

Abstract
The synthesis, characterization, and self-assembly of a series of biocompatible poly(methyl caprolactone-co-caprolactone)-b-poly(ethylene oxide) amphiphilic block copolymers with variable MCL contents in the hydrophobic block are described. Self-assembly gives rise to polymeric nanoparticles (PNPs) with hydrophobic cores that decrease in crystallinity as the MCL content increases, and their morphologies and sizes show nonmonotonic trends with MCL content. PNPs loaded with the anticancer drug paclitaxel (PAX) give rise to in vitro PAX release rates and MCF-7 GI50 (50% growth inhibition concentration) values that decrease as the MCL content increases. We also show for selected copolymers that microfluidic manufacturing at a variable flow rate enables further control of PAX release rates and enhances MCF-7 antiproliferation potency. These results indicate that more effective and specific drug delivery PNPs are possible through tangential efforts combining polymer synthesis and microfluidic manufacturing.
Introduction
Polymeric nanoparticles (PNPs), including micellar aggregates of block copolymers, are recognized as promising candidates for drug delivery due to their stability, morphological variability, and ease of functionalization.1−20 Efforts to design PNP nanomedicine formulations exhibiting improved bioavailability and selectivity for a range of hydrophobic drugs have focused on variation of both the chemistry10,11,13−17,21−24 and multiscale structure16,25−36 of the polymeric nanocarriers. For example, enhanced solubilization of therapeutic agents in the PNP cores of block copolymer aggregates can be achieved by the addition of pendant chains or functional groups that lead to increased affinity with the hydrophobic blocks.14,15 Further, the various applications of intermolecular (bottom-up)37−41 and external (top-down)42−47 forces to modulate the size, morphology, and internal crystallinity of PNP nanocarriers highlight the numerous and complex relationships between structure and drug delivery function.
Poly(ε-caprolactone)-b-poly(ethylene oxide) (PCL-b-PEO) is an FDA-approved amphiphilic block copolymer that is commonly applied to drug delivery investigations due to the inherent biodegradable and biocompatible properties arising from its component blocks.26,27,48−57 PEO is a hydrophilic polymer that provides water dispersability and delayed physisorption of proteins, which imparts some degree of stealthiness to PNP surfaces.16 PCL is an aliphatic polyester known to solubilize a wide range of hydrophobic drugs;16 moreover, PCL undergoes hydrolytic degradation under physiological conditions, which makes it an appropriate in vivo host for therapeutic molecules.53 The semicrystalline nature of PCL is an inherent structural feature that provides both advantages and disadvantages with respect to drug delivery.16,26,27,34−36 For instance, the presence of crystallites can increase diffusion times in self-assembled PNP cores for slower, more controlled release;26,45−47 however, crystallites also tend to impede drug solubilization, leading to lower loading efficiencies.45−47 These features highlight the need for new copolymers with hydrophobic blocks that are chemically similar to PCL but with attenuated crystallinity.
Poly(4-methyl-ε-caprolactone) and poly(6-methyl-ε-caprolactone) (PMCL) are hydrophobic polymers that are structurally similar to PCL but with a methyl group on each repeat unit that disrupts the ability of the chains to close-pack.58,59 Therefore, in contrast to PCL, both PMCL isomers are amorphous, low-Tg polymers.58−60 The synthesis, properties, self-assembly, and drug delivery applications of amphiphilic block copolymers based on PMCL hydrophobic blocks have been described by several groups.61−70 In addition, Wang et al. reported the copolymerization of 4-methyl-ε-caprolactone (MCL) and ε-caprolactone (CL) to generate hydrophobic P(MCL-co-CL) random copolymers with various ratios of MCL and CL monomers;58 characterization of their physical and biomedical properties (including degradability and biocompatibility) suggested that such copolymers with variable MCL contents could offer interesting and variable properties for biomedical applications, including drug delivery.58 However, to induce amphiphilic self-assembly, hydrophobic random copolymers of this type would need to be joined to a hydrophilic polymer block such as PEO. To the best of our knowledge, the synthesis and characterization of amphiphilic block copolymers possessing P(MCL-co-CL) hydrophobic blocks of variable MCL content have not been reported to date.
In our group, we have applied gas–liquid two-phase microfluidic reactors to the production of a wide range of PNP systems,42−44,71−74 including semicrystalline PCL-b-PEO PNPs for drug delivery.45−47,75 Within these reactors, counter-rotating vortices within the liquid phase enhance mixing and introduce flow-variable high-shear “hot spots” which provide top-down control of multiscale structure,42 including the size,43 morphology,44 and internal crystallinity45 of the resulting PNPs. We have also shown that microfluidic control of PNP structure enables important biomedical properties, including photoresponsivity,74 degradation rate,45,47 drug loading efficiency,45−47 drug release rate,45−47 and in vitro antiproliferation,47 to be tuned and optimized.
A limitation of such top-down microfluidic control of PNP colloids is that the molecular structure and chemistry of the constituent copolymers can play a dominant role in chain packing,35,36 even when external shear forces are applied to direct the self-assembly process. For instance, we have shown that microfluidic preparation of PNPs from semicrystalline PCL-b-PEO leads to increases in PCL crystallinity with increasing shear rate;45,47,75 however, when PNPs of the same copolymer are formed outside of the microfluidic channels in the absence of shear, significant PCL crystallization is still observed.45,47 Such observations strongly suggest that certain structural properties (e.g., low-crystallinity PNP cores) that could yield desirable biomedical functions cannot be achieved by top-down forces on their own.
In this article, we describe the synthesis, characterization, and self-assembly of a unique series of biocompatible block copolymers possessing a hydrophilic PEO block of constant molecular weight and a hydrophobic P(MCL-co-CL) block of variable MLC content and constant molecular weight. Micellar aggregates of poly(methyl caprolactone-co-caprolactone)-b-poly(ethylene oxide) (P(MCL-co-CL)-b-PEO) containing the anticancer drug paclitaxel (PAX) are found to be smaller and less crystalline with higher PAX loading levels compared to those of the equivalent PCL-b-PEO copolymer. Slower release rates and improved MCF-7 antiproliferation potencies are determined as the MCL content of the hydrophobic block increases. Moreover, microfluidic self-assembly of P(MCL-co-CL)-b-PEO is shown to further decrease release rates and enhance MCF-7 antiproliferation effects compared to those of the conventional bulk preparations. These results highlight the potential of combining polymer design/synthesis (chemical control) with microfluidic shear processing (mechanical control), through which new avenues can be opened for optimizing the structure and function of polymeric nanomedicines.
Results and Discussion
Characterization of Copolymers
From the 1H NMR spectra in Figure 1 and Figure S2 (Supporting Information), number-average molecular weights of the hydrophobic block Mn[P(MCL-co-CL)] and mole fractions of MCL in the hydrophobic block fMCL were determined by measuring the relative intensities of the CL and MCL peaks using the PEO peak (3.65 ppm) as a reference, as the number-average molecular weight of PEO is known (5000 g/mol). When ε-caprolactone and 6-methyl-ε-caprolactone are copolymerized at increasing ratios of MCL to CL monomer, a decrease in the CL methylene peak at 4.06 ppm, and the appearance of peaks at 1.20 and 4.90 ppm, corresponding to the MCL methyl and methylene groups, respectively, is observed (Figure 1). The determined fractions of MCL are consistent with the nominal feed ratios described in the Experimental Section. Equations and sample calculations for PMCL-25 are shown below.
 |
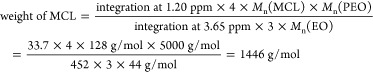 |
Table 1 lists the Mn and fMCL values determined for the various copolymers in the series in addition to polydispersity indexes (PDIs = Mw/Mn) determined from a combination of Mw from GPC data and Mn from 1H NMR data. Also listed in Table 1 are the critical water content (cwc) values of the various copolymers in DMF at a copolymer concentration of 0.33 wt %, based on titration curves shown in Figure S4 (Supporting Information). PMCL-0 has a lower cwc than that of all of the MCL-containing copolymers, with the cwc further increasing slightly as the MCL content of the copolymer increases. With the additional methyl group, PMCL is more hydrophobic than PCL; on the basis of solubility alone, therefore, we would expect the cwc to decrease as the MCL content increases, whereas the opposite trend is observed in Table 1. A possible explanation for this trend is that the decrease in PCL crystallization with the addition of MCL monomer lowers the driving force for micellization, which increases the cwc. This implies that the thermodynamic effects of PCL crystallinity on micelle formation are dominant over hydrophobicity effects.
Figure 1.

Selected 1H NMR spectra of P(MCL-co-CL)-b-PEO block copolymers with various MCL contents: (A) PMCL-0, (B) PMCL-25, and (C) PMCL-100.
Table 1. Copolymer Characteristics and Critical Water Contents.
| copolymer | Mn P(MCL-co-CL)-b-PEOa | Mn P(MCL-co-CL) | fMCL | PDIb | cwc (wt %)c |
|---|---|---|---|---|---|
| PMCL-0 | 9437 | 4437 | 0 | 1.96 | 7.1 ± 0.1 |
| PMCL-25 | 10 063 | 5063 | 0.26 | 2.55 | 8.6 ± 0.2 |
| PMCL-50 | 9795 | 4795 | 0.49 | 1.95 | 8.8 ± 0.1 |
| PMCL-75 | 9432 | 4432 | 0.72 | 1.77 | 9.4 ± 0.3 |
| PMCL-100 | 9690 | 4690 | 1 | 2.34 | 9.3 ± 0.2 |
Mn(PEO) = 5000 g/mol.
PDI = Mw(GPC)/Mn(NMR).
Errors are standard deviations of three separate cwc measurements.
Effect of MCL Content on Multiscale Structure of PNPs
Aqueous PNP dispersions of all five copolymers with different MCL contents were first prepared by the conventional bulk method of dropwise water addition followed by dialysis (Figure 2). Morphologies and mean PCL core dimensions from TEM data (Figure 2A–E) and effective hydrodynamic diameters from dynamic light scattering (DLS) data (Figure 2F) are listed in Table 2.
Figure 2.

Effect of MCL content on PNP morphology and size. (A–E) TEM images of bulk-prepared PNPs with various MCL contents and (F) plot of Dh,eff vs fMCL from DLS of the various PNP samples shown in (A)–(E). Scale bars are 200 nm.
Table 2. Morphologies,a Mean Core Dimensions,b and Effective Hydrodynamic Diametersc for P(MCL-co-CL)-b-PEO Nanoparticles with Various MCL Contents.
| copolymer | morphologies and mean core dimensions (nm) | Dh,eff (nm) |
|---|---|---|
| PMCL-0 | S (13 ± 2) | 1300 ± 100 |
| C (24 ± 2) | ||
| PMCL-25 | S (22 ± 2) | 230 ± 50 |
| C (17 ± 2) | ||
| PMCL-50 | S (20 ± 1) | 62 ± 9 |
| PMCL-75 | S (23 ± 5) | 60 ± 10 |
| PMCL-100 | S (25 ± 1) | 210 ± 30 |
| C (21 ± 4) |
Morphologies determined by TEM are indicated as S (spheres) and C (cylinders).
Mean dimensions refer to sphere diameters or cylinder widths determined by TEM. Errors represent standard deviations of mean values of three separate nanoparticle preparations under the same conditions.
Effective hydrodynamic diameters determined by DLS using cumulent analysis. Errors represent standard deviations of mean values of three separate nanoparticle preparations under the same conditions.
