

Abstract
Protein structure can be flexible to adopt multiple conformations to house small molecules. In this paper, we have attempted to experimentally figure out how the structure of a transport protein steer the drug–drug competition (DDC) by maintaining the equilibrium distribution of the bound and unbound fractions of drugs. This understanding is an important facet in biophysical and medicinal chemistry to ascertain the effectiveness of drugs. It is important to note that majority of studies involving small-molecule–transport protein interaction aimed at characterizing the binding process, and because these proteins can interact with thousands of molecules, there are hundreds of reports on such interactions. This ultimately led to an impression among the readers that any studies involving serum albumin may not lead to any new findings except for traditional binding explorations. However, in the present paper, we would like to draw the attention of the readers that our findings are very surprising, new, and important, involving the phenomenon of ligand–protein interaction. Here, we have studied two structurally similar drugs methotrexate (MTX) and folic acid (FA), which attempt to bind the primary binding site (subdomain IIA), one at a time, of human serum albumin. Details of binding analyses reveal that when both of the drugs are present, the single-site binding mode of FA prefers to occupy the primary binding site and hence pushes the primary-site-bound MTX to another location (subdomain IIIA), which is the second binding site of MTX. The structural analysis indicates that DDC has occurred in a cooperative fashion so that the loss of the protein secondary structure is minimum when both drugs are bound to the protein, which means that in the case of duo-drug binding, the conventional interaction between the drug and the protein is altered to undergo restoration of the protein structure. This can indeed regulate the DDC by modifying the bound and unbound fractions of MTX in plasma. The present study emphasizes that in vitro structural characterizations of the transport protein provide important information to improve the molecular-level understanding of DDC for further therapeutic applications with combination drug.
Introduction
The characterization of structural flexibility of a protein is one of the very important aspects to ascertain the biological effects, binding site location, binding orientation, and binding kinetics of drugs.1 Protein flexibility permits possible improvement in the affinity between a drug and its target and traces the high-affinity interactions with a small drug.1 It is thought that the protein pre-exists as a set of different conformations in similar energy states. Out of those, one of the conformers selectively binds the therapeutic agent more efficiently, thereby increasing the population of the concerned conformer in the whole bunch, and this indeed guides the conformational modifications, which ultimately alters the nature of interaction.1 Therefore, it is very crucial to understand the quantitative structure–binding relationship in a set of structurally related drugs to identify the intriguing effectiveness of a drug in some selective conditions.
Most of the drugs reversibly bind plasma proteins with substantial binding efficiencies and may produce some significant effects on the overall activity profile of the molecule as well as the protein.2,3 Serum albumin (SA) is the major protein constituent of blood plasma, and it is also found to be considerably distributed in the interstitial fluid of body tissues. SA is often chosen as a model protein in various studies because of its high abundance and smaller size.3,4 It can reversibly bind and deliver a number of relatively insoluble endo- and exogenous compounds through blood circulation.3,5,6 SA can affect pharmacokinetic profile by restricting the unbound drug concentration, thereby altering the distribution and elimination.5,6 Human serum albumin (HSA, Scheme 1) has a limited number of active sites for drug accumulation, and they are subdomains IIA, IIIA, and IB.6,7 Among these, subdomains IIA and IIIA are located in hydrophobic cavities, which are regarded as principal regions for binding of drugs and ligands. Besides, in some cases, the ligand binds subdomain IB, which is referred to as site III.7 As metabolism and transportation of drugs are important components for drug efficiency, the role of a carrier protein always needs to be realized quite well.5 In this context, drug–protein interaction in a binary system (interaction of one type of drug with protein) is very common and very well-documented in the literature. In most of the cases, the respective drugs are found to affect the protein secondary structure by conformational induction.8 This type of information is very useful to ascertain the binding efficiency of the drug. The nature of conformational induction is different for different types of drugs, even for structurally similar drugs. For enzymatic proteins, conformational induction is known to be responsible for displaying agonist and antagonist behaviors. However, it is very inconvenient to understand the nature of interaction and the possible fate of the protein structure in the presence of a cocktail of drugs. Interaction of drugs with protein depends upon various factors, but among them, the drug–drug competition (DDC) for the same binding site can also be very crucial in pharmacokinetic and activity profiles. The competition of two drugs for the same pocket of a protein is sometimes referred to as drug–drug interactions (DDIs), and it is thought that such interactions cause undesired adverse (side) effects, such as toxicity or loss of therapeutic properties.9 Moreover, it is considered that if two drugs bind in close proximity to one another, the stronger binding drug shows a narrow therapeutic effect.10 In many instances, DDI has been purposely utilized to increase the free concentration of the weaker binding drug. For example, the anticoagulant activity of warfarin is known to be enhanced by the coadministered anti-inflammatory drugs, such as phenylbutazone/bucolome.11 Importantly, these drugs bind the same pocket (in subdomain IIA) of HSA, and all of these studies have been performed with respect to the drug-binding efficiency. Therefore, the role of the protein structure to anticipate the multiple-drug binding, more specifically the DDC, needs to be explored in detail.
