

Abstract
Engaging Raman spectroscopy as a primary tool, we investigated the early events of insulin fibrilization and determined the structural content present in oligomer and protofibrils that are formed as intermediates in the fibril formation pathway. Insulin oligomer, as obtained upon incubation of zinc-free insulin at 60 °C, was mostly spherical in shape, with a diameter of 3–5 nm. Longer incubation produced “necklace”-like beaded protofibrillar assembly species. These intermediates eventually transformed into 5–8 nm thick fibers with smooth surface texture. A broad amide I band in the Raman spectrum of insulin monomer appeared at 1659 cm–1, with a shoulder band at 1676 cm–1. This signature suggested the presence of major helical and extended secondary structure of the protein backbone. In the oligomeric state, the protein maintained its helical imprint (∼50%) and no substantial increment of the compact cross-β-sheet structure was observed. A nonamide helix signature band at 940 cm–1 was present in the oligomeric state, and it was weakened in the fibrillar structure. The 1-anilino-8-naphthalene-sulfonate binding study strongly suggested that a collapse in the tertiary structure, not the major secondary structural realignment, was the dominant factor in the formation of oligomers. In the fibrillar state, the contents of helical and disordered secondary structures decreased significantly and the β-sheet amount increased to ∼62%. The narrow amide I Raman band at 1674 cm–1 in the fibrillar state connoted the formation of vibrationally restricted highly organized β-sheet structure with quaternary realignment into steric-zipped species.
Introduction
Insulin is an essential protein that interacts with specific transmembrane cell surface receptors of muscle and adipose cells where it stimulates a complex cell signaling pathway, leading to the transport of glucose across the cell membrane and controls the glucose level in the bloodstream.1 It is produced in pancreatic β-cell,2 and its hexameric assembly formation and coordination to Zn2+ stabilizes insulin in cellular environment.3 It, however, exists as a mixture of hexameric, dimeric, and monomeric states in solution condition,4 and the thermal destabilization causes unwanted aggregation and malfunctioning. The aggregation and formation of amyloid-like fibril of insulin thus have long complicated its manufacture, storage, and clinical formulation.5
It was postulated that the B-chain segment of insulin may be a crucial factor for insulin aggregation and fibrilization.6,7 The crystal structure reveals that insulin contains a large α-helical structure consisting of 51 amino acid residues that are arranged into two polypeptide chains, A (21 residues) and B (30 residues). Three helical segments are formed by the residues, A2–A7, A13–A19, and B10–B19. Two of the helices are in the A-chain joined by a loop from A9 to A12 that brings the N- and C-termini together. The B-chain helix (B10–B19) is followed by a turn at the C-terminal end, and the N-terminal end of the B-chain is unstructured.8 Both the A and B chains of the molecule are linked together by two interchains (between A7 and B7 and between A20 and B19) and one intrachain disulfide bond (A6 and A11).9
Several research groups hypothesized different mechanisms of insulin fiber formation10−16 and examined the aggregates under many circumstances, including low pH, different salt concentration, and hydrophobic interfaces that mimic physiologic condition.3,17 Our earlier studies on insulin, mini-proinsulin, and domain swap mutants explored the secondary structural properties of globular and fibrillar forms using Raman spectroscopy.18 In later works, Hua et al. identified an early intermediate using solution-state NMR spectroscopy and reported that the N-terminal helix of both the A and B chain unfolds and detaches from the core.5 They also suggested that the C-terminal segment of B chain also unfolds to some extent but remains tethered to the core. Structural aspects of insulin aggregates were investigated several times,17,19−21 and the fibril showed a parallel intermolecular β-sheet rich secondary structure.22−25 Many of these studies suggested a three-phase process of matured fibril formation.26 During an initial lag phase, the protein may rearrange and position to start to form oligomeric intermediates and in the subsequent event, start stacking together forming elongated structures, commonly known as protofibrils. After this state, protofibrils can eventually pack, leading to the formation of twisted structures of mature amyloid fibrils. Thus, the formation of these ordered aggregated species is typically followed by a nucleation-dependent polymerization mechanism.27 It was reported that partially distorted non-native insulin monomers in oligomeric geometry play a key role in the kinetics of amyloid fiber formation28 and several studies show that the oligomeric species are the possible cause of cytotoxicity in amyloid disease in general.29 Therefore, uncovering the structural details of these species is very important to understand the nucleation-dependent fibril formation mechanism.30−32
However, structural insights of the early intermediates and their conformational rearrangement are not well understood. Vestergaard et al. through small-angle X-ray scattering and computational modeling suggested that a helical structural nucleus could be the primary elongating unit of insulin amyloid fibrils.21 The shape and size of oligomers and their relative population in the lag phase control the rate of fibril formation. The current investigation attempted to provide proper coherent information about the conformational preferences and rate kinetics throughout the fibril formation pathway. We investigated the early stages of self-assembly and the changes in secondary structural or conformational rearrangement of insulin from native monomer to the fibrillar state by engaging Raman spectroscopy. Raman spectroscopy has been well used in the structural characterization of proteins and protein aggregates.33−35 The technique is highly sensitive to changes in conformation and bonding pattern, and the method is, therefore, particularly used to define the perturbation in the secondary and/or tertiary structural alignments in proteins in different assembly conditions. Here, we provided a definitive proof through Raman measurement that the fibrillation process of insulin involves the formation of a helix-rich intermediate in the lag phase; thereafter, in the maturation state, the helical conformation rapidly converted into the β-sheet rich structure.
Results and Discussion
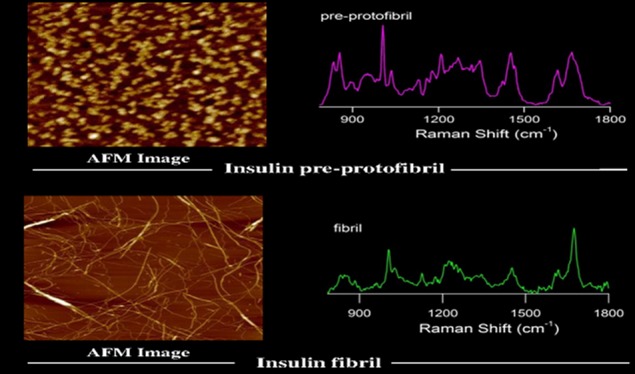
We investigated the early events of self-assembly, associated conformational rearrangement, and changes in the secondary structure of human insulin in the fibril formation pathway, engaging Raman spectroscopy as a primary tool. Figure 1 displays atomic force microscopy (AFM) images of different aggregate species formed in different time intervals upon incubation of human insulin (1.5 mg mL–1 in 20 mM HCl with 50 mM NaCl, pH 1.8) for 3/4 h at 60 °C. For 60 min of incubation, it showed the formation of an oligomeric assembly structure, mostly spherical, with a diameter of 3–5 nm (Figure 1A). Pre-protofibrils were observed after ∼100 min of incubation. The length and height of these pre-protofibrils varied up to several nanometers (Figure 1C). The surface morphology of pre-protofibrils was rough and indicated that no organized structures were yet formed. A more organized protofibrillar species formation was observed for 135 min of incubation, as shown in Figure 1D. The diameter of the aggregated species was 5–8 nm. Further incubation produced a threadlike amyloid fibril. Surface morphology showed a very smooth texture of fibrils and suggested an organized assembly of insulin monomers into a higher ordered assembly structure (Figure 1E,F).
