

Abstract
Membrane-active antibiotics are of great interest in fighting bacterial resistance. α-Mangostin is a membrane-active molecule, but there are no details of its mechanism of action at the atomistic level. We have employed free-energy simulations and microsecond-long conventional molecular dynamics simulations to study the mode of interaction of α-mangostin with a model bacterial membrane and compare it with the mechanisms of three hydrophobic molecules (ciprofloxacin, xanthone, and tetracycline). We find that α-mangostin is thermodynamically more favored to insert into the membrane compared to the other three molecules. Apart from tetracycline, which is largely hydrophilic, the other three molecules aggregate in water; however, only α-mangostin can penetrate into the lipid tail region of the membrane. When it reaches a high concentration in the lipid tail region, α-mangostin can form tubular clusters that span the two head group regions of the membrane, resulting in a large number of water translocations along the transmembrane aggregates. Structure–activity relationship analysis revealed two structural properties that characterize α-mangostin, namely, the two isoprenyl groups and the polar groups present in the aromatic rings, which result in “disruptive amphiphilicity” and hence its excellent membrane activity.
1. Introduction
Antibiotic resistance is a serious healthcare issue globally due to the overuse of antibiotics.1 New antibiotics with novel modes of action are urgently needed. Membrane-active antimicrobials, such as antimicrobial peptides (AMPs), and membrane-active natural products are promising new-generation antibiotics that appear to circumvent the issue of bacterial resistance.2−4 In contrast to conventional antibiotics that target intracellular macromolecules and interfere with bacterial biosynthesis, these new-generation antibiotics target the bacterial membranes and hence have the advantages of rapid killing, low tendency to induce resistance, and a broad antimicrobial spectrum.
Most AMPs are cationic and amphiphilic. The positively charged residues are responsible for steering the peptide toward the anionic bacterial membrane via electrostatic interactions. When adsorbed onto the bacterial membrane, some peptides further penetrate into the lipid tail region of the membrane and form membrane pores, resulting in the loss of membrane potential and release of intracellular components. Depending on the geometry of the pores as well as on the interactions of AMPs with the pores, pores have been described by the barrel-stave or toroidal model. In the barrel-stave model, the membrane does not display significant curvature and the hydration of the membrane remains unchanged, whereas in the toroidal model, the head groups lie along the pore, resulting in significant curvature. Other peptides prefer to locate at the membrane–water interface, and upon reaching a critical concentration, disrupt the bacterial membrane into fragments, and this process is referred to as the carpet mechanism. Molecular dynamics (MD) simulations have been used extensively to examine the modes of interactions of various AMPs with bacterial membranes.5−9 For example, Leontiadou et al. were the first to successfully simulate the process of toroidal pore formation by the peptide magainin and found that unexpectedly the toroidal pore during their simulations displayed high disorder;5 such a dynamical landscape of the toroidal pore was also observed for the peptide melittin.7,8 For AMPs acting via the carpet mechanism, MD simulations have revealed key interactions between peptides and the membrane;6,9 however, the exact process of membrane disruption is still elusive, mainly because membrane disruption processes occur over much longer time scales than are currently accessible to simulations. Although MD simulations have provided exquisite details at the atomistic level to some extent, full details of the mechanisms of actions of these molecules are still lacking, resulting in a paucity of guiding design principles.10
Bacteria are classified into Gram-positive and Gram-negative, depending on their membrane architectures. Gram-positive bacteria consist of an inner membrane and a thick layer of peptidoglycan outside the inner membrane, whereas in Gram-negative bacteria, the inner membrane is surrounded by an outer membrane, which consists of closely packed lipopolysaccharides; this outer membrane forms an additional barrier to exogenous agents, including membrane-active antimicrobials. It has been proposed that the inner membrane is critical in maintaining the stability of the bacterial membrane structure and that the disruption/perturbation of the inner membrane is the rate-limiting step in the action of most membrane-active antimicrobials.10,11 The primary issue in the development of membrane-active antimicrobials is poor selectivity because the difference between the bacterial membrane and human membrane is not strikingly high. Another limitation of AMP antibiotics is low proteolytic stability, which results in a short half-life in the body; however, this can be overcome by using unnatural amino acids.10 Despite these issues, membrane-active antimicrobials are promising potential therapeutics because they do not easily induce resistance.10 Much effort has been put into the development of such molecules targeting the inner membrane, and several membrane-active molecules have entered clinical trials.12−14 Structurally, most of the membrane-active molecules consist of hydrophobic moieties that impart them with the ability to interact favorably with the lipid tails of the bacterial membranes. We have recently developed a series of membrane-active molecules derived from α-mangostin (denoted as mangostin hereafter), which is a xanthone-based natural product isolated from the tropical fruit mangosteen.15 Mangostin displays rapid bactericidal effects and high antimicrobial activity.16,17 Dye-leakage experiments and membrane permeabilization assays confirm its membrane-targeting mode of action.15 The available data have been used to develop a mangostin-based pharmacophore model for the design of membrane-active antimicrobials.18 Modification of mangostin with cationic groups further enhances its selectivity, suggesting that mangostin is a good template for the design of membrane-active antimicrobials. We speculate that the membrane activity of mangostin is attributed to its ability to penetrate into and perturb the bacterial membrane. However, the detailed mechanism of action at an atomic level remains unknown. These molecules have shown efficacy against Gram positives; however, given that they act on the inner membrane, an understanding of their atomistic mechanisms may guide the design of similar molecules to target Gram negatives as well, for which there is an urgent need.
The mangostin molecule consists of three aromatic rings, forming a planar aromatic moiety. It was believed that the membrane activity of mangostin arises from two structural properties: hydrophobicity and the planar core structure. However, there exist other molecules with high hydrophobicity and planarity, such as ciprofloxacin and tetracycline, that do not show any membrane activity and act by entering cells and targeting intracellular proteins. So why is it that molecules with such similar characteristics have such widely different mechanisms of action?
To probe the structural mechanisms underpinning these differences, we examine the interactions of mangostin and the other model hydrophobic molecules (these molecules will also be referred to as hydrophobes in this manuscript) with model bacterial membranes at an atomic level, using molecular simulations. We employ molecular dynamics (MD) simulations with rigorous free-energy calculations to examine the thermodynamic properties of the hydrophobe–membrane systems. We select four hydrophobic molecules, mangostin, xanthone, ciprofloxacin, and tetracycline (Figure 1), as model hydrophobes. All molecules contain aromatic rings and are thought to aggregate in water. Mangostin and xanthone differ only in the lack of the two isoprenyl groups in the latter. It is well established that mangostin is membrane-active,15 whereas xanthone has no membrane activity. The other two molecules, ciprofloxacin and tetracycline, are both Food and Drug Administration-approved antibiotics that act on intracellular targets and apparently have no known membrane activity. We begin our study by first characterizing the aggregation behavior of these molecules in water by computing their association free energies. Next, we characterize the affinity of the four hydrophobes with the membrane by computing the free energies associated with transferring these molecules from the aqueous to the lipid phase. To understand the thermodynamic forces driving membrane penetration, we decomposed the transfer free energies of mangostin and ciprofloxacin into enthalpy and entropy contributions; we chose these two because the other two molecules appear to experience very high free-energy barriers to their translocation across the membrane. Finally, to understand why mangostin is membrane-active, whereas ciprofloxacin is not, we performed conventional MD simulations of the membrane with the hydrophobes at high compound/lipid ratios.
