

Abstract
Five enantiomeric pairs of palladium complexes of 1,2,4-triazole-derived chiral N-heterocyclic carbene ligands were investigated to probe the influence of chirality on the compound’s anticancer activity. Although no chirality-related influence was observed for any of the enantiomeric pair, strong anticancer activity was seen for a particular pair, (1S,2S,5R)-1c and (1R,2R,5S)-1c, which was significantly more active than the benchmark drug cisplatin for human breast cancer cells, MCF-7 (ca. 24–27-fold), and human cervical cancer cells, HeLa (ca. three- to fourfold). Broadening its scope of application, (1R,2R,5S)-1c also exhibited antiproliferative activity against lung cancer (A549), skin cancer (B16F10), and multidrug-resistant mammary tumor (EMT6/AR1) cell lines. Interestingly, (1R,2R,5S)-1c displayed 8- and 16-fold stronger antiproliferative activity toward B16F10 and MCF-7 relative to their respective noncancerous counterparts, L929 (fibroblast skin cells) and MCF10A (epithelial breast cells), thereby upholding the potential of these complexes for further development as anticancer agents. (1R,2R,5S)-1c inhibited tumor-cell proliferation by blocking the cells at the G2 phase. (1R,2R,5S)-1c caused DNA damage in MCF-7 cells, leading to mitochondrial reactive oxygen species production and subsequently cell death. We also present evidence indicating that (1R,2R,5S)-1c induced p53-dependent programmed cell death in MCF-7 cells.
Introduction
Cancer still remains a formidable threat to human society accounting for maximum number of deaths annually worldwide.1 Of the several strategies at disposal for battling the disease, organic2−4 and inorganic compounds5−7 having high anticancer activity play pivotal role in chemotherapy, which may be administered before or after surgery depending upon the line of the treatment.8−11 A variety of such anticancer drugs of organic genre,8,12−14 namely, taxol, podophyllotoxin, camptothecin, and so forth, and of inorganic genre,15,16 such as cis-diamineplatinum(II)dichloride (cisplatin), carboplatin, and oxaliplatin, are thus being routinely prescribed as a part of cancer treatment. However, between these two genres of anticancer drugs, the organic ones are significantly more potent than their inorganic counterparts. For example, the half maximal inhibitory concentration (IC50) range observed for taxol (2.5–4.1 nM)17 is substantially lower than that of the cisplatin (13–36 μM)18 for various cancer cell lines like MCF-7, HeLa, HT-29, and A549 under in vitro conditions.
This interesting observation triggered us to undertake the design and development of transition-metal complexes displaying high anticancer activities at par with the organic genre anticancer drugs. In this regard, while looking into the viability of palladium as a substitute for platinum for designing metal-based anticancer drugs, we have earlier reported a palladium N-heterocyclic carbene (NHC) complex, showing ca. 2–20 times higher activity than the cisplatin under in vitro conditions.19 Moving forward, we aim at further increasing the potency of this palladium complex by lowering its IC50 value by suitable structural modifications. In this context, a recent report of a ruthenium(II)-based anticancer drug, exhibiting a 35-fold increase in potency owing to enhanced solubility arising from a change in counteranion15,20,21 led us to seek similar increase in potency of our palladium complex by enhancing its solubility through incorporation of polar anionic substituents.
It is worth noting that our earlier reported palladium N-heterocyclic carbene complex arrested the cell cycle at the G2/M phase triggering apoptotic cell death19 and thereby indicated a pathway bearing resemblance to that of cisplatin’s, for which the metal center too binds to the nuclear DNA, stopping transcription.16 Hence, given the fact that DNA is chiral by itself as well as expecting our palladium N-heterocyclic carbene complexes to operate by a pathway similar to that of cisplatin’s, we chose to probe the influence of chirality by deciding to study the anticancer properties of the enantiomeric pairs of these palladium N-heterocyclic carbene complexes.
Here in this article, we report a comprehensive anticancer study of five enantiomeric pairs of palladium N-heterocyclic carbene complexes, (1S,2S,5R)-(1–3)b and (1R,2R,5S)-(1–3)b, (1S,2S,5R)-(1c, 3c) and (1R,2R,5S)-(1c, 3c), looking into the influence of chirality on their respective anticancer activities (Figure 1). All of the complexes displayed moderate to strong anticancer activity; however, no effect of chirality on the anticancer activity of the compounds was observed. The complex (1R,2R,5S)-1c was found to be the most active and showed potent antiproliferative activity against various cancer cell lines. It was found to be much more potent than the highly successful anticancer drug, cisplatin. Further, (1R,2R,5S)-1c showed differential antiproliferative activity toward cancer cells and their noncancerous counterparts. (1R,2R,5S)-1c treatment induced DNA damage, inhibited cell-cycle progression at G2 phase, and induced p53-dependent apoptosis in breast cancer cells. The results suggested that complex (1R,2R,5S)-1c has a strong anticancer potential.
Figure 1.

Menthyl-derived chiral palladium complexes of 1,2,4-triazole-based NHC ligands.
Results and Discussion
Five enantiomeric pairs of the palladium N-heterocyclic carbene complexes, centered on a chiral menthyl moiety, were constructed by a sequence of reactions starting from the enantiomers of 1-menthyl-1,2,4-triazoles, (1S,2S,5R)-A22 and (1R,2R,5S)-A, based on our earlier reported protocol for the (1S,2S,5R)-1b22 and (1S,2S,5R)-3b22 complexes. The direct alkylation reaction with alkyl bromides yielded the respective 1,2,4-triazolium bromide salts (1S,2S,5R)-(1–3)a and (1R,2R,5S)-(1–3)a (Scheme 1). The reaction of (1S,2S,5R)-(1–3)a and (1R,2R,5S)-(1–3)a with PdBr2 produced the bromide derivatives (1S,2S,5R)-(1–3)b and (1R,2R,5S)-(1–3)b in 71–94% yields in their enantiopure forms and were easily characterized from their equal and opposite optical rotation values. The incorporation of the polar CF3CO2 moiety was achieved by simple salt metathesis reaction of two of the bromide derivatives, (1S,2S,5R)-(1b, 3b) and (1R,2R,5S)-(1b, 3b), with Ag(CO2CF3), giving the corresponding trifluoroacetate-derived enantiomeric pairs, (1S,2S,5R)-(1c, 3c) and (1R,2R,5S)-(1c, 3c), in 57–66% yields.
Scheme 1. Synthesis of the Menthyl-Derived Chiral 1,2,4-Triazole-Based NHC Complexes of Palladium.

The molecular structures of all of the five enantiomeric pairs of the palladium N-heterocyclic carbene complexes, including the two, (1S,2S,5R)-1b and (1S,2S,5R)-3b, reported earlier22 have been determined by X-ray crystallography, which provided further insights into the absolute configurations of the stereocenters in these complexes (Figures 2, 3, S1–S6, and Table S1). As expected, a square planar geometry was observed for all of the complexes with the two N-heterocyclic carbene ligands being bound to the metal center in a trans fashion, and the anionic bromide and the CF3CO2 ligands occupied the remaining two trans sites. The Pd–Ccarbene distance of ca. 2.008(6)−2.049(11) Å observed in the bromo derivatives, (1S,2S,5R)-2b and (1R,2R,5S)-(1–3)b, compare well with the range of 1.999(10)−2.022(11) Å observed in two of earlier reported bromide enantiomers, (1S,2S,5R)-1b and (1S,2S,5R)-3b,22 and also with that of 2.020(8)−2.047(11) Å of the trifluoroacetate derivatives (1S,2S,5R)-(1c, 3c) and (1R,2R,5S)-(1c, 3c).
Figure 2.

Oak Ridge thermal ellipsoid plot (ORTEP) of 1S,2S,5R-1c with thermal ellipsoids are shown at the 50% probability level. Selected bond lengths (angstrom) and angles (degrees): Pd(1)–C(1) 2.025(4), Pd(1)–C(15) 2.030(4), Pd(1)–O(1) 2.028(3), Pd(1)–O(4) 2.029(3), C(1)–Pd(1)–C(15) 178.65(17), O(1)–Pd(1)–O(4) 179.18(13), C(1)–Pd(1)–O(4) 87.09(14), and C(15)–Pd(1)–O(1) 87.95(14).
Figure 3.

ORTEP of 1R,2R,5S-1c with thermal ellipsoids are shown at the 50% probability level. Selected bond lengths (angstrom) and angles (degrees): Pd(2)–C(3) 2.047(11), Pd(2)–C(4) 2.028(10), Pd(2)–O(7) 2.036(7), Pd(2)–O(5) 2.019(7), C(4)–Pd(2)–C(3) 179.0(5), O(7)–Pd(2)–O(5) 178.5(3), C(4)–Pd(2)–O(7) 92.2(3), and C(3)–Pd(2)–O(5) 92.3(4).
All of the enantiomeric pairs, (1S,2S,5R)-(1–3)b and (1R,2R,5S)-(1–3)b, (1S,2S,5R)-(1c, 3c) and (1R,2R,5S)-(1c, 3c), when evaluated against MCF-7, human breast cancer cells, in culture using sulforhodamine B (SRB) assay, showed moderate to excellent activities displaying IC50 values as low as 550 ± 20 nM (Figure 4 and Table S2). Of these, the enantiomeric pair (1S,2S,5R)-1c and (1R,2R,5S)-1c was the most active, exhibiting extremely low IC50 values of 700 ± 5 and 550 ± 20 nM, respectively, in case of MCF-7 cells, which are significantly lower than those reported for other related palladium N-heterocyclic carbene complexes, namely, trans-[1-benzyl-3-t-butylimidazol-2-ylidene]2PdCl2 (IC50 value: 1 ± 3 μM),19 {[1-(2,4,6-trimethylphenyl)-3-(N-tert-butyl-ethan-2-amine)imidazol-2-ylidene]Pd(allyl)}Cl (IC50 value: 6.47 ± 0.07 μM),23 and the benchmark drug cisplatin (IC50 value: 15 ± 2 μM),19 for the same cancer cell line. Also notable is the fact that the extremely low IC50 values of the enantiomeric pair (1S,2S,5R)-1c and (1R,2R,5S)-1c are ca. 27-fold lower than the benchmark drug cisplatin (IC50 value: 14.9 ± 0.4 μM) in MCF-7 cells (Figures 5a, S90, and Table S3). This observation further supports our approach of making potent anticancer compounds by increasing its solubility, which has been achieved in the enantiomeric pair (1S,2S,5R)-1c and (1R,2R,5S)-1c by the incorporation of two polar CF3CO2 moieties.
Figure 4.

Graphical representation of the anticancer activity of the (NHC)2PdX2-type complexes, where X is Br or OCOCF3 against MCF-7 cells.
Figure 5.

(a) Graphical representation of the comparison of the anticancer activity of the most potent Pd–NHC complexes (1S,2S,5R)-1c and (1R,2R,5S)-1c with cisplatin against MCF-7 and HeLa cell lines. (b) Graphical representation of the comparison of the anticancer activity of (1R,2R,5S)-1c against cancer cell lines, MCF-7 and B16F10, and their respective noncancerous counterparts, MCF10A and L929.
Although all of the complexes displayed anticancer activity against MCF-7 cells, no influence was observed of chirality. Both the enantiomers of all of the five pairs of the enantiomeric palladium complexes exhibited near-equal activities (Figure 4 and Table S2, entries 1–5). The lack of the influence of chirality on the anticancer activity may be due to the larger dimensions of the chiral major (ca. 22 Å) and minor (ca. 12 Å) grooves of DNA24 with respect to the chiral menthyl moieties having cross-sectional diameter of ca. 8 Å in the enantiomeric pairs of the palladium N-heterocyclic carbene complexes. The other possibility for the lack of the influence of chirality may also be due to a different species interacting with the DNA strands other than the chiral palladium N-heterocyclic carbene complexes in its original forms.