Figure 1.

AFM images of insulin aggregates prepared at 60 °C. The pH of the solution was 1.8. (A) Insulin oligomers having 3–5 nm diameter obtained after 60 min of incubation. (B) Pre-protofibrils obtained after 120 min of incubation. (C) Three-dimensional views of pre-protofibrils. (D) Protofibrils of diameter 5–8 nm obtained after 135 min. (E) Thin and thick fibrils of insulin, as obtained after 160 min. (F) Mature fibrils after 180 min of incubation.
Figure 2 shows the Raman spectra of insulin monomer, oligomer, pre-protofibril, protofibrils, and the late-state assembly structure of matured fibril, respectively. Table 1 contains the spectral assignments of characteristic Raman bands of the protein in different assembly conditions. The Raman spectrum of insulin monomer mainly consisted of a broad amide I band at 1659 cm–1, with an associated (shoulder) band at 1676 cm–1 (Figure 2A).18 The bandwidth at half maxima (BWHM) for the band was ∼49 cm–1. This large bandwidth indicated the presence of multiple conformation states of the protein in its monomeric state. The band at 1659 cm–1 was an indication of major helical conformation along with a contribution from the disordered state, as marked at 1676 cm–1.18 The amide III region (1230–1300 cm–1) was also enriched with predominant α-helical marker bands (Figure 2A and Table 2).18,36−38 It was composed of a major shoulder band at 1262 cm–1 that marked the signature of pure helix.36,38 The signature of the extended helix/2.51-helix conformation similar to poly-l-glutamic acid conformation was embarked at 1268 and 1277 cm–1.39 The 1249 cm–1 shoulder was possibly due to mixed vibrational contributions from PPII and 3/10-helix conformation from the protein backbone structure (Table 2).38
Figure 2.

Left panel shows the Raman spectra of insulin monomer, oligomer, and pre-protofibril in the frequency range of 800–1800 cm–1. The right panel presents the curve-fitting analysis of the extended amide I band. Panels (A) and (B), respectively, are the Raman spectra of insulin monomer and curve-fitting analysis to Raman amide I band of monomer. Panels (C) and (D) represent Raman spectra of oligomers and fitted bands, respectively. Raman spectra of pre-protofibrils and its amide I band fitting analysis are shown, respectively, in panels (E) and (F).
Table 1. Curve-Fitting Analysis of Amide I Raman Band Profile of Insulin and Its Aggregates, as Obtained at Different Time Points of Incubationa.
| species
formations |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| α-helix |
organized β-sheet |
loose β-strand, PPII and disordered components |
other
disordered components (undefined components) |
|||||||||
| time (min) | peak (cm–1) | % A | width (cm–1) | peak (cm–1) | % A | width (cm–1) | peak (cm–1) | % A | width (cm–1) | peak (cm–1) | % A | width (cm–1) |
| 0 | 1658 | 54 | 28 | 1676 | 9 | 21 | 1686 | 24 | 27 | 1630 | 13 | 25 |
| 60 | 1659 | 50 | 28 | 1676 | 8 | 19 | 1687 | 28 | 31 | 1637 | 14 | 27 |
| 120 | 1660 | 46 | 27 | 1675 | 12 | 22 | 1687 | 21 | 26 | 1644 | 21 | 35 |
| 135 | 1656 | 37 | 19 | 1673 | 29 | 20 | 1690 | 22 | 26 | 1639 | 12 | 17 |
| 160 | 1656 | 29 | 20 | 1675 | 41 | 19 | 1691 | 16 | 25 | 1639 | 14 | 20 |
| 180 | 1655 | 14 | 18 | 1674 | 62 | 18 | 1690 | 14 | 23 | 1642 | 10 | 16 |
Percentage of areas (% A) and the bandwidth of each fitted component band are shown.
Table 2. Raman Vibrational Bands (cm–1) of Insulin Monomer, Oligomer, Pre-Protofiber, Protofiber, Premature Fiber, and Mature Fibers.
| monomer | oligomer | pre-protofiber | protofiber | premature fiber | fiber | modes of vibration |
|---|---|---|---|---|---|---|
| 830 | 831 | 831 | 831 | 830 | Tyr | |
| 854 | 852 | 852 | 852 | 850 | 843 | Tyr |
| 898 | 898 | 890 | 890 | 885 | 899 | Cα–C stretching |
| 940, 949 | 940, 949 | 940, 950 | 940, 949 | 940, 949 | 940 | helix skeletal, Cα–C stretching |
| 962 | 964 | 960 | 960 | 960, 972 | 973 | skeletal β strand, Cα–C stretching |
| 1004 | 1004 | 1004 | 1004 | 1004 | 1005 | Phe |
| 1033 | 1033 | 1034 | 1032 | 1033 | 1033 | Tyr |
| 1130 | 1133 | 1130 | 1125 | 1125 | 1130 | CH2 symmetric rock + Cα–C stretching |
| 1156, 1177 | 1156, 1178 | 1156, 1175 | 1176 | 1156, 1174 | 1153, 1170 | Phe, Tyr |
| 1205 | 1208 | 1206 | 1208 | 1209 | 1209 | Tyr, Phe |
| 1222 | 1222 | 1222 | type II β-turn, strand, amide III | |||
| 1249 | 1245 | 1250 | 1250 | 1250 | 1254 | poly-l-proline, amide III |
| 1262, 1269 | 1262, 1269 | 1266 | 1270 | 1261, 1269 | 1269 | α-helix, amide III |
| 1277 | 1278 | 1276 | 2.5H1-helix (extended β-strand), amide III | |||
| 1312 | 1312 | left-handed PPII type, amide III | ||||
| 1343 | 1341 | 1344 | 1345 | 1344 | 1330 | Cα–H deformation, pure α-helix, amide III |
| 1421, 1449, 1465 | 1423, 1449, 1463 | 1422, 1449, 1463 | 1419, 1449, 1464 | 1419, 1444, 1454, 1463 | 1442, 1460 | CH2, CH3, and CH deformation and scissoring |
| 1587 | 1588 | 1588 | 1589 | 1588 | 1590 | Phe |
| 1605 | 1607 | 1605 | 1604 | 1603 | Phe aromatic vibration | |
| 1615 | 1617 | 1615 | 1616 | 1615 | 1616 | Tyr aromatic vibration |
| 1659 | 1662 | 1662 | 1657 | 1656 | α-helix, amide I | |
| 1676 | 1676 | 1676 | 1673 | 1676 | 1673 | β-sheet, amide I |
The Raman signature of insulin oligomer is shown in Figure 2C. It exhibited a broad amide I Raman band at 1661 cm–1. A shoulder band appeared at 1676 cm–1 and quite resembled the band appearing for the monomeric state of the protein. It indicated the presence of structural heterogeneity in the oligomeric state similar to that of the monomer. We used a drop coating deposition Raman (DCDR) method to collect the Raman spectrum of monomer and aggregated species.40−42 In this process, 20–30 μL of the incubated sample was applied on a glass coverslip and air-dried. The methods helped to collect better quality Raman spectra of protein samples when solution-state experiments were difficult for several reasons. However, in the event of drying and evaporation, some conversion of monomer to oligomer formation may occur.42 Longer incubation of the protein solution produced pre-protofibril structures. The amide I band appeared at ∼1662 cm–1 (Figure 2E and Table 2). The Raman signature of the protein was noticeably changed upon the transformation of pre-protofibril state to protofibril and subsequent fibril formation. For insulin fibrils, amide I vibration appeared at 1674 cm–1 and the BWHM was ∼21 cm–1 (Figure 3E,F). It indicated the formation of a major cross-β-sheet conformation of the protein in the fibrillar state. The narrow bandwidth indicated structural homogeneity and uniformity.34 A significant reduction in band intensity was observed for the α-helical signature amide I band at 1657 cm–1.