Self-assembly of the PCL-b-PEO copolymer without the MCL monomer (PMCL-0, Figure 2A) gave rise to prominent cylinders along with some small spheres. As the MCL content increased to fMCL = 0.25 (PMCL-25, Figure 2B), a decrease in the length and width of the cylinders and a concomitant increase in the core diameters of the spheres (from 13 to 22 nm) was observed. With a further increase in MCL content (PMCL-50 and PMCL-75, Figure 2C,D, respectively), the cylinders no longer formed and pure spheres of fairly constant core diameter (20–23 nm) were observed by TEM. Finally, PNP formation of the PMCL-b-PEO copolymer without the CL monomer (PMCL-100, Figure 2E) gave rise to the return of the cylindrical morphology, in the form of some short rods (Figure 2E, white arrows) with coexisting spheres of mean core diameter 25 nm.
The initial trend of a decreasing number of cylinders with increasing MCL content is consistent with a decrease in PCL crystallinity as the number of methyl groups in the hydrophobic block is increased. In previous studies of semicrystalline block copolymer self-assembly, it has been shown that low-curvature morphologies such as cylinders and lamellae are generally favored by highly crystalline cores, whereas high-curvature spheres become more prominent as the core crystallinity decreases.36,76 On the other hand, the trend reversal leading to the reappearance of cylinders in the PMCL-100 case is most likely related to the competing effect of increasing hydrophobicity as the MCL content increases.38,39 The dominant effect of high hydrophobicity leading to high interfacial tension in the PMCL-100 sample is supported by the fact that the spherical cores in that sample (Figure 2E) are the largest of all five copolymers (Table 2), suggesting densely packed and highly stretched hydrophobic chains at the core–corona interface. The observed disappearance followed by reappearance of cylinders with increasing MCL content (Figure 2A–E) is also reflected in the corresponding effective hydrodynamic diameters from DLS, which decrease and then increase as fMCL increases (Figure 2F).
We carried out XRD on the series of PNP colloids to confirm the trends in crystallinity suggested by the above morphological study. Percent crystalline PCL values, χPCL, were determined from peak deconvolution of the XRD patterns (Figure 3A–C) and are plotted in Figure 3D as a function of MCL fraction in the hydrophobic block, fMCL. The XRD pattern for the PCL-b-PEO copolymer (fMCL = 0, Figure 3A) provides a baseline for comparison with the MCL-containing copolymers, showing deconvoluted peaks associated with crystalline PEO (blue) and crystalline PCL (red) in addition to an amorphous halo (pink) from which a χPCL value of ∼25% is determined (Figure 3D). In contrast, the deconvolution of XRD profiles of all MCL-containing copolymers suggests no significant contribution from crystalline PCL peaks, leading to χPCL values of ∼0% for all copolymers with fMCL ≥ 0.25 (Figure 3D). This suggests that PCL crystallization in the PNP core is disrupted even with the minimum MCL content in this series. However, visual comparison of the XRD patterns of PMCL-25 (Figure 3B) and PMCL-100 (Figure 3C) in the region between the two PEO peaks (2θ = 29.2 and 35.2) suggests a possible weak contribution from crystalline PCL in the former sample. This is consistent with the TEM data, from which we find that PMCL-25 shows some tendency to form cylinders (Figure 2B) but PMCL-50 (Figure 2C) and PMCL-75 (Figure 2D) do not. We conclude that some crystalline PCL is present in copolymers with fMCL ≤ 0.25 but not in copolymers with fMCL > 0.25; in the fMCL = 0.25 case, the PCL crystallinity is too small to be quantified by XRD (Figure 3B), but is large enough to influence self-assembly behavior (Figure 2B).
Figure 3.
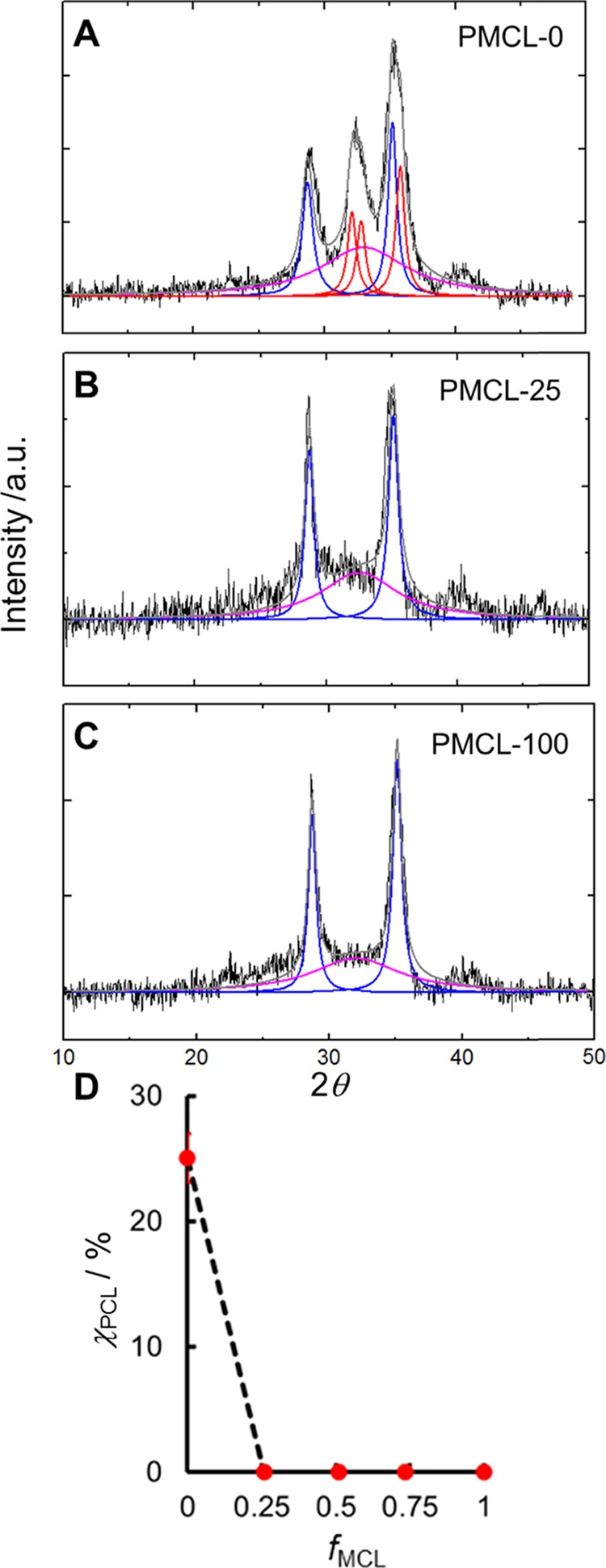
Effect of MCL content on PNP core crystallinity. (A–C) Selected XRD profiles for bulk-prepared PNPs with various MCL contents and (D) corresponding plot of χPCL vs fMCL. Fits show contributions from crystalline PEO (blue), crystalline PCL (red), and amorphous material (pink).
Effect of MCL Content on Drug Delivery Properties of PAX-Loaded PNPs
We next formed PAX-loaded PNPs from each copolymer at a variety of drug-to-polymer loading ratios, r, using the conventional bulk method of dropwise water addition followed by dialysis and centrifugation to remove any unencapsulated drug. Figure 4 shows the TEM data of the PAX-loaded PNPs formed from five different copolymers with variable MCL contents, each with three different loading ratios of r = 0.1, 0.25, and 0.50. For the PMCL-0 copolymer, we find that the morphological effect of the PAX loading ratio is negligible, with all loading ratios forming cylinders and spheres (Figure 4A,F,K), similar to the corresponding PNPs without PAX (Figure 2A). Adding PAX appears to have a more significant morphological effect on the PNPs in which the hydrophobic cores contain at least some MCL repeat units, which is possibly a result of PCL crystallization effects being less dominant in those samples compared to PMCL-0. For example, whereas the PMCL-25 and PMCL-100 PNPs both include cylinders in the absence of PAX (Figure 2B,E), these copolymers form only spheres once a small amount of PAX is added to the formulation (r = 0.1, Figure 4B,E).
Figure 4.

Effect of MCL content and loading ratio r on PNP morphology. (A–O) TEM images of bulk-prepared PAX-loaded PNPs with various MCL contents and prepared with various drug-to-polymer loading ratios, r. Scale bars are 200 nm.
The disruption of the cylinders by PAX addition in the MCL-containing copolymers can be attributed to different effects, depending on the MLC composition and the driving force for cylinder formation. In the case of PMCL-25, it is likely that the plasticizing effect of PAX precludes PCL crystallization and therefore cylinder formation; in the case of PMCL-100, we believe cylinders are prevented by the lowering of interfacial tension associated with solubilization of PAX in the PNP cores. For all four MCL-containing copolymers (PMCL-25, PMCL-50, PMCL-75, and PMCL-100), pure spheres of relatively constant size within experimental error were obtained when the loading ratio was increased from r = 0.1 to 0.5 (Figure 4, Table 3).
Table 3. Morphologies,a Mean Core Dimensions,b and Effective Hydrodynamic Diametersc for PAX-Loaded P(MCL-co-CL)-b-PEO Nanoparticles with Various MCL Contents and Different Drug-to-Polymer Loading Ratios.
| copolymer | drug/polymer loading ratio, r (w/w) | morphologies and mean core dimensions (nm) | Dh,eff (nm) |
|---|---|---|---|
| PMCL-0 | 0.10 | S (16 ± 1) | 1110 ± 90 |
| C (20 ± 3) | |||
| 0.25 | S (13 ± 1) | 1080 ± 90 | |
| C (23 ± 2) | |||
| 0.50 | S (13 ± 3) | 1100 ± 100 | |
| C (20 ± 3) | |||
| PMCL-25 | 0.10 | S (23 ± 2) | 60 ± 10 |
| 0.25 | S (24 ± 1) | 63 ± 6 | |
| 0.50 | S (25 ± 4) | 65 ± 5 | |
| PMCL-50 | 0.10 | S (21 ± 3) | 60 ± 10 |
| 0.25 | S (24 ± 2) | 60 ± 10 | |
| 0.50 | S (23 ± 1) | 61 ± 8 | |
| PMCL-75 | 0.10 | S (25 ± 3) | 60 ± 10 |
| 0.25 | S (26 ± 1) | 60 ± 10 | |
| 0.50 | S (23 ± 2) | 60 ± 9 | |
| PMCL-100 | 0.10 | S (19 ± 2) | 60 ± 10 |
| 0.25 | S (22 ± 2) | 60 ± 10 | |
| 0.50 | S (25 ± 3) | 61 ± 6 |
Morphologies determined by TEM are indicated as S (spheres) and C (cylinders).
Mean dimensions refer to sphere diameters or cylinder widths determined by TEM. Errors represent standard deviations of mean values of three separate nanoparticle preparations under the same conditions.
Effective hydrodynamic diameters determined by DLS using cumulent analysis. Errors represent standard deviations of mean values of three separate nanoparticle preparations under the same conditions.
PAX loading efficiencies (Figure 5A) and corresponding loading levels (Figure 5B) were determined by high-performance liquid chromatography (HPLC) for PNPs of the five copolymers and three different loading ratios. For all five copolymers, loading efficiencies decrease with increasing loading ratio (Figure 5A), which is consistent with the loading levels that remain relatively constant as the loading ratio increases (Figure 5B). Also in Figure 5B, it is apparent that the loading levels at all investigated loading ratios are higher for the MCL-containing copolymers (PMCL-25, PMCL-50, PMCL-75, and PMCL-100) compared to the PMCL-0 case. It should also be noted that the four MCL-containing copolymers, irrespective of MCL content, show identical loading levels within experimental error.
Figure 5.
