,12 The emerging threat of antibiotic resistant biofilm infections has triggered an international push to develop alternative therapeutic platforms capable of eliminating these infections. 13,14
Plant-derived phytochemicals have emerged as an alternative to traditional antibiotic paradigms to combat MDR bacteria, 15,16,17,18,19 providing a potential strategy for avoiding antibiotic tolerance and horizontal gene transfer that dramatically accelerate acquisition of drug resistance. 20,21 Phytochemicals feature low cost 22, biocompatibility 23, and can be effective towards bacterial infections. 24 However, the poor solubility of these hydrophobic oils in aqueous media limit their practical application as antimicrobial agents. 25 Surfactant, nanoparticle, and polymer additives aid in phytochemical delivery by forming oil-in-water emulsions. 26,27,28,29 Furthermore, crosslinking of these emulsions have demonstrated phytochemical stability in even complex media such as serum. 30 However, such crosslinking strategies are non-biodegradable and may persist and accumulate within the body, causing unwanted side effects, such as inflammation and carcinogenesis. 31,32,33
Here, we report crosslinked poly(oxanorborneneimide)-stabilized oil-in-water nanocomposites (X-BNCs) engineered to be biodegradeable in the presence of endogenous biomolecules such as glutathione and esterase enzymes (Scheme 1). These ‘nanosponges’ incorporate disulfide 34 and ester 35,36 crosslinkers that provide long-term stability in aqueous environments while facilitating nanocomposite degradation in biological milieus. We demonstrate the loading of these X-BNCs with carvacrol to provide therapeutics that eradicate Gram negative/positive bacteria including MDR strains. These nanocomposites are stable in serum-containing media, however degrade rapidly in the presence of glutathione or esterase proteins. The potential of X-BNCs as a wound healing therapeutic was demonstrated in an in vitro coculture with biofilms grown on top of mammalian 3T3 fibroblast cells. A 4-log reduction in bacterial colonies was observed with no change in fibroblast cells viability. In stark contrast to antibiotics, X-BNCs do not evoke resistance in bacteria, maintaining their potency against pathogenic E. coli (CD-2) in a 20-cycle serial passage study. Taken together, the efficacy, biodegradability, and stability of these anti-biofilm agents coupled with their lack of resistance accumulation make them a promising therapeutic platform to combat the rising dangers of MDR bacterial infections.
Scheme 1.

(a) Crosslinked PONI-GMT–DTDS structure showing linkage points reactive to endogenous biomolecules. (b) DLS histogram of crosslinked PONI-GMT–DTDS nanosponges loaded with carvacrol. in phosphate buffer saline (150 mM). (c) Chemical structures of PONI-GMT and (d) DTDS. (e) Confocal micrograph of crosslinked micron-sized biodegradable composites. PONI-GMT labeled with TAMRA-X (red fluorescence), and the oil core is loaded with DiO (green fluorescence). A composite morphology is indicated by co-distribution of PONI-GMT with the hydrophobic oil core. Scale bar is 3µm.
Results and Discussion
Generation and Characterization of Nanocomposites.
The X-BNC platform uses a poly(oxanorborneneimide) scaffold bearing guanidine, maleimide, and tetraethyleneglycol monomethyl ether groups (PONI-GMT) and provides a well-controlled and scalable platform. 30 Biodegradability was imparted through use of a dithiol-disulfide (DTDS) crosslinker that is stable > 2 years in storage. (Supporting Information, Synthesis of DTDS). An additional degradation modality was provided using ester-linked maleimide groups to enable thiols of DTDS to crosslink rapidly once the polymers assemble at the oil-water interface. 37 Copolymerization of monomers bearing guanidine, maleimido, and tetreaethylene glycol monomethyl ether units at a 40:10:50 monomer ratio respectively provided the precursor polymer PONI-GMT (Supporting Information, Synthesis of PONI-GMT). The maleimido monomer ratio was kept low to ensure adequate solubility of PONI-GMT in aqueous conditions, necessary for efficient nanocomposite formation.
