

Abstract

The need to develop new alternatives for antimalarial treatment is urgent. Herein, we report the synthesis and antimalarial evaluation of a small library of synthetic 3-alkylpyridine marine alkaloid (3-APA) analogs. First, the compounds were evaluated in vitro against Plasmodium falciparum. The most active compound 5c was selected for optimization of its antimalarial properties. An in silico approach was used based on pure ab initio electronic structure prediction, and the results indicated that a substitution of the hydroxyl group by a fluorine atom could favor a more stable complex with heme at a molecular ratio of 2:1 (heme/3-APA halogenated). A new fluorinated 3-APA analog was synthesized (compound 7), and its antimalarial activity was re-evaluated. Compound 7 exhibited optimized antimalarial properties (P. falciparum IC50 = 2.5 μM), low genotoxicity, capacity to form a more stable heme/3-APA complex at a molecular ratio of 2:1, and conformity to RO5. The new compound, therefore, has great potential as a new lead antimalarial agent.
Introduction
Malaria is a serious infectious disease caused by apicomplexan of the genus Plasmodium, transmitted through blood consumed by female Anopheles mosquitos. Malaria still remains a worldwide public health problem and is responsible for a high degree of morbidity and mortality.1 More than 100 Plasmodium species have been reported, although only 5 are considered as human malaria parasites: Plasmodium falciparum, Plasmodium vivax, Plasmodium malariae, Plasmodium ovale, and Plasmodium knowlesi.2 Infection caused by P. falciparum, as well as being a serious condition, can also evolve to a neurological syndrome known as cerebral malaria.3
Malaria chemotherapy is one of the major strategies for treatment and control of this infection. Therefore, the search for new antimalarial drug candidates has become urgent because of the emergence of parasites resistant against artemisinin-based combination therapy.4 This perilous situation requires efforts not only for the development of new antimalarial compounds but also for the identification of their targets and mechanisms of action.5
A relatively inexpensive method to investigate antimalarial targets that may suggest their possible mechanisms of action is the use of in silico approaches.6 These methods, in association with experimental approaches such as in vitro assays, biophysical techniques to determine drug–target interactions, and genetic toxicology evaluation, could improve the optimization process and lead to new antimalarial drugs.7
In recent years, we have focused our research activity on the synthesis and antimalarial evaluation of 3-alkylpyridine marine alkaloid (3-APA) analogs. Since our first report of antimalarial activity of a small series of 3-APA analogs,8 we have described the mechanism of action of the two most active compounds by employing in silico and biophysical approaches. These 3-APA analogs target hematin and are able to form stable complexes at a molecular ratio of 2:1 (hematin/3-APA).9
In an attempt to improve antimalarial activity, selectivity, and safety, herein, we report the synthesis of a larger library of 3-APA analogs based on our previous studies,8,9 using variations of the side chain length and the type of the functional group attached to the end of this chain. The most active compound was halogenated with fluorine in an attempt to achieve higher target affinity, effective antimalarial activity, increased selectivity, and improved safety.
Results and Discussion
Synthesis
Part I
The synthesis of the 3-APA analogs is shown in Scheme 1. The target compounds 4a–d to 6a–d were obtained in five steps, with moderate to good yields, from easily available starting materials. The synthesis of 1a–d to 5a–d was performed as previously reported by our research group.10 Briefly, the 1,n-diols 1a–d were selectively protected to generate the corresponding monotetrahydropyranyl acetals 2a–d. The monoprotected alcohols 2a–d were mesylated under classical conditions to give compounds 3a–d followed by Williamson etherification under phase-transfer catalysis11 of commercial 3-(pyrid-3-yl)propan-1-ol with the mesylated derivatives 3a–d to give the 3-APA analogs 4a–d. Deprotection of compounds 4a–d with HCl afforded the analogs 5a–d. Finally, the alcohols 5a–d were converted into their respective aldehydes 6a–d by Swern oxidation methodology,12 with yields ranging from 60–95%.
Scheme 1. Synthesis of 3-Alkylpyridine Alkaloid Analogs 4a–d to 6a–d.

Reagents, conditions, and yields: (i) NaHSO4, DHP, dimethyl sulfoxide (DMSO), hexane, 40 °C, 16 h, 74–89%; (ii) MsCl, Et3N, CH2Cl2, rt, 10 h, 77–87%; (iii) 3-pyridinepropanol, NaOH/H2O, Bu4N+Br–, Et2O, rt, 72 h, 57–73%; (iv) MeOH, HCl, rt, 12 h, 71–100%; (v) (COCl)2, Et3N, DMSO, DCM, −60 °C, 25 min, 60–95%.
The antimalarial activity was initially evaluated for the 3-APA analogs 4a–d, 5a–d, and 6a–d (Table 1).
Table 1. In Vitro Antimalarial Activity, Cytotoxicity, and SIs of 3-APA Analogs.
| IC50 (μM) ± SDa |
|||
|---|---|---|---|
| compound | P. falciparum (W2) | Wi-26VA4 | SIb |
| 4a | 53.7 ± 0.7 | >300 | >5.6 |
| 4b | 37.7 ± 0.2 | 37.8 ± 5.0 | 1.0 |
| 4c | 14.0 ± 0.6 | 6.4 ± 0.7 | 0.4 |
| 4d | 8.9 ± 0.4 | 11.3 ± 1.4 | 1.2 |
| 5a | 210.6 ± 12.7 | >400 | 1.9 |
| 5b | >188 | 167.7 ± 10.5 | ND |
| 5c | 14.7 ± 0.2 | 99.1 ± 11.2 | 6.7 |
| 5d | 15.1 ± 0.8 | 34.1 ± 6.5 | 2.2 |
| 6a | 180.2 ± 8.3 | NDc | ND |
| 6b | 52.4 ± 8.9 | ND | ND |
| 6c | 20.3 ± 0.2 | ND | ND |
| 6d | 30.6 ± 2.7 | ND | ND |
| CQd | 0.4 ± 0.066 | >100 | >250 |
SD: standard deviation.
SI: selectivity index.
ND: not determined.
CQ: chloroquine.
