

Abstract
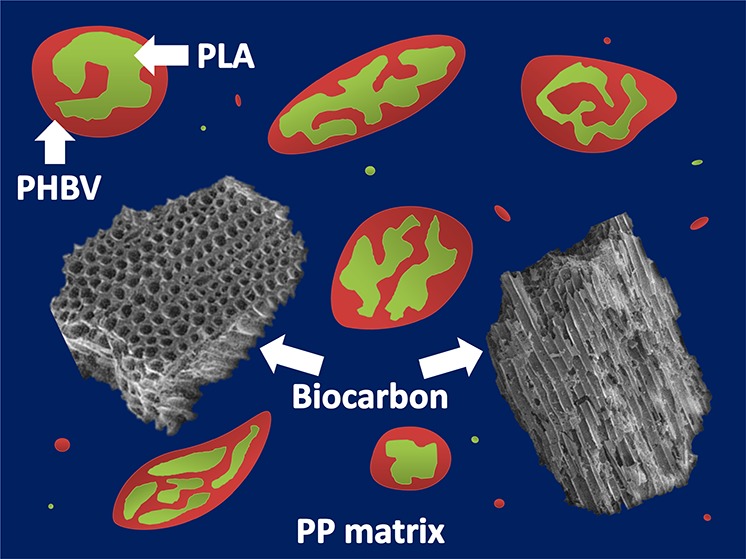
The incorporation of poly(lactic acid) (PLA) and poly(hydroxybutyrate-co-hydroxyvalerate) (PHBV) as a partial biobased polymer substitute for polypropylene (PP) was investigated. The ternary blends were prepared through melt compounding extrusion followed by injection molding techniques with a constant biopolymer ratio of 30 wt %. Further addition of pyrolyzed miscanthus-based carbon was carried out to establish a contrast between talc-filled PP. When the morphology of the biopolymer minor phase was analyzed theoretically using contact angle for interfacial tension and spreading coefficient values along with solubility parameter calculations and via scanning electron microscopy imaging, the core–shell architecture was found with the PHBV encasing the PLA phase. Mechanical testing of the materials showed that only the tensile properties were reduced for all samples, whereas the impact strength was increased above the neat PP. With inclusion of the biobased carbon filler into the blend system, the thermomechanical properties were elevated above that of the PP matrix. The final properties of the multiphase polymeric composites are found to be related to the morphology obtained and inherent properties of the individual constituents.
Introduction
The use of polypropylene (PP) varies widely and can be found in commodity areas like the packaging sector all the way up to engineering applications such as automotive parts. The downside to this plastic, like most others used today, is the disposal issues of such products that end up in landfills or as litter and do not decompose or are incinerated for energy, contributing to greenhouse gas emissions. These current waste options are causing environmental concerns, health hazards, social scrutiny, and economic burdens. To manage the end life of polyolefins and to diminish the negative aspects of plastic waste, there has been an aim to develop biomaterial counterparts.
The use of biodegradable and biobased polymers as a partial component in PP blends can help minimize the impact of discarded products.1 Generally, by blending these types of materials together, the biopolymer properties can be maintained, as they are less prone to degradation during the life span of the product, while also enabling gradual decomposition of the polyolefin through biodisintegration when the material has been discarded.2 Both poly(lactic acid) (PLA) and poly(hydroxybutyrate-co-hydroxyvalerate) (PHBV) have been tested separately in PP blends for these reasons. However, the inclusion of PLA in PP results in poor mechanical strength as there is little compatibility between the polar PLA and nonpolar PP.3 Similarly, PHBV tends to have a minimal effect on the strength of PP because of the poor miscibility of these two polymers.4 Yet, the processing of immiscible polymers in various blends and composites is used as a method to provide a combination of properties that the singular polymers are not able to attain on their own. These multicomponent blends tend to allow for trade-offs in areas such as price, biocontent, balanced property performance, and compostability or recyclability at the end life of the product. Therefore, the polymer ratio in blends can be adjusted to meet specific cost-performance criteria. It is common with these unequal ratios of polymers that the final morphology of the blend will have a minor phase that forms either spheres, elongated ovals, or thin sheet/platelet structures within the major matrix phase.5 Depending on the interfacial tension, viscosity, and elasticity of the polymers being blended as well as the immiscible blend ratio, history, and processing parameters used, differing morphologies can result.6
Other methods to enhance the mechanical properties of these poorly compatible blends are through fillers. Talc is one such filler that is added to polymeric systems for increasing their stiffness and thermal properties. However, with the concept of biocontent being more prevalent nowadays, biobased carbon particles are studied for their reinforcing effects in composites.7 This is due to naturally derived fillers that are readily abundant, which provide less density, have low cost, and remain environmentally benign.8
Therefore, this research study focused on the morphological structures and mechanical properties of the multiphase blends and their composites by using the good stiffness provided by PLA and the thermomechanical stability associated with PHBV in a PP matrix along with a bioderived carbon filler. The aim was focused on augmenting the biobased content of the blend by improving the impact, enhancing the thermal utilization range, minimizing the mechanical performance loss, and providing a less dense filler. The base characteristics of PP were first tested along with the addition of 10 wt % fillers of talc and a biobased carbon separately. A second set of samples of PP with different blend ratios of PLA and PHBV were included at 30 wt % biopolymer additions to analyze the variation in performance of the tertiary blend system while increasing the biobased content of the blend. In the final set of materials, the same fillers were then incorporated into the blend with the optimum overall performance to compare with the base PP composites.
Results and Discussion
Contact Angle Analysis
The contact angles for the three polymers used in this investigation were measured with both polar and nonpolar solvents, water and diiodomethane, respectively, as seen in Table 1. The values obtained with water show that all the samples have a hydrophilic characteristic in them as the contact angle is less than 90°.9 There is however a 20–25° difference observed between PP and the two biopolymers. This greater wettability, smaller contact angle, of PLA and PHBV demonstrate their higher surface energies as compared to PP, with the water coating a larger area of the polymer surface to reduce the surface energy level.10 In the case of the nonpolar solvent, PHBV had the greatest wetting followed by PP and PLA. Only PLA showed a minor change in its contact angle between the two liquids, whereas the contact angles of PP and PHBV reduced as they have more nonpolar regions along the polymer chain.