The most potent enantiomeric pair, (1S,2S,5R)-1c and (1R,2R,5S)-1c, when studied on another cancer cell line, namely, the human cervical cancer cells (HeLa), exhibited IC50 values of 2.6 ± 1.6 and 2.3 ± 1 μM, respectively, which were ca. three- to fourfold more active than that observed for cisplatin (IC50 value: 8.5 ± 0.7 μM) for the same cancer cell line (Figures 5a, S91, and Table S3). Furthermore, a closer look at the IC50 values of all of the palladium N-heterocyclic carbene complexes across the aforementioned two cancer cell lines (Tables S2 and S3) reveals the (1R,2R,5S)-1c complex to be the most potent one, for which the anticancer activity study was further extended to other cancerous cell lines. In this context, (1R,2R,5S)-1c exhibited IC50 values of 5.7 ± 1.6 and 1.3 ± 0.4 μM for human lung cancer (A549) and mouse skin cancer (B16F10) cell lines, respectively (Figures 6, S92, S93, and Table S4). Furthermore, (1R,2R,5S)-1c complex could also inhibit the proliferation of multidrug-resistant EMT6/AR1 cells with an IC50 value of 4.8 ± 0.3 μM, thereby extending the broad scope of acceptability of this (1R,2R,5S)-1c complex against various cancer cell lines (Figures 6, S93, and Table S4).
Figure 6.
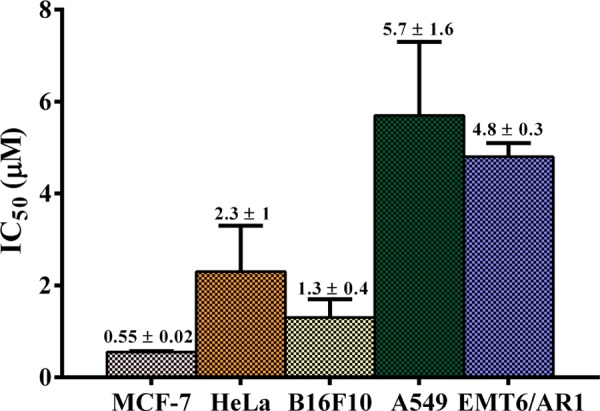
Graphical representation of the anticancer activity of the most potent Pd–NHC complex (1R,2R,5S)-1c against various cancer cell lines, namely, MCF-7, HeLa, B16F10, A549, and EMT6/AR1.
Finally, the selectivity of the (1R,2R,5S)-1c complex was tested with respect to the nontumor cell lines, L929 (mouse fibroblast skin cells) and MCF10A (human epithelial breast cells), and their respective cancerous cells, B16F10 (mouse skin cancer cells) and MCF-7. (1R,2R,5S)-1c showed ca. 8-fold more potency toward B16F10 (IC50 value: 1.3 ± 0.4 μM) relative to L929 (IC50 value: 10.5 ± 0.2 μM) and ca. 16-fold more potency toward MCF-7 (IC50 value: 0.55 ± 0.02 μM) relative to MCF10A (IC50 value: 8.6 ± 0.9 μM) (Figures 5b, S90, S92, and Table S5). These findings thus highlight the potential of this class of palladium N-heterocyclic carbene complexes for further development as anticancer agents.
The mechanism of cytotoxicity of the most potent enantiomeric pair (1R,2R,5S)-1c was investigated through a series of experiments. To examine whether (1R,2R,5S)-1c induces cell death, MCF-7 cells were treated without or with 4 and 8 μM (1R,2R,5S)-1c. The percentage of dead cells was determined by flow cytometry after propidium iodide (PI) staining (Figure 7a). Proportions of 41 and 90% of the total cells were found to be dead when treated with 4 and 8 μM (1R,2R,5S)-1c for 48 h, respectively, whereas only 2% of the total population were dead in control (untreated) cells. On the other hand, 4 μM cisplatin treatment could kill only 8% of the total cells, which was significantly less compared to 4 μM (1R,2R,5S)-1c (Figure 7a and Table 1). (1R,2R,5S)-1c-treated cells could undergo either apoptotic or necrotic cell death. Further, the effect of (1R,2R,5S)-1c on poly(adenosine 5′-diphosphate-ribose) (poly(ADP-ribose)) polymerase-1 (PARP-1) cleavage in cells was elucidated by western blot analysis using anti-PARP antibody. Under cellular stress, PARP-1 aids in DNA repair by adding poly(ADP-ribose) polymers.25 During apoptosis, PARP-1 (125 kDa) is cleaved into 85 kDa fragment by caspases, thus inactivating the protein. Cleaved PARP-1 is thus considered a typical indicator of apoptosis in cells.26 PARP cleavage was observed in MCF-7 cells after 48 h incubation with 4 and 8 μM (1R,2R,5S)-1c, indicating apoptosis in cells (Figure 7b). However, 4 μM cisplatin did not cleave PARP in cells. Cell-death analysis by PI staining and determination of PARP cleavage by western blot showed that (1R,2R,5S)-1c caused apoptosis in cells in a concentration-dependent manner. More interestingly, the effect of (1R,2R,5S)-1c was more pronounced than cisplatin and much lower concentration of (1R,2R,5S)-1c was required to kill cells as compared to cisplatin. Clearly, the complex (1R,2R,5S)-1c is more potent than cisplatin.
Figure 7.

(1R,2R,5S)-1c cleaves PARP and causes apoptosis in MCF-7 cells. (a) (1R,2R,5S)-1c treatment leads to cell death in MCF-7 cells. MCF-7 cells were incubated without and with 4 and 8 μM (1R,2R,5S)-1c for 48 h and analyzed by flow cytometry. Representative images from three independent sets of experiments are shown. (b) (1R,2R,5S)-1c cleaves PARP in MCF-7 cells, indicating apoptosis. MCF-7 cells were treated without and with 4 and 8 μM (1R,2R,5S)-1c for 48 h. PARP cleavage was determined by western blot using anti-PARP immunoglobulin G (IgG). Actin was used as a loading control. The experiment was performed three times. Representative blot is shown.
Table 1. Percentage of Live and Dead MCF-7 Cells after (1R,2R,5S)-1c Treatmenta.
| s. no. | samples | live (%) | dead (%) |
|---|---|---|---|
| 1 | control | 98 ± 1 | 2 ± 1 |
| 2 | 4 μM (1R,2R,5S)-1c | 59 ± 2 | 41 ± 2 |
| 3 | 8 μM (1R,2R,5S)-1c | 10 ± 1 | 90 ± 1 |
| 4 | 4 μM cisplatin | 93 ± 4 | 8 ± 2 |
Percentage of live and dead MCF-7 cells without and with 4 and 8 μM (1R,2R,5S)-1c, and 4 μM cisplatin was determined by flow cytometry. Data are average of three independent set of experiments, and ± indicates standard deviation (SD).
To elucidate the mechanism of cell death, several experiments were performed. Compounds like cisplatin27 induce DNA damage and are known to block cells at the G2/M phase of the cell cycle. As expected, (1R,2R,5S)-1c also showed strong DNA-damaging effects on MCF-7 cells. Exposure of cells to DNA-damaging stress like ionizing radiation or other DNA-damaging agents leads to the generation of double-stranded breaks (DSBs), resulting in the phosphorylation of histone H2A variant H2AX at Ser 139, producing γ-H2AX.28 Phosphorylation of H2AX (γ-H2AX) due to DNA damage is abundant, fast, and can be correlated with each DSB.28 Thus, it is a highly sensitive marker that can be used to determine DNA damage in cells. We determined the DNA-damaging effects of (1R,2R,5S)-1c by assessing the phosphorylation of histone-2AX (γ-H2AX) using immunofluorescence microscopy. Cells treated without and with 4 and 8 μM (1R,2R,5S)-1c and 4 μM cisplatin were stained with γ-H2AX antibody, and DNA was stained with Hoechst. Tremendous increase in the phosphorylation of H2AX (γ-H2AX intensity) was observed in (1R,2R,5S)-1c-treated cells as compared to control cells. However, 4 μM cisplatin-treated cells showed much reduced γ-H2AX intensity compared to 4 μM (1R,2R,5S)-1c-treated cells (Figure 8a,b). The results suggested that (1R,2R,5S)-1c had a stronger DNA-damaging effect on MCF-7 cells than cisplatin.
Figure 8.

(1R,2R,5S)-1c causes DNA damage in MCF-7 cells. (a) MCF-7 cells were treated without and with 4 and 8 μM (1R,2R,5S)-1c for 36 h and immunostaining was performed using anti-γ-H2AX IgG (pink). DNA was stained using Hoechst 33258 (blue). The scale bar is 20 μm. (b) γ-H2AX intensity was calculated using ImageJ software. The experiment was performed three times, and in each case, 500 cells were scored. *p < 0.05 and **p < 0.01 show statistical significance of the data.
DNA damage correlates with the induction of reactive oxygen species (ROS) in cells.29 Interplay between DNA damage response cascade and ROS signaling in cells is responsible for inducing apoptosis following DNA damage.29 DNA damage induced by (1R,2R,5S)-1c led to a dramatic increase in ROS levels in MCF-7 cells. The generation of intracellular ROS was evaluated by ROS assay using 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA).30 H2DCFDA is a cell-permeable molecule. Inside the cells, ROS can oxidize H2DCFDA to fluorescent 2′,7′-dichlorofluorescein,30 which can be detected by flow cytometry. Figure 9a,b shows the percentage of (1R,2R,5S)-1c-treated cells with intracellular ROS generation (2′,7′-dichlorofluorescein diacetate (DCFH-DA)-positive cells) using flow cytometry. Majority of the total cells (72%) were DCFH-DA positive when treated with 30 μM hydrogen peroxide, which was used as a positive control. In cells incubated with 4 and 8 μM (1R,2R,5S)-1c for 24 h, 49 and 74% of the total cells were DCFH-DA positive, respectively (Figure 9b and Table 2). It is well known that dissipation of mitochondrial potential is associated with the mitochondrial production of reactive oxygen species (ROS).31,32 The data suggested that DNA damage induced by (1R,2R,5S)-1c leads to ROS production in cells, causing apoptosis following mitochondrial pathway.
Figure 9.

(1R,2R,5S)-1c generates reactive oxygen species in MCF-7 cells. (a) Flow cytogram showing intracellular ROS generation in MCF-7 cells untreated (negative control) and treated (positive control) with 30 μM H2O2 and 4 and 8 μM (1R,2R,5S)-1c for 24 h. (b) Percentage of DCFH-DA-positive cells were quantified and plotted. **p < 0.01 shows statistical significance of the data.
Table 2. Percentage of DCFH-DA-Positive MCF-7 Cells after (1R,2R,5S)-1c Treatmenta.
| s. no. | samples | percentage of DCFH-DA-positive cells |
|---|---|---|
| 1 | control | 1 ± 0.1 |
| 2 | 4 μM (1R,2R,5S)-1c | 49 ± 11 |
| 3 | 8 μM (1R,2R,5S)-1c | 74 ± 1 |
| 4 | 30 μM H2O2 | 72 ± 4 |
The percentage of DCFH-DA-positive MCF-7 cells without and with 4 and 8 μM (1R,2R,5S)-1c was determined by flow cytometry. H2O2 (30 μM) was used as a positive control. Data are average of three independent set of experiments, and ± indicates SD.
Nuclear DNA damage can cause cell-growth arrest, subsequently leading to apoptosis in cells. To determine the effect of (1R,2R,5S)-1c on the progression of the cell cycle, MCF-7 cells were incubated without or with 2 and 4 μM (1R,2R,5S)-1c for 36 h. Treated cells were incubated with PI and analyzed by flow cytometry. The DNA distribution profiles of different phases of cell cycle showed that (1R,2R,5S)-1c blocks MCF-7 cells at the G2/M phase of the cell cycle (Figure 10a). Proportions of 35 and 50% of the total cells were blocked at G2/M when treated with 2 and 4 μM (1R,2R,5S)-1c for 36 h, respectively, whereas in control cells only 10% of the cells were in the G2/M phase (Figure 10a and Table 3). However, it was not clear from the data whether the cells got arrested in the G2 or M phase of the cell cycle. Further, mitotic cells were visualized under the microscope after staining with Hoechst 33258. MCF-7 cells were incubated without or with 2 and 4 μM (1R,2R,5S)-1c for 36 h, and the number of cells in mitosis were counted on the basis of DNA morphology. The mitotic index (percentage of cells in mitosis) of MCF-7 cells treated with 2 and 4 μM (1R,2R,5S)-1c was found to be similar to that of the control cells. This implies that the cells were arrested at the G2 phase of the cell cycle and not mitosis (Figure 10b,c and Table 3). The results indicated that (1R,2R,5S)-1c arrested cells at the G2 phase and prevented the cells from entering mitosis, which subsequently induced apoptosis in these cells.