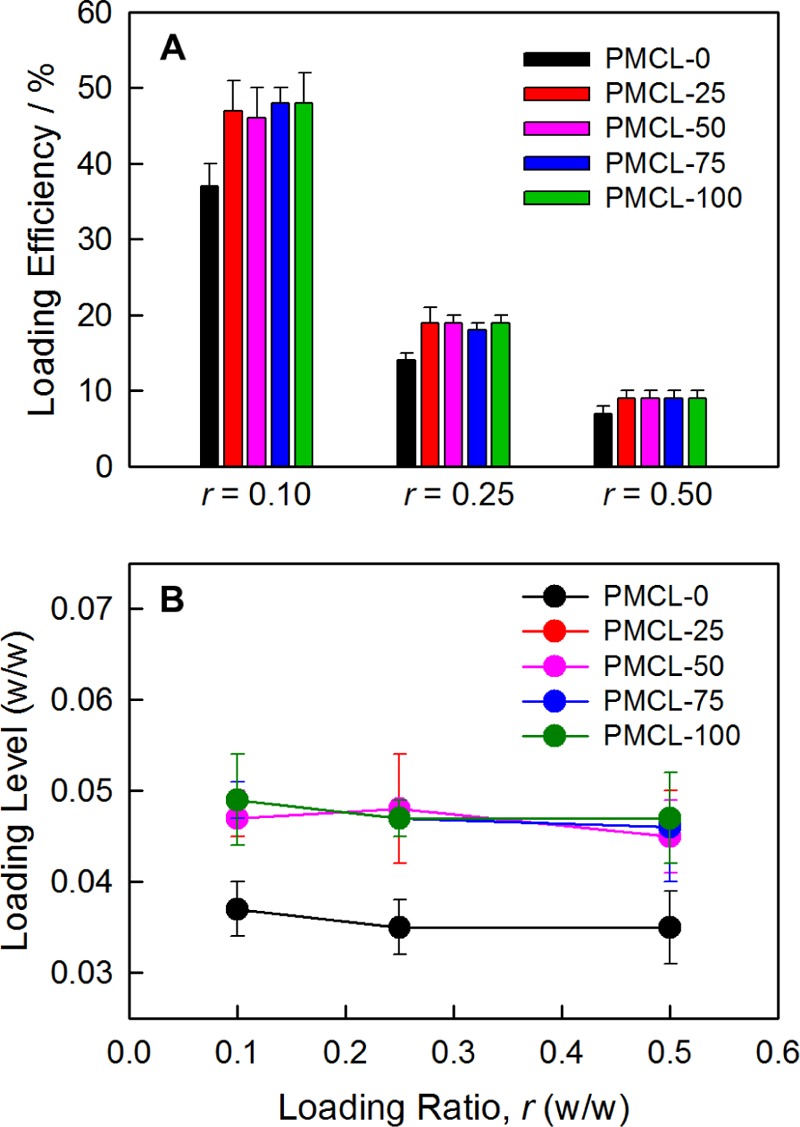
Effect of MCL content and loading ratio r on PAX loading. (A) Loading efficiencies and (B) loading levels for bulk-prepared PAX-loaded PNPs with various MCL contents and prepared with various drug-to-polymer loading ratios, r.
From the above observations, we conclude that all five copolymers form PNPs in which the hydrophobic cores become saturated with PAX at the lowest investigated loading ratio of r = 0.1. Therefore, further addition of PAX beyond r = 0.1 does not increase loading levels (Figure 5B) or change the PNP morphologies or sizes (Table 3), whereas loading efficiencies decrease with increasing r as the amount of unencapsulated drug removed in the centrifugation step increases (Figure 5A). Another conclusion is that the introduction of MCL monomer into the hydrophobic core clearly increases the maximum amount of PAX that can be incorporated into the PNPs, although the saturation level of PNPs with different MCL contents is the same. On the basis of the measured saturated loading levels, we determined that the mole ratio of PAX molecules to copolymer chains within the PNPs is ∼0.4 for PMCL-0 and ∼0.5 for all MCL-containing copolymers.
The effect of copolymerized MCL increasing PAX encapsulation may be due in part to the higher hydrophobicity of the MCL-containing copolymers leading to higher PAX solubilities within the PNP cores. However, we note that although core hydrophobicity should increase monotonically with increasing MCL content, the saturated loading levels do not appear to follow the same trend. Instead, we see a stepwise increase in PAX loading levels between PMCL-0 and PMCL-25, followed by no further increase as the MCL content increases between PMCL-25 and PMCL-100 (Figure 5B). We note that this stepwise increase in PAX loading level with MCL content tracks with the corresponding stepwise decrease in core crystallinity, as measured by XRD (Figure 2F). This suggests that the effect of MLC addition on PAX encapsulation is more strongly related to changes in core crystallinity than to changes in core hydrophobicity. Specifically, the increase in saturated loading levels from 0.004 for PMCL-0 to 0.005 for all MCL-containing copolymers (Figure 5B) can be attributed to the corresponding sharp drop in the percentage of crystalline PCL within the core (Figure 2F), providing a larger amorphous volume in which to accommodate encapsulated PAX.
For in vitro release experiments, PAX-loaded PNPs of all five copolymers prepared at a single loading ratio (r = 0.25) were assessed, and the resulting release profiles are shown in Figure 6A. Release half times, t1/2, were estimated by extrapolation from the various release profiles, and are plotted in the inset to Figure 6A versus MCL content. The release profiles show a clear effect of adding MCL to the hydrophobic block on drug release rates, with PMCL-0 (fMCL = 0) showing the fastest release (t1/2 = ∼50 min), and the release half time increasing modestly but significantly to t1/2 = ∼60 min for PMCL-25 (fMCL = 0.25). This is followed by an apparent (although possibly not statistically significant) gradual increase in release half time from t1/2 = ∼60 to ∼70 min as the MCL content increases from fMCL = 0.25 to 1.
Figure 6.
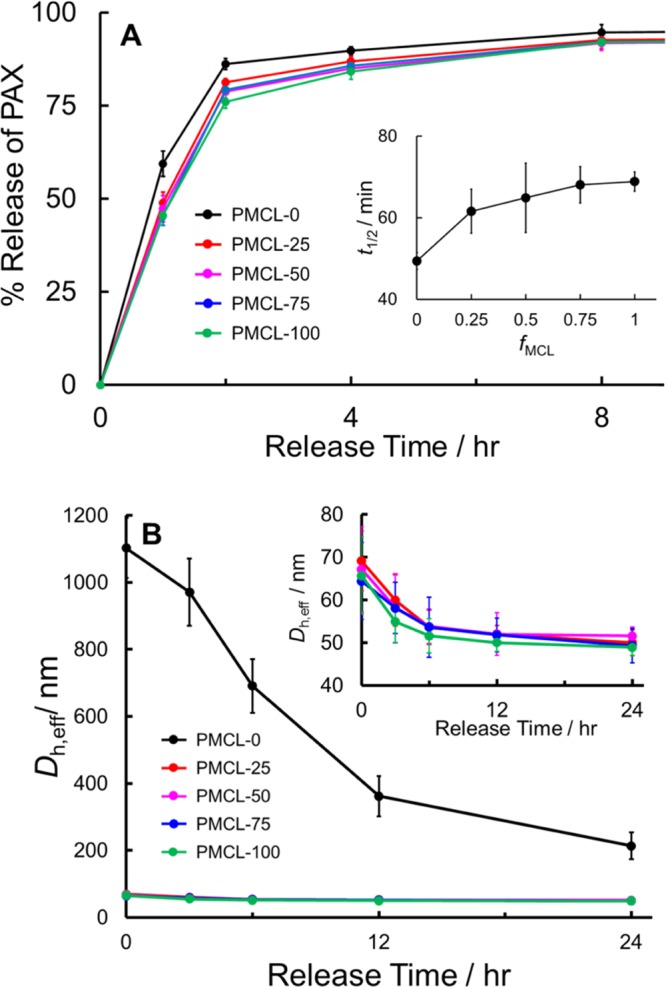
Effect of MCL content on in vitro PAX release. (A) In vitro PAX release profiles for bulk-prepared PAX-loaded PNPs with various MCL contents (r = 0.25) and corresponding plot of t1/2 vs fMCL (inset). (B) Dh,eff, vs release time for samples in (A), showing relative hydrolytic degradation rates during in vitro PAX release; inset shows re-scaled data for MCL-containing copolymers only.
It is at first surprising that the PAX-loaded PNPs prepared from PMCL-0 show the fastest in vitro release, considering that higher crystallinities and larger core volumes should slow down PAX diffusion times to the core–corona interface of the PNPs. However, further insight into this trend comes by considering that an important mechanism of PAX release is the hydrolytic degradation of PNPs at physiological temperature, pH, and ionic strength.16,45 We therefore tracked the hydrolytic degradation of the nanoparticles during the first 24 h of release, by monitoring the effective hydrodynamic diameters by DLS as a function of release time (Figure 6B). We see that over this time period, the mean effective hydrodynamic sizes of the PMCL-0 PNPs drop from ∼1100 to ∼200 nm (by about 80%), which can be attributed to the hydrolytic breakdown of the original cylinders (Figure 4F) into spheres, whereas the PNP sizes of all other copolymers (shown previously to be pure spheres, Figure 4G–J) drop from ∼70 to ∼50 nm (i.e., by about 30%), which can be attributed to a gradual degradation of the spheres. The relatively fast and dramatic increase in surface area via hydrolytic degradation of PMCL-0 compared to the smaller increase in surface area over the same period for the PNPs of all four MCL-containing copolymers (Figure 6C) thus explains the slightly faster release of drug from PMCL-0.
The antiproliferative effects of the PAX-loaded PNPs prepared from three of the five copolymers (PMCL-25, PMCL-50, and PMCL-75; constant loading ratio, r = 0.25) were measured using the MCF-7 cell line. Free PAX was evaluated as a positive control and empty PNPs without PAX prepared from the copolymer PMCL-25 were evaluated as a negative control. For all investigated samples and positive controls, growth inhibition plots were generated for 48, 72, and 96 h incubation times (Supporting Information, Figures S6–S8). Negative controls showed no significant effect of the copolymer alone on cell viability; negligible cell death by empty PNPs was measured up to a polymer concentration of 36.5 ppm (Supporting Information, Figure S9), which was equivalent to the highest polymer concentration applied in doses of PAX-loaded PNPs (∼40 ppm).
Associated GI50 values in Figure 7 show significant differences in antiproliferative effects for the PAX-loaded PNPs prepared from copolymers with different MCL contents. All three PNP formulations show attenuated antiproliferative effects (elevated GI50 values) relative to free PAX; this can be understood in terms of the cell exposure to drug being reduced by encapsulation in the polymer formulations. For both 48 and 96 h incubation times, we found a significant decrease in GI50 value as the MCL content in the hydrophobic block increased. The same clear trend was not found for the 72 h data, perhaps due to the larger relative errors in GI50 values at the intermediate incubation time.
Figure 7.
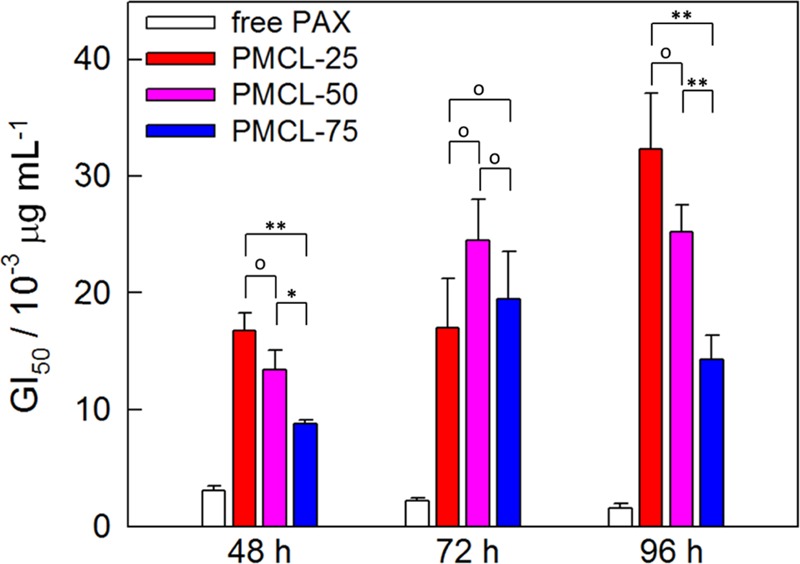
Effect of MCL content on MCF-7 antiproliferation potency. GI50 values for PAX-loaded PNPs with various MCL contents (r = 0.25). Data is shown for incubation times of 48, 72, and 96 h. Connecting brackets indicate statistical comparisons between PNPs generated from copolymers with different MCL contents. Single asterisks indicate p < 0.05; double asterisks indicate p < 0.01; open circles indicate p > 0.05.