Figure 3.

Left panel shows the Raman spectra of insulin protofibril (A), premature fibril (C), and fibril (E) in the frequency range of 500–1800 cm–1. Respective panels (B, D and F) in the right present the curve-fitting analysis of extended amide I band of the Raman spectra. Other experimental and analysis conditions were same as that of Figure 2.
The Raman band patterns and curve-fitting analysis to amide I band often provide ample information related to protein backbone conformation and its stability.18,36,37,43−46 Dong et al. and others performed initial Raman investigation on dimeric and hexameric insulin aggregates, measuring amide I band behavior in different sample conditions.34,47 Our laboratory also derived the structural content of amyloid aggregates of Aβ peptides using the amide I curve-fitting protocol, as suggested by Dong et al. and Maiti et al.34,37 In our current investigation, curve fitting to the Raman amide I showed that ∼54% of residues in insulin monomer preferred α-helical secondary structure and the component band was at 1658 cm–1 (Figure 2B and Table 1).18 A considerable amount (∼24%) of residues remained in the extended/coiled structure, as indicated with a component band at 1686 cm–1.18 β-Sheet content was ∼9% and assigned to a component band at 1676 cm–1. Some contribution of the β-sheet secondary structure could be the result of drying, as the DCDR method was used to collect the Raman spectrum.42,48
Figure 2D depicts the spectral deconvolution (curve fitting) of amide I Raman band of oligomers. In the oligomeric state, the protein maintained its helical imprint (50%) and found no substantial increment of the core-β-sheet secondary structure. The deconvolution of the amide I band in the Raman spectra of the pre-protofibrillar structure is shown in Figure 2F. We noticed some decrease in helical signature in the pre-protofiber state compared to that of the oligomer species. α-Helix signature of the amide III band at 1262 cm–1 was somewhat decreased (Figure 2E and Table 2).36,38,39
Formation of insulin protofiber was identified with the appearance of early β-sheet content as a signature by amide I band at 1673 cm–1 (Figure 3A,B and Table 2). In the protofibril species, the helical content decreased to 37% (the component band at 1656 cm–1), whereas the β-sheet content increased to 29% (the component band at 1673 cm–1, Table 1). The increment of β-sheet was due to the transformation of the helix and extended helix into β-conformational space (Table 1).18Figure 3C,D shows the Raman signature of early fibrils (premature, incubation time 160 min). A considerable amount of loss in amide I helical marker band at 1657 cm–1 in comparison to the β-component band at 1675 cm–1 and thus a significant increase in β-sheet conformation was observed. A substantial shrinkage in the bandwidth was also observed (44 cm–1 for protofiber, 35 cm–1 for premature fiber). A significant gain in the β-sheet component was further established by the broad amide III β-sheet marker band at 1222 cm–1 (Figure 3C and Table 2).36,37,49
We observed a very narrow and sharp vibrational amide I band at 1674 cm–1 (BWHM, 21 cm–1) in the fiber state of insulin, and a significant decrease in α-helical signature amide I band intensity at 1657 cm–1 was observed. It indicated a major cross-β-sheet conformation of the protein in the fibrillar state (Figure 3C,E). In the fiber condition, the helical content was decreased to ∼14% (component band at 1655 cm–1), whereas the β-strand content was increased to ∼62% (component band at 1674 cm–1) and PPII/extended conformation was also decreased to ∼14% with a component band at 1690 cm–1 (Table 1). Amide III also showed a major band at 1222 cm–1 for β-sheet signature and a disappearance of the α-helical band at 1260 cm–1. The Raman bands at 1254 and 1274 cm–1 were the marker for either molted helix or type III β-turn, respectively (Figure 3E and Table 2).38,39Figure 4A graphically summarizes the overall changes in the secondary structural components with time (formation of different intermediate species), as obtained from Raman spectroscopic analysis.
Figure 4.

Changes of different secondary structural contents of incubated insulin over time, as obtained from the curve-fitting analysis of Raman amide I band (Table 1). Panel (A) shows the changes in the secondary structural component (%) against incubation time: helix (orange), β-sheet (brown), loose β-strand, PPII and disordered (light green), and other undefined component (deep green). Panel (B) shows the changes in the Raman intensity of the special helical marker band at 940 cm–1 at different time points of incubation. The Raman intensity at 1449 cm–1 was used as a reference.