Figure 10.

(1R,2R,5S)-1c arrested MCF-7 cells at the G2 phase of the cell cycle. (a) (1R,2R,5S)-1c blocked cells at the G2/M phase of the cell cycle. DNA distribution profiles in different phases of the cell cycle after 36 h treatment with media alone (control) or 2 and 4 μM of (1R,2R,5S)-1c were determined by flow cytometry. Data analysis was done using FlowJo software. (b) (1R,2R,5S)-1c did not arrest cells at mitosis. MCF-7 cells were incubated without and with 2 and 4 μM (1R,2R,5S)-1c for 36 h. Mitotic cells were visualized on the basis of DNA morphology after staining the cells with Hoechst 33258. The scale bar is 20 μm. (c) Mitotic index was calculated from the microscopy data and plotted. The experiment was performed three times, and 1000 cells were scored in each case. *p < 0.05 shows statistical significance of the data.
Table 3. Effect of (1R,2R,5S)-1c on Cell-Cycle Progression of MCF-7 Cellsa.
| percentage of cells in different phases of cell
cycle |
|||||
|---|---|---|---|---|---|
| s. no. | samples | G1 | S | G2/M | mitotic index |
| 1 | control | 60 ± 1 | 28 ± 2 | 10 ± 1 | 2 ± 1 |
| 2 | 2 μM (1R,2R,5S)-1c | 30 ± 2 | 31 ± 2 | 35 ± 2 | 3 ± 1 |
| 3 | 4 μM (1R,2R,5S)-1c | 18 ± 2 | 32 ± 3 | 50 ± 2 | 3.2 ± 1 |
Cell-cycle analysis and mitotic indices of MCF-7 cells without and with 2 and 4 μM (1R,2R,5S)-1c. Cell-cycle analysis was done using flow cytometry. Data were analyzed using FlowJo software. Mitotic index was calculated on the basis of DNA morphology of cells. Data were average of three independent sets of experiments with SD (±), and 1000 cells were scored in each set.
G2 arrest triggers p53 and various other signaling pathways to ensure that apoptosis follows the G2 block in cells.33 Different stress signals, including DNA damage, stabilize cytoplasmic p53 protein and lead to its translocation and accumulation in the nucleus. This leads to transcription activation of p53-dependent genes, such as Bax, p21, and so forth.34 The G2 arrest in MCF-7 cells after (1R,2R,5S)-1c treatment was further confirmed by p53 and p21 staining. MCF-7 cells treated without and with 4 μM (1R,2R,5S)-1c were processed for immunofluorescence using anti-p53 antibody. In the presence of 4 μM (1R,2R,5S)-1c, 78% of the cells showed nuclear accumulation of p53, whereas in control cells, it was only 23% (Figure 11a). Nuclear accumulation of p21, the downstream target gene of p53, was also determined by microscopic analysis of (1R,2R,5S)-1c-treated cells using p21 antibody. Increased (73%) nuclear accumulation of p21 was observed in 4 μM (1R,2R,5S)-1c-treated cells compared to untreated (control) cells (18%) (Figure 11b). The data suggested that nuclear accumulation of p53 led to the activation of p53-responsive genes and subsequent apoptosis in cells. The results indicated that DNA damage induced by (1R,2R,5S)-1c arrested cells at the G2 phase of the cell cycle and followed p53-dependent pathway for cell death.
Figure 11.

(1R,2R,5S)-1c activates p53 pathway and its downstream target p21. (a) (1R,2R,5S)-1c treatment leads to nuclear accumulation of p53. MCF-7 cells were treated without and with 4 and 8 μM (1R,2R,5S)-1c for 36 h, fixed, and processed for immunostaining using anti-p53 IgG. Left panels show DNA stained with Hoechst 33258 (blue), middle panels show the corresponding cells stained with anti-p53 IgG (red), and right panels show merged images. The scale bar is 10 μm. (b) (1R,2R,5S)-1c activates the p21. MCF-7 cells were processed as mentioned above and stained using anti-p21 IgG. Left panels show DNA stained with Hoechst 33258 (blue), middle panels show the corresponding cells stained with anti-p21 IgG (red), and right panels show merged images. The scale bar is 10 μm.
Conclusions
Five enantiomeric pairs of 1,2,4-triazole-based palladium complexes (1S,2S,5R)-(1–3)b and (1R,2R,5S)-(1–3)b, (1S,2S,5R)-(1c, 3c) and (1R,2R,5S)-(1c, 3c) of chiral N-heterocyclic carbene ligands were synthesized and investigated for their anticancer properties. All of the complexes displayed antiproliferative properties against MCF-7 cells in culture. However, the enantiomers of all of the complexes exhibited nearly equal activity, suggesting no influence of chirality on their antiproliferative activity. Furthermore, a particular enantiomeric pair (1S,2S,5R)-1c and (1R,2R,5S)-1c bearing CF3CO2 moieties displayed significantly higher anticancer activity than cisplatin against human breast and cervical cancer cells. It is important to underline that (1R,2R,5S)-1c exhibited very low toxicity toward noncancerous cell lines compared to their cancerous counterpart. The detailed mechanistic study suggests that the (1R,2R,5S)-1c is a highly effective DNA-damaging anticancer agent. (1R,2R,5S)-1c induced the production of intracellular ROS, stalled cells at the G2 phase, and led to p53-dependent programmed cell death (apoptosis). In summary, the biological characterization of the compound (1R,2R,5S)-1c provides compelling evidence to suggest that (1R,2R,5S)-1c is more potent than cisplatin and pointed toward the potential of this type of palladium-based N-heterocyclic carbene complexes for further development as anticancer agents.
Experimental Section
General Procedures
The standard Schlenk techniques were employed for all manipulations. Palladium bromide, (1R)-(−)-menthol, (1S)-(+)-menthol, and silver trifluoroacetate were purchased from Sigma-Aldrich. (1R,2S,5R)-2-isopropyl-5-methylcyclohexyl p-toluenesulfonate22 and (1S,2R,5S)-2-isopropyl-5-methylcyclohexyl p-toluenesulfonate were synthesized by following modified literature procedures.22 The ligand precursors (1S,2S,5R)-1a and (1S,2S,5R)-3a and the palladium N-heterocyclic carbene complexes (1S,2S,5R)-1b and (1S,2S,5R)-3b were prepared according to a literature procedure reported by us.22 Bruker 400 and 500 MHz NMR spectrometers were used for recording the 1H NMR, 13C{1H} NMR, and 19F{1H} NMR spectra. 1H NMR signals are labeled as singlet (s), doublet (d), triplet (t), broad (br), triplet of triplet (tt), doublet of doublet (dd), doublet of quartets (dq), multiplet (m), and septet (sept). PerkinElmer Spectrum One FTIR spectrometer was used to record the infrared spectra. Micromass quad time-of-flight spectrometer and Bruker maxis impact spectrometers were used for the mass spectrometry analysis. Thermo Finnigan FlashEA 1112 series elemental analyzer was used for the analysis of CHNS. Single-crystal X-ray data for compounds (1–3)b, 1c, and 3c were collected on Oxford Diffraction XCALIBUR-S diffractometer and Rigaku Hg 724+ diffractometer, solved using direct method and standard difference map techniques, and refined by full-matrix least-squares procedures on F2. X-ray metrical data are summarized in Table S1. Cambridge Crystallographic Data Center (CCDC)-1435778 (for 1R,2R,5S-1b), CCDC-919872 (for 1S,2S,5R-2b), CCDC-1447721 (for 1R,2R,5S-2b), CCDC-1435777 (for 1R,2R,5S-3b), CCDC-999453 (for 1S,2S,5R-1c), CCDC-1435624 (for 1R,2R,5S-1c), CCDC-999452 (for 1S,2S,5R-3c), and CCDC-1440828 (for 1R,2R,5S-3c) contain the crystallographic data of this article, which can be found free of charge from the Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif.
Synthesis of 1-(1R)-Menthyl-1H-1,2,4-triazole (1R,2R,5S-A)
A mixture of 1,2,4-triazole (5.00 g, 72.4 mmol) and (1S,2R,5S)-2-isopropyl-5-methylcyclohexyl 4-methylbenzenesulfonate (10.0 g, 32.2 mmol) was stirred in dimethylformamide (ca. 100 mL) at 0 °C and NaH was added portionwise (3.00 g, 125 mmol) for 20 min. The mixture was stirred at room temperature for additional 30 min and then refluxed for 24 h. After the reaction, ethyl acetate (EtOAc) (ca. 500 mL) was added to the reaction mixture and was washed with water (ca. 12 × 50 mL). The organic layer was vacuum-dried and crude product was purified by silica gel column chromatography by elution with a petroleum ether/EtOAc mixture (90:10 v/v) to give the product (1R,2R,5S-A) as a colorless solid (2.67 g, 40%). 1H NMR (CDCl3, 400 MHz, 25 °C): δ 8.14 (s, 1H, N–C(5)H–N), 7.86 (s, 1H, N–C(3)H–N), 4.75 (br, 1H, CH3C6H9CH(CH3)2), 2.06–1.00 (m, 9H, CH3C6H9CH(CH3)2 and CH3C6H9CH(CH3)2), 0.87 (d, 3H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2), 0.84 (d, 3H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2), 0.78 (d, 3H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2). 13C{1H} NMR (CDCl3, 125 MHz, 25 °C): δ 151.1 (N–C(5)H–N), 143.6 (N–C(3)H–N), 58.1 (CH3C6H9CH(CH3)2), 46.8 (CH3C6H9CH(CH3)2), 41.1 (CH3C6H9CH(CH3)2), 34.9 (CH3C6H9CH(CH3)2), 29.2 (CH3C6H9CH(CH3)2), 26.5 (CH3C6H9CH(CH3)2), 25.1 (CH3C6H9CH(CH3)2), 22.4 (CH3C6H9CH(CH3)2), 21.2 (CH3C6H9CH(CH3)2), 20.6 (CH3C6H9CH(CH3)2). IR data (KBr pellet) cm–1: 3445 (m), 3109 (w), 2954 (s), 2926 (s), 2868 (s), 2844 (s), 1634 (w), 1505 (m), 1475 (m), 1456 (m), 1443 (m), 1370 (w), 1288 (w), 1270 (m), 1195 (w), 1175 (w), 1136 (m), 1069 (w), 1015 (m), 868 (m), 726 (w), 677 (m). High-resolution mass spectrometry (HRMS) (ES): m/z 230.1637 [M + Na]+, calcd 230.1628. Anal. Calcd for C12H21N3: C, 69.52; H, 10.21; N, 20.27. Found: C, 69.08; H, 9.98; N, 19.38%. [α]D25 −40.9 (c 1.00 in CHCl3).