To understand the observed effect of MCL content on MCF-7 antiproliferation, we first consider that all of the investigated PNP formulations consist of pure spheres (Figure 4G–I, r = 0.25) with core sizes (24–26 nm) and hydrodynamic diameters (60–63 nm) that are remarkably similar for the three investigated copolymers PMCL-25, PMCL-50, and PMCL-75. In addition, loading levels (Figure 5B) and release rates (Figure 6A) are similar for all three copolymers. We conclude that the number, size, and morphology of the PNPs exposed to cells in the antiproliferation assays were effectively the same for the three copolymer formulations, and that the in vitro release profiles were also not significantly different. This leaves us to consider the different mechanical properties of the hydrophobic core surfaces, which should become “stickier” and smoother58 as the MCL content increases. We tentatively propose that changes in the PNP surface properties with increasing MCL content and concomitant enhancements in cellular interactions may be responsible for the observed increases in antiproliferative potency.
Structure and Drug Delivery Properties of PAX-Loaded PNPs Prepared Using Microfluidics
The previous sections have shown that copolymerization of MCL and CL to form amphiphilic block copolymers with composition-variable hydrophobic blocks offers chemical control of structure and function for drug delivery PNPs. In this section, we apply selected MCL-containing copolymers to compare the structure and drug delivery function of PAX-loaded PNPs prepared using the conventional approach of dropwise water addition with those prepared in a two-phase microfluidic reactor at different flow rates. As shown in previous studies from our group,42−47,71−75 these reactors enable particle processing via variable shear. For this comparison, we focus on the PMCL-50 copolymer, although PAX-loaded PNPs of PMCL-25 and PMCL-75 were also prepared in the microfluidic reactor at different flow rates; TEM and DLS data for those samples is presented in the Supporting Information (Figure S10).
Figure 8 shows TEM images of the PAX-loaded PNPs of PMCL-50 (r = 0.25) produced using the bulk preparation method (Figure 8A) and in the microfluidic reactor at different flow rates: Q = 50 μL/min (Figure 8B), Q = 100 μL/min (Figure 8C), and Q = 200 μL/min (Figure 8D). Three of the four PNP samples contain pure spheres, with the exception being the Q = 100 μL/min sample, which contains some cylinders (Figure 8C inset). The effective hydrodynamic diameters of the PNPs, Dh,eff, show a general decrease with increasing flow rate (Figure 8E) with the exception of a sharp increase between Q = 50 and 100 μL/min corresponding to the appearance of cylinders (Figure 8C). The increase in PNP size followed by a decrease with increasing flow rate is attributed to competing mechanisms of microfluidic PNP processing, with shear-induced particle coalescence being dominant at low flow rates and shear-induced particle breakup being dominant at high flow rates.44 Similar nonmonotonic trends in morphology and hydrodynamic size are found for PMCL-75, although not for the PMCL-25 copolymer, which forms only pure spheres of steadily decreasing size as the flow rate increases (Supporting Information, Figure S10).
Figure 8.

Effect of preparation method and flow rate on PNP morphology and size. TEM images of PAX-loaded PNPs of PMCL-50 (r = 0.25) prepared using the bulk preparation method (A) and in the two-phase microfluidic reactor at different flow rates (B–D), and (E) Dh,eff vs Q for the PNPs in (A)–(D); Q = 0 μL/min designates the bulk-prepared PNPs. Scale bars are 200 nm.
Next, loading efficiencies and in vitro release profiles were compared for the bulk and microfluidic preparations of PAX-loaded PNPs of PMCL-50 (r = 0.25, Figure 9). Figure 9A shows that loading efficiency decreased when moving from the bulk to the microfluidic preparation method, as we have previously reported for the preparation of PAX-loaded PCL-b-PEO PNPs,47 but did not change significantly when the flow rate was increased from Q = 50 to 200 μL/min. Comparing in vitro release profiles, Figure 9B and the inset show that PAX release is markedly slower for the microfluidic PNPs prepared at Q = 50 μL/min (t1/2 = ∼90 min) compared to the bulk-prepared PNPs (t1/2 = ∼70 min), and that release rates slow further when the microfluidic flow rate increases to Q = 200 μL/min (t1/2 = ∼120 min).
Figure 9.
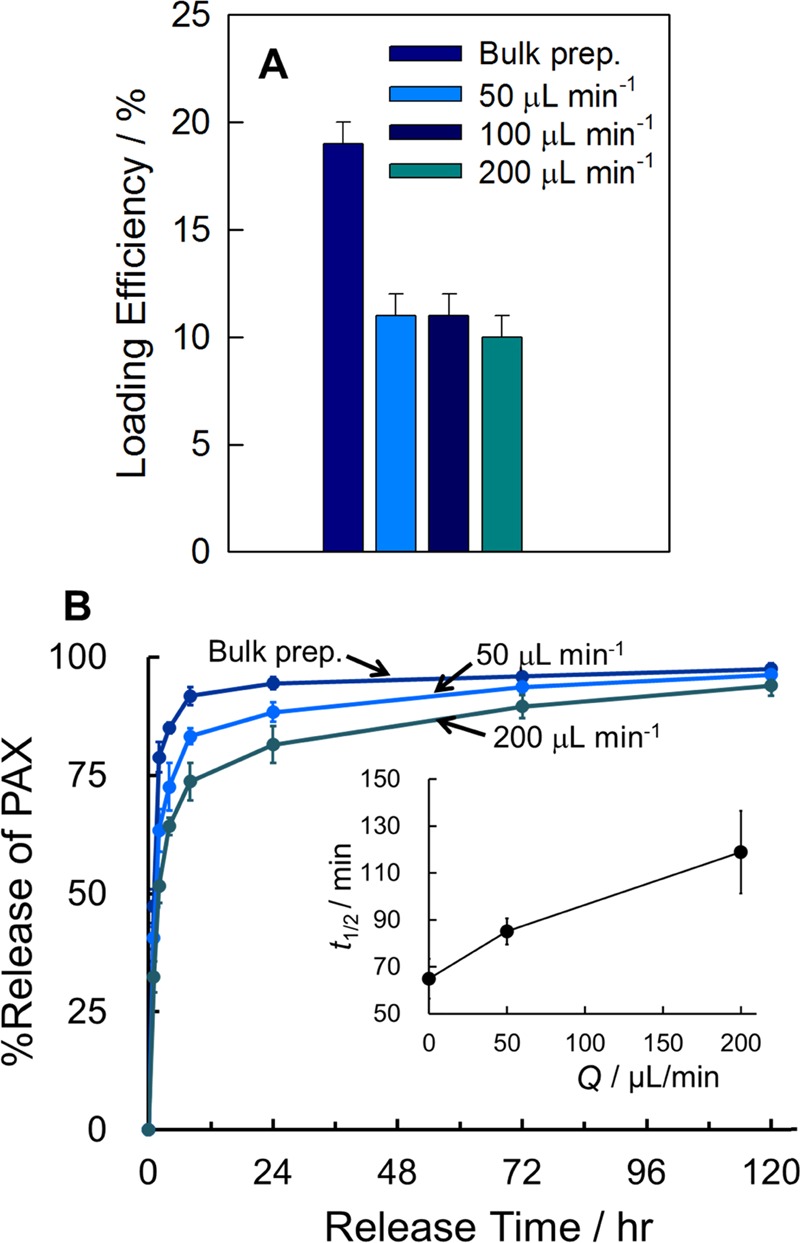
Effect of preparation method and flow rate on PAX loading and in vitro release. (A) Loading efficiencies and (B) release profiles with corresponding plot of t1/2 vs fMCL (inset) for PAX-loaded PNPs of PMCL-50 (r = 0.25) prepared using the bulk preparation method and in the two-phase microfluidic reactor at different flow rates; in the inset to (B), Q = 0 μL/min designates the bulk-prepared PNPs.
Consideration of the series of release profiles in Figure 6A together with that in Figure 9B highlights the merits of combining both chemical synthesis and microfluidic shear processing for optimizing the drug delivery properties of PNP formulations. First, chemical copolymerization of MCL (Figure 6A) increased release half times from t1/2 = ∼50 min (PMCL-0) to t1/2 = ∼70 min (PMCL-50); then, microfluidic processing of PMCL-50 further increased release half times from t1/2 = ∼70 min (bulk preparation) to t1/2 = ∼120 min (microfluidic preparation, Q = 200 μL/min). The final result of coupling chemical and processing control yields PNPs showing extended PAX release over 5 days (Q = 200 μL/min, Figure 9B), which is in sharp contrast to PNPs of bulk-prepared PMCL-0 (Figure 6A) from which ∼90% of PAX is released after only 2 h.
In previous work from our group, increases in PAX release times from PNPs prepared in the two-phase microfluidic reactor at increasing flow rate have been attributed to the increased formation of shear-induced crystallites in the hydrophobic core,45 more homogenous PAX distributions in the core,47 or a combination of both effects.47 Here, we expect that the presence of methyl side groups will preclude core crystallization of PMCL-50, even in the presence of shear processing, although core crystallinities were not measured for the microfluidic-prepared samples of this copolymer. However, we did carry out XRD measurements of the PMCL-25 PNPs without PAX (r = 0) prepared both in the bulk and in the microfluidic reactor at Q = 50 μL/min, and we found no discernible differences in crystallinities in those samples; this suggests that, unlike the PCL-b-PEO copolymers, shear-induced crystallization effects are not strong in the MCL-containing copolymers presented here. From these considerations, we conclude that the slowing down of PAX release for the PNPs prepared at faster flow rates (Figure 9B) is more likely explained by faster mixing times leading to more homogenously distributed PAX molecules within the core; this is in contrast to the bulk-prepared PNPs, where slow water addition is expected to give rise to greater localization of PAX molecules at the core–corona interface with consequential burst release.
Finally, we compare the MCF-7 antiproliferation effects for the bulk and microfluidic preparations of PAX-loaded PNPs of PMCL-50 (r = 0.25). Growth inhibition plots were generated for 48, 72, and 96 h incubation times (Supporting Information, Figures S11–S13). Associated GI50 values in Figure 10 show significant differences in antiproliferative effects for the PAX-loaded PNPs prepared using different preparation methods. Specifically, for all incubation times, a significant decrease in GI50 values is found for the microfluidic preparations relative to the bulk preparation. For the 48 and 96 h incubation times, antiproliferation potencies are not significantly different for the microfluidic preparations at two different flow rates; however, for the 72 h incubation time, a small but significant increase in GI50 is found when the flow rate is increased from Q = 50 to 200 μL/min. The stronger antiproliferation effects for the microfluidic-prepared PNPs compared to those of the bulk-prepared PNPs could be related to the relative decreases in in vitro release times, loading efficiencies, or PNP hydrodynamic sizes, or a combination of these factors.
Figure 10.