In the process of insulin amyloid formation, a significant decrease of helical conformation was prominent, whereas extended conformations (PPII, extended helix) were continued until the formation of mature amyloid fiber (Table 1 and Figure 4A). The special α-helical marker band that refers to the stretching vibration of O=C–Cα–H linkage of the protein backbone near 1340–1345 cm–1 was also clearly observed until the formation of fibrils (Figures 2, 3, and Table 2).38,50 The band intensity of the nonamide helix signature band at 940 cm–1 also decreased substantially after the lag phase completion, as compared to that of the signal at 1434 cm–1 region enlisted for the total CH2 and CH3 deformation and CH2 scissoring of the protein itself (Figures 4B, 2, and 3).36,49 This again suggested that no major secondary structural change happened before the beginning of pre-protofibril formation. Some monomers may transform into an oligomeric state in the DCDR method, and a strict comparison of structural components in monomer and oligomeric samples was not possible. However, our Raman analysis suggested that no major secondary structural changes occurred until the formation of protofibril species. The tertiary realignment could be the major force behind oligomer formation. The 1-anilino-8-naphthalene-sulfonate (ANS) binding study, as discussed later, suggested that a collapse in the tertiary structure and opening of hydrophobic surfaces occurred prior to the formation of protofibril species.
Thioflavin T (ThT) fluorescence assay is often performed to detect the formation of amyloid aggregates enriched with cross-β-sheet structure.51,52 ThT possibly bound to the cradle of cross-β-sheets of amyloid aggregates and attained a suitable conformation that increased its fluorescence yield. Therefore, ThT acts as a chemical probe and reports the formation of stable β-sheet structures in the process of aggregation of many proteins and peptides.53 To determine the content of the compact cross-β-sheet secondary structure in the different states of fibril formation, ThT fluorescence was measured at the different time points of incubation of the protein solution. Figure 5A displays the ThT fluorescence intensity at 485 nm in the presence of different insulin samples incubated for making the aggregates. The oligomers that were formed after 60 min of incubation did not cause major fluorescence enhancement of the ThT solution, suggesting no enhancement of cross-β-sheet structure in the oligomers. The fluorescence enhancement was started in the presence of protein samples incubated for ∼120 min. AFM confirmed that the species formed at this time point of incubation was pre-protofibril. The ThT fluorescence started increasing very rapidly in the presence of pre-protofibrils, and it was the maximum in the presence of fibril solution. Thus, the cross-β-sheet structural transition mainly happened in the late state of the aggregation process.
Figure 5.
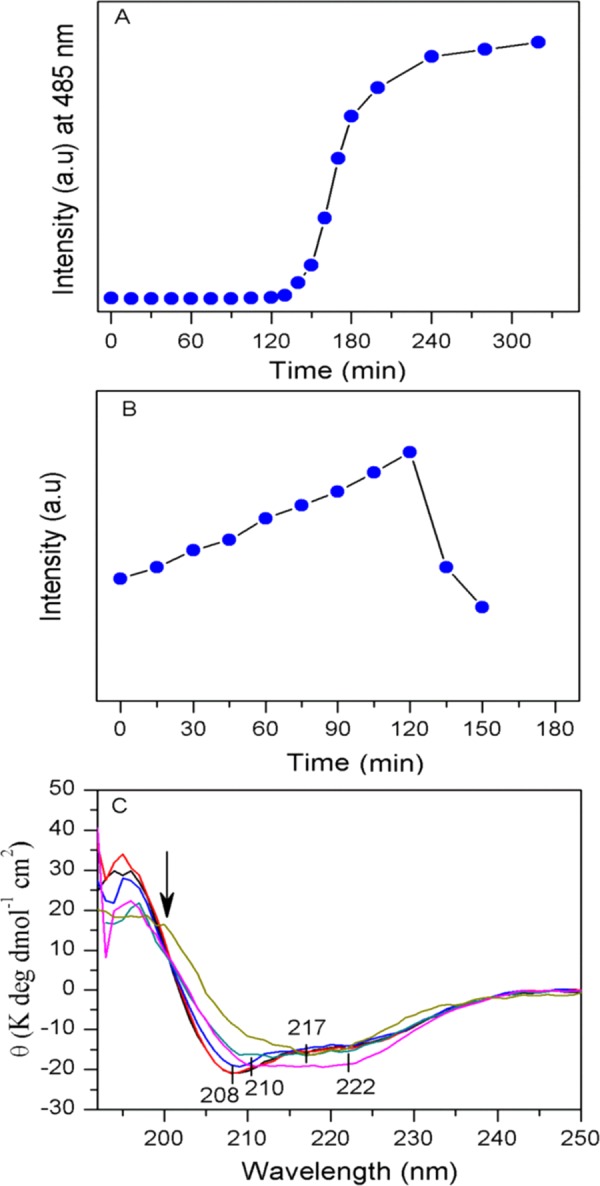
Kinetics of insulin amyloid formation, as monitored by ThT fluorescence assay, ANS binding study, and circular dichroism (CD) spectroscopic measurements of insulin solution incubated for aggregation. (A) ThT fluorescence assay. Each point indicates the ThT fluorescence intensity in the presence of a measured amount of incubated protein solution taken at the different time points of incubation. (B) Fluorescence of 1-anilino-8-naphthalene-sulfonate (ANS) in the presence of a fixed amount of the incubated sample taken at several time points of incubation and added to ANS solution. (C) Far-UV CD spectra of incubated insulin solution at different times: 0 min (black), 60 min (red), 120 min (blue), 135 min (cyan), 160 min (pink), and 180 min (yellow). Details are given in the Materials and Methods section, and the species type may be inferred from Figure 1.
To further investigate the secondary structural transition of insulin in the fibril formation pathway, circular dichroism (CD) spectroscopic analysis was performed. Figure 5C shows the CD spectrum of insulin species at the different time points of incubation. The monomer at 0 min of incubation showed a CD pattern similar to that of native insulin in solution. In the oligomeric state, we observed no significant change in the CD signature. The negative CD signal at 208 nm and at 192 nm started to change in the pre-protofibril condition and indicated that the helical signature started to decrease from the pre-protofibril state. The CD spectra became quite broad (208–222 nm) in the protofibril condition and suggested the coexistence of several protein backbone conformations. The CD spectral signature of insulin fiber showed a strong negative band at 218 nm and defined the presence of the compact β-sheet structure. The deconvolution of very high quality CD spectra often yields secondary structure components.5 However, for the aggregated samples, noise from scattering often disturbs spectrum quality and encounters unexpected errors. Therefore, the deconvolution of our CD spectra was avoided; it simply indicated that a major secondary structural change occurred after oligomer formation.