Synthesis of 1-(1R)-Menthyl-4-(ethyl)-1,2,4-triazolium bromide (1R,2R,5S-1a)
A mixture of 1-(1R)-menthyl-1H-1,2,4-triazole (1R,2R,5S-A) (1.42 g, 6.85 mmol) and ethyl bromide (3.18 g, 29.2 mmol) was refluxed in acetonitrile (ca. 40 mL) for 24 h, after which the solvent was evaporated under vacuum. The residue was washed with hot Et2O (ca. 3 × 10 mL) and vacuum-dried to give the product (1R,2R,5S-1a) as a white solid (0.491 g, 23%). 1H NMR (CDCl3, 400 MHz, 25 °C): δ 11.69 (s, 1H, N–C(5)H–N), 9.69 (s, 1H, N–C(3)H–N), 5.30 (br, 1H, CH3C6H9CH(CH3)2), 4.69 (q, 2H, 3JHH = 7 Hz, CH2CH3), 2.11–1.00 (m, 9H, CH3C6H9CH(CH3)2 and CH3C6H9CH(CH3)2), 1.69 (t, 3H, 3JHH = 7 Hz, CH2CH3), 0.93 (d, 3H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2), 0.88 (d, 3H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2), 0.80 (d, 3H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2). 13C{1H} NMR (CDCl3, 100 MHz, 25 °C): δ 143.6 (N–C(5)H–N), 143.0 (N–C(3)H–N), 62.0 (CH3C6H9CH(CH3)2), 46.3 (CH3C6H9CH(CH3)2), 44.2 (CH2), 40.3 (CH3C6H9CH(CH3)2), 34.4 (CH3C6H9CH(CH3)2), 29.1 (CH3C6H9CH(CH3)2), 25.9 (CH3C6H9CH(CH3)2), 24.3 (CH3C6H9CH(CH3)2), 22.1 (CH3C6H9CH(CH3)2), 21.4 (CH3C6H9CH(CH3)2), 20.5 (CH3C6H9CH(CH3)2), 16.0 (CH2CH3). IR data (KBr pellet) cm–1: 3428 (w), 3091 (w), 3028 (m), 2948 (s), 2867 (m), 1821 (w), 1583 (m), 1510 (w), 1456 (w), 1369 (w), 1330 (w), 1216 (w), 1174 (m), 1009 (w), 991 (w), 913 (w), 632 (m). HRMS (ES): m/z 236.2124 [M – Br]+, calcd 236.2121. Anal. Calcd for C14H26BrN3: C, 53.17; H, 8.29; N, 13.29. Found: C, 53.94; H, 8.39; N, 13.27%. [α]D25 −22.0 (c 1.00 in CHCl3).
Synthesis of 1-(1S)-Menthyl-4-(allyl)-1,2,4-triazolium bromide (1S,2S,5R-2a)
A mixture of 1-(1S)-menthyl-1H-1,2,4-triazole (1S,2S,5R-A) (1.20 g, 5.79 mmol) and allyl bromide (2.81 g, 23.2 mmol) was refluxed in CH3CN (ca. 40 mL) for 24 h, after which the solvent was removed under vacuum, and the solid so obtained was washed with hot Et2O (ca. 3 × 10 mL) and vacuum-dried to give the product 1S,2S,5R-2a as a white solid (1.53 g, 80%). 1H NMR (CDCl3, 400 MHz, 25 °C): δ 11.7 (s, 1H, N–C(5)H–N), 9.27 (s, 1H, N–C(3)H–N), 6.18 (m, 1H, CH2CH=CH2), 5.64 (d, 1H, 3JHH = 17 Hz, CH2CH=CH2), 5.52 (d, 1H, 3JHH = 10 Hz, CH2CH=CH2), 5.37 (d, 2H, 3JHH = 6 Hz, CH2CH=CH2), 5.26 (br, 1H, CH3C6H9CH(CH3)2), 2.08–1.00 (m, 9H, CH3C6H9CH(CH3)2 and CH3C6H9CH(CH3)2), 0.93 (d, 3H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2), 0.88 (d, 3H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2), 0.81 (d, 3H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2). 13C{1H} NMR (CDCl3, 100 MHz, 25 °C): δ 143.3 (N–C(5)H–N), 143.0 (N–C(3)H–N), 129.5 (CH2CH=CH2), 123.8 (CH2CH=CH2), 62.2 (CH3C6H9CH(CH3)2), 50.5 (CH2CH=CH2), 46.3 (CH3C6H9CH(CH3)2), 40.2 (CH3C6H9CH(CH3)2), 34.4 (CH3C6H9CH(CH3)2), 29.1 (CH3C6H9CH(CH3)2), 25.8 (CH3C6H9CH(CH3)2), 24.3 (CH3C6H9CH(CH3)2), 21.3 (CH3C6H9CH(CH3)2), 21.0 (CH3C6H9CH(CH3)2), 20.6 (CH3C6H9CH(CH3)2). IR data (KBr pellet) cm–1: 3434 (m), 3088 (w), 3015 (m), 2951 (s), 2850 (m), 1647 (w), 1573 (m), 1512 (w), 1455 (m), 1441 (m), 1424 (m), 1308 (w), 1289 (w), 1260 (w), 1206 (w), 1176 (m), 1075 (w), 985 (w), 932 (w), 803 (w), 631 (w). HRMS (ES): m/z 248.2122 [M – Br]+, calcd 248.2127. Anal. Calcd for C15H26BrN3: C, 54.88; H, 7.98; N, 12.80. Found: C, 54.91; H, 7.32; N, 12.68%. [α]D25 +21.7 (c 1.00 in CHCl3).
Synthesis of 1-(1R)-Menthyl-4-(allyl)-1,2,4-triazolium bromide (1R,2R,5S-2a)
A mixture of 1-(1R)-menthyl-1H-1,2,4-triazole (1R,2R,5S-A) (0.656 g, 3.16 mmol) and allyl bromide (0.384 g, 3.17 mmol) was refluxed in CH3CN (ca. 40 mL) for 24 h, after which the solvent was removed under vacuum. The residue was repeatedly washed with hot Et2O (ca. 3 × 10 mL) and vacuum-dried to give the product 1R,2R,5S-2a as a white solid (0.865 g, 83%). 1H NMR (CDCl3, 500 MHz, 25 °C): δ 11.7 (s, 1H, N–C(5)H–N), 8.93 (s, 1H, N–C(3)H–N), 6.18 (m, 1H, CH2CH=CH2), 5.64 (d, 1H, 3JHH = 17 Hz, CH2CH=CH2), 5.52 (d, 1H, 3JHH = 10 Hz, CH2CH=CH2), 5.37 (d, 2H, 3JHH = 6 Hz, CH2CH=CH2), 5.26 (br, 1H, CH3C6H9CH(CH3)2), 2.08–1.00 (m, 9H, CH3C6H9CH(CH3)2 and CH3C6H9CH(CH3)2), 0.93 (d, 3H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2), 0.88 (d, 3H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2), 0.81 (d, 3H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2). 13C{1H} NMR (CDCl3, 100 MHz, 25 °C): δ 143.2 (N–C(5)H–N), 143.0 (N–C(3)H–N), 129.5 (CH2CH=CH2), 123.8 (CH2CH=CH2), 62.4 (CH3C6H9CH(CH3)2), 50.6 (CH2CH=CH2), 46.3 (CH3C6H9CH(CH3)2), 40.2 (CH3C6H9CH(CH3)2), 34.4 (CH3C6H9CH(CH3)2), 29.2 (CH3C6H9CH(CH3)2), 25.9 (CH3C6H9CH(CH3)2), 24.3 (CH3C6H9CH(CH3)2), 22.1 (CH3C6H9CH(CH3)2), 21.4 (CH3C6H9CH(CH3)2), 20.6 (CH3C6H9CH(CH3)2). IR data (KBr pellet) cm–1: 3087 (w), 3015 (s), 2951 (s), 2868 (m), 2850 (m), 1574 (m), 1511 (w), 1455 (m), 1441 (m), 1425 (m), 1317 (w), 1207 (w), 1166 (w), 1075 (w), 1000 (w), 986 (w), 932 (w), 894 (w), 870 (w), 631 (m). HRMS (ES): m/z 248.2123 [M – Br]+, calcd 248.2121. Anal. Calcd for C15H26BrN3: C, 54.88; H, 7.98; N, 12.80. Found: C, 54.95; H, 7.39; N, 13.03%. [α]D25 −21.6 (c 1.00 in CHCl3).
Synthesis of 1-(1R)-Menthyl-4-(benzyl)-1,2,4-triazolium bromide (1R,2R,5S-3a)
A mixture of 1-(1R)-menthyl-1H-1,2,4-triazole (1R,2R,5S-A) (0.800 g, 3.86 mmol) and benzyl bromide (0.660 g, 3.86 mmol) was refluxed in CH3CN (ca. 40 mL) for overnight, after which the solvent was removed under vacuum. The residue was washed with hot Et2O (ca. 3 × 10 mL) and vacuum-dried to give the product 1R,2R,5S-3a as a white solid (1.164 g, 80%). 1H NMR (CDCl3, 400 MHz, 25 °C): δ 11.9 (s, 1H, N–C(5)H–N), 8.79 (s, 1H, N–C(3)H–N), 7.68 (dd, 2H, 3JHH = 8 Hz, 4JHH = 2 Hz, C6H5), 7.39 (br, 3H, C6H5), 5.92 (s, 2H, CH2), 4.79 (br, 1H, CH3C6H9CH(CH3)2), 1.99–1.00 (m, 9H, CH3C6H9CH(CH3)2 and CH3C6H9CH(CH3)2), 0.89 (d, 3H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2), 0.85 (d, 3H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2), 0.76 (d, 3H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2). 13C{1H} NMR (CDCl3, 100 MHz, 25 °C): δ 143.1 (N–C(5)H–N), 142.9 (N–C(3)H–N), 132.6 (ipso-C6H5), 130.0 (C6H5), 129.8 (C6H5), 129.6 (C6H5), 62.5 (CH2), 51.8 (CH3C6H9CH(CH3)2), 46.4 (CH3C6H9CH(CH3)2), 40.1 (CH3C6H9CH(CH3)2), 34.4 (CH3C6H9CH(CH3)2), 29.2 (CH3C6H9CH(CH3)2), 25.9 (CH3C6H9CH(CH3)2), 24.2 (CH3C6H9CH(CH3)2), 22.1 (CH3C6H9CH(CH3)2), 21.4 (CH3C6H9CH(CH3)2), 20.5 (CH3C6H9CH(CH3)2). IR data (KBr pellet) cm–1: 3415 (w), 3077 (m), 3006 (s), 2950 (s), 2871 (s), 2850 (s), 1564 (s), 1509 (m), 1497 (w), 1455 (s), 1422 (w), 1403 (w), 1392 (w), 1373 (w), 1322 (w), 1306 (w), 1244 (w), 1213 (m), 1204 (m), 1172 (m), 1160 (s), 1122 (m), 1067 (w), 1008 (w), 983 (m), 937 (w), 913 (w), 874 (w), 821 (m), 774 (w), 724 (m), 706 (s), 651 (s), 472 (w). HRMS (ES): m/z 298.2274 [M – Br]+, calcd 298.2278. Anal. Calcd for C19H28BrN3: C, 60.32; H, 7.46; N, 11.11. Found: C, 59.91; H, 6.66; N, 11.25%. [α]D25 −16.2 (c 1.00 in CHCl3).
