

Abstract
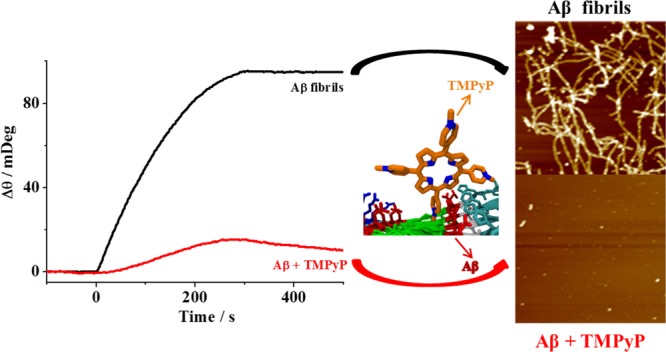
The aggregation or misfolding of amyloid-β (Aβ) is a major pathological hallmark of Alzheimer’s disease (AD). The regulation of Aβ aggregation is thought to be an effective strategy for AD treatment. The capability of a water-soluble porphyrin, 5,10,15,20-tetrakis(N-methyl-4-pyridyl)porphyrin (TMPyP), to inhibit Aβ aggregation and to lower Aβ-induced toxicity was demonstrated. As evidenced by surface plasmon resonance and circular dichroism, TMPyP can not only disrupt Aβ aggregation but also disassemble the preformed Aβ aggregates. The atomic force microscopy imaging proves that TMPyP inhibits the formation of both oligomers and fibrils. Molecular dynamic simulations provide an insight into the interaction between TMPyP and Aβ at the molecular level. The half-maximal inhibitory concentrations of TMPyP acting on the oligomers and fibrils were determined to be 0.6 and 0.43 μM, respectively. As a member of porphyrin family, TMPyP is of rather low cytotoxicity, and the cytotoxicity of the Aβ aggregates was also relieved upon coincubation with TMPyP. The excellent performance of TMPyP thus makes it a potential drug candidate for AD therapy.
Introduction
Alzheimer’s disease (AD) is one of the common neurodegenerative disorders characterized by the accumulation of amyloid-β (Aβ) in brain.1,2 Aβ is composed of 39–42 amino acids and produced by the cleavage of the amyloid precursor protein (APP) by β- and γ-secretases.3 The aggregation of Aβ leads to the formation of fibrillar deposits known as senile plaques. The amyloid aggregates, which are self-assembled from misfolded Aβ, have been presumed to affect the structure and function of neuronal cells and to stimulate cell apoptosis, leading to synaptic dysfunction and neurodegeneration.4,5
The suppression of Aβ production or inhibition of Aβ aggregation is considered as potential strategies for preventing and treating AD.1 The production of Aβ can be suppressed by decreasing the expression of APP6 or inhibiting the activity of β- and γ-secretases.7,8 On the other hand, once generated, Aβ is readily aggregated, and the development of effective inhibitors against Aβ aggregation or drug candidates that dissociate toxic Aβ aggregates plays an important role in AD treatment.9,10 Short peptides (β-sheet breaker peptides)11,12 and small organic molecules8,13−16 have been explored for their inhibitory abilities. For example, short peptides of Aβ(17–21) (LVFFA)11,12 and Aβ(39–42) (VVIA)17 have been shown to reduce Aβ aggregation and to alleviate Aβ-induced neurotoxicity. Small molecules, such as anthocyanins,18 resveratrol,15 carotenoid,16 and tabersonine,8 have proven to inhibit amyloid fibrillation via specific aromatic and hydrophobic interactions. However, peptide-based inhibitors are difficult to cross the blood–brain barrier (BBB) and most small-molecule-based modulators are not suitable for AD therapy due to their high cytotoxicity.9 Developing less cytotoxic and BBB-permeable therapeutic candidates for AD is still a challenge.
Porphyrins possess physiological activity and widely exist in organisms.19 Howlett et al. found that the heme-related porphyrins could inhibit Aβ aggregation, leading to reduced cytotoxicity.20 The half-maximal inhibitory concentration (IC50) of 0.2 μM was attained for the inhibition of Aβ aggregation by ferric dehydroporphyrin IX.21 The perturbation of Aβ aggregation was ascribed to the π–π interactions between the porphyrin ring of heme and the Phe19 residue of Aβ.22 The regulation of Aβ aggregation by photosensitizing meso-tetra(4-sulfonatophenyl)phorphyrin (TPPS) was also reported and TPPS could suppress the neural cell death and synaptic toxicity.23 However, some of the porphyrins are insoluble and not easily absorbed by human bodies. To develop water-soluble drug candidates with good biocompatibility and low cytotoxicity and to further unravel the interaction mechanism between porphyrins and Aβ, the effect of 5,10,15,20-tetrakis(N-methyl-4-pyridyl)porphyrin (TMPyP), a planar and water-soluble cationic porphyrin, on the aggregation properties of Aβ was examined in detail.
