

Abstract
Water-soluble pillar[6]arene (WP6) was used to solubilize camptothecin family antitumor drugs. In the presence of WP6, the solubility of camptothecin (CPT) and 10-hydroxycamptothecin (HCPT) was enhanced by 380 and 40 times, respectively. The solubility enhancement is proved to be the result of the host–guest encapsulation by WP6. WP6 has a low cytotoxicity against normal MC 3T3-E1 cells, whereas the bioactivity of CPT and HCPT is substantially improved as a result of the solubility enhancement.
1. Introduction
Discovered in 1966, 20-(S)-camptothecin (CPT) and its derivatives constitute a family of important antitumor drugs.1 CPT shows powerful antitumor activity by targeting DNA topoisomerase I (TOP I).2 However, CPT suffers from two major limitations: (1) CPT is sparingly soluble in water, limiting its clinical applications via intravenous injection; (2) the biologically active form of CPT (lactone) rapidly undergoes a ring-opening reaction and converts to the biologically inactive form (carboxylate) under neutral conditions (Figure 1a).3 CPT derivatives with improved solubility in water have been developed, including the clinically approved antitumor drugs topotecan and irinotecan (Figure 1b).4,5 Liposomes,6 inorganic nanoparticles,7 hydrogels,8 and prodrug conjugates9 have been developed to formulate and deliver CPT. To achieve the full potential of CPT for cancer treatment, new delivery methods are highly desirable to solubilize this hydrophobic antitumor drug.
Figure 1.

(a) Molecular structure of CPT and its ring-opening reaction; (b) molecular structure of CPT derivatives (modification highlighted in blue); (c) the synthesis of water-soluble pillar[6]arene (WP6).
Pillar[n]arenes (PA[n]s, n = 5–10) are a type of macrocyclic hosts with a unique pillar-shaped architecture.10 Since 2008, the excellent host–guest recognition ability of PA[n]s has attracted great interests.11−18 Water-soluble PA[n]s with either anionic (carboxylate)19 or cationic (amine)20 modifications have been used for drug delivery,21 material assembly,22 and antibiotic applications.23 Among these water-soluble PA[n]s, carboxylated PA[6] (WP6, Figure 1c) is an important macrocyclic host with a large cavity (7.7 Å),24 excellent host–guest recognition ability, and relatively simple synthesis.25 Recent study also demonstrated the satisfactory biocompatibility of WP6.26 There are reports of cyclodextrin and cucurbituril-based molecular containers used to formulate and solubilize hydrophobic pharmaceuticals.27−31 The clinical success of β-cyclodextrin-based Captisol proves that solubility enhancement by host–guest encapsulation has practical use.32,33 Water-soluble PA[n]s have outstanding recognition property and can be easily functionalized for “smart” drug delivery. We envision that WP6 and other water-soluble PA[n]s may be ideal candidates for solubility enhancement applications.
Herein, we report the use of WP6 for the solubility and bioactivity enhancement of CPT and 10-hydroxycamptothecin (HCPT). In this article, we study the WP6-assisted solubility improvement of CPT and HCPT. The recognition behavior of WP6 toward CPT family drugs is investigated. Finally, the bioactivity of WP6-solubilized CPT and HCPT, as well as the biocompatibility of WP6, are tested in vitro.
2. Results and Discussion
2.1. Design and Synthesis
WP6 was chosen as the drug carrier for two reasons: (1) the cavity is large enough to accommodate CPT and its derivatives; (2) PA[6], the precursor to synthesize WP6, is relatively easy to prepare. WP6 was synthesized according to the literary procedures (Figure 1c).25 Starting from inexpensive materials, the condensation reaction of 1,4-diethyoxybenzene and paraformaldehyde delivered PA[6] with a yield of 15%. After removing ethoxy groups with BBr3, phenolic hydroxyl groups were substituted with methyl esters. Finally, the hydrolysis reaction converted esters to carboxylates. The treatment with NaOH yielded WP6 as a sodium carboxylate. The synthesis of WP6 is relatively simple, with low cost, which renders the large-scale preparation and pharmaceutical applications possible. WP6 is highly soluble in water. Its solubility was measured to be 33 mM in NaD2PO4 buffer (10 mM, pD = 7.4). Its excellent solubility in water enables WP6 to be used for solubility enhancement of hydrophobic pharmaceuticals.