Figure 1.

Structures of the four compounds used in the MD simulations.
2. Methods
Molecular dynamics simulations were used to investigate the interactions of the four model compounds with a model bacterial membrane. The membrane was modeled using a mixture of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylglycerol (POPG) lipids in a ratio of 3/1 (POPE/POPG), and the coordinates were taken from our previous studies.6,18,19 The GROMOS 53a6 force field20 was used to model the lipid molecules and the four hydrophobes. The Web server ATB21 was used to generate topologies for the hydrophobes; this server ensures compatibility with the GROMOS family of force fields. As ciprofloxacin contains both amine and carboxyl groups, it is zwitterionic at physiological pH. It has been shown that in the membrane, ciprofloxacin assumes the uncharged form.22 If not otherwise specified, ciprofloxacin is modeled in its zwitterionic form in the simulations without membrane, and in its uncharged form in the membrane simulations. Umbrella sampling and weighted histogram analysis were employed to calculate the association free energies in water and the transfer free energies from the aqueous phase to the lipid phase.23−25 Conventional MD simulations were used to study the aggregation behavior of the four hydrophobes in water and the interactions of the hydrophobes with the membrane at a high compound/lipid ratio.
In umbrella sampling of the association of each hydrophobe in water, two monomer hydrophobe molecules were solvated in simple point-charge (SPC) water26 and the size of each sampling window was set at 0.15 nm, resulting in 24 umbrella windows. Simulations of 50 ns were carried out for each umbrella sampling window for computing the free energies of association of each compound in water, with the last 25 ns used for constructing the potential of mean force (PMF). The reaction coordinate for the free energy of association of each compound was the distance between the centers of masses of the two monomeric units of each compound; a correction term of 2kT ln(r) was added to account for the entropic contributions.27,28 For the free energy of transfer of each compound from water into the membrane, a membrane patch with 72 lipid molecules was used for the umbrella sampling simulations and the size of each sampling window was set at 0.15 nm, resulting in 28 umbrella windows. Simulations in each window were run for at least 200 ns. For the membrane systems, the time scales of membrane undulation are large, thus necessitating a much larger simulation (200 ns) for each umbrella window. It has been shown that the primary error in the PMF calculation of small molecules entering the lipid bilayers from aqueous solution arises from hidden free-energy barriers, which often arise as a result of the formation of lipid defects induced by charged compounds.29 In such cases, complex reaction coordinates are required. However, all four compounds used in this study are uncharged with relatively rigid conformations and hence are unlikely to induce significant membrane defects and will sample the phase space sufficiently, thus resulting in converged PMFs (outlined in Supporting Information). The reaction coordinate was the distance between the center of mass of the compound and the lipid bilayer along the bilayer normal (taken as the z direction); no entropic correction was added because the reaction coordinate is only along the bilayer normal. The error bars of each PMF profile were estimated using the bootstrap method.24
To examine the stability of higher oligomeric states of each compound in water, 16 molecules of each compound were randomly placed in a box of dimensions 7.5 × 7.5 × 7.5 nm3 solvated with SPC water molecules, corresponding to a concentration of 60 mM. Each system was subject to 150 ns of MD simulations. To examine the effects of high compound/lipid ratios on the lipid bilayer, 16 molecules of each compound were initially placed on top of a patch of the membrane made up of 128 lipid molecules. Each simulation was run for 1.0 μs. For ciprofloxacin, an additional simulation of 1.0 μs with 16 ciprofloxacin molecules initially placed in the center of the bilayer was carried out because unlike mangostin, ciprofloxacin did not penetrate into the membrane in the above simulations.
Simulation details are outlined in Tables S1 and S2. During the MD simulations, the covalent bonds involving hydrogen atoms were constrained using the LINCS algorithm,30 enabling a time step of 2 fs to be used. A plain cutoff scheme at 1.4 nm was used to calculate the Lennard-Jones potentials. The short-range electrostatic interactions in real space were cut at a distance of 1.4 nm, whereas the long-range electrostatic interactions in reciprocal space were calculated using the particle-mesh Ewald algorithm. The temperature was maintained at 310 K by coupling the system to a Nosé–Hoover heat bath, and the pressure was maintained at 1 atm using the semi-isotropic Parrinello–Rahman method.31 To decompose the free energies into the associated enthalpy–entropy components, another set of umbrella sampling simulations were also carried out at 323 K for mangostin and ciprofloxacin. The entropy and enthalpy were then calculated following the method of Pettitt32 using the following equations
3. Results and Discussions
3.1. Aggregation in Water
All four compounds investigated in this study consist of large aromatic rings and hence are expected to aggregate in water. This is examined by computing their association free energies in water using umbrella sampling MD simulations (Figure 2). Mangostin, xanthone, and ciprofloxacin show strong tendencies to aggregate as revealed by the large free-energy changes upon association. In contrast, tetracycline shows much less tendency to aggregate, with the computed minimum in the PMF at 0.56 nm and a depth of −9.7 kJ/mol. This value is at least 2-fold lower than for the other three compounds. Although tetracycline has four hydrophobic rings in contrast to the other compounds having only three rings each, the presence of polar groups in tetracycline renders it more hydrophilic and hence much less aggregation-prone than the others. The other three molecules (mangostin, ciprofloxacin, and xanthone) all show strong tendencies to aggregate. In particular, the large hydrophobic isoprenyl group clearly makes mangostin most hydrophobic and hence most aggregation-prone. The association free energies for ciprofloxacin and xanthone are similar, but the xanthone molecules are more planar, enabling them to approach closer to each other, as evidenced by the shorter intermolecular distance of association (0.38 nm). In summary, our computations suggest that the aggregation tendencies of the four molecules studied here follow, in decreasing order: mangostin, ciprofloxacin, xanthone, and tetracycline.
Figure 2.