Surface plasmon resonance (SPR) is highly sensitive to the tiny changes in the refractive index or thickness associated with a biomolecular interaction.24 By immobilizing the capture antibodies that are specific to the Aβ oligomers and fibrils in separate fluidic channels, an SPR assay for real-time monitoring of Aβ aggregation was proposed by our group.25 In this study, the inhibition of Aβ aggregation and dismantling of the preformed Aβ aggregates by TMPyP were investigated by SPR and other methods. The possible binding sites of TMPyP to Aβ were determined by molecular dynamic simulations at the molecular level.
Results and Discussion
TMPyP Inhibits Aβ Aggregation and Dismantles the Aggregates
Amyloid aggregation and the inhibition effect of TMPyP were first monitored by thioflavin-T (ThT) fluorescence assay9 (Figure 1A). As shown by the black curve, the fluorescence intensity of ThT upon incubation with 6.2 μM Aβ increased with the incubation time, and a plateau was reached beyond 48 h. When 6.2 μM TMPyP was coincubated with 6.2 μM Aβ, a much lower fluorescence intensity of ThT was attained (red curve), indicating that TMPyP interfered with ThT binding to amyloid fibrils. Similar trend was obtained in the case of TMPyP alone (blue curve). As can be seen by the magenta curve, the incubation of TMPyP with the preaggregated TMPyP (2 h) leads to a lower fluorescence intensity in comparison with that of Aβ alone (black curve), indicating that TMPyP was capable of dismantling the preformed fibrils. The ThT fluorescence assay is useful for monitoring the progression of fibril formation; however, it is not an effective tool for the detection of soluble and neurotoxic oligomers. Time-dependent SPR sensorgrams at the sensor chips preimmobilized with A11 antibody (Figure 1B) and OC antibody (Figure 1C) were acquired upon injection of the incubated samples. The A11 and OC antibodies were capable of recognizing the oligomers and fibrils of Aβ, respectively.25−27 The Aβ samples with (red curves) and without (black curves) TMPyP were serially flowed over the two channels and the amount of the bound species was measured by examining the difference in the baseline SPR angles before and after the injection. The nonspecific adsorption (2–3 mDeg) was subtracted upon injection of the incubated samples onto the 11-mercaptoundecanoic acid (MUA)-covered sensor chips. In the absence of TMPyP, the SPR signals of 20 mDeg in curve a of Figure 1B and 15 mDeg in curve a of Figure 1C were attained, indicating that the Aβ samples are monomer-dominated at the time point of 0 h, although some oligomers and fibrils were preformed during the storage. Upon incubation of the Aβ samples alone for 6 h, the significantly increased SPR signals in both channels suggest the formation of well-ordered oligomers and fibrils (curve b in Figure 1B,C). Interestingly, the incubation of Aβ with TMPyP for 6 h remarkably decreased the SPR signals (curve c in Figure 1B,C), suggesting that TMPyP could inhibit Aβ oligomerization and fibrillation. Such an inhibition process could be interpreted as follows: TMPyP inhibits the formation of Aβ oligomers, and the lowered levels of the oligomers prevent the fibril formation. Furthermore, we noticed that upon incubation of the preaggregated Aβ (6 h) with TMPyP for 2 h, the smaller SPR signals in curve d than those in curve b of Figure 1B,C indicate that TMPyP could dismantle the mature aggregates of Aβ. Taken together, TMPYP could not only inhibit Aβ aggregation but also disrupt the preformed aggregates.
Figure 1.

(A) Time course of ThT fluorescence assay upon incubation of 6.2 μM Aβ at 37 °C in the absence (black curve) and presence of 6.2 μM TMPyP (red curve). The magenta curve was attained with the addition of 6.2 μM TMPyP into the preaggregated Aβ (2 h). The blue curve corresponds to the case of 6.2 μM TMPyP. Error bars are the standard deviations of the three replicates. (B, C) SPR sensorgrams showing the capture of (B) Aβ oligomers by A11 antibody and (C) Aβ fibrils by OC antibody. Ten micromolar concentration of Aβ was incubated in the absence (curves a and b) and presence (curve c) of TMPyP for 0 h (curve a) and 6 h (curves b and c) in 10 mM phosphate buffer (pH 7.4) at 37 °C. The SPR sensorgram acquired upon incubation of the preaggregated Aβ (6 h) with TMPyP for 2 h is shown as curve d.
Influence of TMPyP on the Secondary Structure of Aβ
Next, we assessed the conformational change of Aβ in the absence (Figure 2A) and presence (Figure 2B) of TMPyP by circular dichroism (CD) spectroscopy. At 0 h, Aβ exhibits natively unstructured conformation, as evidenced by the characteristic peak at 197 nm (curve a in Figure 2A). After incubation for 3 h, the disappearance of the 197 nm peak is accompanied by the appearance of a new negative peak at 215 nm (curve b in Figure 2A), which indicates the formation of β-sheet-structured oligomers.5 However, in the presence of TMPyP, Aβ reserves the unstructured conformation at 0 h (curve a in Figure 2B), and after incubation with TMPyP for 3 h, the negative peak at 215 nm disappears (curve b in Figure 2B). Most dramatically, upon incubation of the preincubated Aβ (3 h) with TMPyP for 2 h, less β-sheet aggregates with much attenuated negative peak at 215 nm were produced (curve c in Figure 2B), suggesting that TMPyP could slow down the formation of the β-sheet structures. Thus, TMPyP serves as a potent inhibitor of the β-sheet formation (i.e., dimerization and oligomerization), as characterized by CD and SPR results. The contention was also supported by UV–vis spectroscopy (Figure 2C). Aβ does not show any absorbance peak over the wavelength range examined (curve a). The incorporation of 10 μM Aβ into 10 μM TMPyP shifted the absorbance peak of TMPyP from 422 to 429 nm (curves b and c), suggesting the π–π interactions between the porphyrin ring and Aβ.28 It has been documented that the porphyrin ring plays an important role in the prevention of Aβ aggregation by heme.22,28
Figure 2.