Antimalarial Activity
Part I
Initially, the first 3-APA derivatives 4a–d, 5a–d, and 6a–d synthesized were evaluated in vitro against P. falciparum (W2 strain), and their cytotoxicity was evaluated against a human fibroblast cell line (WI-26VA4) (Table 1). All studied compounds were moderately active against P. falciparum, with IC50 values ranging from 8.9 to 210.6 μM. The therapeutic potential of the assayed molecules was determined by their selectivity index (SI). Compound 5c exhibited one of the lowest IC50 values of the series, 14.7 μM, and the highest SI, 6.7. At the end of this process, we decided to focus our efforts on optimization of the antimalarial properties of compound 5c.
To improve the antimalarial activity of compound 5c, it was decided to evaluate in silico the substitution of the hydroxyl group by a fluorine atom, leading to the compound numbered as 7 (Scheme 2), aiming to favor acid/base interactions between the fluorine (a hard base) and hard acid Fe3+ from hematin. Metal complexes with Fe3+–F interaction are relatively rare in comparison to those containing other halogens.13 However, there are some reports in the literature describing fluoride Fe3+ porphyrinates complexes.14,15 Furthermore, the incorporation of fluorine into molecules can enhance some of their pharmacological properties, such as increasing their target affinity, changing their lipophilicity, modulating membrane permeability, and thus increasing their potency.7,16,17
Scheme 2. Synthesis of Fluorinated 3-APA Analog 7.

Reagents, conditions, and yields: (i) DAST, DCM, 0 °C, 18 h, 16%.
In Silico Results
The UB3LYP optimized geometry for the 2:1 complex is depicted in Figure 1a. It can be seen from Table 2 that the 2:1 complex formation with ligand 7 is highly energetically favored (ΔE and ΔG are quite negative values) in comparison with compound 5c. This result demonstrates greater chemical affinity of the compound 7 toward the heme group. The replacement of the hydroxyl group by fluorine in 3-APA analog 5c was, therefore, a viable strategy to increase the interactions between the R edge group and [Fe(III)PPIX]. According to the qualitative hard soft acid bases principle,18 the fluorine atom is a hard base and forms stable complexes with acid cations in which simple ionic interactions are dominant. Fe3+ in the heme group has hard acid characteristics such as being an acceptor with a positive charge, having a small size with no easily polarized valence electrons, and tending to bind to hard bases. Furthermore, the energetic results found here are similar to those reported for azide analogs (N3–), from the first generation of 3-APA analogs, which have shown small IC50 values and favorable binding energies.9 Azide also presents hard/borderline base characteristics. Hard acid/base interactions are predominantly electrostatic. This qualitative principle is in line with our theoretical findings because in our previous work,9 we have shown that the driving force responsible for 2:1 complex formation is electronic energy (ΔE). In addition, it is important to highlight that the bonding between hard acids and bases can be properly described by the quantum mechanical calculations applied here.
Figure 1.
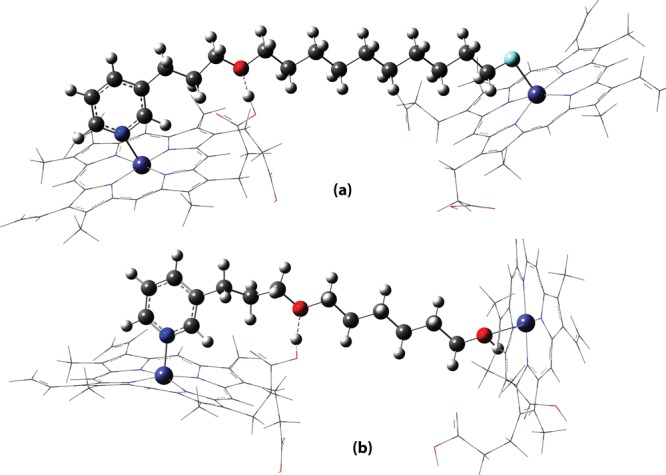
UB3LYP-optimized geometry for (a) compound 7 complex and (b) compound 5a in a 2:1 molar ratio.
Table 2. UB3LYP Binding Energy (ΔE) and Binding Gibbs Free Energy (ΔG) Calculated for the Linking Process of Compounds 5a, 5c, and 7 Analogs and the Heme Group in the 2:1 Molar Ratioa.
| binding
energies (kcal·mol–1) |
||
|---|---|---|
| compound | ΔE | ΔG |
| 5a | –32.5 | –7.4 |
| 5c | –42.8 | –14.6 |
| 7 | –56.9 | –24.7 |
The energies were calculated at 298 K and 1 atm with all values given in kcal·mol–1
On the other hand, the 2:1 complex formed with the compound 5a, which presents the worst antimalarial activity (Table 1), is less energetically favored. This result evidences the weak affinity of the compound 5a for its target and is strongly affected by (i) hard base characteristics and (ii) the short alkyl chain (n = 6). Although the hydroxyl group also presents hard base characteristics, they are less pronounced when compared with fluorine, and the binding strength usually increases as the electrostatic anion characteristic increases. The alkyl chain length also influences the biological activity. This was also observed in our previous work,9 that is, the longer the alkyl chain, the more biologically active the analogs are. The alkyl chain length affects the distance between the analog and both heme sites, influencing the formation of hydrogen bonds and other interactions. The UB3LYP-optimized geometry for the 2:1 complex is depicted in Figure 1b. Cartesian coordinates for the optimized structure of the compound 7 complex (Figure S1), compound 5a complex (Figure S2), and UB3LYP absolute electronic energies (E) calculated for compounds 5a and 7 complexes in a 2:1 molar ratio (Table S1) are presented in the Supporting Information.
Synthesis
Part II
On the basis of our in silico studies, we performed the synthesis of the fluorinated ligand 7. It was synthesized in a 16% yield by diethylamino sulfur trifluoride (DAST) fluorination of the corresponding alcohol 5c.
Antimalarial Activity
Part II
The in vitro antimalarial activity of compound 7 was significantly increased (5.8 fold) compared to compound 5c, with an IC50 of 2.5 μM and an SI value equal to 11.2 against a P. falciparum chloroquine-resistant strain W2. Similar results were achieved against a P. falciparum chloroquine-sensitive strain 3D7 (IC50 of 2.3 μM and an SI value of 12.2) (Table 3).