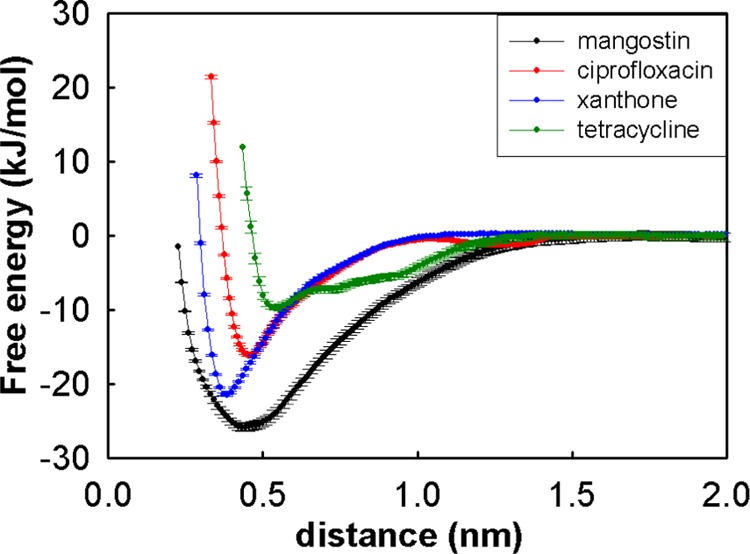
Free energy of association of mangostin, ciprofloxacin, xanthone, and tetracycline in water.
The above calculations were carried out using only two solute molecules. To understand the stability of higher oligomers in water, we carried out conventional MD simulations using 16 molecules of each compound. Typical snapshots obtained after 150 ns are shown in Figure 3. It is observed that mangostin, ciprofloxacin, and xanthone aggregate much more than tetracycline in water (Figure 3a–d). This is in agreement with the free-energy calculations above and we see that tetracycline has low hydrophobicity and exists largely in a monomeric form, interspersed with relatively few oligomers. The almost globular (proteinlike) aggregate of mangostin (Figure 3a) derives from the combined effects of hydrophobicity resulting from stacking of the aromatic rings, the asymmetry induced by the large isoprenyl groups, and the large number of hydrogen bonds between oxygen and the hydroxyl groups on the aromatic rings (Figures 3e and S1). Xanthone is more planar due to the lack of isoprenyl groups and displays strong stacking behavior, resulting in fewer hydrogen bonds between the oxygen and hydroxyl groups on the aromatic rings. Ciprofloxacin, being planar and containing charged groups, forms head-to-tail stacking, and a large number of hydrogen bonds are formed between the amine and the carboxyl groups. This suggests that aromatic stacking and hydrogen bonding are the two forces driving the aggregation of mangostin and ciprofloxacin, whereas hydrophobic stacking largely drives the aggregation of xanthone.
Figure 3.

Snapshots of the aggregates of each compound in water for (a) mangostin, (b) ciprofloxacin, (c) xanthone, and (d) tetracycline. (e) Distribution of the number of hydrogen bonds in each aggregate over the last 80 ns of the simulation; for clarity, water molecules are not shown.
3.2. Transfer Free Energies into Membranes
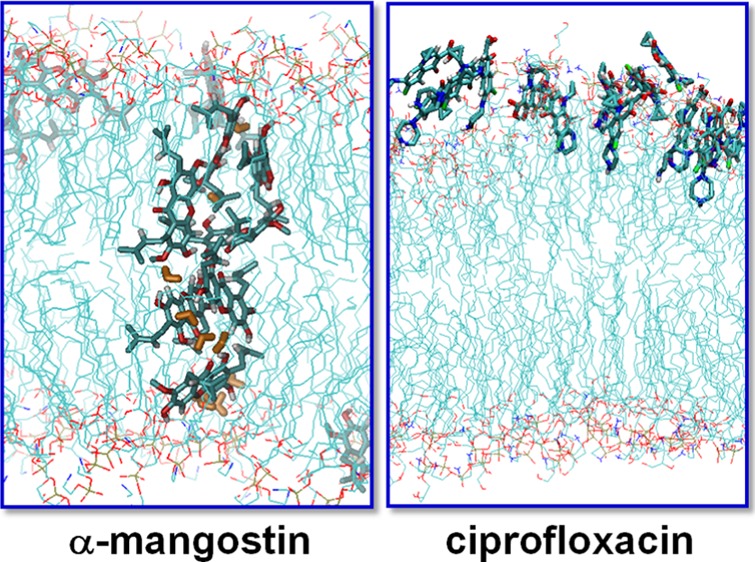
To characterize the membrane affinity of the four hydrophobes, transfer free energies from water to the membrane were calculated using umbrella sampling MD simulations (Figure 4). The free energies of transferring tetracycline into the lipid environment are highly unfavorable. When approaching the membrane from the aqueous phase, the free-energy profile of tetracycline shows a primary minimum of −20 kJ/mol at 2.6 nm. This locates it at the surface of the membrane, where hydrogen bonds with the head groups appear to stabilize the complex. Further penetration leads to rapid increase in the free energy, suggesting unfavorable interactions with the lipid tails. Thus, both the association and transfer free-energy profiles of tetracycline suggest that it is relatively hydrophilic. The other three compounds show favorable free energies of transferring from aqueous phase to the lipid phase, although the free-energy profiles clearly show differences. Mangostin demonstrates the most favorable transfer free energy, followed by ciprofloxacin and xanthone; ciprofloxacin shows more favorable transfer free energies in our work than those reported by Cramariuc and co-workers,22 which may arise from the different force fields and lipid types used in their calculations. In our previous studies,15 we observed spontaneous penetration of mangostin into the lipid tail region of the membrane, which is consistent with the free-energy profile shown in Figure 4. Visualizations show that both ciprofloxacin and mangostin can penetrate into and locate in the lipid tail region of the membrane. However, mangostin is clearly much more favored than ciprofloxacin by ∼15 kJ/mol at the free-energy minimum, clearly reflecting more favorable solvation in the membrane on account of it being more hydrophobic. This can also be seen from the favorable free energy of mangostin at the bilayer center compared to that in bulk water, whereas both ciprofloxacin and xanthone display higher free energies at the bilayer center than in bulk water. The important role of hydrophobicity in the membrane affinity of mangostin can further be seen when its isoprenyl groups are removed as is the case for xanthone; the free-energy difference at the free-energy minimum is ∼20 kJ/mol. Comparison of the position of the free-energy minima reveals that xanthone prefers to remain at the membrane–water interface, whereas ciprofloxacin can penetrate a little and stays just below the charged head groups of the lipid bilayer, thus enabling interactions with the head groups and with solvent water molecules.
Figure 4.