CD spectra of 25 μM Aβ (A) incubated for 0 h (a) and 3 h (b) and (B) coincubated with TMPyP for 0 h (a) and 3 h (b). (B) CD spectrum upon incubation of the preaggregated Aβ (3 h) with TMPyP for 2 h is shown as curve c. (C) UV–vis absorption spectra of (a) 10 μM Aβ preincubated for 3 h, (b) 10 μM TMPyP, (c) 10 μM TMPyP incubated with 10 μM Aβ for 3 h.
Morphology of the Aβ Aggregates with and without TMPyP
The Aβ aggregates with and without TMPyP were clearly resolved from atomic force microscopy (AFM) imaging (Figure 3). Monomeric Aβ is soluble, and no aggregates were formed (Figure 3A). After 12 h incubation, spherical particles with diameters of about 2.5 nm were observed (Figure 3B). With the increase in the incubation time to 36 h, long, mature fibrils of Aβ with the nominal height of 5–6 nm were attained (Figure 3C). However, the morphology of the Aβ aggregates in the presence of TMPyP is totally different from that of Aβ alone. Few spherical particles were formed via incubating Aβ with TMPyP at a concentration ratio of 1:1 for 12 h (Figure 3E). With the elapse of the incubation time to 36 h, the spherical particles were still resolved and no fibrils were obtained (Figure 3F). It is clear that the incorporation of TMPyP prevents the Aβ monomers from further growing into oligomers or fibrils, consistent with our aforementioned experimental results. Note that the incubation time for AFM imaging is much longer than that for SPR and CD characterizations. The conversion of the unstructured Aβ to β-sheet containing structures, such as dimers, is a quick process, and these small Aβ oligomers could be recognized by the antibodies preimmobilized on the SPR chips. Because AFM only images well-formed oligomers and fibrils, the small Aβ oligomers could not be detected by AFM.
Figure 3.

Time-lapse AFM imaging of the oligomerization/fibrillation of 25 μM Aβ in the absence (top) and presence (bottom) of 25 μM TMPyP at 0 h (A, D), 12 h (B, E), and 36 h (C, F).
IC50 of TMPyP Inhibitor
The inhibition assay of TMPyP on the formation of Aβ oligomers (A) and fibrils (B) was conducted (Figure 4). The SPR signals as a function of TMPyP concentrations were measured and the concentration of TMPyP that causes 50% inhibition (IC50) was deduced. The IC50 values of 0.6 and 0.43 μM for TMPyP acting on the oligomers and fibrils, respectively, were attained, being lower than those of the porphyrin derivatives (3.56 and 11.0 μM)29 and other inhibitors, such as hematoxylin (1.6 μM)30 and brazilin (2.3 μM)30. It is worth noting that TMPyP suppresses the formation of both oligomers and fibrils, serving as a novel bifunctional inhibitor of Aβ aggregation. Fe-TMPyP possesses similar but weaker inhibition effect, which indicates that the metal ion center of porphyrins may not play an important role in inhibiting Aβ aggregation. However, several reports indicate that the metal ions, such as Zn2+ and Cu2+, play an important role in AD pathogenesis;31−33 thus, chelating metal ions is another way for the prevention of Aβ aggregation and the cure of AD.
Figure 4.

Dependence of the SPR responses on the concentrations of TMPyP. Ten micromolar concentration of Aβ was incubated with various concentrations of TMPyP for 6 h and the above solutions were flowed over the SPR chips precovered with A11 antibody (A) and OC antibody (B).
TMPyP Alleviates Aβ-Induced Cytotoxicity
The cell cytotoxicity of Aβ in the absence and presence of TMPyP was assessed by cell counting kit-8 (CCK-8) assay34 (Figure 5). Human neuroblastoma SH-SY5Y cells were incubated with Aβ, TMPyP, and the mixture of Aβ with different concentrations of TMPyP for 24 h and the survival rates were determined. It was found that only 71% of the cells remained viable when exposed to 20 μM Aβ for 24 h (lane 2), indicating the cytotoxictity of the Aβ aggregates. However, due to the inhibition effect of TMPyP on Aβ aggregation, reduced cytotoxicity (97%) was obtained when the cells were treated with the mixture of 20 μM Aβ and 20 μM TMPyP (lane 3). The decrease in the concentrations of TMPyP leads to a lowered cell viability (lanes 4–6), and the survival rate of the cells in the case of 20 μM Aβ and 2 μM TMPyP (lane 6) is similar with that in the presence of Aβ alone (lane 2). TMPyP possesses low toxicity and the survival rate is approximately 94% (lane 7). The cell viability assay was consistent with the SPR results in that TMPyP inhibits Aβ aggregation in a dose-dependent manner.