Nanocomposites were fabricated by emulsifying carvacrol oil loaded with DTDS or carvacrol only (non-crosslinked control, NX-NC) into water. Upon emulsification, PONI-GMT assembles and initially stabilizes the oil-water interface. In the presence of DTDS, crosslinking further stabilizing the oil droplets in water. PONI-GMT and DTDS generated nanocomposites (X-BNCs) with a diameter of ~220nm as shown by dynamic light scattering (DLS). We hypothesized the overall charge of X-BNCs would be reversed yielding a positively charged surface. The measured zeta (ζ) potential (Supporting Information, ζ potential of X-BNCs) supported this prediction, reporting a cationic nanocomposite, attributed to guanidine units at or beyond the oil-water interface. Significantly, DLS experiments on stock solutions of X-BNCs that were stored on the bench for one year indicated the composites maintained stability with a minimal change in size (Supporting Information, Long-Term Storage of X-BNCs).
Next, the morphology (core-shell versus nanocomposites) of X-BNCs was established through confocal microscopy of larger micron-sized analogs of the X-BNC nanoemulsions. TAMRA-X (red fluorescence) was conjugated to PONI-GMT (Supporting Information, Synthesis of TAMRA-PONI-GMT), while 3,3-dioctadecyloxacarbocyanine (DiO, green fluorescence) was loaded within the oil. As shown in Scheme 1e, both green and red fluorescence were co-distributed across the microparticle, indicating the morphology adopts a composite “sponge” architecture. Given that previous reports have observed norbornene-based polymers (Mn’s ~ 100,000 g/mol) adopt length scales of ~ 40 nanometers, 38 it is reasonable to suggest that PONI-GMT would exist in a composite morphology in the X-BNCs nanoemulsions. This notion is further supported in literature as carvacrol and other phytochemicals are miscible with glycols, 39 such as the high density of tetraethylene glycol units found on PONI-GMT.
Stability and Degradability of X-BNCs.
Macromolecular vehicles need to be stable to deliver therapeutic payloads yet degrade to avoid vehicle accumulation over time. 40 After characterizing the morphology of X-BNCs, we next probed the colloidal stability of the composites via monitoring particle size by dynamic light scattering (DLS). 41 As shown in Figure 1a, when X-BNCs were incubated with 10% serum media for 2 hours, an increase in X-BNCs size (~25 nm) was observed, suggesting negatively charged serum proteins adsorb onto the positively charged X-BNCs surface. Notably, no evidence of X-BNC destabilization/aggregation was observed even at longer incubation times (e.g. 6 hours). However, as a control, non-crosslinked analogues using the same formulation minus DTDS showed essentially no stability in serum (Supporting Information, Serum Stability of NX-NCs).
Figure 1.

Stability and degradability of X-BNCs. DLS size distribution changes of X-BNCs when incubated with (a) 10% FBS media for two hours or (b) physiologically relevant biomolecules (glutathione and lipase) in PBS, showing degradation from disulfide cleavage and hydrolysis.
We next explored the degradability of X-BNCs in the presence of glutathione (GSH) and the ester-hydrolyzing enzyme porcine liver esterase (PLE). Using physiologically relevant concentrations of both biomolecules, X-BNCs were incubated for 24 hours with either 10mM GSH or 35 µM PLE (1 U/µL) with PBS as a control. Figure 1b shows the size of X-BNCs remained the same after 24 hours in PBS, however the size increased significantly in GSH/PLE solutions, indicating degradation of the nanocomposites structure with concomitant generation of agglomerated structures through an Ostwald ripening-like process. Taken together, the results indicate the crosslinked composite framework within X-BNCs are robust in serum yet biodegrade in the presence of chosen biological environments.
Antimicrobial Activity of X-BNCs Against Biofilms.
We focused our antimicrobial efforts on highly refractory biofilms, where the efficacy of traditional antibiotic therapy is significantly compromised relative to planktonic pathogens. 42 We investigated the ability of X-BNCs to penetrate EPS followed by quantitative analysis of their therapeutic efficacy towards enclosed pathogenic bacteria. X-BNC penetration into biofilms formed by red fluorescent protein (RFP)-expressing E. coli was tracked by loading 3,3-dioctadecyloxacarbocyanine (DiO, green fluorescence) within the oil core. As shown in Figure 2, X-BNCs readily penetrate and diffuse throughout the biofilm, with fluorescence colocalizing with enclosed bacteria. The data demonstrates X-BNCs deliver their payload efficiently, reaching enclosed pathogens deep within the films matrix.