Table 1. Contact Angles and Calculated Total Surface Tension (γ) and Its Polar (γp) and Dispersive (γd) Components for the Three Polymers.
| contact
angle (deg) |
surface tension@20 °C (mN·m–1) |
||||
|---|---|---|---|---|---|
| polymer | water | diiodomethane | γ | γp | γd |
| PP | 86.82 ± (0.12) | 59.13 ± (0.017) | 31.27 | 3.82 | 27.45 |
| PLA | 62.66 ± (0.086) | 63.10 ± (0.017) | 41.58 | 18.41 | 23.18 |
| PHBV | 67.46 ± (0.29) | 46.34 ± (0.033) | 43.93 | 10.81 | 33.12 |
After conducting the surface tension calculations, the total surface tension for biopolymers is approximately 10 mN·m–1 higher than that of PP, as seen in Table 1. This difference is correlated with the intermolecular attractions present in the chain structure of the polymers. For PP chains, weak van der Waals forces provide the majority of the polyolefins attraction between the chains, whereas the biopolymers contain dipole regions and hydrogen bonding that creates a greater affinity for neighboring polymer chains that increase the surface energy of the thermoplastic.11 This is evident when the surface tension is separated into the polar and dispersive components, with PP having a very small polar part but a dispersive constituent similar to the other two polymers.
Through the implementation of the harmonic and geometric mean equations, the lowest interfacial tension values calculated from either method were obtained for the combination of PLA and PHBV, as shown in the results of Table 2. By contrast, PP and PLA showed the highest interfacial tension, demonstrating the immiscibility of these two polymers. However, PHBV has a surface tension similar to both PP in terms of the dispersive component and PLA with respect to the polar component, making it more suited as a partially miscible polymer for both materials. Therefore, PHBV works as an intermediate compatibilizer between the two immiscible portions of the ternary blend.
Table 2. Interfacial Tension and Spreading Coefficient at 20 and 180 °C Extrapolation Using the Harmonic and Geometric Mean Equations.
| |
spreading
coefficient |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| interfacial
tension at 20 °C (mN·m–1) |
20 °C |
180
°C |
|||||||
| equation | PP/PLA | PP/PHBV | PLA/PHBV | PLA/PHBV | PHBV/PLA | PP/(PLA or PHBV) | PLA/PHBV | PHBV/PLA | PP/(PLA or PHBV) |
| harmonic | 9.94 | 3.87 | 3.73 | –9.79 | 2.33 | –10.08 | –10.67 | 1.40 | –9.64 |
| geometric | 5.64 | 2.05 | 1.89 | –5.48 | 1.70 | –5.79 | –5.94 | 1.22 | –5.56 |
To further establish the phase morphology of the system, the spreading coefficient model can be used, which is utilized for three-phase systems. This involves rewriting the generalized Harkins equation into the alternative form tested by Hobbs et al.12
| 1 |
where λij is the spreading coefficient of i over j and γ is the interfacial tension between two of the polymers. When λij is positive, i will coat the j component, and when negative, it will not, while only in the instance where both combinations are negative, the minor phases will disperse separately as droplets in the matrix or none of the polymers will form a complete layer at the interface of the others.13 The case where the last two options are possible is dependent on the sign of the third spreading coefficient associated with the major phase between both minor phases.
In this ternary blend, the tendency of PLA to encapsulate PHBV and vice versa was calculated for the minor phases along with the PP matrix spreading coefficient, as seen in Table 2. The resultant values for the spreading coefficients of λPLA/PHBV and λPHBV/PLA at 20 °C are negative and positive, respectively. This implies that PHBV will tend to encapsulate the PLA phase inside the PP matrix. The values for the third spreading coefficient related to the PP matrix were negative, which refers to the nonwetting of the matrix on both the minor phases and allows the dominant influence of PHBV to completely wet PLA. However, to provide a more predictive model of the ternary blend during the processing conditions, the surface tensions were extrapolated to the melt blending temperature. This was done via the temperature coefficient of −0.06 mN·m–1·K–1, which is commonly used in the literature.14 Upon extrapolation, the same trend was seen in the spreading coefficient with PHBV encasing PLA at 180 °C. This means that the morphology should not undergo any major changes during cooling and retain its matrix–core–shell dispersed phase structure.
Solubility Parameter Method
Upon implementation of the model criteria from the Hoftyzer–van Krevelen and Hoy approaches, the total solubility parameters were summed and compared alongside the Hildebrand and Hansen values obtained from the literature, as displayed in Table 3. The resulting outcome of the calculated values from the Hoftyzer–van Krevelen and Hoy methods shows that PLA and PHBV have a value similar to one another, whereas the value of PP is lower than that of PLA and PHBV. Because of the close solubility parameter of the two biopolymers, there is a likelihood of good mixing between them. The magnitude of the difference between the solubility parameters can be used to distinguish the miscibility, with Δδ < 2 MPa1/2 having good miscibility, and Δδ > 10 MPa1/2 being immiscible, with partial miscibility between these regimes.15 From this, all of the differences between the PLA and PHBV solubility parameters are less than 2 MPa1/2, making them miscible, whereas, for PP, all solubility differences show signs of partial miscibility, with the lowest differences occurring between PP and PHBV. This again emphasizes that PHBV is better suited to act as an intermediate between the PLA and PP polymers in the ternary blend with miscibility characteristics between both the components.
Table 3. Solubility Parameters (δ) in MPa1/2.
Mechanical Properties
The neat PP along with the ternary blends and composites containing biocarbon and talc was tested for their mechanical performance. The tensile and flexural properties including strength and modulus are displayed in Figures 1 and 2, respectively. The neat PP showed a tensile strength of 43 MPa, which was greater than all other samples tested by 5 MPa or more. The addition of the fillers to the system caused a reduction in the strength as there is minimal contribution of the biocarbon and talc to the polymer under stress.
Figure 1.
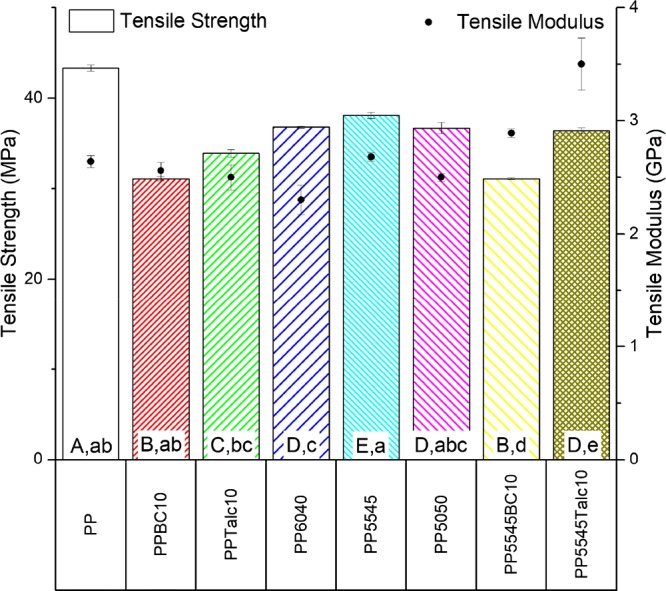
Tensile strength and moduli of the PP blends and composites (samples with arithmetic means that do not share a letter are significantly different; uppercase: tensile strength and lowercase: tensile modulus).