Table 3. In Vitro Antimalarial Activity, Cytotoxicity, and SIs of Halogenated 3-APA.
| IC50 (μM) ± SDa |
SIb |
||||
|---|---|---|---|---|---|
| compound | P.f. W2c | P.f. 3D7d | Wi-26VA4 | P.f. W2 | P.f. 3D7 |
| 7 | 2.5 ± 0.1 | 2.3 ± 0.15 | 28.2 ± 3.7 | 11.2 | 12.2 |
| CQe | 0.45 ± 0.08 | 0.18 ± 0.05 | >100 | >250 | >250 |
SD: standard deviation.
SI: selectivity index.
P.f. W2: P. falciparum strain W2.
P.f. 3D7: P. falciparum strain 3D7.
CQ: chloroquine.
This improvement in its drug-likeness is in agreement with a recently established set of criteria for the development of compounds with activity against malaria.19–21 Some of these criteria were met by compound 7 such as the following: (i) there is preliminary knowledge of the structure–activity relationship; (ii) there is a cutoff in vitro potency around 1–2 μM; (iii) the SI is greater than 10-fold using a human cell line; and (iv) compound 7 was synthesized in 5 steps. In addition, concerning physicochemical properties, we analyzed conformity to the “rule of five” as a predictor of effectiveness in oral therapy (Table 4). No violations of this rule were observed on replacement of the hydroxyl group by a fluorine atom in compound 7.
Table 4. Physicochemical Properties Estimated for Compounds 5c and 7.
| compound |
|||
|---|---|---|---|
| property | recommended values | 5c | 7 |
| molecular weight (g/mol) | ≤500 | 293.4 | 295.4 |
| H-bond donors | ≤5 | 1 | 0 |
| H-bond acceptors | ≤10 | 2 | 2 |
| clog P | ≤5 | 4.49 | 3.85 |
| number of rule of five violations | 0 | 0 | |
On the basis of these data, the next step was to investigate the binding of the heme group with compound 7.
Biophysical Heme Binding Study
Mass Spectrometry Studies
To experimentally prove the interaction between the new fluorinated derivative and hematin, a mixture containing both molecules was prepared and injected into a mass spectrometer to check for the presence of complexes. Mass spectrometry (MS) is a very useful technique for the evaluation of molecular recognition events because of the clear response provided in the mass/charge relationship. In a typical spectrum where there are host and guest molecules, one can observe peaks corresponding to the individual molecules and their complexes. Another very useful feature of this technique is that it is able to furnish the binding stoichiometry, its determination being one of the great challenges in the supramolecular association field.
On the basis of our previous report,9 the 2:1 complexes are energetically about three times more stable than the 1:1 complexes. Thus, it is mandatory to consider the 2:1 molar ratio to appropriately describe their antimalarial activity. During the erythrocytic cycle of P. falciparum, large amounts of hemoglobin are degraded inside its digestive vacuole. The hemoglobin degradation rate is of the order of magnitude of 0.06 fmol/h per vacuole.16 This consumption is considered high, much more than they require for protein biosynthesis.17 To mimetize this condition experimentally, we prepared a solution containing compound 7 (5.0 μmol/L) and hematin (30.0 μmol/L) in 40% DMSO/H2O. We used six times the concentration of hematin in relation to the compound 7, consequently increasing the concentration of the ternary complex (compound 7/hematin 1:2) in solution to observe its peak, although the experiment amounts of compounds were limited by the typical concentration ranges (μM/L) used in MS in contrast to higher concentrations of hematin found in vivo. And following this logic, it was possible to observe the peak of the second complex (2:1) when using a medium with a higher hematin concentration in relation to compound 7.
In Figure 2, there are three important peaks in the main spectrum. The two peaks at 296.2411 and 616.1809/694.1947 correspond to the ligand ([M + H]+) and hematin ([M]+, [M + DMSO]+). At 911.4117 ([M]+), there is a peak corresponding to the 1:1 complex of ligand–hematin. A more detailed inspection of the spectrum revealed a small peak at 1562.5519 ([M + Cl]+), which corresponds to the mass of two hematin molecules and one ligand, thus representing the 1:2 complex (inset data in Figure 2). The low intensity of this peak is in agreement with the low concentration found for this complex because of its low second association constant.
Figure 2.

Mass spectra [electrospray ionisation time-of-flight mass spectrometry (TOF)] of solutions containing ligand and heme showing (A) peaks corresponding to the complex association (compound 7—[M + H]+ = 296.2411 (calculated = 296.2390); hematin—[M]+ = 616.1809 (calculated = 616.1773); hematin–DMSO [M]+ = 694.1947 (calculated = 694.1912); and 1:1 complex M+ = 911.4117 (calculated = 911.4084); inset: 1:2 complex [M + Cl]+ = 1562.5519 (calculated = 1562.5546) and (B) MS–MS spectra of 1:1 complex ions from solutions containing ligand and heme.
Isolation of the 911.4117 peak and its submission to soft fragmentation furnished a spectrum with peaks corresponding to hematine, thus confirming that the complex was formed noncovalently by these host–guest molecules (Figure 2B). Besides that, the UV–vis experiment (Jobs plot) confirmed the interaction of these molecules and the stoichiometry of complex formation (Figure S3, Supporting Information).
The antimalarial development chain can be hampered by a number of different variables, one of which is the inherent toxicity of the compounds in humans.22 Thus, an important process in the preclinical stage of drug development is genetic toxicology evaluation.
Genotoxicity Evaluation
To complete the analyses in this study, the genotoxic effects of compound 7 were evaluated by applying the comet assay, the cytokinesis-block micronucleus (CBMN) assay and the Ames assay. The comet assay is a relatively simple method for measuring DNA damage at the level of individual cells.23 The micronucleus assay permits the detection of fixation of chromosomal mutations induced by clastogenesis (DNA breakages) or aneugenesis (chromosomal losses).24 The Ames assay identifies agents able to induce gene mutations by additions, deletions, or substitutions of bases in the DNA structure.25 The combination of these methodologies in genetic toxicological studies is an important tool for early safety evaluation in the drug development process.26 These assays offer a reliable set of data for the evaluation of the carcinogenicity of drug candidates.
As observed in Figure 3, three concentrations of compound 7 were assessed in comet assays in the RKO-AS45-1 human cell line, and only the highest concentration analyzed (17.7 μM) was genotoxic, with a mean score that was significantly different from the negative control (PBS). It is interesting to highlight that this genotoxic concentration is about seven times higher than the IC50 defined for compound 7 against P. falciparum (2.5 μM).