2.2. Solubility Enhancement
The solubility enhancement assay was performed with poorly soluble drugs CPT and HCPT. Although CPT is barely soluble (5 μM) in phosphate-buffered saline (PBS), HCPT is slightly more soluble (29 μM). To determine the solubility enhancement, a known concentration of WP6 was stirred with excess solid drug for 3 h in NaD2PO4 buffer (10 mM, pD 7.4). The excess insoluble drug was removed with centrifugation. The concentration of the drug in the supernatant was determined by 1H NMR integration of drug resonances versus 1,3,5-benzenetricarboxylic acid, subsequently added as internal reference (Figures S1–S12). The solubility enhancement assay was performed in triplicate. Phase solubility diagrams are shown in Figure 2. WP6 substantially improves the solubility of both drugs (380 times for CPT and 40 times for HCPT). The solubility reaches 1.9 mM for CPT and 1.2 mM for HCPT in the presence of 10 mM WP6, which is soluble enough for dosing via intravenous injection. Both solubility phase diagrams are referred to as AP type plots, signifying the presence of a higher order complex (e.g. (WP6)2-CPT).33
Figure 2.
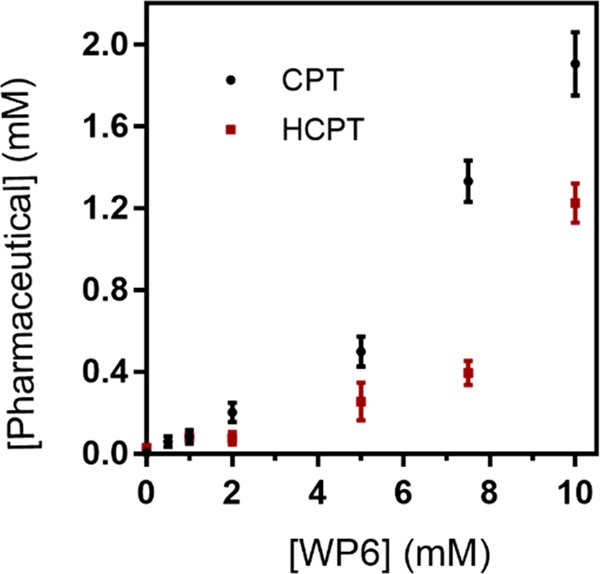
Phase solubility diagrams for CPT (black) and HCPT (red) with WP6. Error bars are derived from triplicates.
2.3. Host–Guest Chemistry
We investigated the host–guest interaction between WP6 and CPT family pharmaceuticals. The intrinsic solubility of CPT and HCPT is too low to determine the host–guest chemistry at neutral pH. Instead, a basic solvent of 0.1 M NaOD in D2O was used to improve the solubility of CPT (Figure S13). The 1H NMR spectrum recorded for equal molarities of WP6 and CPT shows a broadening of CPT aromatic resonances, which indicates the host–guest interaction between WP6 and CPT (carboxylate form). To investigate the encapsulation under neutral condition, topotecan (TPT), a water-soluble and clinically approved CPT family drug, was used to study its binding with WP6 at pH 7.4. Figure 3a shows the 1H NMR spectra recorded for TPT, WP6, and mixtures of WP6 and TPT (1:2, 1:1, and 1:0.5). Aromatic proton a and methyl proton g undergo significant upfield shifts in the presence of WP6, which indicates the encapsulation of TPT. The resonances of upfield-shifted aromatic protons are broadened in the presence of excess TPT, without the observation of free topotecan resonances, demonstrating a fast exchange on the chemical shift time scale. TPT has a (CH3)2NCH2– substituent, which enhances its electrostatic interaction with WP6. Parts of the TPT aromatic system are encapsulated inside the cavity, which implicates that hydrophobicity and π–π stacking are also the causes of encapsulation. Unlike TPT, there is no electrostatic interaction between WP6 and CPT at basic pH. We believe that the driving force for the host–guest interaction between WP6 and CPT (carboxylate form) under basic condition is hydrophobicity and π–π stacking.