Figure 2.
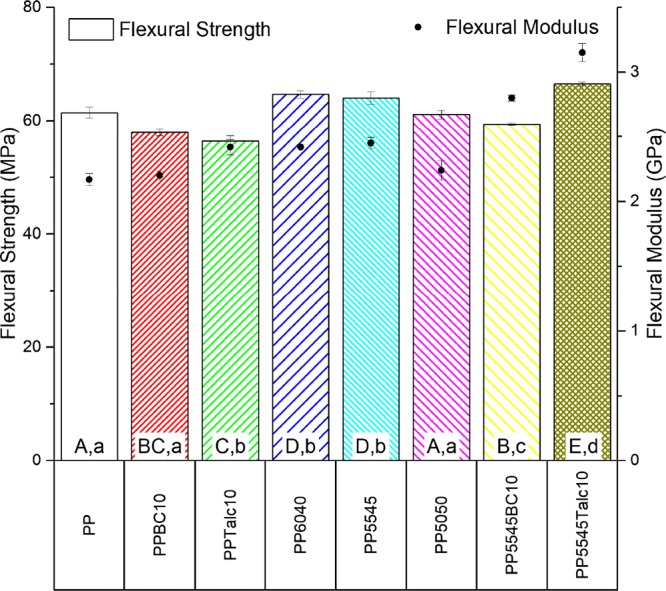
Flexural strength and moduli of the PP blends and composites (samples with arithmetic means that do not share a letter are significantly different; uppercase: flexural strength and lowercase: flexural modulus).
To correlate the lack of stress transfer between the matrix and the filler, the Nicolais–Narkis model was tested. Assuming the fillers are spherical, the model can predict the ratio of the tensile strengths of the matrix and composite systems. For the situation where there is no adhesion between the materials and the filler is only causing a reduction in cross-sectional area, the equation is19
| 2 |
where σc and σm are the tensile strengths of the composite and matrix, respectively, with the filler volume fraction represented by φf. When analyzing the ratio for 10 wt % fillers to PP, the calculated σc/σm ratio was 20, with a 13% reduction in strength for the biocarbon and talc. However, the actual measurements had a lower ratio with a 28% loss in strength when biocarbon was included and 22% for talc. This discrepancy is due to the irregular shape of the biocarbon and platelike structure of the talc, neither of which is completely spherical. In the case of the ternary blends with fillers, the biocarbon showed a similar loss from the measured value of 18% to the theoretical value of 21% based on eq 2. By contrast, the talc did not undergo a significant loss in the quaternary composite with the strength reduction of only 4%, with the calculated value from eq 2 estimated to be 14%.
When considering the ternary blends in relation to the neat PP, all samples showed a loss in the tensile strength. It may be reasoned that the poor miscibility of the biopolymers, PLA and PHBV, into the PP matrix does not provide good stress transfer, thus reducing the tensile strength. PHBV on its own has a tensile strength of 35 MPa which will decrease the performance of the ternary blend. Though PLA has a higher tensile strength than PP, the lack of compatibility between PP and PLA leads to poor mechanical performance. Out of the three ternary blends, the one containing 55 wt % PLA in the biopolymer portion showed the optimum strength and modulus. Once the fillers were incorporated into this ternary blend, the modulus was increased above that of the neat PP because of the high stiffness of the carbon and mineral components.
When analyzing the flexural properties of the blends and composites, a similar trend is observed. The fillers cause a loss of flexural strength but not to the extent seen in the tensile data. The difference may arise from the fact that flexural tests during three-point bending combine tension on the bottom of the test bar and compression on the top of the test bar. The compressive strength of the material will be higher with the fillers, as both the biocarbon and talc are denser than the PP matrix but will ultimately have a lower tensile component. The flexural strength of the ternary blends containing a greater portion of PLA showed a minor increase over PP. The benefit of including PLA is seen here with its high flexural strength of 95 MPa which is more than offsetting the PHBV components’ flexural strength of 57 MPa. That is why the greater content of PLA in the ternary blend improved both the flexural strength and modulus. Again, once the fillers were included in the ternary system, the modulus was further increased. However, in the case of the talc addition, the two-dimensional (2-D) plate structure enhanced the flexural strength by 4%.
To characterize the toughness of the samples, notched Izod impact testing was conducted. During these measurements, all of the blends and composites had complete breaks and were evaluated after specimen toss energy correction when less than 27 J·m–1. The resultant data can be seen in Figure 3 for the impact properties of the materials. All samples showed enhanced impact strength capabilities over the neat PP. The inclusion of the fillers provided an improvement in the notch impact. The well-dispersed particles in the PP matrix are responsible for imparting a more uniformly distributed impact energy.20 Other aspects that the fillers provide toward enhancement of the impact strength are the crack pinning, which augments fracture energy, and particle pull-out.21 The minor difference between the biocarbon and the talc composite impact strengths may be attributed to the variation in particles’ aspect ratios, with the greater aspect ratio of the platy structure of the talc, causing a larger stress concentration compared to the biocarbon and reducing the mobility of the molecular chains during impact.21,22
Figure 3.
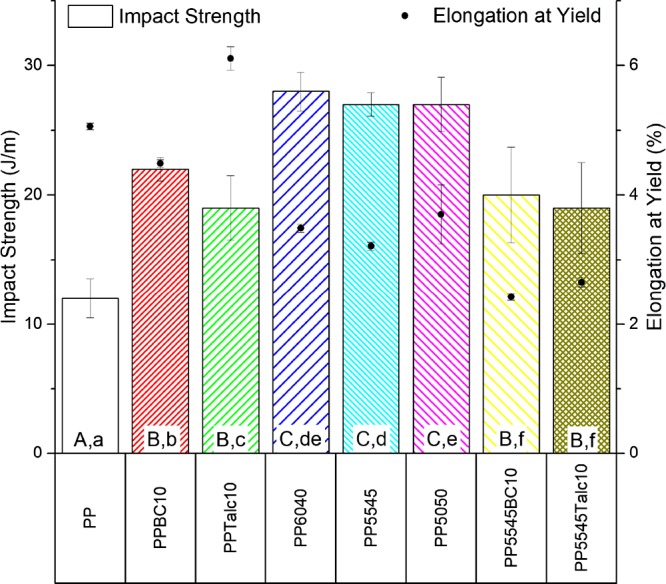
Impact strength and elongation at yield of the PP blends and composites (samples with arithmetic means that do not share a letter are significantly different; uppercase: impact strength and lowercase: elongation at yield).