Figure 3.

Mean and SD of the score values obtained in three independent experiments employing the comet assay with compound 7 in the RKO-AS45-1 human cell line. P < 0.05 when compared with the negative control (phosphate buffered saline, PBS).
The CBMN assay was also performed using the RKO-AS45-1 cell line, and the results are observed in Figure 4. In this assay, compound 7 was evaluated in concentrations ranging from 2.5 to 17.7 μM. It was observed that the frequency of micronuclei was altered only at concentrations higher than the IC50 of compound 7 against P. falciparum. None of the concentrations assessed affected the nuclear division index (NDI) values, indicating that the proliferation rate was not altered by these treatments.
Figure 4.

Frequency of cells containing micronuclei in 1000 binucleated cells observed in CBMN assay performed with the RKO-AS45-1 cell line exposed for 3 h to compound 7.
The combination of the results observed in comet assay and in CBMN assay demonstrate that the compound 7 can induce chromosomal mutations at higher concentrations not only by the induction of chromosomal breakages but also by the induction of chromosomal losses. However, these effects were observed only at concentrations higher than IC50 defined to P. falciparum.
Besides, the Ames assay, performed with the strains TA98 and TA100 of Salmonella typhimurium, with and without metabolic activation, shows that compound 7 is not able to induce gene mutations (Figure 5) in these conditions.
Figure 5.

Mean of revertants and SD observed after the treatment with different concentrations of compound 7 in Ames assay performed with TA98 and TA100 strains of S. typhimurium with (+S9) and without (−S9) metabolic activation.
Conclusions
In conclusion, we performed the synthesis of a new halogenated 3-APA analog 7 with optimized antimalarial properties such as increased activity against P. falciparum and an increased SI. In addition, despite the structural modifications, compound 7 retained its molecular target, forming a more stable complex with heme.
Materials and Methods
In Vitro Schizonticidal Activity of the 3-APA Analogs against P. falciparum
P. falciparum chloroquine-resistant (W2) and chloroquine-sensitive (3D7) strains were maintained in continuous culture using human red blood cells in RPMI 1640 medium supplemented with human plasma.27 Human red blood cells and human plasma were provided by Foundation of Hemotherapy and Hematology of Minas Gerais (Fundação Hemominas).
The parasites were synchronized using sorbitol treatment,28 and the parasitemias were evaluated microscopically with Giemsa-stained blood smears.
The antimalarial activity was determined using an ELISA antiHRPII assay.29 Infected red blood cells were plated in a 96-well plate at 0.05% parasitemia and 1.5% hematocrit. Different concentrations of the drugs were added in triplicate, and twelve drug-free wells were used as controls (six frozen after 24 h as the HRPII background). After the incubation (72 h), the plate was frozen and thawed twice, and an ELISA using antiHRPII antibodies was performed.
The results were expressed as the mean of the half-maximal inhibitory dose (IC50) of three assays with different drug concentrations performed in triplicate, compared with drug-free controls. Curve fitting was performed using OriginPro 8.0 software (Origin Lab. Corporation, Northampton, MA, USA).
In Vitro Cytotoxicity Test
The noncancerous human lung fibroblast cell line WI-26-VA4 (ATCC CCL-95.1) was used to assess the cell viability after each chemical treatment employing the MTT colorimetric assay.30 Briefly, 1 × 106 cells were plated in 96-well microplates with RPMI 1640 medium supplemented with fetal bovine serum (FBS) and penicillin–streptomycin antibiotics. Then, microplates were incubated overnight at 37 °C, 5% CO2, followed by the treatment with each compound solubilized in DMSO 0.1% (v/v). Negative control groups were constituted of cells without treatment. Five serial dilutions (1:10) were made from a stock solution (10 mg·mL–1) using RPMI supplemented with 1% FBS. After 48 h of incubation, cell viability was evaluated by discarding the medium and adding 100 μL of MTT 5%, followed by 3 h of incubation. Then, the supernatant was discarded, and the insoluble formazan product was dissolved in DMSO. The optical density (OD) of each well was measured using a microplate spectrophotometer at 550 nm. The OD in untreated control cells was defined as 100% cell viability. All assays were performed in triplicate. The SI of the 3-APA analogs was calculated as SI = IC50 WI-26-VA4/IC50P. falciparum (strain W2 or 3D7).
Alkaline Comet Assay
The alkaline comet assay (single-cell gel electrophoresis assay) was performed according to Olive and Banáth31 with adaptations. Briefly, RKO-AS45-1 (ATCC CRL-2579) cells were seeded in 24-well plates (2 × 105 cells/well) in complete medium, and the treatments were performed after 24 h. The cells were exposed to different concentrations of compound 7 (5.9, 11.8, and 17.7 μM) for 3 h in culture medium without serum. The cells of the positive control group were treated with methyl methanesulfonate (MMS—120 μM), and the negative control group was treated with PBS.
The quantification of chromosomal damages was achieved by visual scoring, and the comets were classified from 0 (no damage) to 4 (maximum damage).32 For each treatment, 100 comets were analyzed, and the calculation of the scores was performed employing the equation Score = 0(C0) + 1(C1) + 2(C2) + 3(C3) + 4(C4), where C0–C4 are the numbers of comets in each classification of damage.
Three independent experiments were performed, and a mean of the scores obtained was calculated for each treatment. In the statistical analysis, ANOVA (analysis of variance) was performed followed by the Tukey–Kramer multiple comparisons posttest with a significance level of 0.05.
CBMN Assay
To assess the potential of compound 7 to induce chromosomal mutations in vitro, the CBMN assay was performed in the RKO-AS45-1 (ATCC CRL-2579) human cell line. The procedures were carried out as described by Fenech,33 with adaptations.24 Briefly, 2.5 × 105 cells/well were seeded in 24-well plates in complete medium, and the treatments were performed after 24 h. MMS (400 μM) was used as the positive control, and the negative control group received culture medium without serum as treatment. Compound 7, diluted in culture medium without serum, was evaluated in four different concentrations (2.5, 5.9, 11.8, and 17.7 μM), and three independent experiments were conducted under these conditions.