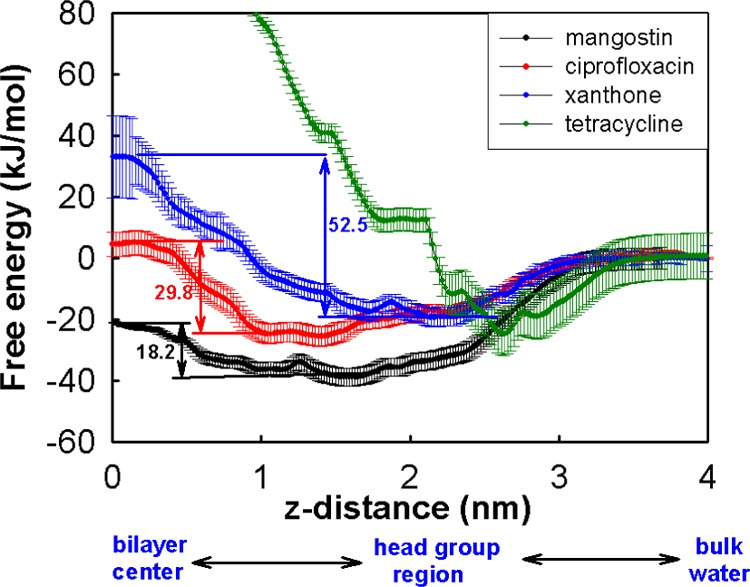
Transfer free energy of each compound from water to a model bacterial membrane. Values next to the bidirectional arrows are the barriers between the free energy at the minima and at the bilayer centers.
Another significant feature that is evident in Figure 4 concerns the free-energy barriers of different hydrophobic compounds as they cross the membrane center. The barrier is defined as the free-energy difference between the free-energy minimum and the free energy at the bilayer center, which relates to the membrane permeability of a molecule. Again, mangostin shows the lowest free-energy barrier (18.2 kJ/mol), followed by ciprofloxacin (29.8 kJ/mol) and xanthone (52.5 kJ/mol). The high free-energy penalty suggests that neither ciprofloxacin nor xanthone like to localize at the center of the bilayer, in contrast to mangostin. The extent of hydrophobic interactions afforded by the presence of the large isoprenyl groups in mangostin is sufficient to compensate for the loss of stabilizing interactions needed by the polar groups; the other two molecules do not have such compensating hydrophobic groups. As a result, ciprofloxacin or xanthone can accumulate only at low concentrations in the lipid tail region of the membrane. In comparison, the much lower free-energy barrier for mangostin enables it to translocate easily across the bilayer center and can accumulate in high concentrations in both leaflets of the membrane. The large numbers of mangostin molecules in the interior of the membrane could potentially induce significant membrane perturbations. Because the membrane activity of mangostin is concentration-dependent, these results offer a plausible molecular detail underlying the membrane activity of mangostin, in contrast to ciprofloxacin and xanthone.
3.3. Enthalpy–Entropy Decomposition of Mangostin and Ciprofloxacin
Mangostin has been shown to have antimicrobial activity with an MIC in the micromolar range.15 We have so far established that the antimicrobial activity of mangostin likely arises from its ability to induce membrane perturbations as a result of its high hydrophobicity combined with its planar structure. Surprisingly, ciprofloxacin, with a planar structure and relatively high hydrophobicity, is not membrane-active. Although we have seen above that the isoprenyl groups of mangostin are the major drivers of its increased solvability in the membrane, we would like to understand the forces that drive the thermodynamic profiles seen above. We decompose the above free-energy profiles of mangostin and ciprofloxacin into the associated enthalpy and entropy contributions. This requires the calculations of the free-energy profiles for both compounds at an increased temperature, which we chose to be 323 K (Figure S2); the corresponding enthalpy and entropy contributions were then obtained using the method of Pettitt et al.32 The method assumes that S is independent of temperature, which strictly speaking, does not hold true for biological systems. However, the two temperatures we chose are 310 and 323 K, which are well above the phase-transition temperatures of POPE (298 K) and POPG (269 K).33,34 Given the small temperature interval (13 K), the entropy weakly depends on temperature. Nevertheless, such approximation provides a qualitative understanding of the enthalpy and entropy contributions to the transfer free energy.
As the molecules approach the membrane surface (2 nm from the lipid bilayer center in Figure 5), their adsorption on the membrane surface is enthalpy-driven. This likely arises from two effects: (i) the direct interactions between the polar groups of mangostin and of ciprofloxacin with the charged head groups of the membrane and (ii) the large hydrophobic moieties of the molecules in the aqueous phase exclude the surrounding water molecules, thus “forcing” them into re-forming strong interwater hydrogen bonds.35 It is known that the hydrophobic effect can be either entropically or enthalpically driven, depending on the length scale of the hydrophobe. When the size of the hydrophobe is small, the hydrophobic association is entropically driven, whereas it is enthalpically driven for large hydrophobes.36 It is reported that the crossover occurs when the size is around that of neopentane.37 It is further reported that molecules with planar curvatures display more significant dewetting than molecules with positive curvatures.38 Considering the size and the planar curvatures of both mangostin and ciprofloxacin, the adsorption of both molecules onto the membrane surface is an enthalpically driven process, in which water molecules in the hydration shell re-form hydrogen bonds during desolvation.
Figure 5.
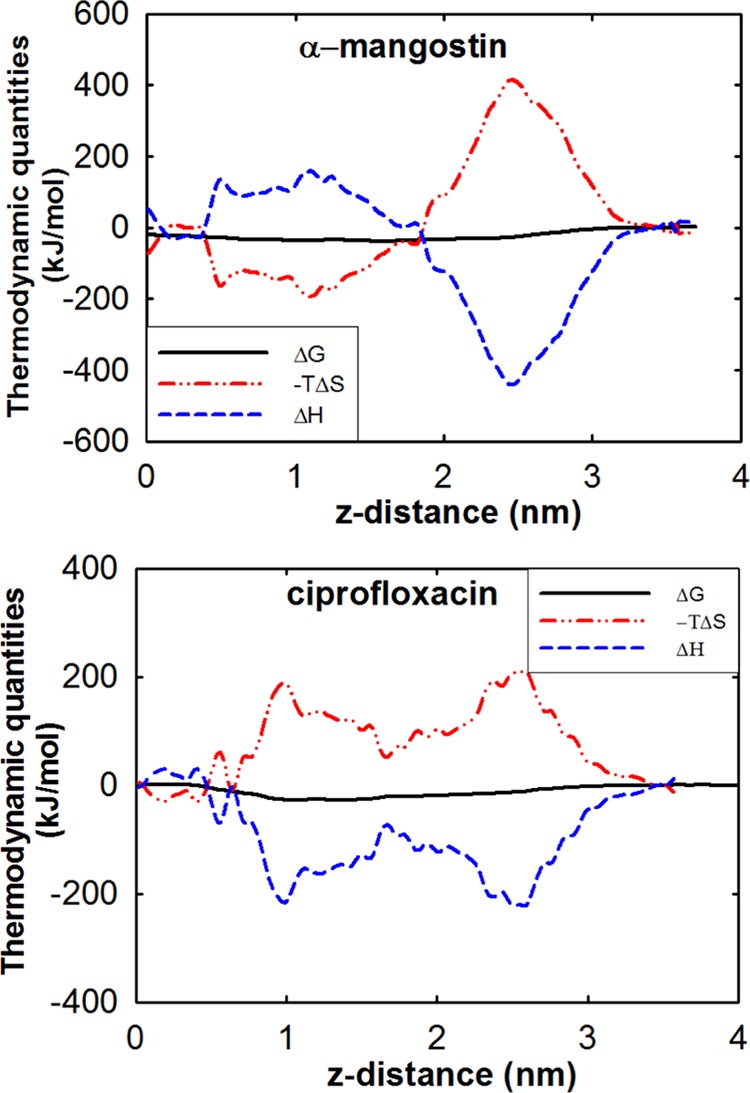
Enthalpic and entropic contributions to the total free energy for mangostin and ciprofloxacin translocating across the membrane.