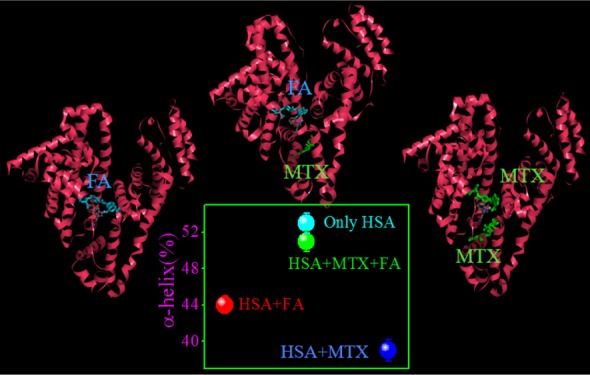
Scheme 1. Molecular Structures of (a) MTX and (b) FA and (c) Crystal Structure of HSA (PDB ID 1AO6) with Possible Binding Locations.

In this regard, the drugs have been chosen for exploration of the structural fate of protein are folic acid (FA) and methotrexate (MTX), a common pair of supplementary drugs having a quite similar structure (Scheme 1). These drugs are known to be the main competitors for the same binding site in subdomain IIA of SA.12,13 Therefore, we can expect to observe a DDC here. MTX has been widely used as a disease-modifying antirheumatic drug in the treatment of rheumatoid arthritis (RA) and also used in various types of cancers, such as leukemia, lymphoma, and bone cancer. MTX interferes with the supply of folates by inhibiting the action of dihydrofolate reductase. At high doses, it has a considerable effect on the purine and pyrimidine biosyntheses, contributing to its usefulness in many malignancies, and at low doses (<20 mg/week), it is also beneficial in treating RA. However, the mechanism of action of MTX is not yet understood fully. Folate antagonism and folate deficiency are the major adverse effects of low-dose MTX in patients with RA. FA or folinic acid is often coadministered with MTX as a folate supplementation to minimize adverse effects (e.g., stomatitis, gastrointestinal intolerance, bone marrow toxicity, and abnormal liver function tests). Again, Whittle et al. proposed that folate supplements do not appear to significantly reduce the effectiveness of MTX in the treatment of RA.14 These supplemental FA offsets can be useful for reducing the risk of cardiovascular problems of RA patients, but they promote the possibility of risk factor of hyperhomocysteinemia through abnormally increasing the level of homocysteine in blood. Furthermore, MTX toxicity and efficacy with the FA dose still remain debatable, and the effect of different doses of FA is also unknown. Now, with respect to this study, the drug having a stronger force for adherence can reside on the binding site longer and should be responsible for the induced structural change of the receptor protein. As a result, the interaction will be modulated through the alteration of the equilibrium distribution of the bound and unbound ligands. In this context, it would be very fascinating to assess the changes in the binding characteristics of the drug in the presence of the carrier protein SA and the competitive binding aspects of the drugs to be housed in the same protein pocket. In this regard, the understanding of the structural modification of receptors caused by DDC can have immense biochemical relevance.
Results and Discussion
Herein, the steady-state absorption spectra of MTX and FA have been recorded as a function of HSA concentration. At pH 7.4, FA shows two distinct peaks at around 280 and 350 nm, whereas MTX shows three peaks at around 258, 303, and 372 nm.15Figure 1 indicates that with increasing HSA, FA shows a substantial bathochromic shift (by ∼21 nm) in the absorbance spectra. However, in the case of MTX, its absorption spectra are very little red-shifted (by ∼2 nm) (the inset of Figure 1). This type of observations can easily be explained by the protonation/deprotonation of dissociable groups available in the drug molecule. Many of these dissociations are directly reflected in the ultraviolet absorbance changes, and depending upon the pH, MTX and FA may exist in different ionic forms.15 FA generally exhibits four prototropic forms (bicationic, cationic, neutral, and anionic), whereas MTX shows only three forms (bicationic, cationic, and neutral). Because of the lack of amide moieties in MTX, not much change in the absorption bands has been observed in between the pH values 7.4 and 9.8 (Figure S1, Supporting Information). However, a substantial shift in the absorbance maxima (from 350 to 370 nm) is observed in the case of FA, and as its pKa is around 8.38, the transition from the neutral to the anionic form is expected to occur above pH 8.5. This is the pH range in which the assigned dissociation of acidic proton of the amide group of pterin occurs. These results clearly indicate that FA gets incorporated into the protein pocket, accompanying a shift of its prototropic equilibrium from the neutral to the anionic form.