After 3 h of treatment, cells were washed, and fresh complete medium containing cytochalasin-B (3.0 μg/mL) was added for 24 h. Next, cells were processed for the slides confection. Slides were stained with 4′,6-diamidino-2-phenylindole—1 μg/mL diluted in PBS for cytogenetic analysis under a fluorescent microscope (Zeiss, Axioscope A1) with an excitation filter of 365 nm and a barrier filter of 445/450 nm in a blind test, 1000 binucleated cells were analyzed for each treatment, and cells containing 1–3 micronuclei were scored,34 following the criteria for the identification of micronuclei described in a previous study.35 For statistical analysis, ANOVA was performed followed by the Tukey’s multiple comparison posttest with a significance level of 0.05.
The influence of compound 7 on cell proliferation was assessed by calculating the NDI in the same slides prepared for the CBMN assay. Three hundred cells with a well-preserved cytoplasm were counted using fluorescence microscopy, as described above. The NDI was calculated according to Fenech33 and Eastmond and Tucker,36 using the equation NDI = (M1 + 2(M2) + 3(M3) + 4(M4))/N, where M1–M4 are the numbers of cells with 1, 2, 3, and 4 nuclei, respectively, and N is the total number of analyzed cells.
Ames/Salmonella Mutagenicity Assay
The microsuspension Ames assay was performed using the highly sensitive protocol described by Kado et al.37 with adaptations.38 Briefly, overnight cultures of S. typhimurium strains TA98 and TA100 (1 × 109 cells/mL) were 10× concentrated by centrifugation (4000g, 4 °C) and treated with five concentrations of compound 7 (ranging between 6.7 and 30.9 μM for TA100 and 16.7 and 77.5 μM for TA98). The mixtures were incubated for 90 min at 37 °C without shaking. In tests with metabolic activation, the S9 fraction (MOLTOX, Inc.) was added before incubation. After this time, 2 mL of top agar (0.6% agar, 0.5% NaCl, 0.5 mM l-histidine, 0.5 mM biotin, pH 7.4) was added to the mixture and poured on to a plate containing minimal agar (1.5% agar, Vogel–Bonner E medium, containing 10% glucose). The frequency of his+ revertant colonies was manually counted after incubation for 48 h (37 °C).
DMSO (10 μL/plate) was used as a negative control for both strains. The positive controls used for this assay in the absence and presence of the S9 fraction, respectively, were 4-nitro-o-phenylenediamine and 2-aminoanthracene (2AA) for TA98 and sodium azide (SA) and 2AA for TA100.
The SALANAL software was employed for statistical analysis, adopting the Bernstein model.39 The equation mutagenicity index (MI) = Rev Trat/Rev Control was used to calculate the MI for each concentration assessed, where “Rev Trat” is the frequency of revertants per plate in treated groups and “Rev Control” is the frequency of revertants in the negative control group. Only treatments that induce an MI ≥ 2 were considered mutagenic in this study.40
Synthesis
General Synthesis Methods
General Procedure for the Synthesis of the Aldehydes
To a solution of oxalyl chloride (2.2 equiv) in DCM cooled at −60 °C, a solution of DMSO (4.4 equiv) was added in DCM. This solution was stirred for 10 min at −60 °C, followed by the addition of the alcohol 5a–d (1.0 equiv) solution in DCM. After 15 min of stirring this reaction mixture, triethylamine (5.0 equiv) was added, and then the solution was left to warm to room temperature. The reaction mixture was washed with brine, and the organic layer was dried (Na2SO4), filtered, and evaporated under reduced pressure. The residue obtained was purified by column chromatography (SiO2, EtOAc) to yield the pure aldehydes 6a–d.
6-(3-(Pyridin-3-yl)propoxy)hexanal6a. Yield 71% (0.212 g, 0.90 mmol), Rf = 0.45 (EtOAc), yellow oily product: IR (KBr): ν̅ 2935, 2860, 2802, 1722, 1456, 1423, 1114, 950, 829 cm–1. 1H NMR (CDCl3, 400 MHz): δ 1.29–1.34 (m, 2H), 1.57–1.71 (m, 4H), 1.85–1.92 (m, 2H), 2.44–2.48 (m, 2H), 2.71 (t, J = 7.4 Hz, 2H), 3.39–3.42 (m, 4H), 7.22–7.25 (m, 1H), 7.53 (d, J = 7.7 Hz, 1H), 8.44–8.46 (m, 2H), 9.78 (s, 1H) ppm. 13C NMR (CDCl3, 100 MHz): δ 21.86, 25.81, 29.47, 29.60, 30.90, 43.80, 69.48, 70.60, 123.36, 136.14, 137.33, 146.97, 149.61, 202.64 ppm. HRMS (m/z): [M + H]+ calcd for C14H22NO2+, 236.1606; found, 236.1641.
8-(3-(Pyridin-3-yl)propoxy)octanal6b. Yield 60% (0.447 g, 1.70 mmol), Rf = 0.59 (EtOAc), yellow oily product: IR (KBr): ν̅ 2933, 2856, 2830, 1722, 1456, 1423, 1114, 956, 850 cm–1. 1H NMR (CDCl3, 400 MHz): δ 1.31–1.34 (m, 6H), 1.52–1.62 (m, 4H), 1.83–1.90 (m, 2H), 2.38–2.43 (m, 2H), 2.69 (t, J = 7.9 Hz, 2H), 3.35–3.40 (m, 4H), 7.21–7.26 (m, 1H), 7.53 (d, J = 7.7 Hz, 1H), 8.42–8.44 (m, 2H), 9.73 (s, 1H) ppm. 13C NMR (CDCl3, 100 MHz): δ 21.72, 21.92, 25.93, 28.77, 29.01, 29.12, 29.44, 30.85, 43.79, 69.32, 70.88, 123.43, 136.49, 137.55, 146.50, 149.26, 202.74 ppm. HRMS (m/z): [M + H]+ calcd for C16H26NO2+, 264.1964; found, 264.1957.