Figure 5.
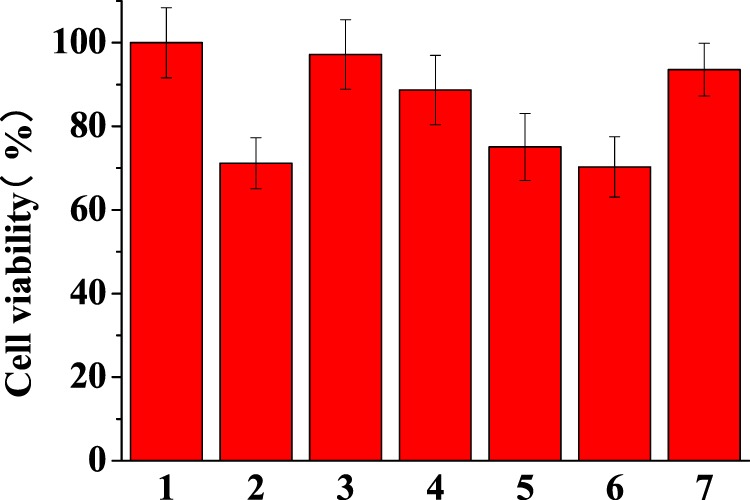
CCK-8 viability assay of human neuroblastoma SH-SY5Y cells treated with 20 μM Aβ (lane 2), 20 μM TMPyP (lane 7), and the mixture of 20 μM Aβ with different concentrations of TMPyP (lanes 3, 4, 5, and 6 correspond to 20, 10, 4, and 2 μM TMPyP, respectively) for 24 h. The SH-SY5Y cells in the cell media were taken as 100% viable (lane 1).
Binding Modes of TMPyP to Aβ
To gain a better understanding of the inhibition mechanism, we performed all-atom molecular dynamic simulations to study the binding modes of TMPyP to the Aβ pentamer (Figure 6). It can be clearly seen that TMPyP does not disturb the structural integrity of the Aβ pentamer within 30 ns. TMPyP preferentially binds to the N-terminus and the salt-bridge region of Aβ. These results suggest that the attachment of TMPyP to the Aβ pentamer hinders the elongation and lateral aggregation of the pentamer.
Figure 6.
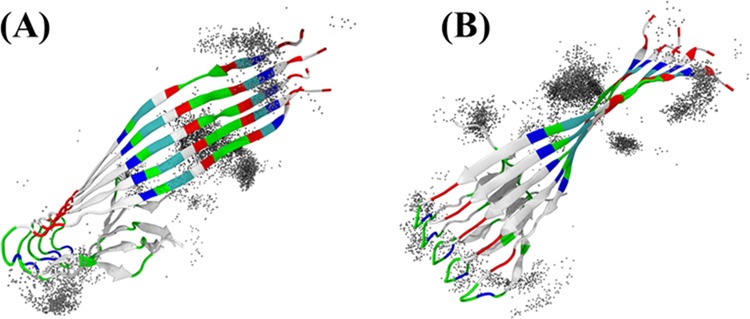
Distribution of TMPyP molecules bound to an Aβ(1–42) pentamer in (A) top and (B) side views. TMPyP molecules within 5 Å from the pentamer are shown as gray spheres.
Possible Binding Sites of TMPyP to Aβ
The inhibition mechanism and the possible binding sites were further demonstrated by molecular dynamic simulations (Figure 7). The Aβ pentamer–TMPyP complex was clustered into different structural groups using a root-mean-square deviation of 10 Å as the cutoff value, and highly populated binding sites at Ser8-His13 (region 1, 31.6%), Phe4-Ser8 (region 2, 10.6%), Asn27-Ile31 (region 3, 7.4%), Ala30-Leu34 (region 4, 6.2%), Ser8-Val12 (region 5, 6.2%), and Val39-Ala42 (region 6, 5.8%) were attained. TMPyP preferentially binds to the aromatic residues of Aβ, such as Phe4, Phe6, and Tyr10 through π–π interactions (regions 1, 2, and 5), which interferes with the ordered stacking of the β-sheets for the formation of large oligomers and fibrils.35,36 Because the hydrophobic region at the C-terminus of Aβ is capable of initiating the self-assembly of Aβ,37 the binding of TMPyP to the hydrophobic pocket (regions 3, 4, and 6) thus interferes with its aggregation (consistent with our AFM and SPR results). Furthermore, TMPyP binds to Asn27-Ile31 of Aβ (region 3), disrupting the β-sheet structure stabilized by the salt-bridge region (Asp23-Lys28) (supported by our CD data).38,39 Taken together, the insertion of TMPyP into the hydrophobic and salt-bridge regions not only inhibits the self-aggregation of Aβ but also disrupts the β-sheet formation. As a result, the elongation of the Aβ aggregates is largely prevented.
Figure 7.