Figure 1.
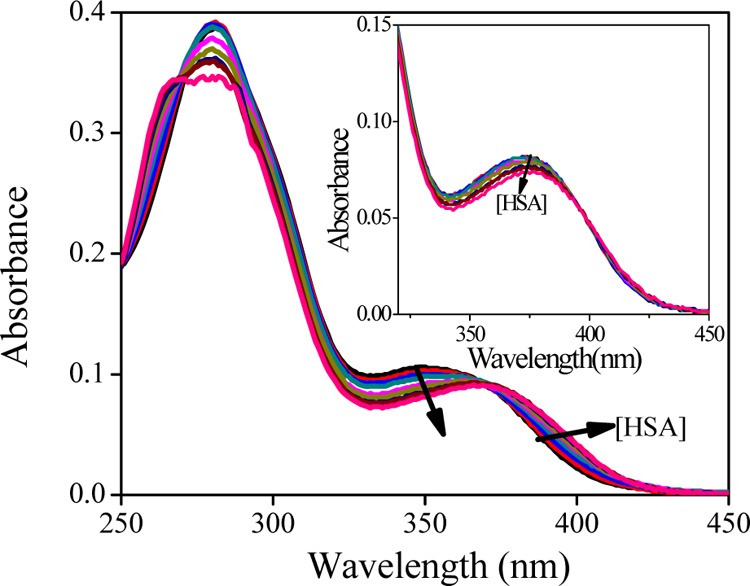
Absorption spectra of FA (10 μM) in the presence of HSA (0–100 μM) at pH 7.4. The inset shows the absorption spectra of MTX (10 μM) in the presence of HSA (0–100 μM).
Now, in the excited electronic state with increasing protein concentration, the fluorescence of the drugs (MTX and FA) is progressively quenched without a shift of the emission maxima (Figure S2, Supporting Information). It has already been observed elsewhere4 that the prototropic behavior of the drug molecule has a significant role in drug–protein interaction in the ground state. We have also explored the fluorescence emission of MTX and FA at various pH values (Figure S3, Supporting Information). At pH 7.4, FA shows a considerably high fluorescence intensity, but with further increase of pH, no additional change is observed. Therefore, it may be stated that the ground-state prototropic properties do not appear to have a considerable role in the excited-state properties here and the observed quenching of emission is the signature of the formation of a HSA–drug complex. With increasing concentration of HSA, the extent of quenching of FA is found to be more than that of MTX. Moreover, the steady-state anisotropy study furnishes the similar findings, where at the point of saturation, the anisotropy values are 0.22 and 0.25 for MTX and FA, respectively (Figure S4, Supporting Information). This clearly indicates that FA experiences a more rigid environment than MTX, which is presumably due to its higher binding affinity compared to MTX. Excited-state lifetime measurement also highlights the possible binding nature in the micro-heterogeneous pocket of protein.16 Here, the average lifetime (τavg) values of both MTX and FA progressively decrease with protein concentration, and an enhancement of nonradiative channels indicates the possible involvement of surrounding protein residues with MTX and FA (Supporting Information). The biexponential rotational correlation time (θr) of FA in the presence of HSA indicates the existence of an equilibrium between two prototropic forms in solution, and a high θr value (5.39 ns) of the slow component reveals that the anionic form has greater binding power than the neutral form, whereas MTX remains mostly in the neutral form as it is unable to get converted to the anionic form in the presence of HSA. This form is housed loosely in the protein cavity (as evident from a low θr value, 1.34 ns) as compared to the anionic form of FA (2.28 ns). These results indicate the higher binding affinity of FA over MTX with HSA (Supporting Information). We have determined the binding stoichiometry for both the drugs using the Job’s method,17 and it is found to be 1:2.33 for the MTX–HSA complex and 1:1 for the FA–HSA complex (Figure S7, Supporting Information). These results suggest that HSA can simultaneously bind two MTX molecules, whereas occupied site by FA in HSA is only one.
As we have seen that the properties of MTX and FA are greatly modulated in the presence of HSA, either way, it is also important to understand the changes of spectral properties of the protein, namely, the fluorescence of tryptophan (Trp) and tyrosine (Tyr) in the presence of these drugs. HSA consists of only one Trp residue, and this Trp is located in the important subdomain IIA ligand-binding site at amino acid position 214. As the understudied drugs in a binary system bind subdomain IIA, the selective excitation at 295 nm can extract the information on the change in the binding blueprints nearer to that region. We have studied the combined behavior of Trp and Tyr (λex = 280 nm) in the presence of these drugs as well.