Further penetration of the hydrophobes into the lipid tail region of the membrane depends on their interactions with the lipid tails (enthalpic) and on maximizing their sampling of the conformational space (entropic). Further penetration of ciprofloxacin into the lipid tails is entropically unfavorable as it is difficult to create a cavity in the lipid tail region due to unfavorable transfer free energies. In contrast, the large isoprenyl groups that help solvate mangostin in the lipid tail region of the membrane would result in its increased mobility. As a result, it is easy to find a cavity large enough to accommodate the mangostin molecule in the lipid tail region, thus driving the process entropically. To examine the changes in motilities, we define two angles, θ and ϕ, to characterize the orientation of each molecule, where θ is the angle between the long axis of the molecule and the bilayer normal and ϕ is the vector normal to the aromatic plane and the bilayer normal. The probability distributions of θ and ϕ at different distances from the bilayer center were calculated (Figure S3). Although the probability distribution of ϕ is similar for both molecules, the distribution of θ is quite different with a much broader distribution associated with mangostin. In particular, at certain distances (e.g., distance = 1.05 nm), mangostin showed two peaks, corresponding to two favorable orientations, whereas ciprofloxacin assumes only one predominant orientation at all distances. This suggests that in the lipid tail region the molecular entropy of mangostin is higher than that of ciprofloxacin. Moreover, the position of the peaks in the distribution profiles of θ for mangostin locates at larger angles than that for ciprofloxacin, suggesting that mangostin tilts more than ciprofloxacin. The higher tilt angle of mangostin is expected to induce large perturbations of adjacent lipid chains, resulting in greater disorder in the lipid chains surrounding the mangostin molecule. In contrast, the smaller tilt angle suggests that ciprofloxacin fits well and forms relatively tight packing with the lipid tails, resulting in less perturbation and stronger nonbonding interactions with the surrounding lipid chains.
3.4. Interactions of Hydrophobes with Membranes at a High Compound/Lipid Ratio
Although the transfer free-energy profiles have provided us with a thermodynamic understanding of the membrane affinity for mangostin and ciprofloxacin, these calculations are focused only on one molecule of each compound, which corresponds to a compound/lipid ratio of 1/72. It is known that membrane perturbation/disruption is concentration-dependent and usually occurs at high compound/lipid ratios, namely, above a critical concentration. To understand the concentration dependence of the membrane activities of mangostin and ciprofloxacin, we performed microsecond-long MD simulations using 16 molecules of each species with 128 lipids, corresponding to a compound/lipid ratio of 1/8. These molecules were initially placed at the membrane surface. The conformations at the end of each simulation after 1 μs are shown in Figures 6 and 7. The ciprofloxacin molecules were found to locate on the membrane surface and form aggregates by stacking together (Figure 6a). During the 1.0 μs MD simulations, only one molecule of ciprofloxacin fully penetrates into the lipid tail region and stays just beneath the head groups, whereas several molecules partially penetrate into the membrane, consistent with the free-energy results. It may be argued that the inability of ciprofloxacin to penetrate into the membrane may be due to the unfavorable initial configuration (e.g., inappropriate orientations and velocities of the 16 ciprofloxacin molecules) used in the MD simulations. To rule out such possibilities, we performed a second replica of the MD simulation for another 1.0 μs with the 16 ciprofloxacin molecules initially placed at the bilayer center. The simulation results (Figure 6b) show that as expected, all ciprofloxacin molecules move to the membrane surface, staying on either the water–lipid interface or just below the head group region of the membrane, consistent with the free-energy calculations.
Figure 6.

Snapshots of 16 ciprofloxacin molecules with a membrane patch of 128 lipid molecules after 1 μs MD simulation with (a) the 16 ciprofloxacin molecules initially placed on top of the membrane and (b) the 16 ciprofloxacin molecules initially placed at the bilayer center.
Figure 7.

(a) Snapshots of 16 mangostin molecules with a membrane patch of 128 lipid molecules after 1 μs MD simulation showing the formation of transmembrane aggregates. Water molecules that penetrated into the lipid tail region are denoted as orange sticks. (b) Number of intermolecular hydrogen bonds formed between the mangostin molecules along the 1 μs MD simulation.
In the simulation of the 16 mangostin molecules initially placed on the membrane surface, all mangostin molecules penetrate into the lipid tail region of the two leaflets of the membrane and remain stabilized. At about 600 ns, some of the mangostin molecules were observed to aggregate inside the membrane, forming a tubular cluster that spans the two head group regions of the membrane (Figure 7a), similar to the structures of transmembrane channels. Unlike the mangostin aggregate in water, which is driven by both aromatic stacking and hydrogen bonding, the mangostin aggregate in the lipid tail region is mainly driven by hydrogen bonding between mangostin molecules and of course with the head groups/water molecules at the membrane–water interface (Figure 7b). The formation of the hydrogen bonds significantly reduces the unfavorable free energies of introducing the polar groups in the lipid tail region and stabilizes the aggregate. The formation of the transmembrane aggregate has two effects: (i) it induces lipid defects around the mangostin aggregate and destabilizes the membrane–water interface; (ii) the polar groups of the mangostin molecules in the transmembrane aggregates attract water molecules, resulting in a large number of water translocations across the membrane, forming transient water channels along the tubular cluster, suggesting that the membrane becomes leaky. In addition, due to the incorporation of a large number of mangostin molecules in the lipid tail region, the membrane undergoes expansion, with the area per lipid increasing by 10%; in contrast, in the case of ciprofloxacin, the area per lipid increases by only 5% (Figure S4).