10-(3-(Pyridin-3-yl)propoxy)decanal6c. Yield 80% (0.238 g, 0.82 mmol), Rf = 0.64 (EtOAc), yellow oily product: IR (KBr): ν̅ 2927, 2854, 2800, 1728, 1454, 1423, 1114, 952, 829 cm–1. 1H NMR (CDCl3, 400 MHz): δ 1.29–1.34 (m, 10H), 1.51–1.62 (m, 4H), 1.83–1.93 (m, 2H), 2.40–2.44 (m, 2H), 2.71 (t, J = 7.9 Hz, 2H), 3.37–3.43 (m, 4H), 7.20–7.23 (m, 1H), 7.52 (d, J = 7, 7 Hz, 1H), 8.44–8.46 (m, 2H), 9.76 (s, 1H) ppm. 13C NMR (CDCl3, 100 MHz): δ 21.98, 24.50, 24.59, 25.68, 26.10, 26.18, 29.06, 29.51, 29.69, 29.90, 29.93, 30.99, 43.90, 69.43, 70.92, 123.30, 135.95, 136.04, 147.21, 149.91, 202.91 ppm. HRMS (m/z): [M + H]+ calcd for C18H30NO2+, 292.2232; found, 292.2273.
12-(3-(Pyridin-3-yl)propoxy)dodecanal6d. Yield 95% (0.140 g, 0.44 mmol), Rf = 0.70 (EtOAc), yellow oily product: IR (KBr): ν̅ 2924, 2852, 2804, 1728, 1451, 1423, 1111, 989, 835 cm–1. 1H NMR (CDCl3, 400 MHz): δ 1.26–1.34 (m, 14H), 1.53–1.62 (m, 4H), 1.89–1.98 (m, 2H), 2.41–2.45 (m, 2H), 2.91 (t, J = 7.5 Hz, 2H), 3.38–3.45 (m, 4H), 7.70–7.78 (m, 1H), 8.12 (d, J = 7.0 Hz, 1H), 8.54–8.73 (m, 2H), 9.77 (s, 1H) ppm. 13C NMR (CDCl3, 100 MHz): δ 21.98, 24.50, 24.59, 25.68, 26.10, 26.18, 26.79, 29.06, 29.33, 29.43, 29.60, 29.62, 29.94, 29.98, 30.18, 30.32, 43.83, 68.74, 71.13, 125.80, 140.32, 142.51, 143.45, 148.52, 202.96 ppm. HRMS (m/z): [M + H]+ calcd for C20H34NO2+, 320.2590; found, 320.2584.
General Procedure for the Synthesis of the Fluorinated Product
To a solution of compound 5c (1.0 equiv) in DCM cooled to 0 °C, DAST (1.1 equiv) was added. The reaction mixture was allowed to warm at room temperature and stirred for 16 h. Subsequently, water was added to quench the DAST excess. After that, the reaction mixture was washed with a diluted solution of sodium bicarbonate (5 w/v %) and water. The organic layer was dried (Na2SO4), filtered, and evaporated under reduced pressure. The residue obtained was purified by column chromatography (SiO2, EtOAc) to yield the pure compound 7c.
3-(3-((10-Fluorodecyl)oxy)propyl)pyridine7. Yield 16% (0.013 g, 0.043 mmol), Rf = 0.68 (hexane/EtOAc 1:1), yellow oily product: IR (KBr): ν̅ 3320, 3170, 2928, 2855, 1456, 1425, 1117, 713 cm–1. 1H NMR (CDCl3, 400 MHz): δ 1.33–1.38 (m, 12H), 1.55–1.57 (m, 2H), 1.65–1.73 (m, 2H), 1.87–1.91 (m, 2H), 2.70 (t, J = 7.6 Hz, 2H), 3.38–3.42 (m, 4H), 4.43 (dt, 2JHF = 47.2 Hz, 3JHH = 6.0 Hz, 2H), 7.19–7.22 (m, 1H), 7.51 (d, J = 7.6 Hz, 1H), 8.43–8.46 (m, 2H) ppm. 13C NMR (CDCl3, 100 MHz): δ 25.09, 25.14, 26.18, 29.19, 29.43, 29.45, 29.49, 29.73, 30.28, 30.47, 30.98, 69.42, 71.06, 84.22 (d, J1 = 162.9 Hz), 123.25, 135.88, 137.23, 147.28, 149.98 ppm. HRMS (m/z): [M + H]+ calcd for C18H30FNO+, 296.2384; found, 296.2411.
MS Investigation of the Heme–Ligand Complex
The binding of ligands with heme was studied using a high-resolution quadrupole-TOF electrospray mass spectroscopy (Bruker-Compact). Solutions of mixture of ligands/heme were prepared in Milli-Q water/DMSO 6:4, with concentrations of 5–30 μmol/L, and injected into the mass spectrometer. The spectra were recorded in a positive mode.
In Silico Study
Density functional theory calculations were performed to obtain, at the molecular level, structure and energetic properties regarding the binding process of the compounds 5a, 5c, and 7 analogs with the heme group [Fe(III)PPIX] in a 2:1 molar ratio. These three analogs were chosen because of their very distinct biological activities.
In the present work, we used the same theoretical methodology, that is, UB3LYP with a mixed basis set, as described in our recent paper.9 The basis set used for all oxygen, fluorine, nitrogen, and iron atoms was 6-31G(d,p). For the atoms belonging to the CHn groups, a minimal STO-3G basis set was assigned. The UB3LYP binding energy (ΔE) and the Gibbs free energy (ΔG) related to this linking process were obtained. All theoretical calculations were performed using the Gaussian 09 quantum mechanical package.41
Acknowledgments
The authors would like to acknowledge financial support from FAPEMIG (APQ-01597-14, APQ-01598-15, APQ-01254-15), and CNPq (471909/2013-0, 478629/2013-3, 481502/2013-0, 402733/2016-9). The authors would also like to thank ESPEC-UEL/FINEP and LAMM-UEL/FINEP (CT INFRA 2009-01.10.0534.01 and CT-INFRA-2011-01.13.0049.00) for the spectroscopic and MS experiments. This study is part of a project involving the Rede Mineira de Química (RQ-MG) supported by FAPEMIG (REDE-00010-14).The authors also thank the Cellular Biology Service from the Ezequiel Dias Foundation for donation of the human cell lines.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b01302.