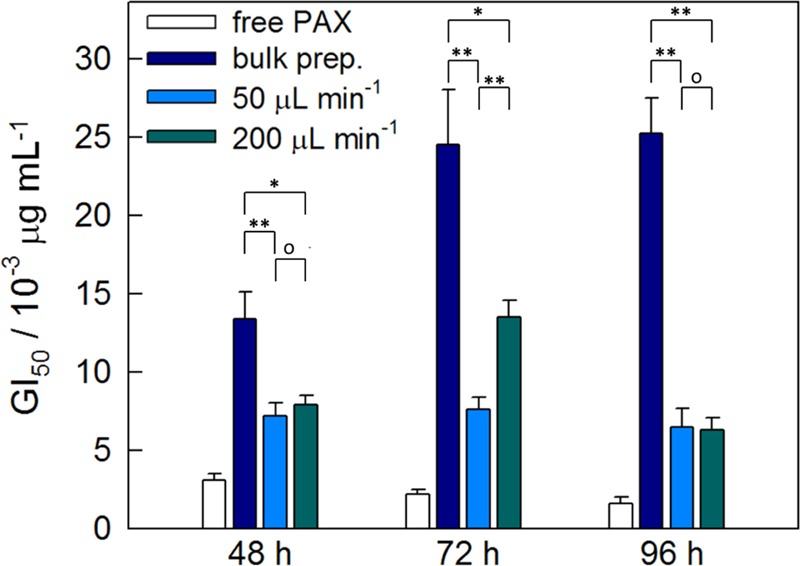
Effect of preparation method and flow rate on MCF-7 antiproliferation potency. GI50 values for PAX-loaded PNPs of PMCL-50 (r = 0.25) prepared using the bulk preparation method an in the two-phase microfluidic reactor at different flow rates. Data is shown for incubation times of 48, 72, and 96 h. Connecting brackets indicate statistical comparisons between PNPs generated by different methods. Single asterisks indicate p < 0.05; double asterisks indicate p < 0.01; open circles indicate p > 0.05.
Similar to our previous discussion of in vitro release times, we find that a combination of chemistry and processing enables optimization of PNP antiproliferation effects. First, increasing the MCL content in the hydrophobic block via chemical synthesis was shown to effect a steady decrease in 48 and 96 h GI50 values (Figure 7); next, microfluidic processing of a selected copolymer (PMCL-50) led to a further decrease in GI50 compared to that of the bulk preparation method (Figure 10). Taken together, we conclude that a combination of high MCL content (chemical control) and microfluidic manufacturing (processing control) should provide routes to PNPs with optimum potency for MCF-7 antiproliferation.
Conclusions
New biocompatible block copolymers and improved nanoparticle manufacturing methods define two fronts in the concerted development of better polymeric nanomedicines. In this study, we applied tandem efforts on both fronts, bringing to bear both chemical and processing approaches to the drug delivery problem. First, the synthesis, characterization, and self-assembly of a series of biocompatible P(MCL-co-CL)-b-PEO amphiphilic block copolymers with variable MCL contents in the hydrophobic block were described. The hydrophobic cores of the resulting PNPs were less crystalline as the MCL content increased, and the morphologies and sizes showed nonmonotonic trends with MCL content due to the competing effects of core crystallinity and core hydrophobicity. Loading the anticancer cargo drug PAX into PNPs of the self-assembled block copolymers showed that in vitro drug release rates decreased and MCF-7 antiproliferation effects increased as the MCL content increased. Next, the effects of microfluidic manufacturing at variable flow rate on the structure and drug delivery function of the PAX-loaded PNPs was explored, indicating that shear processing enables further control of release rates and enhances antiproliferation potency. Along with showing specific routes to interesting biomolecular nanomaterials, these results highlight the merits of developing more effective and specific drug delivery PNPs through tangential efforts combining both polymer synthesis and microfluidic manufacturing.
Experimental Section
Materials
2-Methylcyclohexanon (99.0%), potassium peroxymonosulfate (Oxone), sodium bicarbonate (99.0%), calcium hydride (95%), stannous octonate (95%), PEG methyl ether (MePEG-OH, MW ∼5000), and ε-caprolactone (97%) were all purchased from Sigma-Aldrich and used for the synthesis of copolymers. NaCl (Bio Basic Canada, 99.9%), KCl (Caledon, 99.0%), Na2HPO4 (Bio Basic Canada, 98.0%), and KH2PO4 (Caledon, 99.0%) were used to prepare phosphate buffered saline (PBS, pH = 7.4). Methanol (Fisher Chemical, 99.9%), methylene chloride (Fisher Chemical, 99.9%), toluene (Caledon, 99.5%), ethyl ether (VWR, 99%), dimethylformamide (DMF, Caledon, 99.8%), acetonitrile (Caledon, HPLC grade), t-butyl methyl ether (Sigma-Aldrich, HPLC grade), and dichloromethane (DCM, EMD, HPLC grade) were used as received without further purification. Paclitaxel (99.0%) was purchased from Polymed Inc.
Synthesis of 6-Methyl-ε-Caprolactone (MCL)
The synthesis of the MCL monomer was modified from ref (60). To a 100 mL round-bottom flask, 2-methylcyclohexanone (0.721 g, 6.43 mmol), methanol (20 mL), water (20 mL), and sodium bicarbonate (3 g, 36 mmol) were added. The vessel was vigorously stirred with a Teflon-coated magnetic stir bar. Oxone (4 g, 13 mmol, ∼2 equiv) was added in two portions—the second added 10 min after the first. After the addition of Oxone, vigorous gas evolution occurred over 20 min, abated, and ceased after ∼1 h. The reaction was allowed to stir for 6 h, followed by filtration, and was then extracted with methylene chloride. The organic phase was concentrated under vacuum. A total of 0.82 g was recovered (99% crude yield). The monomer was purified by fractional vacuum distillation (the distillate temperature was 55 °C at 0.98 Torr) from calcium hydride and stored over 3 Å activated molecular sieves. Additional purification of 6-methyl-ε-caprolactone was needed to produce monomodal high molecular weight PMCL by passing the distilled monomer through a column of activated alumina under nitrogen. The 1H NMR spectra of the resulting MCL product is shown in Figure S1 (Supporting Information).
Synthesis of P(MCL-co-CL)-b-PEO Copolymers
The copolymers of P(MCL-co-CL)-b-PEO with different ratios of ε-caprolactone (CL) and 6-methyl-ε-caprolactone (MCL) were synthesized via ring opening polymerization of ε-CL and ε-MCL in the presence of MePEG-OH as the macroinitiator and tin(II) 2-ethylhexanoate (stannous octoate; Sn(Oct)2) as the catalyst. The polymerization was carried out under a nitrogen atmosphere in a flame-dried round-bottom flask equipped with a magnetic stirrer bar. In brief, following azeotropic distillation in toluene, MePEG-OH (5 g, 1 mmol) was dissolved in a 75 mL volume of dried toluene; CL (3.63 g, 31.8 mmol), MCL (1.36 g, 10.6 mmol), and 0.7 wt % Sn(Oct)2 relative to the total mass of lactones were added to the reaction vessel. The polymerization was carried out at 110 °C for 24 h with continuous stirring. The copolymers were isolated by precipitation in cold ether and dried under vacuum. The copolymers were then characterized by 1H NMR and gel permeation chromatography (GPC). By changing the feed ratio of MCL and CL, copolymers with different MCL/CL ratios were synthesized. The molecular weight and MCL fraction, defined as the number-averaged weight of MCL in a copolymer chain over the number-averaged weight of the hydrophobic block, were calculated from the 1H NMR spectra. GPC characterization (Supporting Information, Figure S3) was done by Advanced Polymer Materials Inc. with chloroform as eluent and a series of PEO standards. PDI values (Mw/Mn) were calculated from Mn from NMR and Mw from GPC. Copolymers with nominal MCL fractions of fMCL = 0, 0.25, 0.50, 0.75, and 1 are referred to as PMCL-0, PMCL-25, PMCL-50, PMCL-75, and PMCL-100 in the text. Copolymer characteristics are tabulated in Table 1.
Critical Water Content Determination
Critical water contents were determined for solutions of the various copolymers in DMF at an initial copolymer concentration of 0.33 wt % by dropwise water addition and static light intensity measurement. Light scattering experiments were performed on a Brookhaven Instruments photocorrelation spectrometer equipped with a BI-200SM goniometer, a BI-9000AT digital autocorrelator, and a Melles Griot He–Ne Laser (632.8 nm) with a maximum power output of 75 mW, at a scattering angle of 90° and a temperature of 23 °C. For each copolymer, a 0.66 wt % solution in DMF was prepared and allowed to equilibrate overnight. This stock solution was then filtered through 2 × 0.45 μm teflon syringe filters (VWR) in series and then diluted to a final concentration of 0.33 wt % using filtered DMF (2 × 0.20 μm Teflon syringe filter, National Scientific). To initiate PNP formation, filtered deionized water (2 × 0.20 μm Nylon syringe filters, National Scientific) was added in successive 0.03–0.06 g quantities to this solution. After each addition of water, the solution was vortexed to help mixing, and then equilibrated for 15 min before the scattered light intensity was measured. From the resulting plots of scattered light intensity versus water concentration, cwc values were determined by extrapolation of the steep increase in intensity to the baseline. Titration plots and cwc determinations for each copolymer are shown in Figure S4 (Supporting Information); the resulting cwc values for each copolymer are tabulated in Table 1 and were used as reference values for selecting water contents for the bulk and microfluidic PNP preparations.
Bulk Preparation of PAX-Loaded P(MCL-co-CL)-b-PEO PNPs
PNPs of each block copolymer containing various quantities of PAX were formed by the conventional method of dropwise water addition. For these experiments, ∼3 g of 0.33 wt % copolymer solutions in DMF with various PAX/polymer (w/w) loading ratios (r = 0, 0.10, 0.25, and 0.50) were prepared and equilibrated overnight. Deionized water was added dropwise at a rate of 80 μL/min to a target water content of cwc + 10 wt % using a syringe pump with moderate stirring. After reaching the target water content, PNPs were immediately quenched into a 10× excess (by volume) of deionized water, followed by 12 h dialysis (6–8 kD MWCO dialysis membrane, Spectrum Laboratories) against deionized water to remove residual DMF. Then, unencapsulated drug precipitate was removed from the dialyzed PNPs by centrifugation at 16 000g for 10 min. Unless otherwise stated, all samples were prepared in triplicate under the specified conditions.
Microfluidic Reactor Fabrication
Negative masters were fabricated on silicon wafers (Silicon Materials) using the negative photoresist SU-8 100 (Microchem). A 150 μm thick SU-8 film was spin-coated at 2000 rpm onto the silicon wafer and heated at 65 °C for 12 min and then at 95 °C for 50 min. After the wafer was cooled, a photomask was placed directly above and the wafer was exposed to UV light for 100 s. Then, the UV-treated film was heated at 65 °C for 1 min and then at 95 °C for 20 min. Finally, the silicon wafer was submerged in SU-8 developer (Microchem) and rinsed with isopropanol until all unexposed photoresist was removed.