Molecular dynamic simulations of the possible binding regions of TMPyP with Aβ: Ser8-His13 (region 1, 31.6%), Phe4-Ser8 (region 2, 10.6%), Asn27-Ile31 (region 3, 7.4%), Ala30-Leu34 (region 4, 6.2%), Ser8-Val12 (region 5, 6.2%), and Val39-Ala42 (region 6, 5.8%). Shown in the parenthesis are the percentages of the total population.
Conclusions
Via the specific recognition of the preimmobilized A11 and OC antibodies to the oligomers and fibrils, respectively, an SPR assay of the inhibition of Aβ aggregation by TMPyP has been proposed. TMPyP not only inhibits Aβ aggregation but also disassembles the preformed Aβ aggregates. The SPR results were further confirmed by CD, AFM, and molecular dynamic simulation characterizations. The inhibition effect might be ascribed to the π−π interactions between Aβ and TMPyP and the insertion of TMPyP into the hydrophobic and salt-bridge regions of Aβ, which interferes its aggregation and disrupts the β-sheet structure stabilized by the salt-bridge region. The IC50 values for TMPyP acting on the oligomers and fibrils were estimated to be 0.6 and 0.43 μM, respectively, being lower than those of the porphyrin derivatives and other inhibitors, such as hematoxylin and brazilin. As a member of porphyrin family, TMPyP possesses low cytotoxicity, and the cytotoxicity of the Aβ aggregates was relieved upon the incorporation of TMPyP. Thus, TMPyP not only inhibits Aβ aggregation but also alleviates amyloid-induced cytotoxicity, providing a new insight for the development of porphyrin-based therapeutic drugs for neurodegenerative diseases.
Experimental Section
Chemicals and Reagents
MUA, ethanolamine (EA), K2HPO4, KH2PO4, NaOH, N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS), and TMPyP were acquired from Sigma-Aldrich (St. Louis, MO). Aβ(1–42) was obtained from Bachem (Torrance, CA). Monoclonal antibodies against oligomers and fibrils of Aβ (A11 and OC, respectively) were obtained from Millipore Inc. (Dedham, MA). All of the reagents were of analytical grade and used without further purification. Unless otherwise stated, all of the stock solutions were prepared daily with deionized water treated with a water purification system (Simplicity185; Millipore Corp., Billerica, MA).
Solution Preparation
To effectively inhibit the aggregation of Aβ(1–42), lyophilized Aβ(1–42) samples were dissolved in 10 mM NaOH solution. Upon sonication for 1 min, the solution was centrifuged at 13 000 rpm for 30 min to remove any insoluble particles, and the supernatants were pipetted out and diluted by phosphate-buffered saline (PBS, 100 mM phosphate, pH 7.4). Aβ(1–42) or the mixture of Aβ(1–42) and TMPyP was incubated at 37 °C for different time periods before assay. OC and A11 antibodies were prepared with PBS buffer and their concentrations were maintained at 2 nM. TMPyP was dissolved in 20 mM NaOH solution. MUA and EA were dissolved in ethyl alcohol and water, respectively. EDC/NHS solution was prepared by mixing 0.4 M EDC and 0.1 M NHS in water before the activation of MUA self-assembled monolayers (SAMs).
SPR Detection
SPR assay was performed on a BI-3000 system (Biosensing Instrument Inc., Tempe, AZ). Au films coated onto BK7 glass slides were annealed in a hydrogen flame to eliminate surface contaminants. The treated Au films were immersed in 500 μM MUA solution for 24 h and the SAMs were gently rinsed with ethanol, water, and dried under nitrogen. For antibody immobilization, 200 μL EDC/NHS solution was injected onto the sensor chip for activation of the carboxyl groups on MUA, followed by the injection of A11 antibody on channel 1 and OC antibody on channel 2. Both channels were treated with 1 M EA to block the empty sites. The incubated Aβ(1–42) or the mixture of Aβ(1–42) and TMPyP was injected into the flow cell at a flow rate of 20 μL/min.
CD Characterization
CD spectra were collected on a J-815 spectropolarimeter (Jasco Inc., Tokyo) from 260 to 190 nm at a 0.5 nm interval at room temperature. Each spectrum is the average of six scans.
AFM Imaging
AFM images of Aβ incubated with and without TMPyP for a predetermined time period were obtained on a NanoIR2 system (Anasys Instruments Corporation, Santa Barbara, CA). The scanning speed was 0.5 Hz.
CCK-8 Assay
Human neuroblastoma SH-SY5Y cells with a density of 1 × 105 cells/well were seeded in triplicate in 0.1 mL culture medium in a 96-well plate that was incubated in a humidified atmosphere containing 5% (v/v) CO2 at 37 °C overnight. Next, 20 μM Aβ(1–42) or the mixture of 20 μM Aβ(1–42) with various concentrations of TMPyP was added to the cells and kept for 24 h. The SH-SY5Y cells in the cell media were used as the control. After washing and pipetting 100 μL culture medium containing 10 μL CCK-8 solution into each well, the wells were incubated for 1 h for formazan formation. The absorbance of the suspensions was measured on a microplate reader. The cell viability was attained by the percentage absorbance of Aβ(1–42)-treated groups or Aβ(1–42)/TMPyP-treated groups relative to that of the control group.