4. Discussions
Traditional antibiotics are thought to work by entering cells by active or passive transport mechanisms and then somehow engage their intended targets. However, the emergence of resistance to these molecules, largely witnessed by single-point mutations in the targets, has driven the focus of research on membrane-targeting molecules. These are fast-acting bactericidals and are thought to perturb the bacterial membrane, thus circumventing or averting the emergence of resistance.2 Atomic-level insights into the modes of actions of these molecules on the membranes are essential for developing molecules with higher therapeutic potentials. We have identified one such molecule called mangostin.15 Comparing it with traditional antibiotics tetracycline39 and ciprofloxacin, we see that all three share similar features governed by hydrophobic ring systems and decorated with different functional groups. However, we know that only mangostin is membrane-active and this inspired us to investigate their differential interactions with a model bacterial membrane using MD simulations. Computations of the free energies of association in water for each of these molecules suggest that although tetracycline consists of a larger aromatic moiety indicative of easy dimerization, the actual tendency to dimerize is quite low (10 kJ/mol), indicating that the presence of polar groups in tetracycline (Figure 2) renders it a weak hydrophobe. It is not surprising therefore that the computed transfer free energy of tetracycline from the aqueous phase to the lipid phase is not very favorable. The other three hydrophobic compounds, despite consisting of only three hydrophobic rings each, all show much higher hydrophobicity and membrane affinity, which likely arises from the presence of fewer number of polar groups. To further understand the partitioning of each compound into the membrane, we compared the octanol–water partition coefficient log P of each compound. Because of the lack of experimental data, the log P values of mangostin and xanthone are calculated from the ZINC database.40 The log P values for mangostin, ciprofloxacin, xanthone, and tetracycline are 6.32, −0.55, 2.27, and 0.036, respectively.41,42 Except for ciprofloxacin, the log P values of the other three compounds are in qualitative agreement with the transfer free energies in Figure 4. We note that ciprofloxacin is predominantly zwitterionic in aqueous solution; the negative log P for ciprofloxacin refers to the zwitterionic form, which is not relevant here because only the uncharged form is used in the membrane simulations in this study. As only the uncharged forms of molecules are believed to penetrate across membranes,22 the actual log P for the uncharged forms will be much higher.
However, an examination of Figures 2 and 4 reveals larger differences in the association free energies of the compounds in water and their transfer free energies into lipids. For example, the most favorable association free energies of mangostin and xanthone in water differ only by 5 kJ/mol, but their transfer free energies into the lipid phase differ by ∼20 kJ/mol at the corresponding free-energy minimum. This discrepancy arises because the two graphs represent different properties of the molecules. In water, the interaction energy between two monomers is dominated by hydrophobic stacking and hydrogen bonding, whereas the transfer free energies report on the interaction between the compound and the lipid molecules. Although these two free energies are both related to the hydrophobicity of the particular molecule, their association in water is reflective of their solubilities and the transfer into the membrane is a measure of their membrane affinities.
Comparison of the transfer free energies of mangostin and xanthone revealed that the two isoprenyl groups in the mangostin molecule contribute significantly to its membrane affinity. Prenylation has been observed to enhance the membrane affinity of many other molecules. For example, prenylated isoflavanoids show enhanced membrane partitioning compared to their nonprenylated analogues.43 Even for macromolecules such as proteins, it was found that many membrane proteins contain lipid chains such as farsenyl or geranyl groups to facilitate their localization into/at membranes.44,45 This suggests that prenylation can be an efficient strategy to enhance the affinity of a hydrophobic moiety for the membrane.
It is known that among the four molecules studied here, only mangostin is membrane-active. Thermodynamic analysis of the penetration of mangostin into the membrane shows that once solvated into the lipid tail region, mangostin is very mobile compared to ciprofloxacin, with entropy playing a significant role. MD simulations further show that a large number of mangostin molecules can penetrate into the membrane and form tubular aggregates in the lipid tail region spanning the two head groups. In contrast to its hydrophobic aggregation in water, this membrane-spanning aggregate of mangostin molecules is stabilized by intermolecular hydrogen bonds between the molecules; interactions between the terminal mangostin molecules and the head groups of the membrane further stabilize this assembly. The transmembrane aggregate of mangostin perturbs the bilayer stability as follows: (i) it results in local heterogeneity in the membrane, affecting the fluidity of the lipids; (ii) it perturbs the surrounding lipid chains; and (iii) the presence of many polar groups in the transmembrane aggregate leads to a transient water channel. In contrast, xanthone, ciprofloxacin, or tetracycline, at low compound/lipid ratios, cannot favorably penetrate the membrane. At high compound/lipid ratios, only mangostin accumulates inside the membrane and forms the membrane-disrupting oligomers. The other molecules clearly cross the membrane; however, detailed mechanisms of cellular entry of these molecules remain unclear.
Mangostin’s unique mechanism of adsorption–penetration–aggregation is different from that attributed to most AMPs. Mangostin has a small molecule size with high hydrophobicity, which endows it with a favorable transfer free energy from aqueous to the membrane phase. As a result, mangostin accumulates to a high concentration in the lipid tail region and forms membrane-spanning aggregates. The transmembrane aggregate resembles the barrel-stave model in terms of the membrane curvature. However, the mangostin aggregates do not induce membrane pores, although it results in water translocation across the membrane. Moreover, unlike most AMPs that are positively charged, mangostin is uncharged, and is not favored to stay at the membrane surface and hence does not function through the carpet mechanism. The lack of strong electrostatic interactions with the head groups of the membrane results in a low selectivity of 5.7,16 suggesting that adding positively charged moieties to mangostin can potentially reduce the activity (and hence toxicity) toward human membranes. Indeed, we have shown that cationic analogues of mangostin significantly enhance the selectivity for bacterial membranes.16−18
To develop principles for the design of membrane-active molecules, it is important to understand the structure–activity relationships of molecules such as mangostin. From the structural point of view, mangostin has several properties that characterize it as a membrane-active molecule, including (i) a high membrane affinity resulting from the presence of two isoprenyl groups (both ciprofloxacin and xanthone lack the isoprenyl groups and show no penetration into the lipid tail region of the membrane); (ii) a planar structure, which favors penetration into the lipid tail region as it requires a smaller cavity compared to that required by bulky globular groups; (iii) the presence of several polar groups enabling it to form intermolecular hydrogen bonds and subsequently transmembrane aggregates: this is an important property because without those polar groups, a molecule will solubilize in a lipid environment rather than form aggregates. This “disruptive amphiphilicity” of the transmembrane aggregate is thought to destabilize the membranes.46 On the other hand, tetracycline contains more polar groups than mangostin, but it cannot penetrate into the membrane because of the lack of lipid probes, such as the isoprenyl groups. Together our findings suggest that membrane activity may be enhanced in a molecule by engineering in “disruptive amphiphilicity”. This would stem from ensuring that the molecular scaffold combines strong hydrophobicity to solvate into the lipid tail regions of the membranes with a sufficient number of polar groups that can engage in hydrogen bonds with other monomers upon penetration into the lipid tail regions and with the lipid head groups.