Now, with increasing concentration of MTX or FA in the presence of HSA, the fluorescence intensity (λex = 280 and 295 nm) is progressively quenched with a slight blue shift in the emission maxima (Figure S8, Supporting Information). This characteristic quenching is due to the incorporation of a drug molecule in the protein pocket. From this, we can estimate the binding constants of the two drugs with HSA, and the binding constant has been determined by the modified Scatchard method as follows:12,18
| 1 |
where r is the number of ligand molecules to one protein molecule; [Lf] is the free (unbound) ligand concentration; n is the number of binding sites in a protein molecule (binding stoichiometry), which corresponds to the mean number of drug molecules bound in the independent class of drug-binding sites in an albumin molecule; and Ka is the binding constant. The binding plots (Figure 2a) suggest that binding of MTX with HSA takes place by involving two types of binding sites, whereas FA binds a single site. Table 1 highlights that the binding constants (Ka) for the MTX–HSA complex are equal to (1.64 ± 0.11) × 105 for the first binding site and (0.16 ± 0.02) × 105 M–1 for the second binding site with stoichiometries of (0.36 ± 0.04) and (2.21 ± 0.14), respectively. Maciążek-Jurczyk et al. also concluded similar binding patterns of the MTX–HSA complex in a different drug cocktail pair, and they have observed a quite high binding affinity in the first binding site.12 They proposed that subdomain IIA is the higher affinity site for MTX, and a decrease of Ka with increasing the number of other drug molecules (phenylbutazone) has also been observed for the MTX–HSA complex.12
Figure 2.

Scatchard plot of the binding of (a) MTX and (b) FA with HSA for a two-component system at 298 K.
Table 1. Parameters of Competitive Binding Analysis for HSA with MTX and FA at 298 K.
| system | fixed [drug] (μM) | KI (M–1) | KII (M–1) | nI | nII |
|---|---|---|---|---|---|
| HSA–[FA]–MTX | 0 | (1.64 ± 0.11) × 105 | (0.16 ± 0.02) × 105 | 0.36 ± 0.04 | 2.21 ± 0.14 |
| 2 | (1.74 ± 0.14) × 105 | (0.24 ± 0.02) × 105 | 0.41 ± 0.05 | 1.73 ± 0.12 | |
| 10 | (0.78 ± 0.09) × 105 | (0.43 ± 0.03) × 105 | 0.58 ± 0.05 | 1.20 ± 0.09 | |
| 30 | (0.75 ± 0.06) × 105 | 0.56 ± 0.06 | |||
| HSA–[MTX]–FA | 0 | (0.87 ± 0.08) × 105 | 1.02 ± 0.07 | ||
| 2 | (0.79 ± 0.07) × 105 | 1.04 ± 0.06 | |||
| 10 | (0.74 ± 0.09) × 105 | 1.12 ± 0.07 | |||
| 30 | (0.71 ± 0.07) × 105 | 1.01 ± 0.05 |
The one-site binding of FA is primarily associated in subdomain IIA (Figure 2b)13 with Ka and n being (0.87 ± 0.08) × 105 M–1 and 1.02 ± 0.07, respectively (Table 1). This indicates that MTX shows a higher binding affinity with a small bound fraction in subdomain IIA, whereas FA binds only to this site of HSA.
Again, excitation at 280 nm provides very similar information about the binding processes (Figure S9, Supporting Information). The binding constants of MTX are found to be around (1.66 ± 0.12) × 105 M–1 for the first binding site and (0.16 ± 0.02) × 105 M–1 for the second site with stoichiometries of 0.34 and 2.39, respectively. On the other hand, FA shows single-site (n ≈ 1.00) occupancy with a binding constant of (0.86 ± 0.04) × 105 M–1. These results clearly suggest that Tyr residues do not participate or have only minimal effects in the interaction process. Modulation of binding efficiencies of one drug molecule in the presence of another structurally similar drug or a second drug molecule can be of great significance in the pharmacokinetic behavior of the concerned drug in relation to drug stability and sustained delivery.9Table 1 and Figure S10, Supporting Information indicate that the binding affinities of the MTX–HSA complex in the presence of FA differ substantially from the binary system (absence of FA), whereas the FA–[MTX]–HSA (in the ternary system, the concentration of MTX is fixed) complex does not differ much from the corresponding binary system. Now, in the case of the MTX–[FA]–HSA system, n of MTX for the first binding site increases slightly (from 0.36 to 0.58) with a decrease in the Ka value [from (1.64 ± 0.11) × 105 to (0.78 ± 0.09) × 105]. On the other hand, in the second site, the binding constant increases (from (0.16 ± 0.02) × 105 to (0.75 ± 0.06) × 105 M–1) gradually with a considerable decrease in the binding stoichiometry (from n = 2.21 ± 0.14 to 0.56 ± 0.06). From the binding characteristics of the FA–[MTX]–HSA complex, shown in Table 1, it is evident that MTX (even at a higher concentration of 30 μM) shows a nominal effect on the FA–HSA binding affinities and also the stoichiometry does not alter much. Therefore, FA binds stronger with HSA in subdomain IIA (also evident from previous studies), and this is not affected by MTX. Because in the presence of FA, the binding of the MTX–HSA complex gets decreased, this implies that one site (subdomain IIA) is arriving at saturation by FA and this causes difficulty for MTX to form a complex with HSA. In fact, FA blocks the principal binding site (subdomain IIA) of MTX and forces it to occupy the other site nearer to subdomain IIA (seems to be subdomain IIIA).12 It has been previously reported that subdomain IIA is primarily regarded as the anion receptor pocket, and many exogenous ligands bind there in the form of anionic species from the neutral state via pKa shifting.4,19 In the previous section, we have mentioned that HSA binds FA principally in the form of folate (the anionic form) and MTX in the neutral form. Therefore, it is evident that the formation of folate is playing the key role here for its encapsulation over MTX in subdomain IIA. On the other hand, because the subdomain IIIA pocket is generally surrounded by hydrophobic residues, this binding process is principally manifested by π–π type of interaction between the drug and the protein via formation of a sandwichlike complex. Furthermore, we have also performed docking analysis to understand the binding location and associated forces between drugs and HSA (discussed in detail in the Supporting Information). The docking results aptly support the experimental findings, where in a ternary system, FA and MTX are found to bind subdomains IIA and IIIA, respectively, and it is found to bind nearer to Trp 214 (∼4–10 Å). In subdomain IIA, the presence of some charged amino acid side chains around FA confers the stabilization of anionic folate, whereas in the IIIA pocket, predominance of hydrophobic residues craves that the binding mode for MTX should be, by and large, π–π type.