Molecular Dynamic System Construction
A complete structure of Aβ pentamer was modeled to probe the TMPyP binding sites and modes. Briefly, the initial monomer coordinates of an Aβ(17–42) peptide were derived from the NMR structure (protein data bank code 2BEG) and then the unresolved N-terminus (residues 1–16) was constructed and reassembled to Aβ(17–42) to yield a full-length Aβ(1–42) monomer with the β hairpin structure. The whole structure of Aβ monomer consists of two antiparallel β-strands (residues Val1-Ser26 and Ile31-Ala42) linked by a U-bend (residues Asn27-Ala30). The Aβ pentamer was constructed manually by stacking five monomers in parallel and registered form according to the NMR structure. To eliminate the potential collisions and gain a more reasonable initial structure, the oligomer was subjected to a molecular mechanics optimization. Five TMPyP molecules were initially and randomly placed around the relaxed Aβ pentamer with a minimal distance of 10 Å to allow them to land on optimal binding sites. The Aβ pentamer–TMPyP complex was then immersed in a cuboid box filled with TIP3P water molecules, leaving at least a 12 Å buffering zone away from any boundary of the box. Each system was neutralized by adding Cl– counter ions.
Molecular Dynamic Simulation
Simulation of the Aβ pentamer–TMPyP complex containing explicit water molecules and counter ions was performed using the AmberTools14 program (sander module) with the Amber ff14SB force field for protein and generalized AMBER force field for ligands. Restrained electrostatic potential charges for TMPyP were obtained with the PyRED server using Gaussian 09 (rev. D.01). The simulation begins with a three-phase energy minimization. In the beginning, 10 kcal/mol Å2 elastic constant was used to constrain the heavy atoms of the solute for 2000 cycles, followed by another 2000 cycles with the same position constraints on the backbone atoms of Aβ; finally, the whole system was relaxed without any constrain for 4000 cycles. After energy minimization, the system was gradually heated from 50 to 310 K in 50 ps and equilibrated at 310 K for 150 ps to adjust the size and density. Finally, 30 ns production molecular dynamic runs were conducted to examine the mutual dynamics and binding events between Aβ and TMPyP. A time step of 2 fs was used to integrate the equations of motion, permitted by constraining the fast stretching of the hydrogen atoms covalently linked to the heavy atoms. The Coulomb potentials were handled by the smooth particle-mesh Ewald method with a direct space cutoff of 9.0 Å. The same threshold value was also used for the truncation of the Lennard–Jones potentials, whereas long-range analytic corrections were applied to the energy and pressure. Molecular dynamic trajectories were saved every 2 ps for subsequent analyses. All of the calculations were performed on the MolDesigner Molecular Simulation Platform.
Acknowledgments
The authors thank the financial support of this work by the National Natural Science Foundation of China (Nos. 21575166, 21375150, and 21602054); the Chinese National Key Basic Research Program (No. 2014CB744502); and China Postdoctoral Science Foundation (No. 2015M580696).
Author Contributions
Y.F. and L.W. collected the SPR data and CD spectra. X.Y. and D.W. performed the UV–vis and AFM characterizations. Y.X. and Z.W. conducted the cytotoxicity experiments. X.Y. and J.W. directed the project, analyzed the results, and wrote the manuscript. H.T., Q.L., and Z.Z. gave some useful comments.
The authors declare no competing financial interest.
References
- Hardy J.; Selkoe D. J. Medicine - The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Selkoe D. J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [DOI] [PubMed] [Google Scholar]
- Wolfe M. S.; Xia W.; Ostaszewski B. L.; Diehl T. S.; Kimberly W. T.; Selkoe D. J. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature 1999, 398, 513–517. 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- Bokvist M.; Lindström F.; Watts A.; Gröbner G. Two types of Alzheimer’s beta-amyloid (1–40) peptide membrane interactions: Aggregation preventing transmembrane anchoring versus accelerated surface fibril formation. J. Mol. Biol. 2004, 335, 1039–1049. 10.1016/j.jmb.2003.11.046. [DOI] [PubMed] [Google Scholar]