Microfluidics chips were fabricated from poly(dimethyl siloxane) (PDMS) using a SYLGARD 184 silicon elastomer kit (Dow Corning). For fabrication of all PDMS chips, the elastomer and curing agent were mixed at a 7:1 ratio and degassed under vacuum. The resulting mixture was poured over a clean negative master chip in a Petri dish and further degassed until all remaining air bubbles were removed. PDMS was heated at 85 °C until cured (∼20 min), and then peeled from the negative master; holes were punched through the reservoirs of the resulting PDMS chip to allow for the insertion of tubing. A thin PDMS film (substrate layer) was also made on a glass slide by spin-coating a 20:1 elastomer/curing agent mixture followed by curing. The substrate layer was then permanently bonded to the base of the microfluidic reactor (channel layer) after both components were exposed to oxygen plasma for 45 s. The resulting reactor (Figure S5) has a set channel depth of 150 μm and consists of a sinusoidal mixing channel 100 μm wide and a sinusoidal processing channel 200 μm wide, which is identical to the reactor described in previous publications from our group.74,75
Flow Delivery and Control
Pressure-driven flow of liquids to the reactor inlet was provided using 1 mL gastight syringes (Hamilton, Reno, NV) mounted on syringe pumps (Harvard Apparatus, Holliston, MA). The microfluidic chip was connected to the liquid syringes via 1/16th in. (OD) Teflon tubing (Scientific Products and Equipment, ON). Gas flow was introduced to the chip via an Ar tank regulator and a downstream regulator (Johnston Controls) for fine adjustments. The chip was connected to the downstream regulator through a 1/16th in. (OD)/100 μm (ID) Teflon tube (Upchurch Scientific, Oak Harbor, WA). The liquid flow rate (Qliq) was programmed via the syringe pumps and the gas flow rate (Qgas) was fine-tuned via the downstream pressure regulator to set the nominal total flow rates (Q) of 50, 100, and 200 μL/min described in the main text. Because of the compressible nature of the gas and the high gas/liquid interfacial tension, discrepancies arise between the nominal (programmed) and actual values of Qgas, Qgas/Qliq, and the total flow rate (Qtotal). Therefore, actual values of Qgas, Qgas/Qliq, and Qtotal = Qgas + Qliq for each microfluidic experiment (Table S1) were calculated from the frequency of bubble formation and the average volume of gas bubbles, determined from image analysis of the mean lengths of liquid and gas plugs, Lliq and Lgas, respectively, under a given set of flow conditions. This method of flow calculation has been previously described by our group.43 For all experiments, the relative gas-to-liquid flow ratio Qgas/Qliq ∼1 and all actual Qtotal values were within 10% of the nominal Q values reported in the main text.
Visualization of the gas bubbles and liquid plugs within the microfluidic reactor was achieved using an upright optical microscope (Omax) with a 10× objective lens. Images were captured using a 2.07 megapixel PupilCam camera (Ken-A-Vision) and mean lengths of liquid and gas plugs were determined from the images using image analysis software (ImageJ).
Microfluidic Preparation of PAX-Loaded P(MCL-co-CL)-b-PEO PNPs
For the microfluidic preparation of the PNPs, the following three fluid streams were combined to form gas-segmented liquid plugs within the reactor: (1) 1.0 wt % P(MCL-co-CL)-b-PEO solution in DMF with PAX/polymer loading ratio r = 0.25, (2) pure DMF, and (3) DMF/water. The flow rates of the three liquid streams were equal for all runs and the water content of the DMF/water stream was selected to yield steady state on-chip concentrations of 0.33 wt % copolymer and cwc + 10.0 wt % water.
For each PNP preparation, the sample was collected from the chip into vials containing a 10× excess by volume of deionized water, followed by 12 h dialysis (6–8 kD MWCO dialysis membrane, Spectrum Laboratories) against deionized water to remove residual DMF. Then, unencapsulated drug precipitate was removed from the dialyzed PNPs by centrifugation at 16 000g for 10 min. Unless otherwise stated, all samples were prepared in triplicate under the specified conditions.
Transmission Electron Microscopy
Negatively stained samples for TEM imaging were prepared by depositing a drop of block copolymer PNP dispersion (typically ∼0.1 mg/mL) on a carbon-coated 300-mesh copper TEM grid followed by a drop of 1 wt % uranyl acetate aqueous solution as a negative staining agent. Excess liquid was immediately removed using lens paper, followed by drying of the remaining liquid under ambient conditions. Imaging was performed on a JEOL JEM-1400 transmission electron microscope, operating at an accelerating voltage of 80 kV and equipped with a Gatan Orius SC1000 CCD camera.
For each copolymer and flow condition, morphologies and mean PNP sizes were determined based on triplicate preparations; for each preparation, morphology and size analysis was carried out using at least three images taken in different regions of the grid. Mean dimensions for each condition were determined from a total of 450 spheres and 150 cylinders. Averaging and statistical analysis of dimensions from TEM images were conducted using ImageJ software.
Dynamic Light Scattering
Effective hydrodynamic diameters of PAX-loaded PNPs were determined using dynamic light scattering (DLS). DLS measurements were carried out using a Brookhaven Instruments Zeta-Pals Analyzer equipped with a solid state Laser (660 nm) with a maximum power output of 35 mW. All DLS measurements of PAX-loaded PNPs were performed in pure water at an experimental temperature of 25 °C and at a scattering angle of 90°.
For DLS sample preparation, PAX-loaded PNPs were transferred to precleaned polystyrene cuvettes, and then diluted with deionized water filtered through two nylon syringe filters in series with nominal pore sizes of 0.2 μm (National Scientific Company) if necessary. For each PNP preparation, mean effective hydrodynamic sizes were determined from three measurements of the autocorrelation function using cumulent analysis.
X-ray Diffraction
For XRD sample preparation, water was removed from dialyzed PNPs by rotary evaporation at 25 °C until solid films were obtained. The resulting films were then scraped as a powder onto the XRD specimen holder. X-ray diffraction measurements were performed on a Rigaku Miniflex diffractometer with a Cr source (KR radiation, λ = 2.2890 Å), operating at 30 kV and 15 mA with a resolution of 0.05° (2θ) and a scan speed of 1°/min. X-ray diffraction profiles were collected for 2θ ranging from 10 to 50°.
Peak deconvolution was done using Origin Pro 2015. Two characteristic reflections for both crystalline PCL and crystalline PEO were identified and used to fix the positions of four Lorentzian peak contributions to the fit;45 another small Lorentzian peak contribution was used to account for a small shoulder on the more intense PCL peak to obtain a good fit. Thus, XRD data was fit to a sum of 6 Lorentzian functions: 3 peaks assigned to crystalline PCL (2θ = 32.5, 32.7, and 35.7), 2 peaks assigned to crystalline PEO (2θ = 29.2 and 35.2), and 1 peak (no fixed position) assigned to incoherent scattering from amorphous copolymer (amorphous halo). Areal peak contributions from the three components (crystalline PCL, crystalline PEO, and amorphous copolymer) were then determined by integration and the percentage of crystalline PCL (relative to the total copolymer mass), χPCL, was calculated using
Paclitaxel Loading Efficiency Determination
PAX loading efficiencies of PAX-loaded PNPs were determined by high-performance liquid chromatography-mass spectrometry (HPLC-MS). Water was removed from ∼2 g of a gravimetrically determined quantity of PNP dispersion by rotary evaporation at 25 °C; then ∼0.2 g of a gravimetrically determined quantity of acetonitrile was added to the resulting solid and the mixture was vortexed for 2 min to ensure complete dissolution of drug. HPLC-MS (Ultimate 3000, Thermo Scientific) with a C18 column (Phenomenex Luna 5u C18), a constant eluent composition of 65/35 acetonitrile/water (v/v) with 1 v/v % formic acid, and a diode array detector (DAD) set at 227 nm, was then used to quantify the concentration of drug in the resulting solutions. Sample injection volumes were 50 μL, and the flow rate was 1 mL/min. A calibration curve for the DAD was generated by analysis of 7 standards containing different known PAX concentrations in acetonitrile. Quantities of PAX in the various dissolved PNP solutions were determined and loading efficiencies and loading levels were calculated for each sample using the following equations.
In Vitro PAX Release Kinetics
Experiments were carried out to monitor the in vitro release of PAX from PAX-loaded PNPs using HPLC-MS. In a typical experiment, after centrifugation to remove precipitated drug, ∼2 g of a gravimetrically determined quantity of dialyzed PAX-loaded PNPs were transferred to a 5 mL Float-A-Lyzer tube (SpectrumLabs, MWCO 100 kDa) for each predetermined time. These tubes were then placed in a 5 L beaker of the release medium, consisting of ∼4 L of PBS; throughout the release experiments, the release medium was constantly stirred using magnetic stirring and maintained at physiological temperature (37 °C), and was changed every 2 days. At each predetermined time (t = 1, 2, 4, 8, 24, 72, and 96 h), the solution of a single tube was transferred to a vial and then dried by rotary evaporation at 25 °C. After that, ∼0.2 g of a gravimetrically determined quantity of acetonitrile was added. The resulting solution was injected for HPLC-MS analysis and PAX was detected by DAD. Percentages of PAX released were calculated relative to the determined mass of PAX in the PNPs at the t = 0 release time. Reported release percentages at each release time are averages determined from triplicate preparations under the specified conditions.
Hydrolytic Degradation of PAX-Loaded P(MCL-co-CL)-b-PEO PNPs
During the in vitro release of PAX into PBS (pH = 7.4, 37°C), aliquots of PAX-loaded PNPs were removed at four different release times (t = 3, 6, 12, and 24 h) for analysis by DLS, to monitor hydrolytic degradation of the PNPs.
Cell-Culture and Antiproliferation Assay
MCF-7 cells were grown to ∼95% confluence in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum in a 75 cm2 tissue culture flask, and maintained at 37 °C with 5% CO2 in a tissue culture incubator. Cells were then trypsinized, collected, and pelleted by centrifugation (3 min at 160g). The cell pellet was then resuspended in DMEM media, and the cell concentration was determined using a hemocytometer. After the initial cell concentration was determined, the suspension was diluted to 1.0 × 105 cells/mL. Next, a multichannel pipette was used to fill a 96-well plate with 100 μL/well of the diluted cell suspension. The cell-loaded plates were then incubated for 24 h at 37 °C under an atmosphere of 5% CO2.
According to calculated loading levels, aqueous stock dispersions of each investigated sample of PAX-loaded PNPs were prepared by either diluting or concentrating the original dispersions, such that 100× dilution of the stock would result in a working PAX concentration of 2 μg/mL. After 24 h of cell incubation, 6.5 μL aliquots of stock dispersions of aqueous PAX-loaded PNPs or 6.5 μL of free PAX dissolved in dimethylsulfoxide were diluted in 643.5 μL of DMEM media to obtain a working PAX concentration of 2 μg/mL. Serial dilutions were carried out, and then 100 μL of each diluted stock was added to the appropriate well of the 96-well plate (containing ∼1.0 × 104 cells in 100 μL of media, as described above), to generate a range of different concentrations for analysis (1, 0.308, 0.095, 0.029, 0.0090, 0.0028, and 8.5 × 10–4 μg/mL PAX). The treated cells were incubated for 48, 72, or 96 h at 37 °C under a 5% CO2 atmosphere. To determine cell viability, 20 μL of CellTiter-Blue was added to each well after the predetermined incubation time was complete. After the addition of CellTiter-Blue, the 96-well plates were incubated for 4 h (5% CO2, 37 °C), and then fluorescence (λex = 560 nm; λem = 590 nm emission) readings were recorded on a 96-well plate reader. Percent growth was calculated for each well, based upon the following formula
where S is the sample reading (cells + drug + media), Bt is the average reading for the untreated population of cells (cells + media), and B0 is the average reading of the untreated population of cells at the beginning of the experiment (cells + media at t = 0). In the above equation, the numerator represents cell proliferation in the presence of drug and the denominator represents cell proliferation in the absence of drug. Percentage growth data sets were fit using XLfit (IDBS) and by forcing fits through 100% growth at low PAX concentrations. Reported GI50 values were determined from a single PNP preparation, with each concentration being tested 3 times (Figures S6–S8 and S11–S13 for percentage growth data). Standard errors (used as error bars in Figures 6 and 9) were obtained from the goodness of fit for each percentage growth plot, using the statistical package in XLfit. These same standard errors were then used to calculate the p-values used to determine statistical significance for comparisons between measured growth inhibition results.
Acknowledgments
We are grateful to the Natural Sciences and Engineering Research Council of Canada, NSERC, for financial support. We acknowledge Dr. Patrick Nahirney and the UVic EM lab (Department of Biology) for the continued use of their TEM. We also thank Natkritta Hüppe for her contributions to drawing the chemical structures.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b00829.