Now, we have investigated how the dual-drug binding affects the protein structure in contrast to single-drug-binding processes. For that, we have used infrared (IR) (Figure S14, Supporting Information) and UV circular dichroism (CD) spectroscopic methods as a function of composition of MTX and FA in a mixture in the presence of HSA. The observed α-helical content (from IR and CD studies) of free HSA is ∼53%, which is in good agreement with the reported results.20 From IR studies, it is found that with the addition of MTX and FA, the α-helical content of HSA has been substantially decreased (from 53 to 39 and 44% for FA and MTX, respectively), following some alteration of the β-sheet content (from 16 to 31 and 29% for FA and MTX, respectively) or the coil structure (from 8 to 5 and 22% for FA and MTX, respectively) and in the presence of cocktail of both drugs, these percentages are not much affected (change of the α-helical content from 53 to 51%) (Figure 3). Likewise, in the CD studies, the α-helical content is found to decrease (from 52.71 to 50.83 and 52.19% for MTX and FA, respectively) with the addition of drugs and the extent of loss of the secondary structure is more in the case of MTX than that in FA, although FA unfolded the protein secondary structure in a lesser extent (Table S4, Supporting Information). In connection with dual binding, we varied the FA and MTX concentrations in terms of mole fraction over a fixed HSA concentration. It is found that at the same molar ratio of these drugs, ternary system imposes less effect (the change in the α-helical content is only ∼0.3%) on the protein secondary structure than the binary system (the inset of Figure 3). The difference in the values of α-helical content of the drug–protein complex of IR and CD methods may be due to the different sampling concentration and dissimilar analysis procedure. These results indicate that here HSA can retain its original structure by partial refolding or preventing the structural loss (unfolding) in the presence of cocktail of drugs. It is previously reported that competitive drug binding has a strong impact on the protein structure.21 However, in most of the cases, competitive drug binding significantly modifies the protein structure, and in many instances, it accompanies the impairment of the activity of the bio-macromolecule. Herein, the results clearly report the refurbishment/reconstruction of the HSA secondary structure in the presence of a drug cocktail, where FA facilities MTX to occupy a different site (subdomain IIIA), maintaining the helical content (in a cooperative manner) intact, which is very much like a free unperturbed protein or in a less perturbed form. In other words, here, DDC signifies the drug-mediated partial refolding (repair) of the protein structure, where one drug eliminates other drug from its active binding site via maintaining the equilibrium distribution of bound and unbound fractions.10 This type of structural reconstruction due to dual binding reports a new challenging aspect of conformational adaptation of HSA through DDC. It is reported that the conformational/structural flexibility allows the protein to adopt multiple conformations of an approximately similar energy to accommodate a variety of ligands or drugs.1,22 The number of domains of a protein is very limited toward binding of drugs, and in most of the cases to accumulate the drug, the more affinity conformer of the protein can be responsible. This may lead to the structural changeover of HSA. However, in the presence of cocktail of drugs, the residues of HSA face many types of interaction, and here the protein gets a chance to attain more preferable conformation, by manipulating this interaction through changing the binding affinity of drugs. In other words, out of many possible conformations in solution, the most potent conformation will automatically be selected by the cocktail of drugs, which is as per the right energetics. Furthermore, a recent report depicts the binding pattern of diclofenac (Dic) and naproxen (Nps) with equine SA, where both drugs hold two binding sites with one common location (the preferable binding site) and in competitive binding mode Nps occupy the most preferable binding site without affecting the other site of Dic.23 This infers that combined application of both drugs may result in increased concentration of free Dic in plasma. Figure 4a shows that with increasing the amount of fixed FA, the bound MTX concentration decreases with gradually increasing concentration of the unbound form of MTX. Also, the binding stoichiometry of MTX is found to decrease substantially (Figure 4b). Hence, the present study paves the utility of combination-drug delivery, where the effective unbound concentration of MTX will increase with increasing the concentration of folate in plasma.