When examining the ternary blends of PP/PLA/PHBV, they all have a 15 J·m–1 increase over PP. Each of the three neat polymers has a similar impact strength, with PP, PLA, and PHBV having values of 12, 15, and 14 J·m–1, respectively. Therefore, the morphology of the minor phases within the PP matrix may cause obstructions in the crack propagation through the PP matrix, allowing for absorption of impact energy through pull-outs and further energy requirements for crack initiation of the minor phases. There will also be a greater toughening of the blend system as the compatibility of the PP and PLA phases is improved due to PHBV acting as an intermediary for stress transfer between the polymers.23
Looking at the impact strength of the quaternary composites, the performance is reverted to that of the PP composites. There is a larger standard deviation present in these as the fillers disturb the morphology of the minor phases in the blend system. The strengthening is now dominated by the fillers once again, and the same factors mentioned previously would apply.
The percent elongation at yields for the tensile tests are also shown in Figure 3. It is evident that in the presence of the biocarbon the elongation is reduced, resulting from its brittle nature, poor compatibility, wide particle size distribution, and restriction of polymer chains.24 In the case of talc addition, the 2-D platelet structure of talc provides easier chain sliding during testing, increasing the elongation at yield, as the small particle size of the talc is known to elevate the elongation marginally.25 For the ternary blends, the reduction in elongation comes mainly from the low elongation of PLA and PHBV being 2.2 and 1.5%, respectively. Then, the resulting quaternary composites have even lower elongation at break as the particles now cause imperfections throughout the system along with the low elongation of the blend.
Thermomechanical Analysis
Heat Deflection Temperature
The analysis of the heat deflection temperature (HDT) is a way to determine the processing temperature boundaries for molded parts and the thermal limits for a product. It is a measure of the temperature where a material undergoes a 0.25 mm flexural deflection under a specified load and relies on the temperature dependence associated with the flexural modulus.26 The HDTs of the eight samples discussed in this paper are presented in Figure 4. For the neat PP sample, an HDT of 114 °C was measured. When the fillers of biocarbon or talc were used, the composite samples showed similar HDTs with no change when considering the standard deviation. The ternary blends on the other hand had a reduced HDT <100 °C. PLA having a measured HDT of 57 °C caused reduced thermal stiffness at elevated temperatures, as can be seen when increasing PLA weight percentages in the blend system. PHBV was used in part to offset this as it has a measured HDT of 143 °C, though it did not follow the rule of mixtures well as PP5545 resulted in the experimental value of 94 °C when it is calculated to be approximately 108.5 °C, which could be a result of the morphology of the blend system. Next, when looking at the quaternary composites, the HDTs are heightened for both instances from the blend. The biocarbon inclusion had a greater improvement with a 24% increase from the ternary blend and returned to the range of the PP/biocarbon composite sample having an HDT of 117 °C. The increased HDT values observed for the quaternary composites may be attributed to the increased stiffness as seen in the mechanical data with the variation between the two samples associated with differences in the filler shape, size, orientation, aspect ratio, adhesion, and size distribution.27
Figure 4.
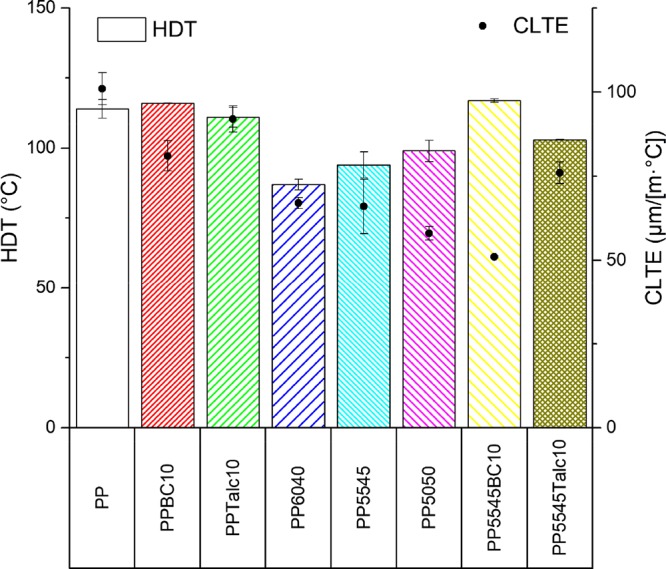
HDT and CLTE of the PP blends and composites.
Thermomechanical Analysis
A common trait of polymeric materials is the coefficient of linear thermal expansion (CLTE). By analyzing this property, a minimal expansion temperature range may be determined to reduce the likelihood of thermal shock or very precise dimensional fluctuations and allow for implementation in interfaces with similar CLTE.28 The thermal stability of most isotropic polymers is enhanced with the inclusion of inorganic fillers as they have low expansion coefficients as compared to the polymer matrix.29 In the case of the PP composites tested, the same trend is evident for the addition of biocarbon and talc samples, as seen in Figure 4. The neat PP had a CLTE of 101 μm/(m·°C) in the temperature range of 25–50 °C, similar to that of 93 μm/(m·°C) in a slightly cooler range of 0–30 °C found in the literature.30 The reduction in the CLTE value was more apparent in the case of biocarbon addition, with 20 μm/(m·°C) reduction or 20% decrease, whereas talc only reduced the CLTE by half as much as the biocarbon. Next, when analyzing the ternary blend systems, the thermal stability was improved further with a CLTE value from 58 to 67 μm/(m·°C) with increasing PHBV content. The reasoning behind this reduction is related to the low CLTE of the biopolymers, with PLA and PHBV measured to have 61 and 54 μm/(m·°C), respectively. Therefore, with more PHBV present in the PP5050 blend, the lower the CLTE out of the three ternary blends. Another factor in the reduction of CLTE for the ternary blend systems may come as a result of the minor phases impeding the PP matrix from expanding, with PLA being a rigid obstacle below its Tg and being surrounded with PHBV. When the fillers are used in the ternary blend system, only the biocarbon could reduce the CLTE, whereas the talc caused no change within the standard deviation. The quaternary composite-containing biocarbon showed a CLTE of 51, which is a reduction of 23% compared to the ternary blend PP5545 and a 50% reduction over the neat PP. This demonstrates the superior thermal stability performance of the biocarbon over the talc filler.