Figure 3.

(a) 1H NMR spectra recorded for (1) WP6, (2) topotecan, and mixtures of WP6 and topotecan, with ratios of 1:2 (3), 1:1 (4), and 1:0.5 (5) (400 MHz, room temperature, 10 mM NaD2PO4, pD 7.4, [WP6] = 1 mM). (b) The molecular structure of dyes used in this paper, and (c) UV–vis spectra from the titration of proflavine (10 μM) with WP6 (0 – 72 μM) at pH 7.4 (PBS).
The stability of the complex formed by WP6 and the pharmaceutical is critical for the clinical use. We evaluated the complex stability by measuring the binding constant Ka. Phosphate-buffered saline (PBS) was used to mimic the neutral physiological environment (pH 7.4). Because CPT and HCPT are poorly soluble in water, direct 1H NMR titration could not be used to determine the value of Ka. Instead, we used the indicator displacement assay.34 We first measured the value of Ka for the complex of WP6 and dye. Two dyes, proflavine and neutral red, were used in the study (Figure 3b). The value of Ka for the complex of WP6 and dye was determined by direct UV–vis titration (Figures 3c and S15a). The existence of the isosbestic point shows a 1:1 binding stoichiometry for both dyes. We also used the Job plot to confirm that the binding stoichiometry for proflavine is 1:1 (Figure S16). The Ka value for the complex of WP6 and proflavine/neutral red was calculated to be (1.4 ± 0.1) × 105 M–1 and (1.3 ± 0.1) × 105 M–1, respectively (Figures S14b and S15b). We then used the indicator displacement assay by fluorescence or UV–vis spectroscopy to determine the Ka value for the complex of WP6 and CPT family antitumor drugs. The Ka value was determined to be (8.4 ± 1.0) × 106 M–1 for CPT, (1.2 ± 0.2) × 105 M–1 for HCPT, and (1.3 ± 0.1) × 104 M–1 for TPT (Figures S17–S19). The Ka value for the complex of WP6 and CPT/HCPT falls in the range of 105–107 M–1 in PBS, which indicates that WP6 could form a stable complex with CPT/HCPT during injection.
2.4. In Vitro Study
We then investigated the bioactivity of WP6-solubilized CPT and HCPT in vitro. It is crucial for the WP6 carrier to be biocompatible for any drug delivery application. The cytocompatibility of WP6 was checked in a normal cell line MC 3T3-E1 using a Cell Counting Kit-8 assay, and the results are presented in Figure S20. Very little cytotoxicity was observed for the WP6 systems with various concentrations. Even if the concentration of WP6 was as high as 500 μM, the cell viability was still above 90%. Hence, WP6 has very good biocompatibility and shows great potential for biomedical applications.