Figure 2.

Confocal image stacks and penetration profile of E. coli DH5α biofilm after 1-hour treatment with X-BNCs loaded with DiO. Scale bars are 40µm and are not representative of the biofilm depth. Each projected z-stack image (a) is spaced by 1.3 µm at a 5o angle from the biofilms x-plane. Both the overlay and biofilm depth fluorescence graph (b) indicates X-BNCs completely penetrates the biofilm, colocalizing with bacteria that expresses red fluorescent protein.
Next, the antimicrobial activity of X-BNCs against multiple pathogenic Gram-negative and Gram-positive biofilms were evaluated. Four pathogenic bacterial strains of clinical isolates, Staphylococcus aureus (CD-489, a methicillin-resistant strain), Pseudomonas aeruginosa (CD-1006), Escherichia coli (CD-2), and Enterobacter cloacae (E. cloacae, CD-1412) complex were chosen to be tested as their associated infections are typically common in hospital-related settings. 43,44 As shown in Figure 3, X-BNCs effectively eliminated bacterial cells in all four-biofilm species within 3 hours. Notably, both Gram positive (S. aureus) and Gram negative (P. aeruginosa, E. coli, and E. cloacae complex) bacteria can be treated with X-BNCs, highlighting their broad-spectrum activity even in a biofilm matrix setting.
Figure 3.

Viability of one-day-old Gram-negative/positive biofilms after a three-hour treatment with X-BNCs and the individual components as controls at different emulsion concentrations (Displayed as mM and v/v % of emulsion). The results shown are averaged triplicates, and the error bars indicate the standard deviation.
Selective Killing of Biofilms in a Coculture Model.
Beyond treating biofilms on surfaces, eliminating these bacterial infections on human tissue and organs is a greater challenge and more relevant to patients who suffer from skin ulcers, burn injuries, or wound trauma. A fundamental issue associated with these infections is their ability to interfere with the host’s tissue regeneration process. We evaluated the efficacy of X-BNCs as a topical treatment by using an in vitro co-culture model comprised of a biofilm grown on top of mammalian fibroblasts. P. aeruginosa and NIH 3T3 fibroblast cells were selected to build this co-culture model, since P. aeruginosa is widely associated with skin infections and fibroblast cells are critical during wound healing. 45,46,47 The co-cultures were treated with X-BNCs for 3 hours, washed, and the viabilities of bacteria and fibroblast cells were determined. As shown in Figure 4, a 4-fold log reduction (~99.5%) in biofilm colonies was observed at a X-BNCs concentration of 16 v/v %., while 3T3 fibroblast viability remained uncompromised. Significantly, little change in fibroblast viability was observed at 32 v/v %, where a 6-log unit reduction in bacteria was observed. This selective toxicity to biofilm bacteria makes the X-BNC platform promising for addressing wound biofilms.
Figure 4.

Viability of 3T3 fibroblast cells and P. aeruginosa biofilms in the coculture model after treating X-BNCs at different emulsion concentrations for three hours. 3T3 fibroblast cell viabilities are shown as a line. Bar plots represent log10 of colony-forming bacteria units in biofilms. Each result is an average of three experiments, and the error bars designate the standard deviations.
Bacterial Resistance Towards Antibiotics Vs. X-BNCs.