Scanning Electron Microscopy Morphological Analysis
Scanning electron microscopy (SEM) was performed on the Izod impact fractured surfaces of the PP-based materials. For the neat PP, there are no distinguishable features present as the material undergoes a brittle fracture with a smooth surface, as seen in Figure 5. In the case of the PP composite system, the biocarbon is randomly distributed throughout the matrix with varying particle dimensions. In the biocarbon-containing samples, the macropores (≤10 μm) of the biocarbon are visible with PP penetrating into them, which was also observed in the quaternary composites. A good dispersion of talc in PP was also found (not presented). In the case of the ternary blends, the minor bioplastic phase was found in the form of spheres or elongated globules throughout the PP matrix with a wide range of sizes. The droplet-shaped minor phase is similar to the sea–island morphology, which is determined from the components’ viscosity and volume ratios.31 To distinguish between minor phases of PLA and PHBV in the blend, etching was performed to remove the PLA component. The resulting holes and voids observed were in the location of PLA. As seen in Figure 6, the etched sample of the PP5050 ternary blend shows cavities within the minor PHBV phase. This finding correlates well with the solubility and spreading coefficient calculations previously mentioned. An illustration of the ternary blend morphology is also presented to depict the core–shell characteristic of the system. The same topography was seen for all ternary blends. Upon addition of the fillers into the ternary blend, the size of the minor phase is disturbed and tends toward smaller domains. The fillers themselves are confined within the PP matrix, whereas the biopolymer core–shell configuration is retained, as seen with the etched-out surface showing the same hollowed-out PHBV.
Figure 5.

Scanning electron micrographs of the impact fracture surfaces of (a) PP, (b) PPBC10, (c) PP5545, (d) etched PP5545, (e) PP5545BC10, and (f) etched PP5545BC10.
Figure 6.

SEM images of the (a) unetched and (b) etched PP5050 ternary blends, with (c) a schematic of the morphology.
Conclusions
The morphology, mechanical, and thermomechanical properties of the PP/PLA/PHBV ternary blends and composites containing biobased carbon and talc were examined. The findings showed that the spreading coefficient determined through the contact angle and solubility parameter values coincided well with the structure observed for the ternary blend seen under SEM imaging. The minor biopolymer inclusions formed a core–shell arrangement with PHBV coating the PLA phase, within the PP matrix. On the basis of the statistical analysis, the ternary blend showed a reduction in the tensile performance but was contrasted by the improved flexural and impact properties above the neat PP when the ratio of PLA to PHBV was greater for the 30 wt % biopolymer component of the blend. Once the fillers of biocarbon and talc were present in the quaternary composite, the moduli of the samples were enhanced above both PP and the ternary blends. In the case of the biocarbon specifically, the HDT and CLTE values were ameliorated above all other samples, providing the most thermally stable specimens and a maximum biobased content of 37 wt %. The resulting partially biobased composites analyzed in this study may provide further insights into the advancement of cost-performance balance materials, whereas the use of the miscibility models to quickly predict the compatibility between the polymers remains an asset when researching new blend interactions.
Experimental Section
Materials
Three injection grade polymers were utilized in this investigation including 1120H homopolymer PP provided by Pinnacle Polymers (USA), Ingeo 3251D PLA, a product of NatureWorks LLC (USA), and ENMAT Y1000P PHBV with 3% HV32 content acquired from TianAn Biological Materials Co., Ltd. (China). Along with these polymers, two types of particulate fillers were used as received from their respective suppliers. The first one is Mistron Vapor R talc from Imerys Talc (Canada), with a mean size range of 1.7–2.2 μm, a surface area of 13.4 m2·g–1, and a density of 2.8 g·cm–3 (technical data sheet). The second one is a biobased carbon produced from the pyrolysis of miscanthus grass at 700 °C supplied by Competitive Green Technologies (Canada), with particles ≤400 μm33 having an average size range of 20–75 μm,34 an ash content of ∼10%,35 a carbon content >80%, a surface area of 100–200 m2·g–1, and a density of 1.4 g·cm–3.
Specimen Preparation
The biopolymers PLA and PHBV were dried at 80 °C in an industrial oven for a minimum of 4 h prior to processing, whereas the biocarbon was dried at 105 °C for 24 h to remove excess moisture. The PP polymer was kept under room-temperature conditions and used as received, along with the talc powder. Processing of the samples was performed with a 15 mL microcompounder DSM Xplore with a corotating twin screw extruder (Netherlands). At a screw speed of 100 rpm and a temperature of 180 °C, the materials were added according to the weight ratios of the different formulations. The retention time was set to 2 min before being injection-molded at 30 °C in a DSM Xplore 12 mL microinjection molding machine. Tensile, flexural, and impact bars were prepared for further analysis. Neat PP, PLA, and PHBV samples were also prepared under the same conditions for use in contact angle measurements and comparison of mechanical results.
Characterization
The base polymer for the blends was PP, which acted as a baseline for the other samples under investigation. The PP matrix component was set at 70 wt % of the blend ratio with the biopolymer component making up the other 30 wt % of the blend, whereas the inclusion of the filler was kept at a constant ratio of 10 wt % for the composite samples. See Table 4 for the complete formulation breakdown, where the abbreviation of the samples is used to distinguish the ratio of the biopolymer component of PLA/PHBV within the PP matrix. An example is PP5545 which refers to PP/(PLA/PHBV) having a 70/(55/45) wt %. Upon filler addition, such as biocarbon, to this same blend, it becomes PP5545BC10 where the ratio is [70/(55/45)]/10 wt % for [PP/(PLA/PHBV)]/biocarbon.
Table 4. Composition of Polymeric Test Samples.
| formulation (wt %) | PP | PPBC10 | PPTalc10 | PP6040 | PP5545 | PP5050 | PP5545BC10 | PP5545Talc10 |
|---|---|---|---|---|---|---|---|---|
| PP | 100 | 90 | 90 | 70 | 70 | 70 | 63 | 63 |
| PLA | 18 | 16.5 | 15 | 14.85 | 14.85 | |||
| PHBV | 12 | 13.5 | 15 | 12.15 | 12.15 | |||
| biocarbon | 10 | 10 | ||||||
| talc | 10 | 10 |
Contact Angle Measurements
The ramé-hart standard goniometer contact angle equipment model 260-U1 was used to test extruded samples containing only the neat polymers PP, PLA, or PHBV. The contact angles were measured by the static sessile drop technique, where drops of liquid were microsyringed onto the sample. The tests were conducted at room temperature (∼20 °C) with an injection-molded sample, and a drop of either deionized water or diiodomethane was used as the polar and nonpolar liquid, respectively. See Table 5 for the total surface free energies and related components for the two liquids. With the DROPimage software (version 2.8.05), the contact angles were calculated with the circle method using the captured images. A total of three repetitions were performed on each sample to obtain an average and standard deviation.