Cartesian coordinates for the optimized structure of the compound 7 complex, Cartesian coordinates for the optimized structure of the compound 5a complex, UB3LYP absolute electronic energies (E) calculated for compounds 5a and 7 complexes in a 2:1 molar ratio, and UV–vis experiments (PDF)
Author Contributions
The manuscript was written with the contributions of all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Nkumama I. N.; O’Meara W. P.; Osier F. H. A. Changes in Malaria Epidemiology in Africa and New Challenges for Elimination. Trends Parasitol. 2017, 33, 128–140. 10.1016/j.pt.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossati A.; Bargiacchi O.; Kroumova V.; Zaramella M.; Caputo A.; Garavelli P. L. Climate, environment and transmission of malaria. Infez. Med. 2016, 24, 93–104. [PubMed] [Google Scholar]
- Tiberti N.; Latham S. L.; Bush S.; Cohen A.; Opoka R. O.; John C. C.; Juillard A.; Grau G. E.; Combes V. Exploring experimental cerebral malaria pathogenesis through the characterisation of host-derived plasma microparticle protein content. Sci. Rep. 2016, 6, 37871. 10.1038/srep37871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corey V. C.; Lukens A. K.; Istvan E. S.; Lee M. C. S.; Franco V.; Magistrado P.; Coburn-Flynn O.; Sakata-Kato T.; Fuchs O.; Winzeler E. A. A broad analysis of resistance development in the malaria parasite. Nat. Commun. 2016, 7, 11901. 10.1038/ncomms11901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrows J. N.; Duparc S.; Gutteridge W. E.; Hooft van Huijsduijnen R.; Kaszubska W.; Macintyre F.; Mazzuri S.; Möhrle J. J.; Wells T. N. C. New developments in anti-malarial target candidate and product profiles. Malar. J. 2017, 16, 26. 10.1186/s12936-016-1675-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner-Adams T. S.; Sumanadasa S. D. M.; Fisher G. M.; Davis R. A.; Doolan D. L.; Andrews K. T. Defining the targets of antiparasitic compounds. Drug Discovery Today 2016, 21, 725–739. 10.1016/j.drudis.2016.01.002. [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C. Advancing the drug discovery and development process. Angew. Chem., Int. Ed. 2014, 53, 9128–9140. 10.1002/anie.201404761. [DOI] [PubMed] [Google Scholar]
- Hilário F. F.; de Paula R. C.; Silveira M. L. T.; Viana G. H. R.; Alves R. B.; Pereira J. R. C. S.; Silva L. M.; de Freitas R. P.; de Pilla Varotti F. Synthesis and evaluation of antimalarial activity of oxygenated 3-alkylpyridine marine alkaloid analogues. Chem. Biol. Drug Des. 2011, 78, 477–482. 10.1111/j.1747-0285.2011.01154.x. [DOI] [PubMed] [Google Scholar]
- Ribeiro-Viana R. M.; Butera A. P.; Santos E. S.; Tischer C. A.; Alves R. B.; Pereira de Freitas R.; Guimarães L.; Varotti F. P.; Viana G. H. R.; Nascimento C. S. Revealing the Binding Process of New 3-Alkylpyridine Marine Alkaloid Analogue Antimalarials and the Heme Group: An Experimental and Theoretical Investigation. J. Chem. Inf. Model. 2016, 56, 571–579. 10.1021/acs.jcim.5b00742. [DOI] [PubMed] [Google Scholar]
- Gonçalves A.; de Lima A.; da Silva Barbosa M.; de Camargos L.; de Oliveira J.; de Souza Barbosa C.; Villar J.; Costa A.; Silva I.; Silva L.; de Pilla Varotti F.; dos Santos F.; Viana G. Synthesis and Biological Evaluation of Novel 3-Alkylpyridine Marine Alkaloid Analogs with Promising Anticancer Activity. Mar. Drugs 2014, 12, 4361–4378. 10.3390/md12084361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman H. H.; Dubois R. A. An improved Williamson ether synthesis using phase transfer catalysis. Tetrahedron Lett. 1975, 16, 3251–3254. 10.1016/s0040-4039(00)91417-5. [DOI] [Google Scholar]
- Mancuso A. J.; Swern D. Activated Dimethyl Sulfoxide: Useful Reagents for Synthesis. Synthesis 1981, 165–185. 10.1055/s-1981-29377. [DOI] [Google Scholar]
- Zhang Y.; Su J.; Li Q.; Li W.; Liang G.; Li H.; Ma H.; Lin Q.; Yao H.; Wei T. Novel Fluorescent Chemosensor for Detection of F– Anions Based on a Single Functionalized Pillar[5]arene Iron(III) Complex. Chin. J. Chem. 2016, 34, 1263–1267. 10.1002/cjoc.201600588. [DOI] [Google Scholar]
- Meininger D. J.; Muzquiz N.; Arman H. D.; Tonzetich Z. J. A convenient procedure for the synthesis of fluoro-iron(III) complexes of common synthetic porphyrinates. J. Porphyrins Phthalocyanines 2014, 18, 416–423. 10.1142/s108842461450014x. [DOI] [Google Scholar]
- Sil D.; Kumar A.; Rath S. P. Diiron(III)-μ-Fluoro Bisporphyrins: Effect of Bridging Ligand on the Metal Spin State. Chem.—Eur. J. 2016, 22, 11214–11223. 10.1002/chem.201600786. [DOI] [PubMed] [Google Scholar]
- Liang T.; Neumann C. N.; Ritter T. Introduction of fluorine and fluorine-containing functional groups. Angew. Chem., Int. Ed. 2013, 52, 8214–8264. 10.1002/anie.201206566. [DOI] [PubMed] [Google Scholar]
- Gillis E. P.; Eastman K. J.; Hill M. D.; Donnelly D. J.; Meanwell N. A. Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58, 8315–8359. 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]
- Pearson R. G. Hard and Soft Acids and Bases. J. Am. Chem. Soc. 1963, 85, 3533–3539. 10.1021/ja00905a001. [DOI] [Google Scholar]
- Katsuno K.; Burrows J. N.; Duncan K.; van Huijsduijnen R. H.; Kaneko T.; Kita K.; Mowbray C. E.; Schmatz D.; Warner P.; Slingsby B. T. Hit and lead criteria in drug discovery for infectious diseases of the developing world. Nat. Rev. Drug Discovery 2015, 14, 751–758. 10.1038/nrd4683. [DOI] [PubMed] [Google Scholar]