The number of antibiotic-resistant bacteria and their associated infections is increasing globally, leading to cases where infections have become untreatable. Although efforts to discover novel antibiotic classes to slow the progression of antibiotic resistance is ongoing, 48 developing alternative therapeutic platforms where bacteria cannot develop resistance towards must take precedence. We hypothesized that the membrane disruption induced by the antimicrobial phytochemical payload of X-BNCs would sidestep normal bacterial defense adaptations, preventing accumulated resistance. We tested this hypothesis by subjecting planktonic bacteria to X-BNCs and a negative control antibiotic (ciprofloxacin, enzyme inhibitor) in the presence of propidium iodide (PI, Supporting Information, Membrane Disruption Study). PI is not permeant to live bacterial cells, however easily diffuses through membrane-compromised cells, generating fluorescence upon intercalating with DNA. X-BNC treatment quickly generated PI fluorescence, indicating their action mechanism compromises bacterial membrane integrity. Ciprofloxacin is an enzyme inhibitor antibiotic and therefore does not act on bacterial membranes, generating no observed fluorescence. Next, pathogenic E. coli (CD-2) was passaged in the presence of X-BNCs, or three commercially available antibiotics, ciprofloxacin (quinolone class), ceftazidime (β-lactam class), and tetracycline (tetracycline class). Briefly, we subjected the bacteria to the sub-minimum inhibitory concentrations (66% of MIC) of the antimicrobial agent. The resulting bacterial populations for each individual therapeutic was defined as the first passage, harvested, and their respective MICs evaluated. The subsequent passage was derived by exposing the previous passage with the 66% MIC of each respective therapeutic dosage. As shown in Figure 5a, no resistance was generated towards X-BNCs even after 20 serial passages (~1,300 bacterial generations). Meanwhile, E. coli rapidly developed resistance towards each of the antibiotics, with respective MIC increases of 33,000, 4,200, and 256-fold where the drugs reached their solubility limit in media. Going further, biofilms were grown with the 20th serial passage and subjected to X-BNCs for 3 hours, where CD-2 E. coli was still susceptible to nanoemulsion treatment (Figure 5b). These results indicate the mechanism of X-BNCs mitigates the onset of resistance in both planktonic and biofilm settings.
Figure 5.

Accumulated resistance of pathogenic E. coli (CD-2) in both plankton and biofilm settings. (a) Resistance development of planktonic species during serial passaging in the presence of sub-MIC dosing’s of antimicrobials. The y-axis indicates the increase in dosage as compared to the initial bacterial cells (0th passage) and the figure is representative of three independent experiments. (b) Derived E. coli cells from 20 serial passages of sub-MIC X-BNCs dosing was grown into a biofilm and subjected to a three-hour treatment of X-BNCs at different emulsion concentrations. The results indicate that evolved pathogenic E. coli remain susceptible to X-BNCs.
Conclusion
In summary, we report the construction, characterization, and antimicrobial potential of a biodegradable crosslinked polymer-stabilized oil-in-water nanocomposite. These nanoemulsions maintain stability in serum yet degrade in the presence of selected biomolecules, a necessary attribute to avoid vehicle accumulation over time. Furthermore, the nanocomposites are highly effective against both Gram negative and positive bacteria biofilms, with no observed toxicity to mammalian fibroblast cells. In stark contrast to traditional antibiotics, bacteria were unable to accumulate resistance towards our nanoemulsions whether the bacteria were planktonic or in biofilms. The therapeutic polymer-based phytochemical nanoemulsion we present is a highly promising antimicrobial platform with the potential to impact treatment of wound biofilms and other difficult bacterial infections.
Acknowledgements
Support from the NIH (GM077173) and the Manning Innovation Fund is acknowledged. Clinical samples obtained from the Cooley Dickinson Hospital Microbiology Laboratory (Northampton, MA) were kindly provided by Dr. Margaret Riley. The microscopy data was gathered in the Light Microscopy Facility and Nikon Center of Excellence at the Institute for Applied Life Sciences, UMass Amherst with support from the Massachusetts Life Sciences Center.
Footnotes
Supporting Information
Experimental procedures, biofilm and co-culture formation, charge – stability – storage of X-BNCs.