1-Anilino-8-naphthalene-sulfonate (ANS) binds to the exposed hydrophobic patches/surfaces of proteins when the surface opens up. The bound ANS produces enhanced fluorescence, and thus it enables to monitor the relative hydrophobic surface exposure during unfolding and refolding events, such as protein aggregation. To realize the tertiary collapse and exposure of the hydrophobic surfaces of structured insulin, we also performed ANS binding to the protein as monomer (0 min of incubation) and when it formed different aggregates in the process of fibril formation. A gradual increase in the ANS fluorescence in the presence of incubated insulin solution was observed (Figure 5B). However, the fluorescence dropped quickly in the presence of insulin samples incubated for more than 120 min and for the protein sample in the exponential growth phase of fibril formation. It appeared that the hydrophobic compact core of the protein slowly loosened and opened up, as time progressed, from its compact globular fold in the initial phase of aggregation. This indicated a tertiary collapse of the globular structure, without much changing its secondary conformation (as confirmed by CD, Raman, and ThT fluorescence results). The exposure of hydrophobic surfaces allows ANS molecule to access the hydrophobic surfaces. Interestingly, ANS fluorescence dropped suddenly for the samples incubated for more than 120 min when actual protofibril formation started. The protein molecules in amyloid fibrils are highly organized and attained a compact cross-β sheet structure, as indicated in Raman and CD spectra. This again caused reduction of exposed hydrophobic surfaces as the molecules strongly associated to form fiber with a β-sheet structure. In the intermediate states (lag phase), the protein domain rearrangements and gradual melting/unwinding of the tertiary structure may occur and this may cause more and more exposure of hydrophobic surfaces. ANS could bind these exposed surfaces and enhance its fluorescence yield. These exposed hydrophobic surfaces eventually, however, associate strongly and get organized in the steric zipper mode54,55 to form cross-β sheet rich amyloid-like insulin fiber.
Conclusions
Protein misfolding and aggregation processes are extensively studied leading edges in biochemistry as well as in molecular medicine for more than half a century. As such, protein aggregation and formation of the oligomeric state and fibrillar structure are linked to several pathologies, such as systemic amyloidosis, type II diabetes, and neurodegenerative disorders. However, in the trajectory of protein aggregation and fibril formation, the transition of protein monomer to oligomeric geometry is one of the most crucial events, from both the pharmacological and biochemical viewpoint. Our investigation on insulin fibrillization suggested that the tertiary structural collapse was a dominating event in the lag phase and subsequent oligomerization whereas minimal alteration occurred in its core secondary structure. Eventually, the exposed hydrophobic domains rapidly transformed into very organized cross-β-sheet rich fibrillar structure and in this late event of fibrillization, a remarkable secondary and tertiary realignment occurred in the polypeptide backbone.
Materials and Methods
Materials
Zinc-free Insulin was purchased from Sigma Chemical Company (91077C). MilliQ water (Millipore Ltd., Bedford, MA) was used in all experiments, and the pH of the solution was controlled by using HCl. Thioflavin T and 1-anilinonapthalene-8-sulfonic acid were obtained from Sigma. Stock solutions of thioflavin T (ThT) were prepared in MilliQ water, and concentration was checked using a molar extinction coefficient of 36 000 M–1 cm–1 at 412 nm. ThT solution was stored in dark at 4 °C.
Preparation of Insulin Aggregates
Insulin solution for the aggregation study was prepared by dissolving zinc-free insulin in 20 mM HCl (pH 1.8) with 50 mM NaCl. The protein concentration was ∼1.5 mg mL–1, and the final concentration was checked by absorption measurement at 276 nm.4 Each time, the freshly prepared protein solution was incubated for several hours at 60 °C without agitation and the formation of aggregate species due to incubation was followed by ThT assay and AFM using aliquots withdrawn at different time points of incubation. The integrity of the protein solution was checked by gel electrophoresis of the incubated samples. The gel image (Figure S2) indicated that some degradation and chemical modification might have occurred for longer incubation. The previous investigation also observed some changes in the chemical modification of insulin upon incubation for a longer time.18,56
AFM Imaging
AFM images were recorded in Pico plus 5500 AFM (Agilent Technologies, Tempe, AZ) with a piezo scanner over the range of 9 μm2. After incubation of the protein solution, aliquots were diluted 10–50 times and 10 μL of the diluted aliquots was drop casted onto a freshly cleaved mica surface. The sample was dried gently using air flow. Microfabricated silicon cantilevers of 225 μm length, with a nominal spring force constant of 21–98 N m–1 were used for imaging. The cantilever oscillation frequency was turned into the resonance frequency of 150–300 kHz. Images were processed by flattening using Pico view software (Molecular Imaging Corporation).
Raman Spectroscopy
A unique drop coating deposition Raman (DCDR) method was used to collect the Raman spectrum of monomer and aggregated species. This method was used by others in the field.40−42 Twenty to thirty microliters of the incubated insulin solution at different time points of incubation was placed on a glass coverslip and air-dried. Raman data were collected in the backscattering geometry with an STR Raman microscope (AIRIX Corp, Japan) under ambient condition (24 °C). A 633 nm He–Ne laser (model LGK 7665 P18, LASOS, Germany) with ∼1 mW of laser power at the sample was used for excitation. The laser was focused onto the sample through the microscope (Olympus BX51M, Japan) using 50× objective, and Raman scattering was collected using a 500 mm focal length triple grating monochromator equipped with an air-cooled charge-coupled device detector. The spectra were recorded with a typical accumulation time of 100 s. The wave numbers of the Raman band were calibrated with the silica wafer focused under the 50× objective, and the spectral resolution was ∼1 cm–1. The spectral data so obtained were processed with GRAMS/A1 software.
Curve Fitting of the Amide I Profile
Amide I band of Raman spectra is a characteristic band to investigate the changes in the secondary structure of protein.45,57,58 The band fitting of the Raman amide I band was performed by using the Levenberg–Marquardt nonlinear least-squares process, as applied in the Curve Fit Ab routine of GRAMS/AI 9.02 software.59 The band region of 1575–1720 cm–1 was fitted assuming four asymmetrical component bands that represented different structural conformations of insulin: α-helical band at 1655 cm–1 with spectral window (1650–1660 cm–1), β-sheet component band near 1673 cm–1 (1670–1675 cm–1), polyproline II or loose β strands and disordered structure near 1685 cm–1 (1680–1690 cm–1), and disordered structure or vibronic coupling band near 1637 cm–1 (1630–1645 cm–1).47 During amide I band fitting, three different bands at ∼1585, ∼1604, and ∼1615 cm–1 for the ring vibrational mode of phenylalanine and tyrosine were also included.37,43 Component bands were fitted by 15–40 cm–1 bandwidth features; Gaussian and Lorentzian functions were allowed for homogenous and heterogeneous broadening. The standard error for peak positions and peak widths were <5 cm–1 for well characteristic components; this typically introduced an uncertainty of ∼10% in the measurement of area under each fitted curve.
Thioflavin T (ThT) Fluorescence Spectroscopy Assay
ThT fluorescence assays were performed to investigate insulin amyloid fibrillation. At room temperature, 10 μL of the incubated insulin sample at the different time points of incubation was individually added to 700 μL of ThT solution (25 μM) and mixed properly. The solution was excited at 440 nm and emission was recorded at 485 nm on a VARIAN CARY Eclipse fluorescence spectrometer using a 1 cm path length quartz cuvette. The integration time and slit widths were fixed at 1 s and 5 nm, respectively. ThT in buffer without protein was used as a baseline.