To determine the antitumor activity of solubilized drugs in the WP6 systems, the cytotoxicity of WP6 systems containing CPT or HCPT against a tumor cell line Hela was also examined, and the results are shown in Figure 4. Because of the good biocompatibility of WP6 itself, the treatment of WP6 systems alone with Hela cells had no significant effect on the cell viability. In sharp contrast, both the CPT-loaded WP6 system and the HCPT-loaded WP6 system exhibited an increased cytotoxicity with drug concentration. As shown in Figure 4, with a drug concentration of 5 μM, the viability of Hela cells reached 10 and 40% when treated with CPT-loaded WP6 and HCPT-loaded WP6, respectively. Calculated by the SPSS method, the half maximal inhibitory concentration (IC50) of the CPT-loaded WP6 system was 0.1 μM, whereas that of the HCPT-loaded WP6 system was 0.5 μM. We also treated Hela cells with CPT or HCPT alone and calculated the IC50 to be 0.8 and 1.3 μM, respectively. It is obvious that the IC50 of WP6-solubilized CPT or HCPT is lower compared to that of the free drug. We also determined the influence of WP6 encapsulation toward bioactivity of the water-soluble antitumor drug TPT (Figure S21). The bioactivity of WP6-encapsulated TPT was slightly higher compared to that of the free drug at 5 μM.
Figure 4.
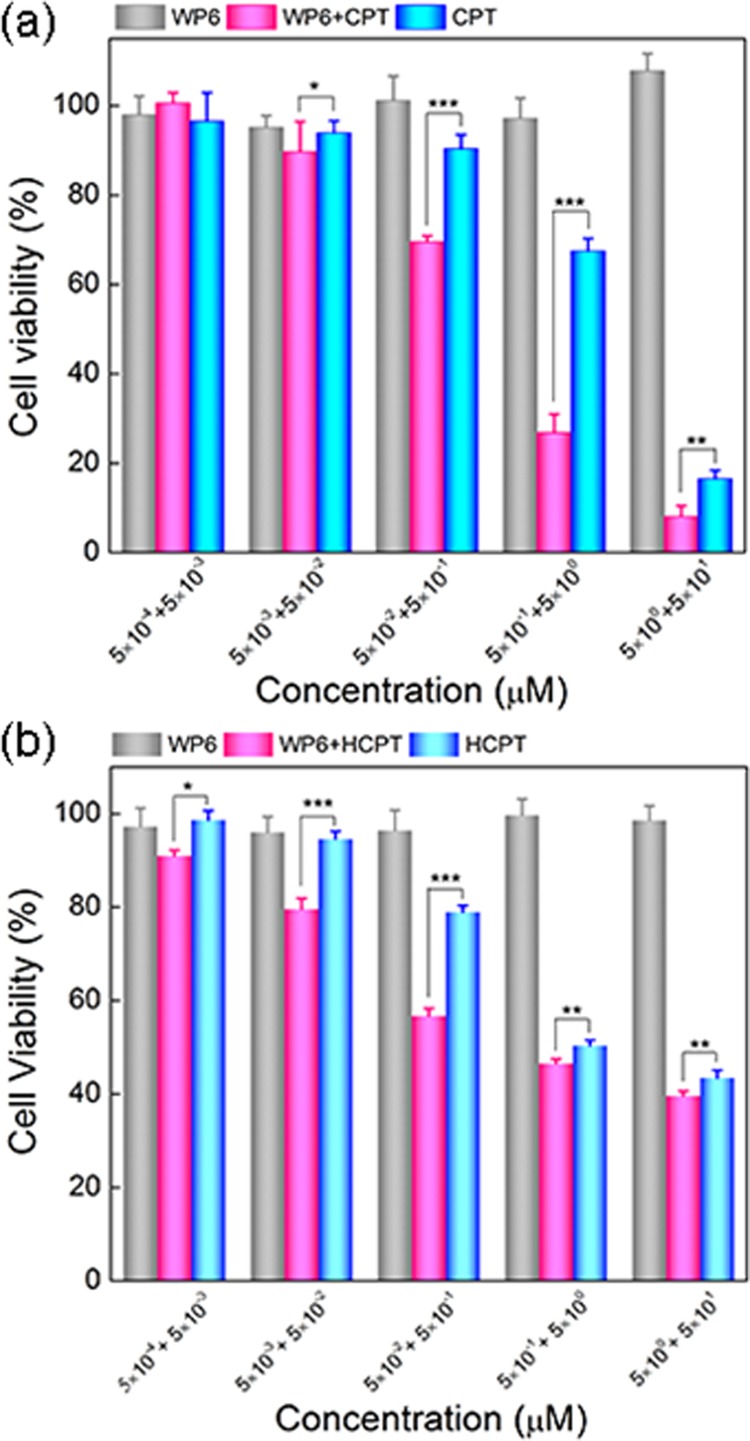
(a) In vitro cytotoxicity of WP6, CPT-loaded WP6, and free CPT against Hela cells as a function of CPT concentration. (b) In vitro cytotoxicity of WP6, HCPT-loaded WP6, and free HCPT against Hela cells as a function of HCPT concentration. The two values in the horizontal coordinate indicate the concentration of the drug (front) and WP6 (back), respectively. The blank control with culture medium only was set as 100% cell viability. Each point represents the mean ± SD; n = 6. Significant differences between the groups of free HCPT and HCPT-loaded WP6 determined by the Student’s t test are marked as the following criterion: *p < 0.05, **p < 0.01, ***p < 0.001.