References
- (1).Nathwani D; Raman G; Sulham K; Gavaghan M; Menon V Antimicrob. Resist. Infect. Control 2014, 3, 32–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Morales E; Cots F; Sala M; Comas M; Belvis F; Riu M; Salvadó M; Grau S; Horcajada JP; Montero MM; Castells X BMC Health Serv. Res 2012, 12, 122–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Tam VH; Rogers CA; Chang K-T; Weston JS; Caeiro J-P; Garey KW Antimicrob. Agents Chemother 2010, 54, 3717–3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Drenkard E; Ausubel FM Nature 2002, 416, 740–743. [DOI] [PubMed] [Google Scholar]
- (5).Schierle CF; De la Garza M; Mustoe TA; Galiano RD Wound Repair Regen. 2009, 17, 354–359. [DOI] [PubMed] [Google Scholar]
- (6).Sanchez CJ; Mende K; Beckius ML; Akers KS; Romano DR; Wenke JC; Murray CK BMC Infect. Dis 2013, 13, 47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Wu H; Moser C; Wang H; Høiby N; Song Z-J; Hoiby N; Song Z-J Int. J. Oral Sci 2014, 7, 1–7. [Google Scholar]
- (8).Anderl JN; Franklin MJ; Stewart PS Antimicrob. Agents Chemother 2000, 44, 1818–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Tseng BS; Zhang W; Harrison JJ; Quach TP; Song JL; Penterman J; Singh PK; Chopp DL; Packman AI; Parsek MR Environ. Microbiol 2013, 15, 2865–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Szomolay B; Klapper I; Dockery J; Stewart PS Environ. Microbiol 2005, 7, 1186–1191. [DOI] [PubMed] [Google Scholar]
- (11).Walters MC; Roe F; Bugnicourt A; Franklin MJ; Stewart PS Antimicrob. Agents Chemother 2003, 47, 317–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Olsen I Eur. J. Clin. Microbiol. Infect. Dis 2015, 34, 877–886. [DOI] [PubMed] [Google Scholar]
- (13).Stewart PS; William Costerton J Lancet 2001, 358, 135–138. [DOI] [PubMed] [Google Scholar]
- (14).del Pozo JL; Patel R Clin. Pharmacol. Ther 2007, 82, 204–209. [DOI] [PubMed] [Google Scholar]
- (15).Cowan MM Clin. Microbiol. Rev 1999, 12, 564–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Djeussi DE; Noumedem JAK; Seukep JA; Fankam AG; Voukeng IK; Tankeo SB; Nkuete AHL; Kuete V BMC Complement. Altern. Med 2013, 13, 164–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Monte J; Abreu CA; Borges A; Simões CL; Simões M Pathogens 2014, 3, 473–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ahmad I; Beg AZ J. Ethnopharmacol 2001, 74, 113–123. [DOI] [PubMed] [Google Scholar]
- (19).Simoes M; Bennett RN; Rosa EAS; Simões M; Bennett RN; Rosa EAS Nat. Prod. Rep 2009, 26, 746–757. [DOI] [PubMed] [Google Scholar]
- (20).O’Connell KMG; Hodgkinson JT; Sore HF; Welch M; Salmond GPC; Spring DR Angew. Chemie Int. Ed 2013, 52, 10706–10733. [DOI] [PubMed] [Google Scholar]
- (21).Madsen JS; Burmølle M; Hansen LH; Sørensen SJ FEMS Immunol. Med. Microbiol 2012, 65, 183–195. [DOI] [PubMed] [Google Scholar]
- (22).Maia MF; Moore SJ Malar. J 2011, 10, S11–S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Freire Rocha Caldas G; Araújo AV; Albuquerque GS; Silva-Neto J. da C.; Costa-Silva JH; de Menezes IRA; Leite ACL; da Costa JGM; Wanderley AG Evidence-Based Complement. Altern. Med 2013, 2013, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Nazzaro F; Fratianni F; De Martino L; Coppola R; De Feo V Pharmaceuticals 2013, 6, 1451–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Chen H; Davidson PM; Zhong Q Appl. Environ. Microbiol 2013, 80, 907–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Chang Y; McLandsborough L; McClements DJ J. Agric. Food Chem 2013, 61, 8906–8913. [DOI] [PubMed] [Google Scholar]