Circular Dichroism (CD) Spectroscopy
A JASCO J-815 spectropolarimeter (Jasco) equipped with a Peltier temperature control unit which was set to 25 °C with an accuracy of ±0.1 °C was used to measure the protein’s secondary structure at different times of incubation. The data acquisition interval time was 2 s. Ten microliters of the incubated protein solution each time was mixed with 300 μL of 20 mM HCl with 50 mM NaCl, and the final concentration of the protein became 10 μM. Spectra were recorded using a 0.1 mm path length cell, with a 50 nm min–1 scan speed. Three scans were averaged, and the buffer background was subtracted.
ANS Binding Assay
ANS is a specific fluorescence probe that binds to the hydrophobic area of a protein and shows a blue shift in its emission maximum along with a substantial increase in the fluorescence intensity. CARY Eclipse, VARIAN spectrofluorometer was used to measure the ANS fluorescence in the presence of insulin samples at different time points of incubation. The final concentration of insulin and ANS were 5 and 10 μM, respectively. The sample was excited at 370 nm. The emission range was fixed to 400–600 nm, and the integration time and slit widths were 1 s and 5 nm, respectively.
Acknowledgments
S.D. acknowledges CSIR-network project BSC0113, BSC0115, and BSC0121 for funding support. A.R. acknowledges UGC for the research fellowship. The authors thank T. Murganandan and J. Mandal for recording AFM and CD measurement and also acknowledge other central instrumental facilities. The Raman instrument was bought under DBT, New Delhi grant (GAP-299) to Dr. N.C.M. We are also greatly thankful to Animesh Mandal for reading the manuscript and helping in some of the experiments. We also thank Dr. Sandhya R. Dungdung and Prasanta Ghosh for help in some of the experiments.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b01776.
Figure S1, Raman spectrum of human insulin in its solid state; Figure S2, sodium dodecyl sulfate polyacrylamide gel electrophoresis of insulin solutions taken at different time points of incubation; and Table S1, Raman vibrational bands of insulin monomer as air-dried, solid powder, and in the aqueous solution state (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Michael J.; Carroll R.; Swift H. H.; Steiner D. F. Studies on the Molecular Organization of Rat Insulin Secretory Granules. J. Biol. Chem. 1987, 262, 16531–16535. [PubMed] [Google Scholar]
- Blundell T. L.; Cutfield J. F.; Cutfield S. M.; Dodson E. J.; Dodson G. G.; Hodgkin D. C.; Mercola D. A. Three-Dimensional Atomic Structure of Insulin and Its Relationship to Activity. Diabetes 1972, 21, 492–505. [DOI] [PubMed] [Google Scholar]
- Brange J.; Andersen L.; Laursen E. D.; Meyn G.; Rasmussen E. Toward Understanding Insulin Fibrillation. J. Pharm. Sci. 1997, 86, 517–525. 10.1021/js960297s. [DOI] [PubMed] [Google Scholar]
- Nielsen L.; Khurana R.; Coats A.; Frokjaer S.; Brange J.; Vyas S.; Uversky V. N.; Fink A. L. Effect of Environmental Factors on the Kinetics of Insulin Fibril Formation: Elucidation of the Molecular Mechanism. Biochemistry 2001, 40, 6036–6046. 10.1021/bi002555c. [DOI] [PubMed] [Google Scholar]
- Hua Q.-x.; Weiss M. A. Mechanism of insulin fibrillation the structure of insulin under amyloidogenic conditions resembles a protein-folding intermediate. J. Biol. Chem. 2004, 279, 21449–21460. 10.1074/jbc.M314141200. [DOI] [PubMed] [Google Scholar]
- Colletier J.-P.; Laganowsky A.; Landau M.; Zhao M.; Soriaga A. B.; Goldschmidt L.; Flot D.; Cascio D.; Sawaya M. R.; Eisenberg D. Molecular Basis for Amyloid-β Polymorphism. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 16938–16943. 10.1073/pnas.1112600108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong D.-P.; Fink A. L. Independent Heterologous Fibrillation of Insulin and Its B-Chain Peptide. Biochemistry 2005, 44, 16701–16709. 10.1021/bi051658y. [DOI] [PubMed] [Google Scholar]
- Dodson G.; Steiner D. The Role of Assembly in Insulin’s Biosynthesis. Curr. Opin. Struct. Biol. 1998, 8, 189–194. 10.1016/S0959-440X(98)80037-7. [DOI] [PubMed] [Google Scholar]
- Mayer J. P.; Zhang F.; DiMarchi R. D. Insulin Structure and Function. Biopolymers 2007, 88, 687–713. 10.1002/bip.20734. [DOI] [PubMed] [Google Scholar]
- Langmuir I.; Waugh D. F. Pressure-Soluble and Pressure-Displaceable Components of Monolayers of Native and Denatured Proteins. J. Am. Chem. Soc. 1940, 62, 2771–2793. 10.1021/ja01867a046. [DOI] [Google Scholar]
- Waugh D. F. A Fibrous Modification of Insulin. I. The Heat Precipitate of Insulin. J. Am. Chem. Soc. 1946, 68, 247–250. 10.1021/ja01206a030. [DOI] [Google Scholar]
- Bouchard M.; Zurdo J.; Nettleton E. J.; Dobson C. M.; Robinson C. V. Formation of Insulin Amyloid Fibrils Followed by FTIR Simultaneously with CD and Electron Microscopy. Protein Sci. 2000, 9, 1960–1967. 10.1110/ps.9.10.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Librizzi F.; Carrotta R.; Spigolon D.; Bulone D.; San Biagio P. L. α-Casein Inhibits Insulin Amyloid Formation by Preventing the Onset of Secondary Nucleation Processes. J. Phys. Chem. Lett. 2014, 5, 3043–3048. 10.1021/jz501570m. [DOI] [PubMed] [Google Scholar]
- Nielsen L.; Frokjaer S.; Brange J.; Uversky V. N.; Fink A. L. Probing the Mechanism of Insulin Fibril Formation with Insulin Mutants. Biochemistry 2001, 40, 8397–8409. 10.1021/bi0105983. [DOI] [PubMed] [Google Scholar]