Synthesis of trans-[1-(1R)-Menthyl-4-(ethyl)-1,2,4-triazol-5-ylidene]2PdBr2 (1R,2R,5S-1b)
A mixture of 1-(1R)-menthyl-4-(ethyl)-1,2,4-triazolium bromide (1R,2R,5S-1a) (0.474 g, 1.50 mmol), PdBr2 (0.200 g, 0.751 mmol), and Et3N (0.606 g, 5.99 mmol) in CH3CN (ca. 50 mL) was refluxed for 12 h. The solvent was removed under vacuum, and the crude product was purified by column chromatography using petroleum ether/EtOAc (85 15 v/v) to give the product 1R,2R,5S-1b as a light yellow solid (0.434 g, 78%). Both the 1H NMR and 13C{1H} NMR spectra showed the presence of two isomers, trans-syn and trans-anti.22 The major to minor isomer ratio was 3.5:2. 1H NMR (CDCl3, 400 MHz, 25 °C): (major) δ 7.87 (s, 2H, N–C(3)H–N), 5.70 (br, 2H, CH3C6H9CH(CH3)2), 4.78 (dq, 2H, 3JHH = 7 Hz, CH2CH3), 4.46 (dq, 1H, 3JHH = 7 Hz, CH2CH3), 2.59–1.00 (m, 18H, CH3C6H9CH(CH3)2 and CH3C6H9CH(CH3)2), 1.73 (t, 6H, 3JHH = 7 Hz, CH2CH3), 1.12 (d, 6H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2), 0.85 (d, 6H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2), 0.74 (d, 6H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2). (Minor) δ 7.87 (s, 2H, N–C(3)H–N), 5.61 (br, 2H, CH3C6H9CH(CH3)2), 4.74 (dq, 2H, 3JHH = 7 Hz, CH2CH3), 4.42 (dq, 2H, 3JHH = 7 Hz, CH2CH3), 2.59–1.00 (m, 18H, CH3C6H9CH(CH3)2 and CH3C6H9CH(CH3)2), 1.71 (t, 6H, 3JHH = 7 Hz, CH2CH3), 1.09 (d, 6H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2), 0.84 (d, 6H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2), 0.72 (d, 6H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2). 13C{1H} NMR (CDCl3, 125 MHz, 25 °C): (major) δ 172.2 (Pd–NCN), 140.7 (N–C(3)H–N), 60.73 (CH3C6H9CH(CH3)2), 47.7 (CH3C6H9CH(CH3)2), 44.2 (CH2CH3), 41.6 (CH3C6H9CH(CH3)2), 35.5 (CH3C6H9CH(CH3)2), 29.1 (CH3C6H9CH(CH3)2), 26.2 (CH3C6H9CH(CH3)2), 24.3 (CH3C6H9CH(CH3)2), 23.4 (CH3C6H9CH(CH3)2), 22.6 (CH3C6H9CH(CH3)2), 20.3 (CH3C6H9CH(CH3)2), 16.1 (CH2CH3). (Minor) δ 172.17 (Pd–NCN), 140.5 (N–C(3)H–N), 60.7 (CH3C6H9CH(CH3)2), 47.6 (CH3C6H9CH(CH3)2), 44.1 (CH2CH3), 41.3 (CH3C6H9CH(CH3)2), 35.5 (CH3C6H9CH(CH3)2), 29.0 (CH3C6H9CH(CH3)2), 26.2 (CH3C6H9CH(CH3)2), 24.2 (CH3C6H9CH(CH3)2), 23.3 (CH3C6H9CH(CH3)2), 22.6 (CH3C6H9CH(CH3)2), 20.3 (CH3C6H9CH(CH3)2), 16.0 (CH2CH3). IR data (KBr pellet) cm–1: 3433 (w), 3126 (w), 2950 (s), 2925 (s), 2870 (m), 2848 (m), 1539 (w), 1456 (m), 1373 (w), 1262 (w), 1213 (w), 1097 (w), 1007 (w), 982 (w), 845 (w), 710 (w). HRMS (ES): m/z 657.2302 [M – Br]+, calcd 657.2308. Anal. Calcd for C28H50PdBr2N6: C, 45.63; H, 6.84; N, 11.40. Found: C, 46.06; H, 6.45; N, 11.62%. [α]D25 +29.9 (c 1.00 in CHCl3).
Synthesis of trans-[1-(1S)-Menthyl-4-(allyl)-1,2,4-triazol-5-ylidene]2PdBr2 (1S,2S,5R-2b)
A mixture of 1-(1S)-menthyl-4-(allyl)-1,2,4-triazolium bromide (1S,2S,5R-2a) (0.495 g, 1.51 mmol), PdBr2 (0.200 g, 0.751 mmol), and Et3N (0.606 g, 5.99 mmol) in CH3CN (ca. 50 mL) was refluxed for 12 h. The solvent was removed under vacuum, and the crude product was purified by column chromatography using petroleum ether/EtOAc (88:12 v/v) to give the product 1S,2S,5R-2b as a light yellow solid (0.408 g, 71%). Both the 1H NMR and 13C{1H} NMR spectra showed the presence of two isomers, trans-syn and trans-anti. The major to minor isomer ratio was 3.5:2. 1H NMR (CDCl3, 400 MHz, 25 °C): (major) δ 7.86 (s, 2H, N–C(3)H–N), 6.24 (m, 2H, CH2CH=CH2), 5.73 (br, 2H, CH3C6H9CH(CH3)2), 5.45 (m, 4H, CH2CH=CH2), 5.30 (m, 2H, CH2CH=CH2), 5.07 (dq, 2H, 2JHH = 15 Hz, 3JHH = 6 Hz, CH2CH=CH2), 2.59–0.88 (m, 18H, CH3C6H9CH(CH3)2 and CH3C6H9CH(CH3)2), 1.14 (d, 6H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2), 0.83 (d, 6H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2), 0.74 (d, 6H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2). (Minor) 7.86 (s, 2H, N–C(3)H–N), 6.24 (m, 2H, CH2CH=CH2), 5.72 (br, 2H, CH3C6H9CH(CH3)2), 5.40 (m, 4H, CH2CH=CH2), 5.30 (m, 2H, CH2CH=CH2), 5.04 (dq, 2H, 2JHH = 15 Hz, 3JHH = 6 Hz, CH2CH=CH2), 2.59–0.88 (m, 18H, CH3C6H9CH(CH3)2 and CH3C6H9CH(CH3)2), 1.08 (d, 6H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2), 0.83 (d, 6H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2), 0.71 (d, 6H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2). 13C{1H} NMR (CDCl3, 100 MHz, 25 °C): (major) δ 172.3 (Pd–NCN), 140.9 (N–C(3)H–N), 132.2 (CH2CH=CH2), 120.8 (CH2CH=CH2), 60.7 (CH3C6H9CH(CH3)2), 51.7 (CH2CH=CH2), 47.6 (CH3C6H9CH(CH3)2), 41.6 (CH3C6H9CH(CH3)2), 35.5 (CH3C6H9CH(CH3)2), 29.1 (CH3C6H9CH(CH3)2), 26.2 (CH3C6H9CH(CH3)2), 24.3 (CH3C6H9CH(CH3)2), 23.4 (CH3C6H9CH(CH3)2), 22.6 (CH3C6H9CH(CH3)2), 20.3 (CH3C6H9CH(CH3)2). (Minor) δ 172.3 (Pd–NCN), 140.9 (N–C(3)H–N), 132.2 (CH2CH=CH2), 120.5 (CH2CH=CH2), 60.7 (CH3C6H9CH(CH3)2), 51.7 (CH2CH=CH2), 47.5 (CH3C6H9CH(CH3)2), 41.4 (CH3C6H9CH(CH3)2), 35.5 (CH3C6H9CH(CH3)2), 29.1 (CH3C6H9CH(CH3)2), 26.2 (CH3C6H9CH(CH3)2), 24.1 (CH3C6H9CH(CH3)2), 23.3 (CH3C6H9CH(CH3)2), 22.6 (CH3C6H9CH(CH3)2), 20.3 (CH3C6H9CH(CH3)2). IR data (KBr pellet) cm–1: 3436 (m), 3123 (m), 3051 (w), 3017 (w), 2955 (s), 2867 (s), 2848 (s), 2722 (w), 1640 (w),1538 (s), 1459 (w), 1432 (m), 1388 (w), 1369 (w), 1350 (w), 1331 (m), 1285 (w), 1245 (m), 1227 (w), 1201 (w), 1176 (m), 1139 (w), 1007 (w), 984 (m), 931 (w), 918 (m), 871 (w), 844 (w), 787 (m), 717 (w), 666 (m) 614 (w), 562 (w). HRMS (ES): m/z 681.2299 [M – Br]+, calcd 681.2308. Anal. Calcd for C30H50PdBr2N6: C, 47.35; H, 6.62; N, 11.04. Found: C, 47.72; H, 6.56; N, 11.13%. [α]D25 −41.7 (c 1.00 in CHCl3).
Synthesis of trans-[1-(1R)-Menthyl-4-(allyl)-1,2,4-triazol-5-ylidene]2PdBr2 (1R,2R,5S-2b)
A mixture of 1-(1R)-menthyl-4-(allyl)-1,2,4-triazolium bromide (1R,2R,5S-2a) (0.495 g, 1.51 mmol), PdBr2 (0.200 g, 0.751 mmol), and Et3N (0.606 g, 5.99 mmol) in CH3CN (ca. 50 mL) was refluxed for 12 h. Subsequent removal of the solvent under vacuum gave a yellow solid, which was purified by column chromatography using petroleum ether/EtOAc (88:12 v/v) to give the product 1R,2R,5S-2b as a light yellow solid (0.427 g, 75%). Both the 1H NMR and 13C{1H} NMR spectra showed the presence of two isomers, trans-syn and trans-anti. The major to minor isomer ratio was 3.5:2. 1H NMR (CDCl3, 400 MHz, 25 °C): (major) δ 7.86 (s, 2H, N–C(3)H–N), 6.24 (m, 2H, CH2CH=CH2), 5.73 (br, 2H, CH3C6H9CH(CH3)2), 5.45 (m, 4H, CH2CH=CH2), 5.30 (m, 2H, CH2CH=CH2), 5.07 (dq, 2H, 2JHH = 15 Hz, 3JHH = 6 Hz, CH2CH=CH2), 2.59–0.88 (m, 18H, CH3C6H9CH(CH3)2 and CH3C6H9CH(CH3)2), 1.14 (d, 6H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2), 0.83 (d, 6H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2), 0.74 (d, 6H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2). (Minor) 7.86 (s, 2H, N–C(3)H–N), 6.24 (m, 2H, CH2CH=CH2), 5.72 (br, 2H, CH3C6H9CH(CH3)2), 5.40 (m, 4H, CH2CH=CH2), 5.30 (m, 2H, CH2CH=CH2), 5.04 (dq, 2H, 2JHH = 15 Hz, 3JHH = 6 Hz, CH2CH=CH2), 2.59–0.88 (m, 18H, CH3C6H9CH(CH3)2 and CH3C6H9CH(CH3)2), 1.08 (d, 6H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2), 0.83 (d, 6H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2), 0.71 (d, 6H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2). 13C{1H} NMR (CDCl3, 100 MHz, 25 °C): (major) δ 172.36 (Pd–NCN), 140.91 (N–C(3)H–N), 132.23 (CH2CH=CH2), 120.76 (CH2CH=CH2), 60.73 (CH3C6H9CH(CH3)2), 51.75 (CH2CH=CH2), 47.61 (CH3C6H9CH(CH3)2), 41.61 (CH3C6H9CH(CH3)2), 35.48 (CH3C6H9CH(CH3)2), 29.12 (CH3C6H9CH(CH3)2), 26.19 (CH3C6H9CH(CH3)2), 24.30 (CH3C6H9CH(CH3)2), 23.37 (CH3C6H9CH(CH3)2), 22.61 (CH3C6H9CH(CH3)2), 20.33 (CH3C6H9CH(CH3)2). (Minor) δ 172.36 (Pd–NCN), 140.82 (N–C(3)H–N), 132.19 (CH2CH=CH2), 120.47 (CH2CH=CH2), 60.73 (CH3C6H9CH(CH3)2), 51.67 (CH2CH=CH2), 47.47 (CH3C6H9CH(CH3)2), 41.37 (CH3C6H9CH(CH3)2), 35.48 (CH3C6H9CH(CH3)2), 29.09 (CH3C6H9CH(CH3)2), 26.19 (CH3C6H9CH(CH3)2), 24.14 (CH3C6H9CH(CH3)2), 23.28 (CH3C6H9CH(CH3)2), 22.61 (CH3C6H9CH(CH3)2), 20.25 (CH3C6H9CH(CH3)2). IR data (KBr pellet) cm–1: 3445 (w), 3123 (w), 2953 (s), 2925 (s), 2869 (s), 2846 (m), 1645 (w),1538 (m), 1453 (m), 1444 (m), 1370 (m), 1202 (w), 1009 (w), 983 (m), 938 (m), 919 (w), 845 (w), 787 (m), 717 (w), 667 (m) 562 (w). HRMS (ES): m/z 681.2301 [M – Br]+, calcd 681.2308. Anal. Calcd for C30H50PdBr2N6: C, 47.35; H, 6.62; N, 11.04. Found: C, 47.75; H, 6.37; N, 11.13%. [α]D25 +39.6 (c 1.00 in CHCl3).