Figure 3.
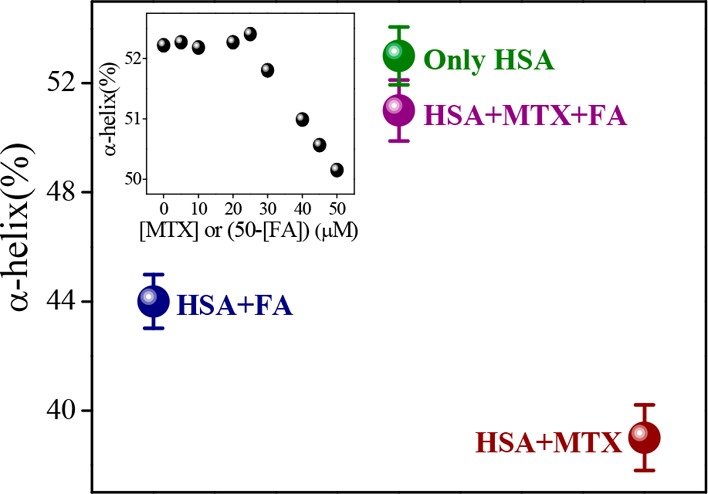
Change in α-helical content (%) of HSA as a function of composition of MTX and FA mixture from IR study. The inset indicates the change in α-helical content (%) of HSA with variation of the composition of MTX and FA from CD study.
Figure 4.

Change in (a) bound MTX concentration and (b) the number of binding sites (n) in the presence of various amounts of fixed FA.
Now, it is important to look into the possible implication of delivery of MTX–FA as a combination drug in the treatment of RA. Researchers suggest that in many ways, FA supplementations are required for patients with RA during treatment with MTX.24−27 However, American College of Rheumatology has not strongly recommended the FA or folinic acid supplementation to prevent the complications with MTX in the therapy of RA.25 As FA and MTX both compete for the same pocket of the transporter to get absorbed from the gastrointestinal tract and cellular uptake, it is likely that the reversal of anti-inflammatory effects of MTX by high doses of FA results in the diminished MTX uptake in those patients, so patients taking FA are advised to skip their FA doses for a period around the day that they take their MTX.25 However, the present study suggests that SA can transport these drugs simultaneously and more importantly folate in plasma helps to maintain the bound fraction of MTX. Therefore, from the above results, we can state that combination of these drugs should be useful in the aforementioned contexts.
Conclusions
In summary, the present work explores the nature of competitive binding of a drug pair in terms of structural modification of HSA. For that, we have used MTX and FA, a well-known supplementary drug pair for the treatment of RA. The results provide a comprehensive insight of duo-binding feature with respect to single-drug binding by refurbishing the protein structure toward the less perturbed state. It signifies how the protein structure governs the drug binding in the presence of a cocktail of drugs and hence maintains the bound and unbound fractions. The results suggest that MTX prefers to bind within two locations simultaneously, whereas FA is found to bind in only subdomain IIA. In this site, these two drugs show strong binding affinities with different stoichiometries. However, FA binds as folate via shifting the prototropic equilibrium, providing better rigidity at the site of interaction than MTX. In the ternary system, the binding of MTX is found to be largely affected in the presence of FA. In that case, FA holds the primary binding site (subdomain IIA) strongly and forces MTX to leave the high-affinity binding site subdomain IIA. Subsequently, MTX prefers to occupy the second site with increasing the binding efficiency, followed by a decrease in the number of bound drug fractions. As a consequence, the elimination of excess MTX is facilitated as a free fraction, which can be important for curing RA. Interestingly, it has been found that single-drug binding causes slight unfolding of the protein structure and in the presence of both the drugs, the altered protein secondary structure for single-drug binding is substantially repaired/restored in reference to the free protein. This result indicates that in the presence of a cocktail of drugs, protein modifies the interactions in such a way that the flexible nature of this protein molecule allows the retention of the secondary structure from the perturbed state by regulating the DDC.