1H NMR data of monomer; additional characterization data for copolymers (1H NMR, GPC, XRD, critical water content data); schematic of microfluidic reactor; complete MCF-7 antiproliferative data for PAX-loaded PNPs; percentage cell death for empty (control) PNPs; additional TEM images and DLS sizes; table of actual flow rates; table of morphologies and DLS sizes for PNPs prepared by various methods (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Allen C.; Maysinger D.; Eisenberg A. Nano-engineering block copolymer aggregates for drug delivery. Colloids Surf., B 1999, 16, 3–27. 10.1016/S0927-7765(99)00058-2. [DOI] [Google Scholar]
- Discher D. E.; Ortiz V.; Srinivas G.; Klein M. L.; Kim Y.; Christian D.; Cai S.; Photos P.; Ahmed F. Emerging Applications of Polymersomes in Delivery: from Molecular Dynamics to Shrinkage of Tumors. Prog. Polym. Sci. 2007, 32, 838–857. 10.1016/j.progpolymsci.2007.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branco M. C.; Schneider J. P. Self-assembling materials for therapeutic delivery. Acta Biomater. 2009, 5, 817–831. 10.1016/j.actbio.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyrrell Z. L.; Shen Y. Q.; Radosz M. Fabrication of micellar nanoparticles for drug delivery through the self-assembly of block copolymers. Prog. Polym. Sci. 2010, 35, 1128–1143. 10.1016/j.progpolymsci.2010.06.003. [DOI] [Google Scholar]
- Gong J.; Chen M. W.; Zheng Y.; Wang S. P.; Wang Y. T. Polymeric micelles drug delivery system in oncology. J. Controlled Release 2012, 159, 312–323. 10.1016/j.jconrel.2011.12.012. [DOI] [PubMed] [Google Scholar]
- Kataoka K.; Harada A.; Nagasaki Y. Block copolymer micelles for drug delivery: Design, characterization and biological significance. Adv. Drug Delivery Rev. 2012, 64, 37–48. 10.1016/j.addr.2012.09.013. [DOI] [PubMed] [Google Scholar]
- Parveen S.; Misra R.; Sahoo S. K. Nanoparticles: a boon to drug delivery, therapeutics, diagnostics and imaging. Nanomedicine 2012, 8, 147–166. 10.1016/j.nano.2011.05.016. [DOI] [PubMed] [Google Scholar]
- Lee J. S.; Feijen J. Polymersomes for drug delivery: Design, formation and characterization. J. Controlled Release 2012, 161, 473–483. 10.1016/j.jconrel.2011.10.005. [DOI] [PubMed] [Google Scholar]
- Kwon G. S.; Kataoka K. Block copolymer micelles as long-circulating drug vehicles. Adv. Drug Delivery Rev. 2012, 64, 237–245. 10.1016/j.addr.2012.09.016. [DOI] [Google Scholar]
- Rosler A.; Vandermeulen G. W. M.; Klok H. A. Advanced drug delivery devices via self-assembly of amphiphilic block copolymers. Adv. Drug Delivery Rev. 2012, 64, 270–279. 10.1016/j.addr.2012.09.026. [DOI] [PubMed] [Google Scholar]
- Elsabahy M.; Wooley K. L. Design of polymeric nanoparticles for biomedical delivery applications. Chem. Soc. Rev. 2012, 41, 2545–2561. 10.1039/c2cs15327k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Chan H. F.; Leong K. W. Advanced materials and processing for drug delivery: The past and the future. Adv. Drug Delivery Rev. 2013, 65, 104–120. 10.1016/j.addr.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas J.; Mura S.; Brambilla D.; Mackiewicz N.; Couvreur P. Design, functionalization strategies and biomedical applications of targeted biodegradable/biocompatible polymer-based nanocarriers for drug delivery. Chem. Soc. Rev. 2013, 42, 1147–1235. 10.1039/C2CS35265F. [DOI] [PubMed] [Google Scholar]
- Lu Y.; Park K. Polymeric micelles and alternative nanonized delivery vehicles for poorly soluble drugs. Int. J. Pharm. 2013, 453, 198–214. 10.1016/j.ijpharm.2012.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad Z.; Shah A.; Siddiq M.; Kraatz H. B. Polymeric micelles as drug delivery vehicles. RSC Adv. 2014, 4, 17028–17038. 10.1039/C3RA47370H. [DOI] [Google Scholar]
- Oltra N. S.; Nair P.; Discher D. E. From Stealthy Polymersomes and Filomicelles to “Self” Peptide-Nanoparticles for Cancer Therapy. Annu. Rev. Chem. Biomol. Eng. 2014, 5, 281–299. 10.1146/annurev-chembioeng-060713-040447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun T.; Zhang Y. S.; Pang B.; Hyun D. C.; Yang M.; Xia Y. Engineered nanoparticles for drug delivery in cancer therapy. Angew. Chem., Int. Ed. 2014, 53, 12320–12364. 10.1002/anie.201403036. [DOI] [PubMed] [Google Scholar]
- Yokoyama M. Polymeric micelles as drug carriers: their lights and shadows. J. Drug Targeting 2014, 22, 576–583. 10.3109/1061186X.2014.934688. [DOI] [PubMed] [Google Scholar]
- Prabhu R. H.; Patravale V. B.; Joshi M. D. Polymeric nanoparticles for targeted treatment in oncology: current insights. Int. J. Nanomed. 2015, 10, 1001–1018. 10.2147/IJN.S56932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulbrich K.; Hola K.; Subr V.; Bakandritsos A.; Tucek J.; Zboril R. Targeted Drug Delivery with Polymers and Magnetic Nanoparticles: Covalent and Noncovalent Approaches, Release Control, and Clinical Studies. Chem. Rev. 2016, 116, 5338–5431. 10.1021/acs.chemrev.5b00589. [DOI] [PubMed] [Google Scholar]
- Park E. K.; Kim S. Y.; Lee S. B.; Lee Y. M. Folate-conjugated methoxy poly(ethylene glycol)/poly(epsilon-caprolactone) amphiphilic block copolymeric micelles for tumor-targeted drug delivery. J. Controlled Release 2005, 109, 158–168. 10.1016/j.jconrel.2005.09.039. [DOI] [PubMed] [Google Scholar]
- Jiang J.; Tong X.; Zhao Y. A new design for light-breakable polymer micelles. J. Am. Chem. Soc. 2005, 127, 8290–8291. 10.1021/ja0521019. [DOI] [PubMed] [Google Scholar]
- Jiang J. Q.; Tong X.; Morris D.; Zhao Y. Toward photocontrolled release using light-dissociable block copolymer micelles. Macromolecules 2006, 39, 4633–4640. 10.1021/ma060142z. [DOI] [Google Scholar]
- Blanco E.; Shen H.; Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. 10.1038/nbt.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K.; Tang X.; Zhang J.; Lu W.; Lin X.; Zhang Y.; Tian B.; Yang H.; He H. B. PEG-PLGA copolymers: Their structure and structure-influenced drug delivery applications. J. Controlled Release 2014, 183, 77–86. 10.1016/j.jconrel.2014.03.026. [DOI] [PubMed] [Google Scholar]
- Letchford K.; Liggins R.; Wasan K.; Burt H. In vitro human plasma distribution of nanoparticulate paclitaxel is dependent on the physicochemical properties of poly(ethylene glycol)-block-poly(caprolactone) nanoparticles. Eur. J. Pharm. Biopharm. 2009, 71, 196–206. 10.1016/j.ejpb.2008.08.003. [DOI] [PubMed] [Google Scholar]
- Glover A. L.; Nikles S. M.; Nikles J. A.; Brazel C. S.; Nikles D. E. Polymer micelles with crystalline cores for thermally triggered release. Langmuir 2012, 28, 10653–10660. 10.1021/la300895c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai S.; Vijayan K.; Cheng D.; Lima E. M.; Discher D. E. Micelles of different morphologies--advantages of worm-like filomicelles of PEO-PCL in paclitaxel delivery. Pharm. Res. 2007, 24, 2099–2109. 10.1007/s11095-007-9335-z. [DOI] [PubMed] [Google Scholar]
- Geng Y.; Dalhaimer P.; Cai S.; Tsai R.; Tewari M.; Minko T.; Discher D. E. Shape effects of filaments versus spherical particles in flow and drug delivery. Nat. Nanotechnol. 2007, 2, 249–255. 10.1038/nnano.2007.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairley N.; Hoang B.; Allen C. Morphological Control of Poly(ethylene glycol)-block-poly(eta-caprolactone) Copolymer Aggregates in Aqueous Solution. Biomacromolecules 2008, 9, 2283–2291. 10.1021/bm800572p. [DOI] [PubMed] [Google Scholar]
- Venkataraman S.; Hedrick J.; Ong Z.; Yang C.; Ee P.; Hammond P.; Yang Y. The effects of polymeric nanostructure shape on drug delivery. Adv. Drug Delivery Rev. 2011, 63, 1228–1246. 10.1016/j.addr.2011.06.016. [DOI] [PubMed] [Google Scholar]
- Gaumet M.; Vargas A.; Gurny R.; Delie F. Nanoparticles for drug delivery: The need for precision in reporting particle size parameters. Eur. J. Pharm. Biopharm. 2008, 69, 1–9. 10.1016/j.ejpb.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Truong N. P.; Whittaker M. R.; Mak C. W.; Davis T. P. The importance of nanoparticle shape in cancer drug delivery. Expert Opin. Drug Delivery 2015, 12, 129–142. 10.1517/17425247.2014.950564. [DOI] [PubMed] [Google Scholar]
- Du Z. X.; Xu J. T.; Fan Z. Q. Micellar morphologies of poly(epsilon-caprolactone)-b-poly(ethylene oxide) block copolymers in water with a crystalline core. Macromolecules 2007, 40, 7633–7637. 10.1021/ma070977p. [DOI] [Google Scholar]
- Du Z. X.; Xu J. T.; Fan Z. Q. Regulation of micellar morphology of PCL-b-PEO block copolymers by crystallization temperature. Macromol. Rapid Commun. 2008, 29, 467–471. 10.1002/marc.200700795. [DOI] [Google Scholar]
- Rizis G.; van de Ven T. G. M.; Eisenberg A. Crystallinity-driven morphological ripening processes for poly(ethylene oxide)-block-polycaprolactone micelles in water. Soft Matter 2014, 10, 2825–2835. 10.1039/c3sm52950a. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Eisenberg A. Multiple Morphologies of “Crew-Cut” Aggregates of Polystyrene-b-poly(acrylic acid) Block Copolymers. Science 1995, 268, 1728–1731. 10.1126/science.268.5218.1728. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Eisenberg A. Multiple Morphologies and Characteristics of “Crew-Cut” Micelle-like Aggregates of Polystyrene-b-poly(acrylic acid) Diblock Copolymers in Aqueous Solutions. J. Am. Chem. Soc. 1996, 118, 3168–3181. 10.1021/ja953709s. [DOI] [Google Scholar]
- Yu Y. S.; Eisenberg A. Control of morphology through polymer-solvent interactions in crew-cut aggregates of amphiphilic block copolymers. J. Am. Chem. Soc. 1997, 119, 8383–8384. 10.1021/ja9709740. [DOI] [Google Scholar]