Table 5. Surface Tension (γ) and the Polar (γp) and Dispersive (γd) Components (in mN·m–1) for Water and Diiodomethanea.
| liquid | γ | γp | γd |
|---|---|---|---|
| water | 72.8 | 51.0 | 21.8 |
| diiodomethane | 50.8 | 0.4 | 50.4 |
Values obtained from ref (36).
From the contact angles measured, the surface tensions of the three polymers may be calculated. This is done using Young’s equation37
| 3 |
where γs and γl are the surface tensions at the equilibrium of the solid–vapor and liquid–vapor, respectively, γsl is the solid–liquid interfacial tension, and θ is the measured contact angle. This concept can be extended further by using the Owens–Wendt–Rabel–Kaelble (OWRK) model that splits the surface tension into two intermolecular forces that act at the interface.38−40 The two components are a dispersive and polar portion that makes up the sum of the surface tension values. The OWRK equation is then written as
| 4 |
where the superscripts d and p refer to the dispersive and polar components of the surface tension, respectively, and θ is the measured contact angle converted to radians. On the basis of the contact angles measured for the two liquids and their known surface tension values (γl, γld, and γl), see Table 5, eq 4 becomes a calculation of two equations with two unknowns (γsd and γs). The contributing components of the surface tension and the total sum (γs = γsd + γs) for the polymers were then obtained.
The interfacial tension between each of the polymers was then calculated to provide further insights into the most probable morphology. Both the harmonic mean equation41,42
| 5 |
and the geometric mean equation
| 6 |
where the subscripts i and j refer to two separate polymers were used to identify the magnitude of interfacial tension between the three combinations of polymers.
Solubility Parameter Calculation
To provide a greater understanding of the miscibility of the ternary blend, estimation of the solubility parameter (δ) for each of the three polymers was done. This is a technique that describes the cohesive forces of materials and is a good predictor in the case of miscibility for polymers and solvents.43 The initial method of calculating the solubility behavior was developed by Hildebrand and further modified by splitting the parameter into a polar, dispersive, and hydrogen-bonding component by Hansen.44 This adaption of the solubility parameter can then be approximated using the Hoftyzer–van Krevelen and Hoy methods45 based on the individual groups of the polymer chain.
Therefore, to determine the solubility parameters of the polymers, the Hoftyzer–van Krevelen and Hoy approximation techniques were used. These methods involve summing up the contributions of the functional groups within the chemical structure to the dispersive, polar, and hydrogen-bonding components. In the case of the polymers used in this investigation, the average molecular weights (Mw) were chosen to establish the degree of polymerization from the monomer units. The Mw of PLA, PHBV, and PP used were 55 000,46 240 000,47 and 170 00048 g·mol–1, respectively.
Mechanical Testing
For the measurement of the mechanical strength and stiffness of the material, the Instron 3382 Universal testing machine was utilized. Under room-temperature conditions, type IV tensile bars were tested at a rate of 5 mm/min, according to ASTM D638. Similarly, the flexural specimens were tested at a rate of 1.4 mm/min with a span length of 52 mm following procedure A of ASTM D790. All tensile and flexural results were evaluated with Bluehill software.
A TMI Monitor Impact tester was used for the data acquisition of the notched Izod impact strengths of the samples conforming to method C of ASTM D256 with a 6.78 J pendulum. The impact bars were prenotched with a motorized notching cutter, 40 h prior to testing. To provide averages and standard deviations, five of each sample type were measured.
For statistical analysis of the mechanical data, a one-way analysis of variance (significance level α = 0.05) was performed using Minitab 17 software, on the data of the individual samples. A Tukey pairwise comparison test was conducted with the groupings determined with 95% confidence intervals, showing means that do not share the same letter as being significantly different.
Thermomechanical Analysis
A dynamic mechanical analyzer, DMA Q800 instrument (TA Instruments), was used to conduct measurements for the HDT of the specimens. The ASTM D648 standard was followed, with a load of 0.455 MPa in a three-point bending configuration, where the heating rate was set at 2 °C/min starting at 30 °C until a 250 μm displacement was obtained.
For the CLTE, a thermomechanical analyzer, TMA Q400 (TA Instruments), was set up in accordance with ASTM E831. The samples were tested with an expansion probe normal to the injection flow direction. The testing conditions were set at a heating rate of 5 °C/min, from 20 to 60 °C, with a 0.1 N probe force. The CLTE was then calculated in the linear range of 25–50 °C to remain below the nonlinear region associated with the glass-transition temperature of PLA.
The Universal Analysis 2000 software v. 4.5A (TA Instruments) was used to analyze the thermomechanical data from both tests.
Microscopy Imaging
To provide a visual image of the morphology of various samples, a scanning electron microscope Phenom ProX (Phenom-World BV) was used. The impact samples fractured surfaces were used as the image region. The samples were tested with and without etching for 4 h in tetrahydrofuran, to dissolve away PLA from the blends. The SEM settings were kept constant with an accelerating voltage of 5 kV, a 1500× magnification, and a charge reduction stage.
Acknowledgments
This research is financially supported by the Ontario Ministry of Agriculture, Food, and Rural Affairs (OMAFRA)–New Directions Research Program (project # 049528); the Natural Sciences and Engineering Research Council (NSERC), Canada Discovery grant (project # 400320 and # 401111); the Ontario Research Fund, Research Excellence Program; and Round-7 (ORF-RE07) from the Ontario Ministry of Research and Innovation, currently known as the Ontario Ministry of Research, Innovation and Science (MRIS) (project # 052644 and # 052665).
The authors declare no competing financial interest.