- Goldberg D. E.; Slater A. F.; Cerami A.; Henderson G. B. Hemoglobin degradation in the malaria parasite Plasmodium falciparum: an ordered process in a unique organelle. Proc. Natl. Acad. Sci. U.S.A. 1990, 87, 2931–2935. 10.1073/pnas.87.8.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew V. L.; Tiffert T.; Ginsburg H. Excess hemoglobin digestion and the osmotic stability of Plasmodium falciparum-infected red blood cells. Blood 2003, 101, 4189. 10.1182/blood-2002-08-2654. [DOI] [PubMed] [Google Scholar]
- Mishra M.; Mishra V. K.; Kashaw V.; Iyer A. K.; Kashaw S. K. Comprehensive review on various strategies for antimalarial drug discovery. Eur. J. Med. Chem. 2017, 125, 1300–1320. 10.1016/j.ejmech.2016.11.025. [DOI] [PubMed] [Google Scholar]
- Bausinger J.; Speit G. The impact of lymphocyte isolation on induced DNA damage in human blood samples measured by the comet assay. Mutagenesis 2016, 31, 567–572. 10.1093/mutage/gew021. [DOI] [PubMed] [Google Scholar]
- Araldi R. P.; de Melo T. C.; Mendes T. B.; de Sá Júnior P. L.; Nozima B. H. N.; Ito E. T.; de Carvalho R. F.; de Souza E. B.; de Cassia Stocco R. Using the comet and micronucleus assays for genotoxicity studies: A review. Biomed. Pharmacother. 2015, 72, 74–82. 10.1016/j.biopha.2015.04.004. [DOI] [PubMed] [Google Scholar]
- Mortelmans K.; Zeiger E. The Ames Salmonella/microsome mutagenicity assay. Mutat. Res., Fundam. Mol. Mech. Mutagen. 2000, 455, 29–60. 10.1016/s0027-5107(00)00064-6. [DOI] [PubMed] [Google Scholar]
- Luo Q.; Li Y.; Zhang Z. A systematic assessment of genotoxicity on pivaloylacylation-7ADCA-a wide existing antibiotic impurity. Int. J. Clin. Exp. Med. 2014, 7, 4260–4271. [PMC free article] [PubMed] [Google Scholar]
- Trager W.; Jensen J. Human malaria parasites in continuous culture. Science 1976, 193, 673–675. 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- Lambros C.; Vanderberg J. P. Synchronization of plasmodium falciparum erythrocytic stages in culture. J. Parasitol. 1979, 65, 418–420. 10.2307/3280287. [DOI] [PubMed] [Google Scholar]
- Noedl H.; Wernsdorfer W. H.; Miller R. S.; Wongsrichanalai C. Histidine-rich protein II: a novel approach to malaria drug sensitivity testing. Antimicrob. Agents Chemother. 2002, 46, 1658–1664. 10.1128/aac.46.6.1658-1664.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmichael J.; DeGraff W. G.; Gazdar A. F.; Minna J. D.; Mitchell J. B. Evaluation of a tetrazolium-based semiautomated colorimetric assay: assessment of chemosensitivity testing. Cancer Res. 1987, 47, 936–942. [PubMed] [Google Scholar]
- Olive P. L.; Banáth J. P. The comet assay: a method to measure DNA damage in individual cells. Nat. Protoc. 2006, 1, 23–29. 10.1038/nprot.2006.5. [DOI] [PubMed] [Google Scholar]
- Collins A. R. The Comet Assay for DNA Damage and Repair: Principles, Applications, and Limitations. Mol. Biotechnol. 2004, 26, 249–261. 10.1385/mb:26:3:249. [DOI] [PubMed] [Google Scholar]
- Fenech M. Cytokinesis-block micronucleus cytome assay. Nat. Protoc. 2007, 2, 1084–1104. 10.1038/nprot.2007.77. [DOI] [PubMed] [Google Scholar]
- Gomes C. C.; Moreira L. M.; Santos V. J. S. V.; Ramos A. S.; Lyon J. P.; Soares C. P.; Santos F. V. Assessment of the genetic risks of a metallic alloy used in medical implants. Genet. Mol. Biol. 2011, 34, 116–121. 10.1590/s1415-47572010005000118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titenko-Holland N.; Windham G.; Kolachana P.; Reinisch F.; Parvatham S.; Osorio A. M.; Smith M. T. Genotoxicity of malathion in human lymphocytes assessed using the micronucleus assay in vitro and in vivo: a study of malathion-exposed workers. Mutat. Res., Genet. Toxicol. Environ. Mutagen. 1997, 388, 85–95. 10.1016/s1383-5718(96)00140-4. [DOI] [PubMed] [Google Scholar]
- Eastmond D. A.; Tucker J. D. Identification of aneuploidy-inducing agents using cytokinesis-blocked human lymphocytes and an antikinetochore antibody. Environ. Mol. Mutagen. 1989, 13, 34–43. 10.1002/em.2850130104. [DOI] [PubMed] [Google Scholar]
- Kado N. Y.; Langley D.; Eisenstadt E. A simple modification of the Salmonella liquid-incubation assay. Increased sensitivity for detecting mutagens in human urine. Mutat. Res. Lett. 1983, 121, 25–32. 10.1016/0165-7992(83)90082-9. [DOI] [PubMed] [Google Scholar]
- Gontijo V. S.; Espuri P. F.; Alves R. B.; de Camargos L. F.; dos Santos F. V.; de Souza Judice W. A.; Marques M. J.; Freitas R. P. Leishmanicidal, antiproteolytic, and mutagenic evaluation of alkyltriazoles and alkylphosphocholines. Eur. J. Med. Chem. 2015, 101, 24–33. 10.1016/j.ejmech.2015.06.005. [DOI] [PubMed] [Google Scholar]
- Bernstein L.; Kaldor J.; McCann J.; Pike M. C. An empirical approach to the statistical analysis of mutagenesis data from the Salmonella test. Mutat. Res., Environ. Mutagen. Relat. Subj. 1982, 97, 267–281. 10.1016/0165-1161(82)90026-7. [DOI] [PubMed] [Google Scholar]
- Santos F. V.; Andreo M.; Nasser A. L. M.; Moreira L. M.; Vilegas W.; Cylus I. M. S.; Varanda E. A. Absence of mutagenicity of plants used to treat gastrointestinal disorders. Arch. Biol. Sci. 2013, 65, 191–195. 10.2298/ABS1301191S. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Fox D. J.. Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford CT, 2009.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.