Synthesis of trans-[1-(1R)-Menthyl-4-(benzyl)-1,2,4-triazol-5-ylidene]2PdBr2 (1R,2R,5S-3b)
A mixture of 1-(1R)-menthyl-4-(benzyl)-1,2,4-triazolium bromide (1R,2R,5S-3a) (0.568 g, 1.50 mmol), PdBr2 (0.200 g, 0.751 mmol), and Et3N (0.606 g, 5.99 mmol) in CH3CN (ca. 50 mL) was refluxed for 12 h. The solvent was then evaporated under vacuum, and the crude mass obtained was purified by column chromatography using petroleum ether/EtOAc (88:12 v/v) to give the product 1R,2R,5S-3b as a light yellow solid (0.511 g, 79%). Both the 1H NMR and 13C{1H} NMR spectra showed the presence of two isomers, trans-syn and trans-anti.22 The major to minor isomer ratio was 3.5:2. 1H NMR (CDCl3, 400 MHz, 25 °C): (major) δ 7.65 (s, 2H, N–C(3)H–N), 7.53 (br, 2H, C6H5), 7.45 (br, 4H, C6H5), 7.35 (br, 4H, C6H5), 5.93 (br, 2H, CH2), 5.67 (br, 2H, CH3C6H9CH(CH3)2), 5.53 (br, 2H, CH2), 2.55–0.88 (m, 18H, CH3C6H9CH(CH3)2 and CH3C6H9CH(CH3)2), 1.17 (d, 6H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2), 0.84 (d, 6H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2), 0.74 (d, 6H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2). (Minor) δ 7.60 (s, 2H, N–C(3)H–N), 7.52 (br, 2H, C6H5), 7.43 (br, 4H, C6H5), 7.33 (br, 4H, C6H5), 5.95 (br, 2H, CH2), 5.69 (br, 2H, CH2), 5.65 (br, 2H, CH3C6H9CH(CH3)2), 2.55–0.88 (m, 18H, CH3C6H9CH(CH3)2 and CH3C6H9CH(CH3)2), 1.02 (d, 6H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2), 0.81 (d, 6H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2), 0.64 (d, 6H, 3JHH = 7 Hz, CH3C6H9CH(CH3)2). 13C{1H} NMR (CDCl3, 100 MHz, 25 °C): (major) δ 172.5 (Pd–NCN), 141.0 (N–C(3)H–N), 134.8 (ipso-C6H5), 129.3 (C6H5), 129.23 (C6H5), 129.20 (C6H5), 60.8 (CH2), 52.8 (CH3C6H9CH(CH3)2), 47.6 (CH3C6H9CH(CH3)2), 41.5 (CH3C6H9CH(CH3)2), 35.4 (CH3C6H9CH(CH3)2), 29.2 (CH3C6H9CH(CH3)2), 26.2 (CH3C6H9CH(CH3)2), 24.2 (CH3C6H9CH(CH3)2), 23.9 (CH3C6H9CH(CH3)2), 22.6 (CH3C6H9CH(CH3)2), 20.3 (CH3C6H9CH(CH3)2). (Minor) δ 172.46 (Pd–NCN), 140.9 (N–C(3)H–N), 134.6 (ipso-C6H5), 129.0 (C6H5), 128.9 (C6H5), 128.8 (C6H5), 60.8 (CH2), 52.76 (CH3C6H9CH(CH3)2), 47.4 (CH3C6H9CH(CH3)2), 41.4 (CH3C6H9CH(CH3)2), 35.44 (CH3C6H9CH(CH3)2), 29.1 (CH3C6H9CH(CH3)2), 26.18 (CH3C6H9CH(CH3)2), 23.99 (CH3C6H9CH(CH3)2), 23.33 (CH3C6H9CH(CH3)2), 22.6 (CH3C6H9CH(CH3)2), 20.1 (CH3C6H9CH(CH3)2). IR data (KBr pellet) cm–1: 3444 (m), 2951 (s), 2919 (s), 2867 (m), 2847 (w), 1634 (w), 1538 (w), 1498 (w), 1455 (m), 1431 (m), 1387 (w), 1369 (m), 1216 (w), 1200 (w), 1008 (w), 985 (w), 938 (w), 719 (s), 660 (w). HRMS (ES): m/z 781.2593 [M – Br]+, calcd 781.2623. Anal. Calcd for C38H54PdBr2N6: C, 53.00; H, 6.32; N, 9.76. Found: C, 53.65; H, 6.21; N, 9.57%. [α]D25 +45.0 (c 1.00 in CHCl3).
Synthesis of trans-[1-(1S)-Menthyl-4-(ethyl)-1,2,4-triazol-5-ylidene]2Pd(OCOCF3)2 (1S,2S,5R-1c)
A mixture of trans-[1-(1S)-menthyl-4-(ethyl)-1,2,4-triazol-5-ylidene]2PdBr2 (1S,2S,5R-1b) (0.230 g, 0.312 mmol) and AgOCOCF3 (0.152 g, 0.687 mmol) was stirred in CH2Cl2 (ca. 20 mL) at room temperature for 4 h. The reaction mixture was filtered through celite, and solvent was evaporated under vacuum. The residue so obtained was further purified by column chromatography using petroleum ether/EtOAc (80:20 v/v) to give the product 1S,2S,5R-1c as a colorless solid (0.152 g, 61%). 1H NMR (CDCl3, 500 MHz, 25 °C): δ 7.93 (s, 2H, N–C(3)H–N), 5.90 (br, 2H, CH3C6H9CH(CH3)2), 4.78 (br, 4H, CH2CH3), 2.09–0.98 (m, 18H, CH3C6H9CH(CH3)2 and CH3C6H9CH(CH3)2), 1.62 (br, 6H, CH2CH3), 1.06 (br, 6H, CH3C6H9CH(CH3)2), 0.74 (d, 6H, 3JHH = 6 Hz, CH3C6H9CH(CH3)2), 0.74 (br, 6H, CH3C6H9CH(CH3)2). 13C{1H} NMR (CDCl3, 100 MHz, 25 °C), δ 168.1 (Pd–NCN), 162.3 (q, 2JCF = 40 Hz, OCOCF3), 140.6 (N–C(3)H–N), 114.1 (q, 1JCF = 288 Hz, OCOCF3), 60.6 (CH3C6H9CH(CH3)2), 47.1 (CH2CH3), 43.9 (CH3C6H9CH(CH3)2), 41.4 (CH3C6H9CH(CH3)2), 35.2 (CH3C6H9CH(CH3)2), 29.5 (CH3C6H9CH(CH3)2), 26.1 (CH3C6H9CH(CH3)2), 24.5 (CH3C6H9CH(CH3)2), 22.2 (CH3C6H9CH(CH3)2), 21.6 (CH3C6H9CH(CH3)2), 20.1 (CH3C6H9CH(CH3)2), 15.5 (CH2CH3). 19F{1H} NMR (CDCl3, 470 MHz, 25 °C) δ −73.79 ( Pd–OCOCF3). IR data (KBr pellet) cm–1: 3125 (w), 3060 (w), 2960 (m), 2924 (w), 2873 (w), 2849 (w), 1696 (s), 1571 (w), 1542 (w), 1458 (w), 1406 (w), 1371 (w), 1262 (w), 1224 (w), 1189 (s), 1147 (m), 1096 (w), 1012 (w), 850 (w), 799 (w), 786 (w), 732 (w). HRMS (ES): m/z 689.2988 [M – OCOCF3]+, calcd 689.2988. Anal. Calcd for C32H50F6N6O4Pd: C, 47.85; H, 6.27; N, 10.46. Found: C, 48.34; H, 5.96; N, 10.71%. [α]D25 +47.8 (c 1.00 in CHCl3).
Synthesis of trans-[1-(1R)-Menthyl-4-(ethyl)-1,2,4-triazol-5-ylidene]2Pd(OCOCF3)2 (1R,2R,5S-1c)
A mixture of trans-[1-(1R)-menthyl-4-(ethyl)-1,2,4-triazol-5-ylidene]2PdBr2 (1R,2R,5S-1b) (0.200 g, 0.271 mmol) and AgOCOCF3 (0.132 g, 0.597 mmol) was stirred in CH2Cl2 (ca. 20 mL) at room temperature for 4 h. The reaction mixture was filtered through a pad of celite, solvent was removed under vacuum, and purified by column chromatography using petroleum ether/EtOAc (80:20 v/v) to give the product 1R,2R,5S-1c as a colorless solid (0.124 g, 57%). 1H NMR (CDCl3, 500 MHz, 25 °C): δ 7.93 (s, 2H, N–C(3)H–N), 5.90 (br, 2H, CH3C6H9CH(CH3)2), 4.78 (br, 4H, CH2CH3), 2,09–0.98 (m, 18H, CH3C6H9CH(CH3)2 and CH3C6H9CH(CH3)2), 1.62 (br, 6H, CH2CH3), 1.06 (br, 6H, CH3C6H9CH(CH3)2), 0.74 (d, 6H, 3JHH = 6 Hz, CH3C6H9CH(CH3)2), 0.74 (br, 6H, CH3C6H9CH(CH3)2). 13C{1H} NMR (CDCl3, 125 MHz, 25 °C), δ 168.5 (Pd–NCN), 162.3 (q, 2JCF = 40 Hz, OCOCF3), 140.8 (N–C(3)H–N), 114.1 (q, 1JCF = 288 Hz, OCOCF3), 60.9 (CH3C6H9CH(CH3)2), 47.5 (CH2CH3), 44.3 (CH3C6H9CH(CH3)2), 41.8 (CH3C6H9CH(CH3)2), 35.6 (CH3C6H9CH(CH3)2), 29.8 (CH3C6H9CH(CH3)2), 26.4 (CH3C6H9CH(CH3)2), 24.8 (CH3C6H9CH(CH3)2), 22.6 (CH3C6H9CH(CH3)2), 22.0 (CH3C6H9CH(CH3)2), 20.4 (CH3C6H9CH(CH3)2), 15.9 (CH2CH3). 19F{1H} NMR (CDCl3, 470 MHz, 25 °C) δ −73.88 (Pd–OCOCF3). IR data (KBr pellet) cm–1: 3445 (m), 3125 (w), 2955 (m), 2926 (m), 2871 (m), 2851 (m), 1695 (s), 1542 (w), 1457 (w), 1406 (w), 1263 (w), 1190 (s), 1149 (s), 1010 (w), 850 (w), 783 (w), 732 (w). HRMS (ES): m/z 689.2988 [M – OCOCF3]+, calcd 689.2988. Anal. Calcd for C32H50F6N6O4Pd: C, 47.85; H, 6.27; N, 10.46. Found: C, 48.26; H, 5.71; N, 10.64%. [α]D25 −48.0 (c 1.00 in CHCl3).
Synthesis of trans-[1-(1S)-Menthyl-4-(benzyl)-1,2,4-triazol-5-ylidene]2Pd(OCOCF3)2 (1S,2S,5R-3c)
A mixture of trans-[1-(1S)-menthyl-4-(benzyl)-1,2,4-triazol-5-ylidene]2PdBr2 (1S,2S,5R-3b) (0.340 g, 0.395 mmol) and AgOCOCF3 (0.192 g, 0.869 mmol) was stirred in CH2Cl2 (ca. 20 mL) at room temperature for 4 h. The reaction mixture was subjected to celite filtration. The solvent was reduced in volume. The residue so obtained was purified by column chromatography using petroleum ether/EtOAc (90:10 v/v) to give the product 1S,2S,5R-3c as a colorless solid (0.241 g, 66%). 1H NMR (CDCl3, 500 MHz, 25 °C): δ 7.63 (br, 2H, N–C(3)H–N), 7.52 (br, 6H, C6H5), 7.30 (br, 4H, C6H5), 6.22 (br, 1H, CH2), 5.99–5.87 (m, 4H, CH2 and CH3C6H9CH(CH3)2), 5.64 (br, 1H, CH2), 2.05–0.62 (m, 18H, CH3C6H9CH(CH3)2 and CH3C6H9CH(CH3)2), 1.12 (br, 6H, CH3C6H9CH(CH3)2), 0.79 (br, 12H, CH3C6H9CH(CH3)2 and CH3C6H9CH(CH3)2). 13C{1H} NMR (CDCl3, 125 MHz, 25 °C): δ 168.8 (Pd–NCN), 162.3 (q, 2JCF = 40 Hz, OCOCF3), 141.1 (N–C(3)H–N), 134.3 (ipso-C6H5), 129.5 (C6H5), 129.1 (C6H5), 129.1 (C6H5), 114.3 (q, 1JCF = 288 Hz, OCOCF3), 60.8 (CH3C6H9CH(CH3)2), 53.0 (CH2), 47.3 (CH3C6H9CH(CH3)2), 41.5 (CH3C6H9CH(CH3)2), 35.2 (CH3C6H9CH(CH3)2), 29.5 (CH3C6H9CH(CH3)2), 26.1 (CH3C6H9CH(CH3)2), 24.5 (CH3C6H9CH(CH3)2), 22.3 (CH3C6H9CH(CH3)2), 21.7 (CH3C6H9CH(CH3)2), 20.3 (CH3C6H9CH(CH3)2). 19F{1H} NMR (CDCl3, 470 MHz, 25 °C) δ −73.69 ( Pd–OCOCF3). IR data (KBr pellet) cm–1: 3114 (w), 3044 (w), 2953 (s), 2929 (m), 2873 (w), 1687 (s), 1574 (w), 1535 (w), 1498 (w), 1456 (w), 1409 (m), 1372 (w), 1305 (w), 1261 (w), 1203 (s), 1183 (s), 1075 (w), 1011 (w), 981 (w), 951 (w), 871 (w), 849 (w), 804 (w), 788 (w), 729 (m), 712 (w), 676 (w). HRMS (ES): m/z 813.3309 [M – OCOCF3]+, calcd 813.3304. Anal. Calcd for C42H54F6N6O4Pd: C, 54.40; H, 5.87; N, 9.06. Found: C, 54.31; H, 5.51; N, 8.65%. [α]D25 +40.2 (c 1.00 in CHCl3).