- Terzi E.; Hölzemann G.; Seelig J. Interaction of Alzheimer beta-amyloid peptide(1–40) with lipid membranes. Biochemistry 1997, 36, 14845–14852. 10.1021/bi971843e. [DOI] [PubMed] [Google Scholar]
- Simons M.; Schwärzler F.; Lütjohann D.; Von Bergmann K.; Beyreuther K.; Dichgans J.; Wormstall H.; Hartmann T.; Schulz J. B. Treatment with simvastatin in normocholesterolemic patients with Alzheimer’s disease: A 26-week randomized, placebo-controlled, double-blind trial. Ann. Neurol. 2002, 52, 346–350. 10.1002/ana.10292. [DOI] [PubMed] [Google Scholar]
- Sambamurti K.; Marlow L.; Canet R.; Pinnix I.; Greig N. Regulation of beta secretase (BACE) activity in Alzheimer’s disease. Neurobiol. Aging 2002, 23, 188–189. [Google Scholar]
- Zhou S.; Zhou H.; Walian P. J.; Jap B. K. Regulation of gamma-secretase activity in Alzheimer’s disease. Biochemistry 2007, 46, 2553–2563. 10.1021/bi602509c. [DOI] [PubMed] [Google Scholar]
- Wong H. E.; Qi W.; Choi H.-M.; Fernandez E. J.; Kwon I. A safe, blood-brain barrier permeable triphenylmethane dye inhibits annyloid-beta neurotoxicity by generating nontoxic aggregates. ACS Chem. Neurosci. 2011, 2, 645–657. 10.1021/cn200056g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkes C. A.; Ng V.; McLaurin J. Small molecule inhibitors of abeta-aggregation and neurotoxicity. Drug Dev. Res. 2009, 70, 111–124. 10.1002/ddr.20290. [DOI] [Google Scholar]
- Tjernberg L. O.; Näslund J.; Lindqvist F.; Johansson J.; Karlström A. R.; Thyberg J.; Terenius L.; Nordstedt C. Arrest of beta-amyloid fibril formation by a pentapeptide ligand. J. Biol. Chem. 1996, 271, 8545–8548. 10.1074/jbc.271.15.8545. [DOI] [PubMed] [Google Scholar]
- Pallitto M. M.; Ghanta J.; Heinzelman P.; Kiessling L. L.; Murphy R. M. Recognition sequence design for peptidyl modulators of beta-amyloid aggregation and toxicity. Biochemistry 1999, 38, 3570–3578. 10.1021/bi982119e. [DOI] [PubMed] [Google Scholar]
- Colvin M. T.; Silvers R.; Ni Q. Z.; Can T. V.; Sergeyev I.; Rosay M.; Donovan K. J.; Michael B.; Wall J.; Linse S.; Griffin R. G. Atomic resolution structure of monomorphic abeta42 amyloid fibrils. J. Am. Chem. Soc. 2016, 138, 9663–9674. 10.1021/jacs.6b05129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kai T.; Zhang L.; Wang X.; Jing A.; Zhao B.; Yu X.; Zheng J.; Zhou F. Tabersonine inhibits amyloid fibril formation and cytotoxicity of abeta(1–42). ACS Chem. Neurosci. 2015, 6, 879–888. 10.1021/acschemneuro.5b00015. [DOI] [PubMed] [Google Scholar]
- Feng Y.; Wang X.-p.; Yang S.-g.; Wang Y.-j.; Zhang X.; Du X.-t.; Sun X.-x.; Zhao M.; Huang L.; Liu R.-t. Resveratrol inhibits beta-amyloid oligomeric cytotoxicity but does not prevent oligomer formation. Neurotoxicology 2009, 30, 986–995. 10.1016/j.neuro.2009.08.013. [DOI] [PubMed] [Google Scholar]
- Katayama S.; Ogawa H.; Nakamura S. Apricot carotenoids possess potent anti-amyloidogenic activity in vitro. J. Agric. Food Chem. 2011, 59, 12691–12696. 10.1021/jf203654c. [DOI] [PubMed] [Google Scholar]
- Hetényi C.; Szabó Z.; Klement T.; Datki Z.; Kortvélyesi T.; Zarándi M.; Penke B. Pentapeptide amides interfere with the aggregation of beta-amyloid peptide of Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2002, 292, 931–936. 10.1006/bbrc.2002.6745. [DOI] [PubMed] [Google Scholar]
- Shih P.-H.; Wu C.-H.; Yeh C.-T.; Yen G.-C. Protective effects of anthocyanins against amyloid beta-peptide-induced damage in neuro-2A cells. J. Agric. Food Chem. 2011, 59, 1683–1689. 10.1021/jf103822h. [DOI] [PubMed] [Google Scholar]
- St. Denis T. G.; Huang Y.-Y.; Hamblin M. R.. Cyclic Tetrapyrroles in Photodynamic Therapy: The Chemistry of Porphyrins and Related Compounds in Medicine; World Scientific, 2010; Vol. 27. [Google Scholar]
- Howlett D.; Cutler P.; Heales S.; Camilleri P. Hemin and related porphyrins inhibit β-amyloid aggregation. FEBS Lett. 1997, 417, 249–251. 10.1016/S0014-5793(97)01290-8. [DOI] [PubMed] [Google Scholar]
- Masuda M.; Suzuki N.; Taniguchi S.; Oikawa T.; Nonaka T.; Iwatsubo T.; Hisanaga S.; Goedert M.; Hasegawa M. Small molecule inhibitors of alpha-synuclein filament assembly. Biochemistry 2006, 45, 6085–6094. 10.1021/bi0600749. [DOI] [PubMed] [Google Scholar]
- Yuan C.; Gao Z. Abeta interacts with both the iron center and the porphyrin ring of heme: Mechanism of heme’s action on abeta aggregation and disaggregation. Chem. Res. Toxicol. 2013, 26, 262–269. 10.1021/tx300441e. [DOI] [PubMed] [Google Scholar]