- Foderà V.; Cataldo S.; Librizzi F.; Pignataro B.; Spiccia P.; Leone M. Self-Organization Pathways and Spatial Heterogeneity in Insulin Amyloid Fibril Formation. J. Phys. Chem. B 2009, 113, 10830–10837. 10.1021/jp810972y. [DOI] [PubMed] [Google Scholar]
- Foderà V.; Librizzi F.; Groenning M.; van de Weert M.; Leone M. Secondary Nucleation and Accessible Surface in Insulin Amyloid Fibril Formation. J. Phys. Chem. B 2008, 112, 3853–3858. 10.1021/jp710131u. [DOI] [PubMed] [Google Scholar]
- Ahmad A.; Uversky V. N.; Hong D.; Fink A. L. Early Events in the Fibrillation of Monomeric Insulin. J. Biol. Chem. 2005, 280, 42669–42675. 10.1074/jbc.M504298200. [DOI] [PubMed] [Google Scholar]
- Huang K.; Maiti N. C.; Phillips N. B.; Carey P. R.; Weiss M. A. Structure-Specific Effects of Protein Topology on Cross-Beta Assembly: Studies of Insulin Fibrillation. Biochemistry 2006, 45, 10278–10293. 10.1021/bi060879g. [DOI] [PubMed] [Google Scholar]
- Jansen R.; Dzwolak W.; Winter R. Amyloidogenic Self-Assembly of Insulin Aggregates Probed by High Resolution Atomic Force Microscopy. Biophys. J. 2005, 88, 1344–1353. 10.1529/biophysj.104.048843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad A.; Millett I. S.; Doniach S.; Uversky V. N.; Fink A. L. Partially Folded Intermediates in Insulin Fibrillation. Biochemistry 2003, 42, 11404–11416. 10.1021/bi034868o. [DOI] [PubMed] [Google Scholar]
- Vestergaard B.; Groenning M.; Roessle M.; Kastrup J. S.; van de Weert M.; Flink J. M.; Frokjaer S.; Gajhede M.; Svergun D. I. A Helical Structural Nucleus Is the Primary Elongating Unit of Insulin Amyloid Fibrils. PLoS Biol. 2007, 5, e134 10.1371/journal.pbio.0050134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manno M.; Craparo E. F.; Podestà A.; Bulone D.; Carrotta R.; Martorana V.; Tiana G.; San Biagio P. L. Kinetics of Different Processes in Human Insulin Amyloid Formation. J. Mol. Biol. 2007, 366, 258–274. 10.1016/j.jmb.2006.11.008. [DOI] [PubMed] [Google Scholar]
- Chatani E.; Inoue R.; Imamura H.; Sugiyama M.; Kato M.; Yamamoto M.; Nishida K.; Kanaya T. Early Aggregation Preceding the Nucleation of Insulin Amyloid Fibrils as Monitored by Small Angle X-Ray Scattering. Sci. Rep. 2015, 5, 15485 10.1038/srep15485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzwolak W.; Loksztejn A.; Galinska-Rakoczy A.; Adachi R.; Goto Y.; Rupnicki L. Conformational Indeterminism in Protein Misfolding: Chiral Amplification on Amyloidogenic Pathway of Insulin. J. Am. Chem. Soc. 2007, 129, 7517–7522. 10.1021/ja066703j. [DOI] [PubMed] [Google Scholar]
- Loksztejn A.; Dzwolak W. Vortex-Induced Formation of Insulin Amyloid Superstructures Probed by Time-Lapse Atomic Force Microscopy and Circular Dichroism Spectroscopy. J. Mol. Biol. 2010, 395, 643–655. 10.1016/j.jmb.2009.10.065. [DOI] [PubMed] [Google Scholar]
- Arosio P.; Knowles T. P. J.; Linse S. On the Lag Phase in Amyloid Fibril Formation. Phys. Chem. Chem. Phys. 2015, 17, 7606–7618. 10.1039/C4CP05563B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tycko R. Progress towards a Molecular-Level Structural Understanding of Amyloid Fibrils. Curr. Opin. Struct. Biol. 2004, 14, 96–103. 10.1016/j.sbi.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Pease L. F. III; Sorci M.; Guha S.; Tsai D.-H.; Zachariah M. R.; Tarlov M. J.; Belfort G. Probing the Nucleus Model for Oligomer Formation during Insulin Amyloid Fibrillogenesis. Biophys. J. 2010, 99, 3979–3985. 10.1016/j.bpj.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campioni S.; Mannini B.; Zampagni M.; Pensalfini A.; Parrini C.; Evangelisti E.; Relini A.; Stefani M.; Dobson C. M.; Cecchi C.; et al. A Causative Link between the Structure of Aberrant Protein Oligomers and Their Toxicity. Nat. Chem. Biol. 2010, 6, 140–147. 10.1038/nchembio.283. [DOI] [PubMed] [Google Scholar]
- Sipe J. D.; Cohen A. S. Review: History of the Amyloid Fibril. J. Struct. Biol. 2000, 130, 88–98. 10.1006/jsbi.2000.4221. [DOI] [PubMed] [Google Scholar]
- Maji S. K.; Wang L.; Greenwald J.; Riek R. Structure-Activity Relationship of Amyloid Fibrils. FEBS Lett. 2009, 583, 2610–2617. 10.1016/j.febslet.2009.07.003. [DOI] [PubMed] [Google Scholar]
- Eisenberg D.; Jucker M. The Amyloid State of Proteins in Human Diseases. Cell 2012, 148, 1188–1203. 10.1016/j.cell.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oladepo S. A.; Xiong K.; Hong Z.; Asher S. A.; Handen J.; Lednev I. K. UV Resonance Raman Investigations of Peptide and Protein Structure and Dynamics. Chem. Rev. 2012, 112, 2604–2628. 10.1021/cr200198a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J.; Wan Z.; Popov M.; Carey P. R.; Weiss M. A. Insulin Assembly Damps Conformational Fluctuations: Raman Analysis of Amide I Linewidths in Native States and Fibrils. J. Mol. Biol. 2003, 330, 431–442. 10.1016/S0022-2836(03)00536-9. [DOI] [PubMed] [Google Scholar]
- Asher S. A. Biochemical Applications of Raman and Resonance Raman Spectroscopies. P. R. Carey, Bernard Horecker Raman Spectroscopy in Biology: Principles and Applications. Anthony T. Tu. Q. Rev. Biol. 1984, 59, 314–315. 10.1086/413923. [DOI] [Google Scholar]
- Roy A.; Chandra K.; Dolui S.; Maiti N. C. Envisaging the Structural Elevation in the Early Event of Oligomerization of Disordered Amyloid β Peptide. ACS Omega 2017, 2, 4316–4327. 10.1021/acsomega.7b00522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiti N. C.; Apetri M. M.; Zagorski M. G.; Carey P. R.; Anderson V. E. Raman Spectroscopic Characterization of Secondary Structure in Natively Unfolded Proteins: Alpha-Synuclein. J. Am. Chem. Soc. 2004, 126, 2399–2408. 10.1021/ja0356176. [DOI] [PubMed] [Google Scholar]