5. Conclusions
In summary, we have examined the aggregation propensity and membrane affinity of four model hydrophobes: mangostin, ciprofloxacin, xanthone, and tetracycline. Both the aggregation propensity and the membrane affinity of these hydrophobes follow the order: mangostin, ciprofloxacin, xanthone, and tetracycline, with tetracycline being the least hydrophobic. MD simulations revealed that only mangostin is able to penetrate into the membrane and accumulate at high concentrations in the lipid tail region. Moreover, large numbers of mangostin molecules can form transmembrane aggregates, which significantly destabilize the membrane. Structurally, the membrane activity of mangostin is attributed to the presence of the two isoprenyl groups and several polar groups on the aromatic rings. These findings may guide the design of new membrane-active antimicrobials.
Acknowledgments
This work was supported by NMRC/TCR/002-SERI/2008/R618, NMRC/TCR/R1018 and NMRC/BNIG/2016/2014. The authors acknowledge BII, NSCC, for providing the computational facilities.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b01759.
Some details of the simulations and results of membrane simulation (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Andersson D. I.; Hughes D. Antibiotic resistance and its cost: is it possible to reverse resistance?. Nat. Rev. Microbiol. 2010, 8, 260–271. 10.1038/nrmicro2319. [DOI] [PubMed] [Google Scholar]
- Hurdle J. G.; O’Neill A. J.; Chopra I.; Lee R. E. Targeting bacterial membrane function: an underexploited mechanism for treating persistent infections. Nat. Rev. Microbiol. 2011, 9, 62–75. 10.1038/nrmicro2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock R. E. W.; Sahl H.-G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551. 10.1038/nbt1267. [DOI] [PubMed] [Google Scholar]
- Fjell C. D.; Hiss J. A.; Hancock R. E. W.; Schneider G. Designing antimicrobial peptides: form follows function. Nat. Rev. Drug Discovery 2012, 11, 37–51. 10.1038/nrd3591. [DOI] [PubMed] [Google Scholar]
- Leontiadou H.; Mark A. E.; Marrink S. J. Antimicrobial Peptides in Action. J. Am. Chem. Soc. 2006, 128, 12156–12161. 10.1021/ja062927q. [DOI] [PubMed] [Google Scholar]
- Li J.; Liu S.; Lakshminarayanan R.; Bai Y.; Pervushin K.; Verma C.; Beuerman R. W. Molecular simulations suggest how a branched antimicrobial peptide perturbs a bacterial membrane and enhances permeability. Biochim. Biophys. Acta, Biomembr. 2013, 1828, 1112–1121. 10.1016/j.bbamem.2012.12.015. [DOI] [PubMed] [Google Scholar]
- Sengupta D.; Leontiadou H.; Mark A. E.; Marrink S.-J. Toroidal pores formed by antimicrobial peptides show significant disorder. Biochim. Biophys. Acta, Biomembr. 2008, 1778, 2308–2317. 10.1016/j.bbamem.2008.06.007. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Chen C. H.; Hu D.; Ulmschneider M. B.; Ulmschneider J. P. Spontaneous formation of structurally diverse membrane channel architectures from a single antimicrobial peptide. Nat. Commun. 2016, 7, 13535 10.1038/ncomms13535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S.; Kar R. K.; Nanga R. P. R.; Mroue K. H.; Ramamoorthy A.; Bhunia A. Accelerated molecular dynamics simulation analysis of MSI-594 in a lipid bilayer. Phys. Chem. Chem. Phys. 2017, 19, 19289–19299. 10.1039/C7CP01941F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Koh J.-J.; Liu S.; Lakshminarayanan R.; Verma C. S.; Beuerman R. W. Membrane Active Antimicrobial Peptides: Translating Mechanistic Insights to Design. Front. Neurosci. 2017, 11, 73. 10.3389/fnins.2017.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaara M.; Viljanen P. Binding of polymyxin B nonapeptide to gram-negative bacteria. Antimicrob. Agents Chemother. 1985, 27, 548–554. 10.1128/AAC.27.4.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi N.; Miller K.; Hobbs J.; Rhys-Williams W.; Love W.; Chopra I. XF-73, a novel antistaphylococcal membrane-active agent with rapid bactericidal activity. J. Antimicrob. Chemother. 2009, 64, 735–740. 10.1093/jac/dkp299. [DOI] [PubMed] [Google Scholar]
- Isaksson J.; Brandsdal B. O.; Engqvist M.; Flaten G. E.; Svendsen J. S. M.; Stensen W. A Synthetic Antimicrobial Peptidomimetic (LTX 109): Stereochemical Impact on Membrane Disruption. J. Med. Chem. 2011, 54, 5786–5795. 10.1021/jm200450h. [DOI] [PubMed] [Google Scholar]
- Mensa B.; Howell G. L.; Scott R.; DeGrado W. F. Comparative Mechanistic Studies of Brilacidin, Daptomycin, and the Antimicrobial Peptide LL16. Antimicrob. Agents Chemother. 2014, 58, 5136–5145. 10.1128/AAC.02955-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh J.-J.; Qiu S.; Zou H.; Lakshminarayanan R.; Li J.; Zhou X.; Tang C.; Saraswathi P.; Verma C.; Tan D. T. H.; et al. Rapid bactericidal action of alpha-mangostin against MRSA as an outcome of membrane targeting. Biochim. Biophys. Acta, Biomembr. 2013, 1828, 834–844. 10.1016/j.bbamem.2012.09.004. [DOI] [PubMed] [Google Scholar]
- Zou H.; Koh J.-J.; Li J.; Qiu S.; Aung T. T.; Lin H.; Lakshminarayanan R.; Dai X.; Tang C.; Lim F. H.; Zhou L.; Tan A. L.; Verma C.; Tan D. T. H.; Chan H. S. O.; Saraswathi P.; Cao D.; Liu S.; Beuerman R. W. Design and Synthesis of Amphiphilic Xanthone-Based, Membrane-Targeting Antimicrobials with Improved Membrane Selectivity. J. Med. Chem. 2013, 56, 2359–2373. 10.1021/jm301683j. [DOI] [PubMed] [Google Scholar]
- Koh J.-J.; Lin S.; Aung T. T.; Lim F.; Zou H.; Bai Y.; Li J.; Lin H.; Pang L. M.; Koh W. L.; et al. Amino acid modified xanthone derivatives: novel, highly promising membrane-active antimicrobials for multidrug-resistant Gram-positive bacterial infections. J. Med. Chem. 2015, 58, 739–752. 10.1021/jm501285x. [DOI] [PubMed] [Google Scholar]
- Li J.; Liu S.; Koh J.-J.; Zou H.; Lakshminarayanan R.; Bai Y.; Pervushin K.; Zhou L.; Verma C.; Beuerman R. W. A novel fragment based strategy for membrane active antimicrobials against MRSA. Biochim. Biophys. Acta, Biomembr. 2015, 1848, 1023–1031. 10.1016/j.bbamem.2015.01.001. [DOI] [PubMed] [Google Scholar]