- Lee B. I.; Lee S.; Suh Y. S.; Lee J. S.; Kim A.-K.; Kwon O.-Y.; Yu K.; Park C. B. Photoexcited porphyrins as a strong suppressor of beta-amyloid aggregation and synaptic toxicity. Angew. Chem., Int. Ed. 2015, 54, 11472–11476. 10.1002/anie.201504310. [DOI] [PubMed] [Google Scholar]
- Homola J. Surface plasmon resonance sensors for detection of chemical and biological species. Chem. Rev. 2008, 108, 462–493. 10.1021/cr068107d. [DOI] [PubMed] [Google Scholar]
- Yi X.; Feng C.; Hu S.; Li H.; Wang J. Surface plasmon resonance biosensors for simultaneous monitoring of amyloid-beta oligomers and fibrils and screening of select modulators. Analyst 2016, 141, 331–336. 10.1039/C5AN01864A. [DOI] [PubMed] [Google Scholar]
- Kayed R.; Head E.; Sarsoza F.; Saing T.; Cotman C. W.; Necula M.; Margol L.; Wu J.; Breydo L.; Thompson J. L.; Rasool S.; Gurlo T.; Butler P.; Glabe C. G. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol. Neurodegener. 2007, 2, 18. 10.1186/1750-1326-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R.; Head E.; Thompson J. L.; McIntire T. M.; Milton S. C.; Cotman C. W.; Glabe C. G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Atamna H. Heme binding to amyloid-beta peptide: Mechanistic role in Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 10, 255–266. 10.3233/JAD-2006-102-310. [DOI] [PubMed] [Google Scholar]
- Hirabayashi A.; Shindo Y.; Oka K.; Takahashi D.; Toshima K. Photodegradation of amyloid beta and reduction of its cytotoxicity to PC12 cells using porphyrin derivatives. Chem. Commun. 2014, 50, 9543–9546. 10.1039/C4CC03791J. [DOI] [PubMed] [Google Scholar]
- Tu Y.; Ma S.; Liu F.; Sun Y.; Dong X. Hematoxylin inhibits amyloid beta-protein fibrillation and alleviates amyloid-induced cytotoxicity. J. Phys. Chem. B 2016, 120, 11360–11368. 10.1021/acs.jpcb.6b06878. [DOI] [PubMed] [Google Scholar]
- Maiti N. C.; Jiang D.; Wain A. J.; Patel S.; Dinh K. L.; Zhou F. Mechanistic studies of Cu(II) binding to amyloid-beta peptides and the fluorescence and redox behaviors of the resulting complexes. J. Phys. Chem. B 2008, 112, 8406–8411. 10.1021/jp802038p. [DOI] [PubMed] [Google Scholar]
- Jiang D.; Men L.; Wang J.; Zhang Y.; Chickenyen S.; Wang Y.; Zhou F. Redox reactions of copper complexes formed with different beta-amyloid peptides and their neuropathalogical relevance. Biochemistry 2007, 46, 9270–9282. 10.1021/bi700508n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise-Scira O.; Xu L.; Perry G.; Coskuner O. Structures and free energy landscapes of aqueous zinc(II)-bound amyloid-beta(1–40) and zinc(II)-bound amyloid-beta(1–42) with dynamics. JBIC, J. Biol. Inorg. Chem. 2012, 17, 927–938. 10.1007/s00775-012-0909-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y.-J.; Chen Y.-Y.; Wang D.-R.; Wei C.; Guo J.; Lu D.-R.; Chu C.-C.; Wang C.-C. Redox/pH dual stimuli-responsive biodegradable nanohydrogels with varying responses to dithiothreitol and glutathione for controlled drug release. Biomaterials 2012, 33, 6570–6579. 10.1016/j.biomaterials.2012.05.062. [DOI] [PubMed] [Google Scholar]
- Gazit E. A possible role for pi-stacking in the self-assembly of amyloid fibrils. FASEB J. 2002, 16, 77–83. 10.1096/fj.01-0442hyp. [DOI] [PubMed] [Google Scholar]
- Tracz S. M.; Abedini A.; Driscoll M.; Raleigh D. P. Role of aromatic interactions in amyloid formation by peptides derived from human amylin. Biochemistry 2004, 43, 15901–15908. 10.1021/bi048812l. [DOI] [PubMed] [Google Scholar]
- Barrow C. J.; Yasuda A.; Kenny P. T. M.; Zagorski M. G. Solution conformations and aggregational properties of synthetic amyloid beta-peptides of Alzheimer’s disease. Analysis of circular dichroism spectra. J. Mol. Biol. 1992, 225, 1075–1093. 10.1016/0022-2836(92)90106-T. [DOI] [PubMed] [Google Scholar]
- Petkova A. T.; Ishii Y.; Balbach J. J.; Antzutkin O. N.; Leapman R. D.; Delaglio F.; Tycko R. A structural model for Alzheimer’s beta-amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 16742–16747. 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W.; Wu Y.-D. A strand-loop-strand structure is a possible intermediate in fibril elongation: Long time simulations of amylold-beta peptide (10–35). J. Am. Chem. Soc. 2005, 127, 15408–15416. 10.1021/ja051699h. [DOI] [PubMed] [Google Scholar]