- Mikhonin A. V.; Ahmed Z.; Ianoul A.; Asher S. A. Assignments and Conformational Dependencies of the Amide III Peptide Backbone UV Resonance Raman Bands. J. Phys. Chem. B 2004, 108, 19020–19028. 10.1021/jp045959d. [DOI] [Google Scholar]
- Mikhonin A. V.; Myshakina N. S.; Bykov S. V.; Asher S. A. UV Resonance Raman Determination of Polyproline II, Extended 2.51-Helix, and β-Sheet Ψ Angle Energy Landscape in Poly-L-Lysine and Poly-L-Glutamic Acid. J. Am. Chem. Soc. 2005, 127, 7712–7720. 10.1021/ja044636s. [DOI] [PubMed] [Google Scholar]
- Sereda V.; Sawaya M. R.; Lednev I. K. Structural Organization of Insulin Fibrils Based on Polarized Raman Spectroscopy: Evaluation of Existing Models. J. Am. Chem. Soc. 2015, 137, 11312–11320. 10.1021/jacs.5b07535. [DOI] [PubMed] [Google Scholar]
- Filik J.; Stone N. Drop Coating Deposition Raman Spectroscopy of Protein Mixtures. Analyst 2007, 132, 544–550. 10.1039/b701541k. [DOI] [PubMed] [Google Scholar]
- Ortiz C.; Zhang D.; Xie Y.; Ribbe A. E.; Ben-Amotz D. Validation of the Drop Coating Deposition Raman Method for Protein Analysis. Anal. Biochem. 2006, 353, 157–166. 10.1016/j.ab.2006.03.025. [DOI] [PubMed] [Google Scholar]
- Apetri M. M.; Maiti N. C.; Zagorski M. G.; Carey P. R.; Anderson V. E. Secondary Structure of α-Synuclein Oligomers: Characterization by Raman and Atomic Force Microscopy. J. Mol. Biol. 2006, 355, 63–71. 10.1016/j.jmb.2005.10.071. [DOI] [PubMed] [Google Scholar]
- Xiong K.; Asher S. A. Circular Dichroism and UV Resonance Raman Study of the Impact of Alcohols on the Gibbs Free Energy Landscape of an α-Helical Peptide. Biochemistry 2010, 49, 3336–3342. 10.1021/bi100176a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuma R. Raman Spectroscopy of Proteins: From Peptides to Large Assemblies. J. Raman Spectrosc. 2005, 36, 307–319. 10.1002/jrs.1323. [DOI] [Google Scholar]
- Tsuboi M.; Suzuki M.; Overman S. A.; Thomas G. J. Intensity of the Polarized Raman Band at 1340–1345 Cm–1 as an Indicator of Protein Alpha-Helix Orientation: Application to Pf1 Filamentous Virus. Biochemistry 2000, 39, 2677–2684. 10.1021/bi9918846. [DOI] [PubMed] [Google Scholar]
- Dong J.; Wan Z. L.; Chu Y. C.; Nakagawa S. N.; Katsoyannis P. G.; Weiss M. A.; Carey P. R. Isotope-Edited Raman Spectroscopy of Proteins: A General Strategy to Probe Individual Peptide Bonds with Application to Insulin. J. Am. Chem. Soc. 2001, 123, 7919–7920. 10.1021/ja011101f. [DOI] [PubMed] [Google Scholar]
- Zhang D.; Xie Y.; Mrozek M. F.; Ortiz C.; Davisson V. J.; Ben-Amotz D. Raman Detection of Proteomic Analytes. Anal. Chem. 2003, 75, 5703–5709. 10.1021/ac0345087. [DOI] [PubMed] [Google Scholar]
- Zheng R.; Zheng X.; Dong J.; Carey P. R. Proteins Can Convert to β-Sheet in Single Crystals. Protein Sci. 2004, 13, 1288–1294. 10.1110/ps.03550404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overman S. A.; Thomas G. J. Amide Modes of the α-Helix: Raman Spectroscopy of Filamentous Virus Fd Containing Peptide 13C and 2H Labels in Coat Protein Subunits. Biochemistry 1998, 37, 5654–5665. 10.1021/bi972339c. [DOI] [PubMed] [Google Scholar]
- LeVine H. Thioflavine T Interaction with Synthetic Alzheimer’s Disease Beta-Amyloid Peptides: Detection of Amyloid Aggregation in Solution. Protein Sci. 1993, 2, 404–410. 10.1002/pro.5560020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naiki H.; Higuchi K.; Hosokawa M.; Takeda T. Fluorometric Determination of Amyloid Fibrils in Vitro Using the Fluorescent Dye, Thioflavin T1. Anal. Biochem. 1989, 177, 244–249. 10.1016/0003-2697(89)90046-8. [DOI] [PubMed] [Google Scholar]
- Biancalana M.; Koide S. Molecular Mechanism of Thioflavin-T Binding to Amyloid Fibrils. Biochim. Biophys. Acta 2010, 1804, 1405–1412. 10.1016/j.bbapap.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaya M. R.; Sambashivan S.; Nelson R.; Ivanova M. I.; Sievers S. A.; Apostol M. I.; Thompson M. J.; Balbirnie M.; Wiltzius J. J. W.; McFarlane H. T.; et al. Atomic Structures of Amyloid Cross-β Spines Reveal Varied Steric Zippers. Nature 2007, 447, 453–457. 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- Chandra B.; Korn A.; Maity B. K.; Adler J.; Rawat A.; Krueger M.; Huster D.; Maiti S. Stereoisomers Probe Steric Zippers in Amyloid-β. J. Phys. Chem. B 2017, 121, 1835–1842. 10.1021/acs.jpcb.6b12332. [DOI] [PubMed] [Google Scholar]
- Nilsson M. R.; Dobson C. M. Chemical Modification of Insulin in Amyloid Fibrils. Protein Sci. 2003, 12, 2637–2641. 10.1110/ps.0360403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M. C.; Lord R. C. Laser-Excited Raman Spectroscopy of Biomolecules. VI. Polypeptides as Conformational Models. J. Am. Chem. Soc. 1974, 96, 4750–4752. 10.1021/ja00822a004. [DOI] [PubMed] [Google Scholar]
- Sane S. U.; Cramer S. M.; Przybycien T. M. A Holistic Approach to Protein Secondary Structure Characterization Using Amide I Band Raman Spectroscopy. Anal. Biochem. 1999, 269, 255–272. 10.1006/abio.1999.4034. [DOI] [PubMed] [Google Scholar]
- Marquardt D. An Algorithm for Least-Squares Estimation of Nonlinear Parameters. J. Soc. Ind. Appl. Math. 1963, 11, 431–441. 10.1137/0111030. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.