Experimental Section
Materials
MTX, HSA (fatty acid-free), and FA were purchased from Sigma-Aldrich and Himedia and used as received. Methanol (UV spectroscopic grade) and NaOH [analytical reagent (AR) grade] were purchased from Spectrochem, India, and Merck, India, respectively, and all other chemicals were of AR grade. Ultrapure Milli-Q water was used throughout the study. All solutions were prepared in 5 mM sodium phosphate buffer of pH 7.4 (±0.1). The pH measurements were made with a precalibrated EUTECH pH 510 ion pH meter.
Sample Preparation
The stock solution of MTX was prepared in methanol, and during the study, methanol was evaporated prior to the addition of buffer solution. Solubility of FA in methanol was quite low, so we prepared the stock solution of FA in water by making its salt by the addition of equivalent amount of NaOH solution.
Instrumentation and Methods
UV–vis absorption spectra were recorded on a Shimadzu UV-2600 absorption spectrophotometer by scanning the solution in the cuvette against the solvent reference in the wavelength range 250–600 nm at 298 K with the help of a TCC-260 thermoelectrically temperature-controlled cell holder.
Steady-state fluorescence spectra were recorded on a HORIBA Jobin Yvon spectrofluorometer (Fluorolog-3) equipped with a temperature-controlled water-cooled cuvette holder kept at 298 K. Two types of quartz cuvettes (1 and 0.4 cm path length) were used for scanning. The experimentally measured fluorescence intensity was corrected for background fluorescence and inner filter effect at both the excitation and emission wavelengths as described elsewhere,28 using the following expression:
| 2 |
Here, Ficor is the corrected fluorescence intensity, Fi is the experimentally measured fluorescence intensity, Fb is the background fluorescence intensity, L is path length of the cuvette used, and Aex and Aem are the absorbances of the sample at the excitation and emission wavelengths for the ith component, respectively.
Steady-state anisotropy (r) measurements were performed with the HORIBA Jobin Yvon spectrofluorometer (Fluorolog-3). The steady-state anisotropy (r0) was expressed as follows:
| 3 |
| 4 |
where IVV and IVH are the emission intensities collected from the sample when the excitation polarizer is oriented vertically and the emission polarizer is placed vertically and horizontally, respectively. G is the correction factor for the instrument (also called G-factor of the instruments) and is estimated by keeping the excitation polarizer horizontal and the emission polarizer vertical and horizontal.
Fluorescence quantum yield (Φ) of MTX and FA was estimated using quinine sulfate as a secondary standard probe (Φ = 0.55 in 0.1 N H2SO4),29 by employing the following equation:
| 5 |
where S and R represent the parameters of the studied sample and reference, respectively; A indicates the area of the fluorescence curve; Abs denotes the absorption of the sample; and n is the refractive index of the medium.
The fluorescence lifetime measurements were performed using a time-correlated single-photon counting (TCSPC) spectrometer from Edinburgh Instrument Ltd. (U.K.). In this present work, the samples were excited at 376 (±10) nm using a picosecond laser diode (EPL-375, pulse width ∼67.9 ps) as an exciting source and a high-speed photomultiplier tube (H10720-01, photosensor module from Hamamatsu) detector was used for collecting signals at a magic angle of 54.7°. All fluorescence decays were analyzed by F900 software from Edinburgh Instruments, with an appropriate decay function considering a χ2 value close to 1, which was an indication of goodness of fit. The amplitude average lifetimes of the samples were estimated by employing a multiexponential decay function, using the following equation:
| 6 |
where ai is the pre-exponential term of ith component of τ.
Time-resolved fluorescence anisotropy decays were also measured with the aforementioned TCSPC setup with the same laser diode. In this process, parallel [I∥(t)] and perpendicular [I⊥(t)] emission polarizations were collected after excitation by a vertical excitation polarizer. Then, the rotational correlation time (θr) can be estimated utilizing the following equations:
| 7 |
| 8 |
Here, r(t) and rlim (limiting anisotropy) represent the anisotropy decay function and the inherent depolarization of the fluorophore, respectively, and air is the pre-exponential term of ith system of θr.
The available crystal structures of HSA (PDB ID: 1AO6)30 in protein data bank were used for docking studies. To get the lowest energy optimized drug–protein structure, we followed the detailed computational method discussed in our earlier report.4 Uncomplexed proteins and their docked conformations (poses) with TZ of lowest energy were used for the calculation of solvent-accessible surface area (SASA) using the Discovery Studio Visualizer 4 from Accelrys Software Inc. The change in SASA (ΔSASA) for a residue was estimated as ΔSASA = SASAprotein – SASAprotein–drug.