- (27).Anwer MK; Jamil S; Ibnouf EO; Shakeel FJ Oleo Sci. 2014, 63, 347–354. [DOI] [PubMed] [Google Scholar]
- (28).Anwer MK; Jamil S; Ibnouf EO; Shakeel F J. Oleo Sci. 2014, 63, 347–354. [DOI] [PubMed] [Google Scholar]
- (29).Donsì F; Annunziata M; Vincensi M; Ferrari G J. Biotechnol. 2012, 159, 342–350. [DOI] [PubMed] [Google Scholar]
- (30).Landis RF; Gupta A; Lee Y-W; Wang L-S; Golba B; Couillaud B; Ridolfo R; Das R; Rotello VM ACS Nano 2017, 11, 946–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Chandra Sekhara Rao G; Satish Kumar M; Mathivanan N; Bhanoji Rao ME Pharmazie 2004, 59, 5–9. [PubMed] [Google Scholar]
- (32).Kamaly N; Yameen B; Wu J; Farokhzad OC Chem. Rev 2016, 116, 2602–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Simone EA; Dziubla TD; Muzykantov VR; Eric A Simone, Thomas D Dziubla, V. R. M Expert Opin. Drug Deliv. 2008, 5, 1283–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Cheng R; Feng F; Meng F; Deng C; Feijen J; Zhong Z J. Control. Release 2011, 152, 2–12. [DOI] [PubMed] [Google Scholar]
- (35).Sanson C; Schatz C; Le Meins J-F; Brûlet A; Soum A; Lecommandoux S Langmuir 2010, 26, 2751–2760. [DOI] [PubMed] [Google Scholar]
- (36).Yang J; Liu F; Yang L; Li S Eur. Polym. J 2010, 46, 783–791. [Google Scholar]
- (37).Pounder RJ; Stanford MJ; Brooks P; Richards SP; Dove AP Chem. Commun 2008, 0, 5158–5160. [DOI] [PubMed] [Google Scholar]
- (38).Sukegawa T; Masuko I; Oyaizu K; Nishide H Macromolecules 2014, 47, 8611–8617. [Google Scholar]
- (39).Jovanka L; Ivana Č; Goran T; Sava P; Slavica S; Tamara C-G; Ljiljana K Rom. Biotechnol. Lett 2011, 16, 6034–6041. [Google Scholar]
- (40).Kataoka K; Harada A; Nagasaki Y Adv. Drug Deliv. Rev 2001, 47, 113–131. [DOI] [PubMed] [Google Scholar]
- (41).Gèze A; Putaux J-L; Choisnard L; Jéhan P; Wouessidjewe DJ Microencapsul. 2004, 21, 607–613. [DOI] [PubMed] [Google Scholar]
- (42).Flemming H-C; Wingender J; Szewzyk U; Steinberg P; Rice SA; Kjelleberg S Nat. Rev. Microbiol 2016, 14, 563–575. [DOI] [PubMed] [Google Scholar]
- (43).Richards MJ; Edwards JR; Culver DH; Gaynes RP Infect. Control Hosp. Epidemiol 2000, 21, 510–515. [DOI] [PubMed] [Google Scholar]
- (44).Jarvis WR; Martone WJ J. Antimicrob. Chemother 1992, 29, 19–24. [DOI] [PubMed] [Google Scholar]
- (45).Serra R; Grande R; Butrico L; Rossi A; Settimio UF; Caroleo B; Amato B; Gallelli L; de Franciscis S Expert Rev. Anti. Infect. Ther 2015, 13, 605–613. [DOI] [PubMed] [Google Scholar]
- (46).Fazli M; Bjarnsholt T; Kirketerp-Møller K; Jørgensen B; Andersen AS; Krogfelt KA; Givskov M; Tolker-Nielsen TJ Clin. Microbiol 2009, 47, 4084–4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Sun BK; Siprashvili Z; Khavari PA Science 2014, 346, 941–945. [DOI] [PubMed] [Google Scholar]
- (48).Ling LL; Schneider T; Peoples AJ; Spoering AL; Engels I; Conlon BP; Mueller A; Schaberle TF; Hughes DE; Epstein S; Jones M; Lazarides L; Steadman VA; Cohen DR; Felix CR; Fetterman KA; Millett WP; Nitti AG; Zullo AM; Chen C; Lewis K Nature 2015, 517, 455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]