The cell assays indicate that WP6 is a biocompatible drug carrier to be used in vitro. By enhancing the solubility of CPT/HCPT, WP6 is able to improve the cytotoxicity of the drugs. Because CPT/HCPT alone has very limited solubility in PBS, they are not soluble enough to be dosed via intravenous injection. With the solubility enhancement of WP6, CPT/HCPT can reach a solubility of more than 1 mM and may be used for chemotherapy in vivo.
3. Conclusions
In summary, water-soluble pillar[6]arene (WP6) dramatically enhances the solubility of CPT and HCPT. The study of host–guest chemistry confirms that WP6 could bind CPT family antitumor drugs with a stable complex formed between WP6 and CPT/HCPT. The bioactivity of CPT and HCPT was substantially improved with WP6 in vitro. The drug carrier WP6 was proven to have a low cytotoxicity and high biocompatibility. Our work demonstrates that WP6 and other water-soluble pillar[n]arenes have a great potential to be used for the formulation of poorly soluble drugs and active pharmaceutical ingredients (APIs).
4. Experimental Section
4.1. General Experimental
All reagents and solvents were purchased from commercial suppliers and were used without further purification. WP6 was prepared according to the procedure in the literature.25,35 Ultraviolet–visible (UV–vis) spectroscopy was performed on a Cary 100 Agilent UV–vis spectrometer. Fluorescence spectra were measured on a Shimadzu RF-1536 spectrometer. NMR spectra were recorded on an AVANCE III 400 spectrometer. Cell viability was measured on a Biotek ELx808 Microplate Reader.
4.2. Intrinsic Solubility of WP6
Excess WP6 was added into NaD2PO4 buffer (10 mM, pD = 7.4). The suspension was stirred at room temperature for 12 h. The excess solid was removed with centrifugation. The concentration of WP6 in the supernatant was determined with 1H NMR spectroscopy by comparing the integral of a known concentration of 1,3,5-benzenetricarboxylic acid as the internal reference. The solubility of WP6 was calculated to be 33 mM.
4.3. Intrinsic Solubility of CPT/HCPT
Excess CPT was added into PBS. The suspension was stirred at room temperature for 6 h. The excess solid was removed with centrifugation. The concentration of CPT was determined with UV–vis spectroscopy on the basis of a standard curve plotted with the UV–vis absorbance of CPT. The solubility of CPT was measured to be 5 μM.
Excess HCPT was added into NaD2PO4 buffer (10 mM, pD = 7.4). The suspension was stirred at room temperature for 3 h. The excess solid was removed with centrifugation. The concentration of HCPT in the supernatant was determined with 1H NMR spectroscopy by comparing the integral of a known concentration of 1,3,5-benzenetricarboxylic acid as internal reference. The solubility of HCPT was measured to be 29 μM.