- Zhang L.; Yu K.; Eisenberg A. Ion-Induced Morphological Changes in “Crew-Cut” Aggregates of Amphiphilic Block Copolymers. Science 1996, 272, 1777–1779. 10.1126/science.272.5269.1777. [DOI] [PubMed] [Google Scholar]
- Zhang L. E.; Eisenberg A. A. Morphogenic Effect of Added Ions on Crew-Cut Aggregates of Polystyrene-b-poly(acrylic acid) Block Copolymers in Solutions. Macromolecules 1996, 29, 8805–8815. 10.1021/ma961376t. [DOI] [Google Scholar]
- Schabas G.; Wang C. W.; Oskooei A.; Yusuf H.; Moffitt M. G.; Sinton D. Formation and shear-induced processing of quantum dot colloidal assemblies in a multiphase microfluidic chip. Langmuir 2008, 24, 10596–10603. 10.1021/la8022985. [DOI] [PubMed] [Google Scholar]
- Wang C. W.; Oskooei A.; Sinton D.; Moffitt M. G. Controlled Self-Assembly of Quantum Dot-Block Copolymer Colloids in Multiphase Microfluidic Reactors. Langmuir 2010, 26, 716–723. 10.1021/la902427r. [DOI] [PubMed] [Google Scholar]
- Wang C. W.; Sinton D.; Moffitt M. G. Flow-Directed Block Copolymer Micelle Morphologies via Microfluidic Self-Assembly. J. Am. Chem. Soc. 2011, 133, 18853–18864. 10.1021/ja2067252. [DOI] [PubMed] [Google Scholar]
- Bains A.; Cao Y. M.; Moffitt M. G. Multiscale Control of Hierarchical Structure in Crystalline Block Copolymer Nanoparticles Using Microfluidics. Macromol. Rapid Commun. 2015, 36, 2000–2005. 10.1002/marc.201500359. [DOI] [PubMed] [Google Scholar]
- Bains A.; Wulff J. E.; Moffitt M. G. Microfluidic synthesis of dye-loaded polycaprolactone-block-poly (ethylene oxide) nanoparticles: Insights into flow-directed loading and in vitro release for drug delivery. J. Colloid Interface Sci. 2016, 475, 136–148. 10.1016/j.jcis.2016.04.010. [DOI] [PubMed] [Google Scholar]
- Bains A.; Cao Y. M.; Kly S.; Wulff J. E.; Moffitt M. G. Controlling structure and function of polymeric drug delivery nanoparticles using microfluidics. Mol. Pharmaceutics 2017, 14, 2595–2606. 10.1021/acs.molpharmaceut.7b00177. [DOI] [PubMed] [Google Scholar]
- Ahmed F.; Discher D. E. Self-porating polymersomes of PEG-PLA and PEG-PCL: hydrolysis-triggered controlled release vesicles. J. Controlled Release 2004, 96, 37–53. 10.1016/j.jconrel.2003.12.021. [DOI] [PubMed] [Google Scholar]
- Allen C.; Yu Y. S.; Maysinger D.; Eisenberg A. Bioconjugate Chem. 1998, 9, 564–572. 10.1021/bc9702157. [DOI] [PubMed] [Google Scholar]
- Allen C.; Yu Y. S.; Eisenberg A.; Maysinger D. Cellular internalization of PCL20-b-PEO44 block copolymer micelles. Biochim. Biophys. Acta, Biomembr. 1999, 1421, 32–38. 10.1016/S0005-2736(99)00108-X. [DOI] [PubMed] [Google Scholar]
- Allen C.; Han J. N.; Yu Y. S.; Maysinger D.; Eisenberg A. Polycaprolactone-b-poly(ethylene oxide) copolymer micelles as a delivery vehicle for dihydrotestosterone. J. Controlled Release 2000, 63, 275–286. 10.1016/S0168-3659(99)00200-X. [DOI] [PubMed] [Google Scholar]
- Lim Soo P.; Luo L.; Maysinger D.; Eisenberg A. Langmuir 2002, 18, 9996–10004. 10.1021/la026339b. [DOI] [Google Scholar]
- Geng Y.; Discher D. E. Hydrolytic degradation of poly(ethylene oxide)-block-polycaprolactone worm micelles. J. Am. Chem. Soc. 2005, 127, 12780–12781. 10.1021/ja053902e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoroghchian P. P.; Li G.; Levine D. H.; Davis K. P.; Bates F. S.; Hammer D. A.; Therien M. J. Bioresorbable Vesicles Formed through Spontaneous Self-Assembly of Amphiphilic Poly(ethylene oxide)-block-polycaprolactone. Macromolecules 2006, 39, 1673–1675. 10.1021/ma0519009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostacolo L.; Marra M.; Ungaro F.; Zappavigna S.; Maglio G.; Quaglia F.; Abbruzzese A.; Caraglia M. In vitro anticancer activity of docetaxel-loaded micelles based on poly(ethylene oxide)-poly(epsilon-caprolactone) block copolymers: Do nanocarrier properties have a role?. J. Controlled Release 2010, 148, 255–263. 10.1016/j.jconrel.2010.08.006. [DOI] [PubMed] [Google Scholar]
- Letchford K.; Burt H. M. Copolymer micelles and nanospheres with different in vitro stability demonstrate similar paclitaxel pharmacokinetics. Mol. Pharmaceutics 2012, 9, 248–260. 10.1021/mp2002939. [DOI] [PubMed] [Google Scholar]
- Chang L.; Wang W. W.; Huang P. S.; Lv Z. S.; Hu F. Q.; Zhang J. H.; Kong D. L.; Deng L. D.; Dong A. J. Photo-crosslinked poly(ethylene glycol)-b-poly(epsilon-caprolactone) nanoparticles for controllable paclitaxel release. J. Biomater. Sci., Polym. Ed. 2013, 24, 1900–1921. 10.1080/09205063.2013.808152. [DOI] [PubMed] [Google Scholar]
- Wang C.; Xiao Y.; Heise A.; Lang M. D. Organometallic and Enzymatic Catalysis for Ring Opening Copolymerization of epsilon-Caprolactone and 4-methyl-epsilon-Caprolactone. J. Polym. Sci., Part A: Polym. Chem. 2011, 49, 5293–5300. 10.1002/pola.25007. [DOI] [Google Scholar]
- Hillmyer M. A.; Tolman W. B. Aliphatic Polyester Block Polymers: Renewable, Degradable, and Sustainable. Acc. Chem. Res. 2014, 47, 2390–2396. 10.1021/ar500121d. [DOI] [PubMed] [Google Scholar]
- Martello M. T.; Hillmyer M. A. Polylactide-Poly(6-methyl-epsilon-caprolactone)-Polylactide Thermoplastic Elastomers. Macromolecules 2011, 44, 8537–8545. 10.1021/ma201063t. [DOI] [Google Scholar]
- Zupancich J. A.; Bates F. S.; Hillmyer M. A. Aqueous dispersions of poly(ethylene oxide)-b-poly(gamma-methyl-epsilon-caprolactone) block copolymers. Macromolecules 2006, 39, 4286–4288. 10.1021/ma060642s. [DOI] [Google Scholar]
- Saito N.; Liu C.; Lodge T. P.; Hillmyer M. A. Multicompartment Micelle Morphology Evolution in Degradable Miktoarm Star Terpolymers. ACS Nano 2010, 4, 1907–1912. 10.1021/nn9016873. [DOI] [PubMed] [Google Scholar]
- Petersen M. A.; Yin L. G.; Kokkoli E.; Hillmyer M. A. Synthesis and characterization of reactive PEO-PMCL polymersomes. Polym. Chem. 2010, 1, 1281–1290. 10.1039/c0py00143k. [DOI] [Google Scholar]
- Lee R.-S.; Hung C.-B. Synthesis and characterization of amphiphilic block copolymers from poly (ethylene glycol) methyl ether and 4-methyl-ε-caprolactone or 4-phenyl-ε-caprolactone. Polymer 2007, 48, 2605–2612. 10.1016/j.polymer.2007.03.018. [DOI] [Google Scholar]
- Lee R. S.; Huang Y. T.; Chen W. H. Synthesis and Characterization of Temperature-Sensitive Block Copolymers from Poly(N-isopropylacrylamide) and 4-Methyl-epsilon-caprolactone or 4-Phenyl-epsilon-caprolactone. J. Appl. Polym. Sci. 2010, 118, 1634–1642. 10.1002/app.32546. [DOI] [Google Scholar]
- Peng K. Y.; Wang S. W.; Lee R. S. Amphiphilic diblock copolymers based on poly(2-ethyl-2-oxazoline) and poly(4-substituted-epsilon-caprolactone): Synthesis, characterization, and cellular uptake. J. Polym. Sci., Part A: Polym. Chem. 2013, 51, 2769–2781. 10.1002/pola.26690. [DOI] [Google Scholar]
- Foster T.; Dorfman K. D.; Davis H. T. Giant biocompatible and biodegradable PEG-PMCL vesicles and microcapsules by solvent evaporation from double emulsion droplets. J. Colloid Interface Sci. 2010, 351, 140–150. 10.1016/j.jcis.2010.05.020. [DOI] [PubMed] [Google Scholar]
- Cabane E.; Malinova V.; Meier W. Synthesis of Photocleavable Amphiphilic Block Copolymers: Toward the Design of Photosensitive Nanocarriers. Macromol. Chem. Phys. 2010, 211, 1847–1856. 10.1002/macp.201000151. [DOI] [Google Scholar]
- Matter Y.; Enea R.; Casse O.; Lee C. C.; Baryza J.; Meier W. Amphiphilic PEG-b-PMCL-b-PDMAEMA Triblock Copolymers: From Synthesis to Physico-Chemistry of Self-Assembled Structures. Macromol. Chem. Phys. 2011, 212, 937–949. 10.1002/macp.201000661. [DOI] [Google Scholar]
- Vasquez D.; Einfalt T.; Meier W.; Palivan C. G. Asymmetric Triblock Copolymer Nanocarriers for Controlled Localization and pH-Sensitive Release of Proteins. Langmuir 2016, 32, 10235–10243. 10.1021/acs.langmuir.6b01803. [DOI] [PubMed] [Google Scholar]
- Wang C. W.; Bains A.; Sinton D.; Moffitt M. G. Flow-Directed Assembly of Block Copolymer Vesicles in the Lab-on-a-Chip. Langmuir 2012, 28, 15756–15761. 10.1021/la303655s. [DOI] [PubMed] [Google Scholar]
- Wang C. W.; Bains A.; Sinton D.; Moffitt M. G. Flow-Directed Loading of Block Copolymer Micelles with Hydrophobic Probes in a Gas-Liquid Microreactor. Langmuir 2013, 29, 8385–8394. 10.1021/la400011n. [DOI] [PubMed] [Google Scholar]
- Wang C. W.; Sinton D.; Moffitt M. G. Morphological Control via Chemical and Shear Forces in Block Copolymer Self-Assembly in the Lab-on-Chip. ACS Nano 2013, 7, 1424–1436. 10.1021/nn305197m. [DOI] [PubMed] [Google Scholar]
- Xu Z. Q.; Yan B.; Riordon J.; Zhao Y.; Sinton D.; Moffitt M. G. Microfluidic Synthesis of Photoresponsive Spool-Like Block Copolymer Nanoparticles: Flow-Directed Formation and Light-Triggered Dissociation. Chem. Mater. 2015, 27, 8094–8104. 10.1021/acs.chemmater.5b03800. [DOI] [Google Scholar]
- Xu Z.; Lu C. H.; Riordon J.; Sinton D.; Moffitt M. G. Microfluidic Manufacturing of Polymeric Nanoparticles: Comparing Flow Control of Multiscale Structure in Single-Phase Staggered Herringbone and Two-Phase Reactors. Langmuir 2016, 32, 12781–12789. 10.1021/acs.langmuir.6b03243. [DOI] [PubMed] [Google Scholar]
- Massey J. A.; Temple K.; Cao L.; Rharbi Y.; Raez J.; Winnik M. A.; Manners I. Self-assembly of organometallic block copolymers: The role of crystallinity of the core-forming polyferrocene block in the micellar morphologies formed by poly(ferrocenylsilane-b-dimethylsiloxane) in n-alkane solvents. J. Am. Chem. Soc. 2000, 122, 11577–11584. 10.1021/ja002205d. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.