References
- Masood F.; Yasin T.; Hameed A. Comparative oxo-biodegradation study of poly-3-hydroxybutyrate-co-3-hydroxyvalerate/polypropylene blend in controlled environments. Int. Biodeterior. Biodegrad. 2014, 87, 1–8. 10.1016/j.ibiod.2013.09.023. [DOI] [Google Scholar]
- Sarasa J.; Gracia J. M.; Javierre C. Study of the biodisintegration of a bioplastic material waste. Bioresour. Technol. 2009, 100, 3764–3768. 10.1016/j.biortech.2008.11.049. [DOI] [PubMed] [Google Scholar]
- Hamad K.; Kaseem M.; Deri F. Rheological and mechanical characterization of poly(lactic acid)/polypropylene polymer blends. J. Polym. Res. 2011, 18, 1799–1806. 10.1007/s10965-011-9586-6. [DOI] [Google Scholar]
- Sadik T.; Massardier V.; Becquart F.; Taha M. Polyolefins/Poly(3-hydroxybutyrate-co-hydroxyvalerate) blends compatibilization: Morphology, rheological, and mechanical properties. J. Appl. Polym. Sci. 2013, 127, 1148–1156. 10.1002/app.37957. [DOI] [Google Scholar]
- Ying-Chen Z.; Hong-Yan W.; Yi-Ping Q. Morphology and properties of hybrid composites based on polypropylene/polylactic acid blend and bamboo fiber. Bioresour. Technol. 2010, 101, 7944–7950. 10.1016/j.biortech.2010.05.007. [DOI] [PubMed] [Google Scholar]
- Afshari M.; Kotek R.; Kish M. H.; Dast H. N.; Gupta B. S. Effect of blend ratio on bulk properties and matrix–fibril morphology of polypropylene/nylon 6 polyblend fibers. Polymer 2002, 43, 1331–1341. 10.1016/s0032-3861(01)00689-9. [DOI] [Google Scholar]
- Ogunsona E. O.; Misra M.; Mohanty A. K. Sustainable biocomposites from biobased polyamide 6,10 and biocarbon from pyrolyzed miscanthus fibers. J. Appl. Polym. Sci. 2017, 134, 44221. 10.1002/app.44221. [DOI] [Google Scholar]
- Kim H.-S.; Kim H.-J. Miscibility and performance evaluation of natural-flour-filled PP/PBS and PP/PLA bio-composites. Fibers Polym. 2013, 14, 793–803. 10.1007/s12221-013-0793-0. [DOI] [Google Scholar]
- Yuan Y.; Lee T. R.. Contact Angle and Wetting Properties; Springer, 2013; pp 3–34. [Google Scholar]
- Janssen D.; De Palma R.; Verlaak S.; Heremans P.; Dehaen W. Static solvent contact angle measurements, surface free energy and wettability determination of various self-assembled monolayers on silicon dioxide. Thin Solid Films 2006, 515, 1433–1438. 10.1016/j.tsf.2006.04.006. [DOI] [Google Scholar]
- Van Oss C.; Good R.; Chaudhury M. The role of van der Waals forces and hydrogen bonds in “hydrophobic interactions” between biopolymers and low energy surfaces. J. Colloid Interface Sci. 1986, 111, 378–390. 10.1016/0021-9797(86)90041-x. [DOI] [Google Scholar]
- Hobbs S.; Dekkers M.; Watkins V. Effect of interfacial forces on polymer blend morphologies. Polymer 1988, 29, 1598–1602. 10.1016/0032-3861(88)90269-8. [DOI] [Google Scholar]
- Harrats C.; Coltelli M.-B.; Groeninckx G.. Features on the Development and Stability of Phase Morphology in Complex Multicomponent Polymeric Systems: Main Focus on Processing Aspects. Polymer Morphology; John Wiley & Sons, Inc: Hoboken, NJ, USA, 2016; pp 418–438. [Google Scholar]
- Zhang K.; Nagarajan V.; Misra M.; Mohanty A. K. Supertoughened Renewable PLA Reactive Multiphase Blends System: Phase Morphology and Performance. ACS Appl. Mater. Interfaces 2014, 6, 12436–12448. 10.1021/am502337u. [DOI] [PubMed] [Google Scholar]
- Forster A.; Hempenstall J.; Tucker I.; Rades T. Selection of excipients for melt extrusion with two poorly water-soluble drugs by solubility parameter calculation and thermal analysis. Int. J. Pharm. 2001, 226, 147–161. 10.1016/s0378-5173(01)00801-8. [DOI] [PubMed] [Google Scholar]
- Chemical Data Retrieval on the Web (CROW) . Polymer Properties Database. http://polymerdatabase.com/polymer%20physics/delta%20Table.html (accessed Jan 27, 2017).
- Abbott S.Chemical Compatibility of Poly(Lactic Acid): A Practical Framework Using Hansen Solubility Parameters. In Poly(Lactic Acid) Synthesis, Structures, Properties, Processing, and Applications; Auras R., Lim L.-T., Selke S. E. M., Tsuji H., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2010; pp 83–96. [Google Scholar]
- Choi J. S.; Park W. H. Effect of biodegradable plasticizers on thermal and mechanical properties of poly(3-hydroxybutyrate). Polym. Test. 2004, 23, 455–460. 10.1016/j.polymertesting.2003.09.005. [DOI] [Google Scholar]
- Maiti S. N.; Sharma K. K. Studies on polypropylene composites filled with talc particles. J. Mater. Sci. 1992, 27, 4605–4613. 10.1007/bf01165994. [DOI] [Google Scholar]
- da Silva A. L. N.; Rocha M. C. G.; Moraes M. A. R.; Valente C. A. R.; Coutinho F. M. B. Mechanical and rheological properties of composites based on polyolefin and mineral additives. Polym. Test. 2002, 21, 57–60. 10.1016/s0142-9418(01)00047-2. [DOI] [Google Scholar]
- McGenity P. M.; Hooper J. J.; Paynter C. D.; Riley A. M.; Nutbeem C.; Elton N. J.; Adams J. M. Nucleation and crystallization of polypropylene by mineral fillers: relationship to impact strength. Polymer 1992, 33, 5215–5224. 10.1016/0032-3861(92)90804-6. [DOI] [Google Scholar]
- Nofar M.; Tabatabaei A.; Park C. B. Effects of nano-/micro-sized additives on the crystallization behaviors of PLA and PLA/CO2 mixtures. Polymer 2013, 54, 2382–2391. 10.1016/j.polymer.2013.02.049. [DOI] [Google Scholar]
- Shi Q.; Chen C.; Gao L.; Jiao L.; Xu H.; Guo W. Physical and degradation properties of binary or ternary blends composed of poly (lactic acid), thermoplastic starch and GMA grafted POE. Polym. Degrad. Stab. 2011, 96, 175–182. 10.1016/j.polymdegradstab.2010.10.002. [DOI] [Google Scholar]