4.4. Concentration of WP6-Solubilized Pharmaceutics
Excess CPT/HCPT was added into a solution of WP6 in NaD2PO4 (10 mM, pD = 7.4) with a known concentration. The suspension was stirred at room temperature for 3 h. The excess solid was removed with centrifugation. The concentration of CPT/HCPT in the supernatant was determined with 1H NMR spectroscopy by comparing the integral with a known concentration of 1,3,5-benzenetricarboxylic acid as the internal reference. Each data point was performed in triplicate to calculate the deviation.
4.5. In Vitro Cytotoxicity of WP6 against Normal MC 3T3-E1 Cells
To determine the biocompatibility of WP6, the cytotoxicity of WP6 against normal MC 3T3-E1 was quantitatively detected by a Cell Counting Kit-8 (CCK-8) assay. MC 3T3-E1 cells were seeded into 96-well plates at 4 × 103/well. Cells were first incubated in the atmosphere containing 0.5% CO2 at 37 °C for 12 h before drug treatment. Next, the medium from the cells was aspirated and fresh medium (200 μL/well) containing WP6 at varying concentrations (5 × 10–3–5 × 102 μM) was added as feed. After 24 h of incubation, the medium of each well was replaced with 200 μL of fresh medium containing 20 μL of CCK-8 solution. Finally, the OD value of each well was detected at a wavelength of 450 nm by a Microplate Reader (Biotek ELx808) after 3 h of incubation. The relative cell viability was calculated to quantify the cytotoxicity, and the control group without treatment of any material except culture medium added was defined as 100% viability.
4.6. In Vitro Cytotoxicity of Combinations between WP6 and CPT/HCPT against Hela Cancer Cells
The cytotoxicity of solubilized CPT/HCPT in the WP6 systems against the Hela cancer cell line was examined by a CCK-8 assay. Hela cells were seeded in 96-plate well (4 × 103/well) and cultured for 12 h. Then, the cells were treated with fresh medium (200 μL/well) containing free CPT/HCPT, pure WP6, and CPT/HCPT-loaded WP6 systems at varying drug concentrations (for CPT/HCPT and CPT/HCPT loaded in WP6: 5 × 10–4–5 × 101 μM; for WP6 carrier: 5 × 10–3–5 × 102 μM) for another 24 h. The detecting and analytical methods were the same as above-mentioned methods. The concentration of the test agent in 50% cell viability was defined as IC50, and the SPSS software (IBM SPSS Statistics 21.0.0.0) was used to calculate the IC50 value.
4.7. Statistical Analysis
All of the cellular experiment results were expressed by their mean ± SD (standard deviation). Comparisons between groups were analyzed by Student’s t test, and a p value less than 0.05 was considered as the criterion of a statistically significant difference.
Acknowledgments
The authors are grateful to Fudan University (start-up grant), Natural Science Foundation of China (grant No. 51503042, 21672042, 51273217, and 21474019), and State Key Project of Research and Development (grant No. 2016YFC1100300) for financial support.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b01032.