- Zhao W.; Róg T.; Gurtovenko A. A.; Vattulainen I.; Karttunen M. Role of phosphatidylglycerols in the stability of bacterial membranes. Biochimie 2008, 90, 930–938. 10.1016/j.biochi.2008.02.025. [DOI] [PubMed] [Google Scholar]
- Oostenbrink C.; Villa A.; Mark A. E.; Van Gunsteren W. F. A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. 10.1002/jcc.20090. [DOI] [PubMed] [Google Scholar]
- Malde A. K.; Zuo L.; Breeze M.; Stroet M.; Poger D.; Nair P. C.; Oostenbrink C.; Mark A. E. An Automated Force Field Topology Builder (ATB) and Repository: Version 1.0. J. Chem. Theory Comput. 2011, 7, 4026–4037. 10.1021/ct200196m. [DOI] [PubMed] [Google Scholar]
- Cramariuc O.; Rog T.; Javanainen M.; Monticelli L.; Polishchuk A. V.; Vattulainen I. Mechanism for translocation of fluoroquinolones across lipid membranes. Biochim. Biophys. Acta, Biomembr. 2012, 1818, 2563–2571. 10.1016/j.bbamem.2012.05.027. [DOI] [PubMed] [Google Scholar]
- Kumar S.; Rosenberg J. M.; Bouzida D.; Swendsen R. H.; Kollman P. A. THE weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992, 13, 1011–1021. 10.1002/jcc.540130812. [DOI] [Google Scholar]
- Hub J. S.; de Groot B. L.; van der Spoel D. g_wham—A Free Weighted Histogram Analysis Implementation Including Robust Error and Autocorrelation Estimates. J. Chem. Theory Comput. 2010, 6, 3713–3720. 10.1021/ct100494z. [DOI] [Google Scholar]
- Torrie G. M.; Valleau J. P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. 10.1016/0021-9991(77)90121-8. [DOI] [Google Scholar]
- Mackerell A. D. Jr. Empirical force fields for biological macromolecules: Overview and issues. J. Comput. Chem. 2004, 25, 1584–1604. 10.1002/jcc.20082. [DOI] [PubMed] [Google Scholar]
- Neumann R. M. Entropic approach to Brownian movement. Am. J. Phys. 1980, 48, 354–357. 10.1119/1.12095. [DOI] [Google Scholar]
- Boresch S.; Karplus M. The Jacobian factor in free energy simulations. J. Chem. Phys. 1996, 105, 5145–5154. 10.1063/1.472358. [DOI] [Google Scholar]
- Neale C.; Pomès R. Sampling errors in free energy simulations of small molecules in lipid bilayers. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2539–2548. 10.1016/j.bbamem.2016.03.006. [DOI] [PubMed] [Google Scholar]
- Hess B.; Bekker H.; Berendsen H. J. C.; Fraaije J. G. E. M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. 10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H. [DOI] [Google Scholar]
- Nosé S.; Klein M. L. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 1983, 50, 1055–1076. 10.1080/00268978300102851. [DOI] [Google Scholar]
- Choudhury N.; Pettitt B. M. Enthalpy–Entropy Contributions to the Potential of Mean Force of Nanoscopic Hydrophobic Solutes. J. Phys. Chem. B 2006, 110, 8459–8463. 10.1021/jp056909r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey A.; Faller R. Examining the Contributions of Lipid Shape and Headgroup Charge on Bilayer Behavior. Biophys. J. 2008, 95, 2636–2646. 10.1529/biophysj.107.128074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navas B. P.; Lohner K.; Deutsch G.; Sevcsik E.; Riske K. A.; Dimova R.; Garidel P.; Pabst G. Composition dependence of vesicle morphology and mixing properties in a bacterial model membrane system. Biochim. Biophys. Acta, Biomembr. 2005, 1716, 40–48. 10.1016/j.bbamem.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Chandler D. Hydrophobicity: Two faces of water. Nature 2002, 417, 491. 10.1038/417491a. [DOI] [PubMed] [Google Scholar]
- Chandler D. Interfaces and the driving force of hydrophobic assembly. Nature 2005, 437, 640–647. 10.1038/nature04162. [DOI] [PubMed] [Google Scholar]
- Huang X.; Margulis C. J.; Berne B. J. Do Molecules as Small as Neopentane Induce a Hydrophobic Response Similar to That of Large Hydrophobic Surfaces?. J. Phys. Chem. B 2003, 107, 11742–11748. 10.1021/jp030652k. [DOI] [Google Scholar]
- Hillyer M. B.; Gibb B. C. Molecular Shape and the Hydrophobic Effect. Annu. Rev. Phys. Chem. 2016, 67, 307–329. 10.1146/annurev-physchem-040215-112316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra I.; Roberts M. Tetracycline Antibiotics: Mode of Action, Applications, Molecular Biology, and Epidemiology of Bacterial Resistance. Microbiol. Mol. Biol. Rev. 2001, 65, 232–260. 10.1128/MMBR.65.2.232-260.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin J. J.; Sterling T.; Mysinger M. M.; Bolstad E. S.; Coleman R. G. ZINC: A Free Tool to Discover Chemistry for Biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kłosińska-Szmurło E.; Pluciński F. A.; Grudzień M.; Betlejewska-Kielak K.; Biernacka J.; Mazurek A. P. Experimental and theoretical studies on the molecular properties of ciprofloxacin, norfloxacin, pefloxacin, sparfloxacin, and gatifloxacin in determining bioavailability. J. Biol. Phys. 2014, 40, 335–345. 10.1007/s10867-014-9354-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colaizzi J. L.; Klink P. R. pH-Partition Behavior of Tetracyclines. J. Pharm. Sci. 1969, 58, 1184–1189. 10.1002/jps.2600581003. [DOI] [PubMed] [Google Scholar]
- Oteiza P. I.; Erlejman A. G.; Verstraeten S. V.; Keen C. L.; Fraga C. G. Flavonoid-membrane Interactions: A Protective Role of Flavonoids at the Membrane Surface?. Clin. Dev. Immunol. 2005, 12, 19–25. 10.1080/10446670410001722168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F. L.; Casey P. J. Protein Prenylation: Molecular Mechanisms and Functional Consequences. Annu. Rev. Biochem. 1996, 65, 241–269. 10.1146/annurev.bi.65.070196.001325. [DOI] [PubMed] [Google Scholar]
- Overmeyer J. H.; Erdman R. A.; Maltese W. A.. Membrane Targeting via Protein Prenylation. In Protein Targeting Protocols; Clegg R. A., Ed.; Humana Press: Totowa, NJ, 1998; pp 249–263. [DOI] [PubMed] [Google Scholar]
- Wimley W. C. Describing the Mechanism of Antimicrobial Peptide Action with the Interfacial Activity Model. ACS Chem. Biol. 2010, 5, 905–917. 10.1021/cb1001558. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.