Synthesis of trans-[1-(1R)-Menthyl-4-(benzyl)-1,2,4-triazol-5-ylidene]2Pd(OCOCF3)2 (1R,2R,5S-3c)
A mixture of trans-[1-(1R)-menthyl-4-(benzyl)-1,2,4-triazol-5-ylidene]2PdBr2 (1R,2R,5S-3b) (0.250 g, 0.290 mmol) and AgOCOCF3 (0.141 g, 0.639 mmol) was stirred in CH2Cl2 (ca. 20 mL) at room temperature for 4 h. The reaction mixture was passed through celite, and the solvent was pumped off under vacuum. Further purification by column chromatography using petroleum ether/EtOAc (90:10 v/v) gave the product 1R,2R,5S-3c as a colorless solid (0.157 g, 58%). 1H NMR (CDCl3, 400 MHz, 25 °C): δ 7.63 (br, 2H, N–C(3)H–N), 7.32 (br, 6H, C6H5), 7.30–7.28 (m, 4H, C6H5), 6.22 (br, 2H, CH2), 5.99–5.87 (m, 4H, CH2 and CH3C6H9CH(CH3)2), 5.64 (br, 2H, CH2), 2.05–0.62 (m, 18H, CH3C6H9CH(CH3)2 and CH3C6H9CH(CH3)2), 1.12 (br, 6H, CH3C6H9CH(CH3)2), 0.79 (br, 12H, CH3C6H9CH(CH3)2 and CH3C6H9CH(CH3)2). 13C{1H} NMR (CDCl3, 100 MHz, 25 °C): δ 168.8 (Pd–NCN), 162.4 (q, 2JCF = 40 Hz, OCOCF3), 141.1 (N–C(3)H–N), 134.3 (ipso-C6H5), 129.5 (C6H5), 129.13 (C6H5), 129.08 (C6H5), 114.3 (q, 1JCF = 288 Hz, OCOCF3), 60.8 (CH3C6H9CH(CH3)2), 53.0 (CH2), 47.2 (CH3C6H9CH(CH3)2), 41.5 (CH3C6H9CH(CH3)2), 35.2 (CH3C6H9CH(CH3)2), 29.9 (CH3C6H9CH(CH3)2), 26.1 (CH3C6H9CH(CH3)2), 24.4 (CH3C6H9CH(CH3)2), 22.3 (CH3C6H9CH(CH3)2), 21.7 (CH3C6H9CH(CH3)2), 20.3 (CH3C6H9CH(CH3)2). 19F{1H} NMR (CDCl3, 470 MHz, 25 °C) δ −73.69 ( Pd–OCOCF3). IR data (KBr pellet) cm–1: 3444 (m), 3114 (w), 2952 (s), 2925 (m), 2873 (m), 2847 (w), 1687 (s), 1536 (w), 1456 (m), 1409 (m), 1375 (w), 1184 (s), 1010 (w), 849 (w), 788 (w), 729 (m), 714 (m), 694 (w). HRMS (ES): m/z 813.3306 [M – OCOCF3]+, calcd 813.3304. Anal. Calcd for C42H54F6N6O4Pd: C, 54.40; H, 5.87; N, 9.06. Found: C, 53.76; H, 5.59; N, 8.72%. [α]D25 −44.4 (c 1.00 in CHCl3).
Antitumor Studies
Materials
cis-Diamineplatinum(II)dichloride (cisplatin), sulforhodamine B (SRB), propidium iodide, mouse monoclonal anti-β actin IgG, Hoechst 33258, and bovine serum albumin (BSA) were purchased from Sigma-Aldrich (St. Louis, MO). Alexa Fluor 555 goat anti-rabbit IgG and fetal bovine serum (FBS) were purchased from Molecular Probes, Invitrogen (Eugene, OR). Rabbit polyclonal anti-PARP-1 IgG, mouse monoclonal anti-p53 IgG, and mouse monoclonal anti-p21 IgG were purchased from Santa Cruz Biotechnology (CA). Horseradish peroxidase (HRP)-linked horse anti-mouse IgG and rabbit monoclonal anti-γ-H2AX IgG were purchased from Cell Signaling Technology. Horseradish peroxidase-linked goat anti-rabbit IgG was purchased from Bio-Rad. SuperSignal West Pico Chemiluminescent Substrate was purchased from Thermo Fisher Scientific. All other reagents were of analytical grade.
Methods
Cell Culture
EMT6/AR1 cells were purchased from Sigma-Aldrich (St. Louis, MO) and were cultured in minimum essential medium containing 1 mg/mL doxorubicin. MCF-7, MCF10A, HeLa, A549, B16F10, and L929 were purchased from National Centre for Cell Science (NCCS), Pune, India. mtrDNA sequence analysis was done by NCCS to confirm the species, and cells were tested free from mycoplasma. MCF-7, HeLa, B16F10, and L929 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) (HiMedia, India). A549 cells were cultured in F-12k medium (Kaighn’s Modification of Ham’s F-12 Medium) (HiMedia, India). MCF10A cells were cultured in 1:1 mixture of DMEM/F-12 medium (HiMedia) supplemented with 20 ng/mL epidermal growth factor, 0.5 μg/mL hydrocortisone, and 10 μg/mL insulin. The media were supplemented with 10% (v/v) FBS, 1.5 g/L sodium bicarbonate, and 1% (v/v) antibiotic–antimycotic solution, as described earlier.35 Cells were grown in a 37 °C incubator in a humidified chamber (Sanyo, Tokyo, Japan) of 5% CO2.
Cell-Proliferation Assay
The palladium N-heterocyclic carbene complexes, (1S,2S,5R)-(1–3)b and (1R,2R,5S)-(1–3)b, (1S,2S,5R)-(1, 3)c and (1R,2R,5S)-(1, 3)c, were dissolved in 100% dimethyl sulfoxide (DMSO) and evaluated for their antiproliferative property in culture. Cells were seeded in 96-well plates at a density of 1 × 104 cells/well. After the cells were attached, they were incubated without or with different concentrations of the complexes at 37 °C for one cell cycle (48 h). The effect of the compounds on the proliferation of the cells was determined using sulforhodamine B assay36,37 by measuring the absorbance at 520 nm using a SpectraMax M2e Microplate reader, as described earlier. IC50 was calculated as the concentration of the complex that inhibited the proliferation of cells by 50% relative to the untreated cells. The inhibitory effect of the most potent complex (1R,2R,5S)-1c on HeLa, A549, B16F10, EMT6/AR1, L929, and MCF10A cells after 24, 22, 20, 15, 14, and 16 h, respectively, was determined by sulforhodamine B assay as described above.
Western Blot Analysis
MCF-7 cells (5 mL) were seeded at a density of 1.5 × 105 cells/mL in 60 mm cell-culture dishes. After cell attachment, they were incubated with 4 and 8 μM (1R,2R,5S)-1c and 4 μM cisplatin for 48 h. The cells were then processed, and the whole cell lysate was isolated as described earlier.38 An equal amount of total protein was resolved on sodium dodecyl sulfate-polyacrylamide gel electrophoresis and electroblotted onto a poly(vinylidene difluoride) membrane (Millipore). Immunoblotting was performed using specific antibodies against PARP-1 and β-actin. The blots were developed by chemiluminescence using HRP-conjugated secondary IgG.
Immunofluorescence Microscopy
MCF-7 (5 × 104 cells/well) cells were seeded on glass cover slip in a 24-well cell-culture plate. After the cells were attached, they were incubated with 4 and 8 μM (1R,2R,5S)-1c and 4 μM cisplatin for 36 h and immunostaining was performed using specific antibodies as described earlier.38,39 Cells were stained with anti-p53 IgG (1:300) and anti-p21 IgG (1:300) for 2 h at 37 °C. All of the antibody dilutions were made in 2% BSA and added after blocking cells with 2% BSA. Primary antibody incubation was followed by Alexa Fluor 568-conjugated sheep anti-mouse secondary IgG (1:400) incubation for 1 h at 37 °C. Hoechst 33258 was used to stain the DNA. To determine DNA damage, cells were immunostained using anti-γ-H2AX IgG (1:200), followed by Alexa Flour 555 goat anti-rabbit secondary IgG (1:400). ImageJ software was used to calculate the fluorescence intensity. A total of 500 cells were scored in each case. For mitotic index (percentage of cells in mitosis) calculation, the cells were scored on the basis of DNA morphology and plotted using graph pad, and 1000 cells were scored in each case.
Reactive Oxygen Species (ROS) Assay
Reactive oxygen species (ROS) production in cells when treated with (1R,2R,5S)-1c was estimated by ROS assay using 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA).40 MCF-7 cells (5 mL) were seeded in 60 mm cell-culture dishes at a density of 1.5 × 105 cells/mL. After cell attachment, they were incubated with 4 and 8 μM (1R,2R,5S)-1c for 24 h. Cells treated with 30 μM hydrogen peroxide were used as a positive control. Following 24 h of incubation, the cells were processed and analyzed using flow cytometry.40
Live–Dead Assay and Cell-Cycle Analysis by Flow Cytometry
MCF-7 cells (5 mL) were seeded in 60 mm cell-culture dishes at a density of 1.5 × 105 cells/mL. Cells were incubated with 4 and 8 μM (1R,2R,5S)-1c for 48 h for live–dead assay. The cells were incubated with 2 and 4 μM (1R,2R,5S)-1c for 36 h for cell-cycle analysis. After the required incubation, cells were processed and analyzed by flow cytometry, as described earlier.41 Cell-cycle data were analyzed using FlowJo software.
Statistical Analysis
Significance test analysis was done using the Student t-test.
Acknowledgments
The authors acknowledge Department of Science and Technology (File No.: EMR/2014/000254), New Delhi, and DAE-SRC fellowship from Board of Research in Nuclear Sciences, Government of India, for financial support of this research. They gratefully acknowledge the Single Crystal X-ray Diffraction Facility, Department of Chemistry, IIT Bombay, India, for the crystallographic characterization data and FACS facility and Centre for Research in Nanotechnology and Science (CRNTS), IIT Bombay, for the flow cytometry data. A.K., A.P.P., and M.K.G. acknowledge CSIR, New Delhi, for research fellowship. Most of the work is covered in Indian patent Application No.: 201721000741 (filed on Jan 7, 2017).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b00688.
1H NMR, 13C{1H} NMR, 19F{1H} NMR, IR, HRMS, and CHN data of 1,2,4-triazolium bromide salts, (1S,2S,5R)-2a and (1R,2R,5S)(1–3)a, and the palladium complexes (1S,2S,5R)-2b, (1R,2R,5S)(1–3)b, (1S,2S,5R)-1c, (1R,2R,5S)-1c, (1S,2S,5R)-3c, and (1R,2R,5S)-3c; X-ray metrical data comparison table; ORTEP plots of (1S,2S,5R)-2b, (1R,2R,5S)(1–3)b (1S,2S,5R)-3c, and (1R,2R,5S)-3c (PDF)
X-ray crystallographic data (CIF)
Author Present Address
# Department of Chemistry, IIT Kanpur, Kanpur 208016, India (M.K.G.).
Author Present Address
⊥ Department of Chemistry, University of Delhi, Delhi 110007, India (A.K.).