Methods for solubility measurement, 1H NMR spectra, determination of the binding constant and supplementary in vitro data (PDF)
Author Contributions
§ Y.L. and X.C. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Wall M. E.; Wani M. C.; Cook C. E.; Palmer K. H.; McPhail A. T.; Sim G. A. J. Am. Chem. Soc. 1966, 88, 3888–3890. 10.1021/ja00968a057. [DOI] [Google Scholar]
- Giovanella B. C.; Hinz H. R.; Kozielski A. J.; Stehlin J. S.; Silber R.; Potmesil M. Cancer Res. 1991, 51, 3052–3055. [PubMed] [Google Scholar]
- Venditto V. J.; Simanek E. E. Mol. Pharmaceutics 2010, 7, 307–349. 10.1021/mp900243b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wani M. C.; Nicholas A. W.; Wall M. E. J. Med. Chem. 1986, 29, 2358–2363. 10.1021/jm00161a035. [DOI] [PubMed] [Google Scholar]
- Natsuka K.; Nakamura H.; Nishikawa Y.; Negoro T.; Uno H.; Nishimura H. J. Med. Chem. 1987, 30, 1779–1787. 10.1021/jm00393a017. [DOI] [PubMed] [Google Scholar]
- Burke T. G.; Staubus A. E.; Mishra A. K.; Malak H. J. Am. Chem. Soc. 1992, 114, 8318–8319. 10.1021/ja00047a069. [DOI] [Google Scholar]
- Wu W.; Li R.; Bian X.; Zhu Z.; Ding D.; Li X.; Jia Z.; Jiang X.; Hu Y. ACS Nano 2009, 3, 2740–2750. 10.1021/nn9005686. [DOI] [PubMed] [Google Scholar]
- Chang G.; Ci T.; Yu L.; Ding J. D. J. Controlled Release 2011, 156, 21–27. 10.1016/j.jconrel.2011.07.008. [DOI] [PubMed] [Google Scholar]
- Yu L.; Chang G. T.; Zhang H.; Ding J. D. Int. J. Pharm. 2008, 348, 95–106. 10.1016/j.ijpharm.2007.07.026. [DOI] [PubMed] [Google Scholar]
- Ogoshi T.; Kanai S.; Fujinami S.; Yamagishi T.; Nakamoto Y. J. Am. Chem. Soc. 2008, 130, 5022–5023. 10.1021/ja711260m. [DOI] [PubMed] [Google Scholar]
- Cao Y.; Hu X.; Li Y.; Zou X.; Xiong S.; Lin C.; Shen Y.; Wang L. J. Am. Chem. Soc. 2014, 136, 10762–10769. 10.1021/ja505344t. [DOI] [PubMed] [Google Scholar]
- Chang Y.; Yang K.; Wei P.; Huang S.; Pei Y.; Zhao W.; Pei Z. Angew. Chem., Int. Ed. 2014, 53, 13126–13130. 10.1002/anie.201407272. [DOI] [PubMed] [Google Scholar]
- Hu W.; Yang H.; Hu W.; Ma M.; Zhao X.; Mi X.; Liu Y. A.; Li J.; Jiang B.; Wen K. Chem. Commun. 2014, 50, 10460–10463. 10.1039/C4CC01810A. [DOI] [PubMed] [Google Scholar]
- Li C.; Zhao L.; Li J.; Ding X.; Chen S.; Zhang Q.; Yu Y.; Jia X. Chem. Commun. 2010, 46, 9016–9018. 10.1039/c0cc03575k. [DOI] [PubMed] [Google Scholar]
- Li S.; Zhang H.; Xu X.; Liu Y. Nat. Commun. 2015, 6, 7590 10.1038/ncomms8590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L.; Nie G.; Tian D.; Deng H.; Jiang L.; Li H. Angew. Chem. 2016, 128, 12905–12908. 10.1002/ange.201603906. [DOI] [PubMed] [Google Scholar]
- Ke C.; Strutt N. L.; Li H.; Hou X.; Hartlieb K. J.; Mcgonigal P. R.; Ma Z.; Iehl J.; Stern C. L.; Cheng C.; Zhu Z.; Vermeulen N. A.; Meade T. J.; Botros Y. Y.; Stoddart J. F. J. Am. Chem. Soc. 2013, 135, 17019–17030. 10.1021/ja407229h. [DOI] [PubMed] [Google Scholar]