- Nan N.; DeVallance D. B.; Xie X.; Wang J. The effect of bio-carbon addition on the electrical, mechanical, and thermal properties of polyvinyl alcohol/biochar composites. J. Compos. Mater. 2016, 50, 1161–1168. 10.1177/0021998315589770. [DOI] [Google Scholar]
- Suh C. H.; White J. L. Talc-thermoplastic compounds: particle orientation in flow and rheological properties. J. Non-Newtonian Fluid Mech. 1996, 62, 175–206. 10.1016/0377-0257(95)01382-2. [DOI] [Google Scholar]
- Jarus D.; Scheibelhoffer A.; Hiltner A.; Baer E. The effect of “skin-core” morphology on the heat-deflection temperature of polypropylene. J. Appl. Polym. Sci. 1996, 60, 209–219. . [DOI] [Google Scholar]
- Takemori M. T. Towards an understanding of the heat distortion temperature of thermoplastics. Polym. Eng. Sci. 1979, 19, 1104–1109. 10.1002/pen.760191507. [DOI] [Google Scholar]
- Kelly A.; Stearn R. J.; McCartney L. N. Composite materials of controlled thermal expansion. Compos. Sci. Technol. 2006, 66, 154–159. 10.1016/j.compscitech.2005.04.025. [DOI] [Google Scholar]
- Holliday L.; Robinson J. Review: The thermal expansion of composites based on polymers. J. Mater. Sci. 1973, 8, 301–311. 10.1007/bf00550148. [DOI] [Google Scholar]
- Kim D. H.; Fasulo P. D.; Rodgers W. R.; Paul D. R. Structure and properties of polypropylene-based nanocomposites: Effect of PP-g-MA to organoclay ratio. Polymer 2007, 48, 5308–5323. 10.1016/j.polymer.2007.07.011. [DOI] [Google Scholar]
- Zou H.; Wang K.; Zhang Q.; Fu Q. A change of phase morphology in poly(p-phenylene sulfide)/polyamide 66 blends induced by adding multi-walled carbon nanotubes. Polymer 2006, 47, 7821–7826. 10.1016/j.polymer.2006.09.008. [DOI] [Google Scholar]
- Martino L.; Berthet M.-A.; Angellier-Coussy H.; Gontard N. Understanding external plasticization of melt extruded PHBV-wheat straw fibers biodegradable composites for food packaging. J. Appl. Polym. Sci. 2015, 132, 41611. 10.1002/app.41611. [DOI] [Google Scholar]
- Behazin E.; Misra M.; Mohanty A. K. Sustainable Biocomposites from Pyrolyzed Grass and Toughened Polypropylene: Structure–Property Relationships. ACS Omega 2017, 2, 2191–2199. 10.1021/acsomega.7b00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan V.; Mohanty A. K.; Misra M. Biocomposites with Size-Fractionated Biocarbon: Influence of the Microstructure on Macroscopic Properties. ACS Omega 2016, 1, 636–647. 10.1021/acsomega.6b00175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behazin E.; Ogunsona E.; Rodriguez-uribe A.; Mohanty A. K.; Misra M.; Anyia A. O. Mechanical, Chemical, and Physical Properties of Wood and Perennial Grass Biochars for Possible Composite Application. BioResources 2016, 11, 1334–1348. 10.15376/biores.11.1.1334-1348. [DOI] [Google Scholar]
- Jańczuk B.; Białlopiotrowicz T. Surface free-energy components of liquids and low energy solids and contact angles. J. Colloid Interface Sci. 1989, 127, 189–204. 10.1016/0021-9797(89)90019-2. [DOI] [Google Scholar]
- Young T. An Essay on the Cohesion of Fluids. Philos. Trans. R. Soc. London 1805, 95, 65–87. 10.1098/rstl.1805.0005. [DOI] [Google Scholar]
- Owens D. K.; Wendt R. C. Estimation of the surface free energy of polymers. J. Appl. Polym. Sci. 1969, 13, 1741–1747. 10.1002/app.1969.070130815. [DOI] [Google Scholar]
- Rabel W. Einige Aspekte der Benetzungstheorie und ihre Anwendung auf die Untersuchung und Veränderung der Oberflächeneigenschaften von Polymeren. Farbe und Lacke 1971, 77, 997–1005. [Google Scholar]
- Kaelble D. H. Dispersion-Polar Surface Tension Properties of Organic Solids. J. Adhes. 1970, 2, 66–81. 10.1080/0021846708544582. [DOI] [Google Scholar]
- Wu S.Interfacial and Surface Tensions of Polymer Melts and Liquids. Polymer Interface and Adhesion; Marcel Dekker: New York, USA, 1982; pp 67–132. [Google Scholar]
- Falsafi A.; Mangipudi S.; Owen M. J.. Surface and Interfacial Properties. In Physical Properties of Polymers Handbook; Mark J. E., Ed.; Springer: New York, USA, 2007; pp 1011–1020. [Google Scholar]
- Greenhalgh D. J.; Williams A. C.; Timmins P.; York P. Solubility parameters as predictors of miscibility in solid dispersions. J. Pharm. Sci. 1999, 88, 1182–1190. 10.1021/js9900856. [DOI] [PubMed] [Google Scholar]
- Hansen C. M. 50 Years with solubility parameters—past and future. Prog. Org. Coat. 2004, 51, 77–84. 10.1016/j.porgcoat.2004.05.004. [DOI] [Google Scholar]
- Van Krevelen D. W.Cohesive Properties and Solubility. Properties of Polymers; Elsevier: New York, USA, 1990; pp 189–226. [Google Scholar]
- Zhang K.; Mohanty A. K.; Misra M. Fully Biodegradable and Biorenewable Ternary Blends from Polylactide, Poly(3-hydroxybutyrate-co-hydroxyvalerate) and Poly(butylene succinate) with Balanced Properties. ACS Appl. Mater. Interfaces 2012, 4, 3091–3101. 10.1021/am3004522. [DOI] [PubMed] [Google Scholar]
- Jost V.; Kopitzky R. Blending of Polyhydroxybutyrate-co-valerate with Polylactic Acid for Packaging Applications—Reflections on Miscibility and Effects on the Mechanical and Barrier Properties. Chem. Biochem. Eng. Q. 2015, 29, 221–246. 10.15255/cabeq.2014.2257. [DOI] [Google Scholar]
- Ferg E. E.; Bolo L. L. A correlation between the variable melt flow index and the molecular mass distribution of virgin and recycled polypropylene used in the manufacturing of battery cases. Polym. Test. 2013, 32, 1452–1459. 10.1016/j.polymertesting.2013.09.009. [DOI] [Google Scholar]