Author Contributions
∥ A.K. and A.N. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Siegel R. L.; Miller K. D.; Jemal A. Cancer statistics, 2016. Ca-Cancer J. Clin. 2016, 66, 7–30. 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- Seyed M. A.; Jantan I.; Bukhari S. N. A.; Vijayaraghavan K. A Comprehensive Review on the Chemotherapeutic Potential of Piceatannol for Cancer Treatment, with Mechanistic Insights. J. Agric. Food Chem. 2016, 64, 725–737. 10.1021/acs.jafc.5b05993. [DOI] [PubMed] [Google Scholar]
- Felth J.; Rickardson L.; Rosén J.; Wickström M.; Fryknäs M.; Lindskog M.; Bohlin L.; Gullbo J. Cytotoxic Effects of Cardiac Glycosides on Colon Cancer Cells, Alone and in Combination with Standard Chemotherapeutic Drugs. J. Nat. Prod. 2009, 72, 1969–1974. 10.1021/np900210m. [DOI] [PubMed] [Google Scholar]
- Casero R. A. Jr.; Woster P. M. Terminally Alkylated Polyamine Analogues as Chemotherapeutic Agents. J. Med. Chem. 2001, 44, 1–26. 10.1021/jm000084m. [DOI] [PubMed] [Google Scholar]
- Farrell N. P. Multi-platinum anti-cancer agents. Substitution-inert compounds for tumor selectivity and new targets. Chem. Soc. Rev. 2015, 44, 8773–8785. 10.1039/C5CS00201J. [DOI] [PubMed] [Google Scholar]
- Page S. Ruthenium compounds as anticancer agents. Educ. Chem. 2012, 49, 26–29. [Google Scholar]
- Gasser G.; Ott I.; Metzler-Nolte N. Organometallic anticancer compounds. J. Med. Chem. 2011, 54, 3–25. 10.1021/jm100020w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg G. M.; Grothaus P. G.; Newman D. J. Impact of Natural Products on Developing New Anti-Cancer Agents. Chem. Rev. 2009, 109, 3012–3043. 10.1021/cr900019j. [DOI] [PubMed] [Google Scholar]
- Narang A. S.; Desai D. S.. Anticancer Drug Development Unique Aspects of Pharmaceutical Development; Springer, 2009; pp 49–92. [Google Scholar]
- van Rijt S. H.; Sadler P. J. Current applications and future potential for bioinorganic chemistry in the development of anticancer drugs. Drug Discov. Today 2009, 14, 1089–1097. 10.1016/j.drudis.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussbaumer S.; Bonnabry P.; Veuthey J.-L.; Fleury-Souverain S. Analysis of anticancer drugs: A review. Talanta 2011, 85, 2265–2289. 10.1016/j.talanta.2011.08.034. [DOI] [PubMed] [Google Scholar]
- Srivastava V.; Negi A. S.; Kumar J. K.; Gupta M. M.; Khanuja S. P. S. Plant-based anticancer molecules: A chemical and biological profile of some important leads. Bioorg. Med. Chem. 2005, 13, 5892–5908. 10.1016/j.bmc.2005.05.066. [DOI] [PubMed] [Google Scholar]
- Lee K.-H. Discovery and Development of Natural Product-Derived Chemotherapeutic Agents Based on a Medicinal Chemistry Approach. J. Nat. Prod. 2010, 73, 500–516. 10.1021/np900821e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberlies N. H.; Kroll D. J. Camptothecin and Taxol: Historic Achievements in Natural Products Research. J. Nat. Prod. 2004, 67, 129–135. 10.1021/np030498t. [DOI] [PubMed] [Google Scholar]
- Mjos K. D.; Orvig C. Metallodrugs in Medicinal Inorganic Chemistry. Chem. Rev. 2014, 114, 4540–4563. 10.1021/cr400460s. [DOI] [PubMed] [Google Scholar]
- Wheate N. J.; Walker S.; Craig G. E.; Oun R. The status of platinum anticancer drugs in the clinic and in clinical trials. Dalton Trans. 2010, 39, 8113–8127. 10.1039/c0dt00292e. [DOI] [PubMed] [Google Scholar]
- Liebmann J. E.; Cook J. A.; Lipschultz C.; Teague D.; Fisher J.; Mitchell J. B. Cytotoxic studies of paclitaxel (Taxol) in human tumor cell lines. Br. J. Cancer 1993, 68, 1104–1109. 10.1038/bjc.1993.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardito S.; Isella C.; Medico E.; Marchì L.; Bevilacqua E.; Hatzoglou M.; Bussolati O.; Franchi-Gazzola R. The Thioxotriazole Copper(II) Complex A0 Induces Endoplasmic Reticulum Stress and Paraptotic Death in Human Cancer Cells. J. Biol. Chem. 2009, 284, 24306–24319. 10.1074/jbc.M109.026583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray S.; Mohan R.; Singh J. K.; Samantaray M. K.; Shaikh M. M.; Panda D.; Ghosh P. Anticancer and antimicrobial metallopharmaceutical agents based on palladium, gold, and silver N-heterocyclic carbene complexes. J. Am. Chem. Soc. 2007, 129, 15042–15053. 10.1021/ja075889z. [DOI] [PubMed] [Google Scholar]
- Hartinger C. G.; Zorbas-Seifried S.; Jakupec M. A.; Kynast B.; Zorbas H.; Keppler B. K. From bench to bedside – preclinical and early clinical development of the anticancer agent indazolium trans-[tetrachlorobis(1H-indazole)ruthenate(III)] (KP1019 or FFC14A). J. Inorg. Biochem. 2006, 100, 891–904. 10.1016/j.jinorgbio.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Hartinger C. G.; Jakupec M. A.; Zorbas-Seifried S.; Groessl M.; Egger A.; Berger W.; Zorbas H.; Dyson P. J.; Keppler B. K. KP1019, a new redox-active anticancer agent – preclinical development and results of a clinical phase I study in tumor patients. Chem. Biodiversity 2008, 5, 2140–2155. 10.1002/cbdv.200890195. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Prakasham A. P.; Gangwar M. K.; Vishnoi P.; Butcher R. J.; Ghosh P. An Efficient Synthetic Approach to trans-(NHC)2Pd(R)Br Type Complexes and Their Use in Suzuki–Miyaura Cross-Coupling Reactions. Eur. J. Inorg. Chem. 2017, 2017, 2144–2154. 10.1002/ejic.201700017. [DOI] [Google Scholar]
- Wang C.-H.; Shih W.-C.; Chang H. C.; Kuo Y.-Y.; Hung W.-C.; Ong T.-G.; Li W.-S. Preparation and characterization of amino-linked heterocyclic carbene palladium, gold, and silver complexes and their use as anticancer agents that act by triggering apoptotic cell death. J. Med. Chem. 2011, 54, 5245–5249. 10.1021/jm101096x. [DOI] [PubMed] [Google Scholar]
- Wing R.; Drew H.; Takano T.; Broka C.; Tanaka S.; Itakura K.; Dickerson R. E. Crystal structure analysis of a complete turn of B-DNA. Nature 1980, 287, 755–758. 10.1038/287755a0. [DOI] [PubMed] [Google Scholar]
- Germain M.; Affar E. B.; D’Amours D.; Dixit V. M.; Salvesen G. S.; Poirier G. G. Cleavage of Automodified Poly(ADP-ribose) Polymerase during Apoptosis: Evidence for Involvement of Caspase-7. J. Biol. Chem. 1999, 274, 28379–28384. 10.1074/jbc.274.40.28379. [DOI] [PubMed] [Google Scholar]
- Soldani C.; Scovassi A. I. Poly(ADP-ribose) polymerase-1 cleavage during apoptosis: An update. Apoptosis 2002, 7, 321–328. 10.1023/A:1016119328968. [DOI] [PubMed] [Google Scholar]
- Mueller S.; Schittenhelm M.; Honecker F.; Malenke E.; Lauber K.; Wesselborg S.; Hartmann J. T.; Bokemeyer C.; Mayer F. Cell-cycle progression and response of germ cell tumors to cisplatin in vitro. Int. J. Oncol. 2006, 29, 471–479. 10.3892/ijo.29.2.471. [DOI] [PubMed] [Google Scholar]
- Mariotti L. G.; Pirovano G.; Savage K. I.; Ghita M.; Ottolenghi A.; Prise K. M.; Schettino G. Use of the γ-H2AX assay to investigate DNA repair dynamics following multiple radiation exposures. PLoS One 2013, 8, e79541 10.1371/journal.pone.0079541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang M. A.; So E.-Y.; Simons A. L.; Spitz D. R.; Ouchi T. DNA damage induces reactive oxygen species generation through the H2AX-Nox1/Rac1 pathway. Cell Death Dis. 2012, 3, e249 10.1038/cddis.2011.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Roper M. G. Measurement of DCF fluorescence as a measure of reactive oxygen species in murine islets of Langerhans. Anal. Methods 2014, 6, 3019–3024. 10.1039/C4AY00288A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamzami N.; Marchetti P.; Castedo M.; Decaudin D.; Macho A.; Hirsch T.; Susin S. A.; Petit P. X.; Mignotte B.; Kroemer G. Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J. Exp. Med. 1995, 182, 367–377. 10.1084/jem.182.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C.-C.; Bratton S. B. Regulation of the Intrinsic Apoptosis Pathway by Reactive Oxygen Species. Antioxid. Redox Signaling 2013, 19, 546–558. 10.1089/ars.2012.4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J.; Zhang L. The transcriptional targets of p53 in apoptosis control. Biochem. Biophys. Res. Commun. 2005, 331, 851–858. 10.1016/j.bbrc.2005.03.189. [DOI] [PubMed] [Google Scholar]
- Takimoto R.; El-Deiry W. S. DNA replication blockade impairs p53-transactivation. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 781–783. 10.1073/pnas.98.3.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai A.; Surolia A.; Panda D. An antitubulin agent BCFMT inhibits proliferation of cancer cells and induces cell death by inhibiting microtubule dynamics. PLoS One 2012, 7, e44311 10.1371/journal.pone.0044311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skehan P.; Storeng R.; Scudiero D.; Monks A.; McMahon J.; Vistica D.; Warren J. T.; Bokesch H.; Kenney S.; Boyd M. R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- Mohan R.; Panda D. Kinetic Stabilization of Microtubule Dynamics by Estramustine Is Associated with Tubulin Acetylation, Spindle Abnormalities, and Mitotic Arrest. Cancer Res. 2008, 68, 6181–6189. 10.1158/0008-5472.CAN-08-0584. [DOI] [PubMed] [Google Scholar]
- Rathinasamy K.; Panda D. Kinetic stabilization of microtubule dynamic instability by benomyl increases the nuclear transport of p53. Biochem. Pharmacol. 2008, 76, 1669–1680. 10.1016/j.bcp.2008.09.001. [DOI] [PubMed] [Google Scholar]
- Asthana J.; Kapoor S.; Mohan R.; Panda D. Inhibition of HDAC6 Deacetylase Activity Increases Its Binding with Microtubules and Suppresses Microtubule Dynamic Instability in MCF-7 Cells. J. Biol. Chem. 2013, 288, 22516–22526. 10.1074/jbc.M113.489328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasidharan S.; Bahadur D.; Srivastava R. Protein-Poly(amino acid) Nanocore–Shell Mediated Synthesis of Branched Gold Nanostructures for Computed Tomographic Imaging and Photothermal Therapy of Cancer. ACS Appl. Mater. Interfaces 2016, 8, 15889–15903. 10.1021/acsami.6b03428. [DOI] [PubMed] [Google Scholar]
- Rai A.; Kapoor S.; Naaz A.; Santra M. K.; Panda D. Enhanced stability of microtubules contributes in the development of colchicine resistance in MCF-7 cells. Biochem. Pharmacol. 2017, 132, 38–47. 10.1016/j.bcp.2017.02.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.