- Sun Y.; Guo F.; Zuo T.; Hua J.; Diao G. Nat. Commun. 2016, 7, 12042 10.1038/ncomms12042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogoshi T.; Ueshima N.; Yamagishi T.; Toyota Y.; Matsumi N. Chem. Commun. 2012, 48, 3536–3538. 10.1039/c2cc30589e. [DOI] [PubMed] [Google Scholar]
- Yakimova L. S.; Shurpik D. N.; Gilmanova L. H.; Makhmutova A. R.; Rakhimbekova A.; Stoikov I. I. Org. Biomol. Chem. 2016, 14, 4233–4238. 10.1039/C6OB00539J. [DOI] [PubMed] [Google Scholar]
- Yu G.; Zhao R.; Wu D.; Zhang F.; Shao L.; Zhou J.; Yang J.; Tang G.; Chen X.; Huang F. Polym. Chem. 2016, 7, 6178–6188. 10.1039/C6PY01402J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G.; Yu W.; Mao Z.; Gao C.; Huang F. Small 2015, 11, 919–925. 10.1002/smll.201402236. [DOI] [PubMed] [Google Scholar]
- Joseph R.; Naugolny A.; Feldman M.; Herzog I. M.; Fridman M.; Cohen Y. J. Am. Chem. Soc. 2016, 138, 754–757. 10.1021/jacs.5b11834. [DOI] [PubMed] [Google Scholar]
- Han C.; Ma F.; Zhang Z.; Xia B.; Yu Y.; Huang F. Org. Lett. 2010, 12, 4360–4363. 10.1021/ol1018344. [DOI] [PubMed] [Google Scholar]
- Yu G.; Xue M.; Zhang Z.; Li J.; Han C.; Huang F. J. Am. Chem. Soc. 2012, 134, 13248–13251. 10.1021/ja306399f. [DOI] [PubMed] [Google Scholar]
- Wheate N. J.; Dickson K.-A.; Kim R. R.; Nematollahi A.; Macquart R. B.; Kayser V.; Yu G.; Church W. B.; Marsh D. J. J. Pharm. Sci. 2016, 105, 3615–3625. 10.1016/j.xphs.2016.09.008. [DOI] [PubMed] [Google Scholar]
- Ma D.; Hettiarachchi G.; Nguyen D.; Zhang B.; Wittenberg J. B.; Zavalij P. Y.; Briken V.; Isaacs L. Nat. Chem. 2012, 4, 503–510. 10.1038/nchem.1326. [DOI] [PubMed] [Google Scholar]
- Yang X.; Wang Z.; Niu Y.; Chen X.; Lee S. M. Y.; Wang R. Med. Chem. Commun. 2016, 7, 1392–1397. 10.1039/C6MD00239K. [DOI] [Google Scholar]
- Dong N.; Xue S.-F.; Zhu Q.-J.; Tao Z.; Zhao Y.; Yang L.-X. Supramol. Chem. 2008, 20, 663–671. 10.1080/10610270701666018. [DOI] [Google Scholar]
- Gavvala K.; Sengupta A.; Hazra P. ChemPhysChem 2013, 14, 532–542. 10.1002/cphc.201200879. [DOI] [PubMed] [Google Scholar]
- Kuok K. l.; Li S.; Wyman I. W.; Wang R. Ann. N. Y. Acad. Sci. 2017, 1398, 108–119. 10.1111/nyas.13376. [DOI] [PubMed] [Google Scholar]
- Okimoto K.; Rajewski R. A.; Uekama K.; Jona J. A.; Stella V. J. Pharm. Res. 1996, 13, 256–264. 10.1023/A:1016047215907. [DOI] [PubMed] [Google Scholar]
- Higuchi T.; Connors K. A. Adv. Anal. Chem. Instrum. 1965, 4, 117–210. [Google Scholar]
- Ma D.; Zavalij P. Y.; Isaacs L. J. Org. Chem. 2010, 75, 4786–4795. 10.1021/jo100760g. [DOI] [PubMed] [Google Scholar]
- Hu X.; Chen Z.; Chen L.; Zhang L.; Hou J.-L.; Li Z.-T. Chem. Commun. 2012, 48, 10999–11001. 10.1039/c2cc36027f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.