CD spectra were recorded on a Jasco-815 automatic recording spectropolarimeter at 298 K at a scan speed of 50 nm/min by averaging three successive scans over a wavelength range of 190–260 nm. A quartz cell having a path length of 0.1 cm was used during the study, and each spectrum was corrected with the baseline for an appropriate buffer solution. Typical CD spectra of HSA exhibit two negative bands at around 208 and 222 nm in the ultraviolet region, recognized as a characteristic band for the α-helical structure.20 CD measurements were performed in the presence of different concentrations of MTX and FA, and the data were expressed in mean residual ellipticity (MRE) in deg cm2 dmol–1, according to the following equation:20
| 9 |
Here, cp is the molar concentration of the protein, nr is the number of amino acid residues of the protein, and l is the path length. The α-helical contents of free and ligand-bound HSA can be calculated from the MRE values at 208 nm using the following equation:
| 10 |
Here, MRE208 is the experimental MRE value of proteins at 208 nm, and the contribution of the MRE value of β form and random coil conformation at that particular point is 4000. Also, at 208 nm, the MRE value of a pure α-helix is 33 000.
Fourier transform infrared (FT-IR) spectroscopy studies were conducted using Thermo Scientific FT-IR spectrometers equipped with a deuterated triglycine sulfate detector and a KBr beam splitter using AgBr windows at room temperature (∼25 °C). For each spectrum, a 64-scan interferogram was collected in a single-beam mode with a 2 cm–1 resolution at room temperature. The reference spectrum was recorded under identical conditions with only the corresponding buffer in the cell. The protein spectra were processed using a previously established protocol.31 The region of 1000–1800 cm–1 in the spectra was baseline-corrected and normalized. The region of 1600–1700 cm–1 corresponding to the amide I band was extracted for analysis. Second-derivative spectra were obtained using a seven-point baseline-corrected Savitzky–Golay derivative function, and the amide I band was area-normalized as described in the literature.31 The secondary structure content of the protein was calculated by curve-fitting analysis of the inverted second-derivative amide I band from 1600 to 1700 cm–1. The fraction of amino acid residues comprising each secondary structural element was proportional to the relative percent area of the associated C=O vibrational bands.
Acknowledgments
M.H. thanks DST SERB-Govt. of India (fund no. SB/S1/PC-041/2013) for financial support. S.P. and D.K.K. thank UGC-India for their individual fellowships. The authors would like to thank the anonymous reviewers for their critical comments and suggestions.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b01437.
Absorption and fluorescence spectra of FA and MTX at various pHs; fluorescence quenching and change of steady-state anisotropy of FA and MTX in the presence of HSA; time-resolved studies of FA and MTX in the presence of HSA; Job’s plot for the HSA–drug complex; fluorescence emission spectra (λex 295 nm) of HSA in the presence of MTX and FA; Scatchard plot (λex 295 and 280 nm) of binding for various fractions of FA and MTX; curve-fitted inverted second-derivative amide I spectrum for HSA and HSA–FA–MTX complex; docking analysis and possible mode of interaction among HSA, FA, and MTX; and change of α-helical contents of HSA with respect to MTX and FA (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Teague S. J. Implications of protein flexibility for drug discovery. Nat. Rev. Drug Discovery 2003, 2, 527–541. 10.1038/nrd1129. [DOI] [PubMed] [Google Scholar]
- Wang H.; Wang Z.; Lu M.; Zou H. Microdialysis sampling method for evaluation of binding kinetics of small molecules to macromolecules. Anal. Chem. 2008, 80, 2993–2999. 10.1021/ac702066x. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Chen X.. Simple bioconjugate chemistry serves great clinical advances: albumin as a versatile platform for diagnosis and precision therapy. Chem.Thereforec. Rev. 2016, 45, 1432–1456. 10.1039/c5cs00158g [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta S.; Panja S.; Halder M. Detailed scenario of the acid–base behavior of prototropic molecules in the subdomain-IIA pocket of serum albumin: results and prospects in drug delivery. J. Phys. Chem. B 2014, 118, 12153–12167. 10.1021/jp5076466. [DOI] [PubMed] [Google Scholar]
- Bertucci C.; Domenici E. Reversible and covalent binding of drugs to human serum albumin: Methodological approaches and physiological relevance. Curr. Med. Chem. 2002, 9, 1463–1481. 10.2174/0929867023369673. [DOI] [PubMed] [Google Scholar]
- Ghuman J.; Zunszain P. A.; Petitpas I.; Bhattacharya A. A.; Otagiri M.; Curry S. Structural basis of the drug-binding specificity of human serum albumin. J. Mol. Biol. 2005, 353, 38–52. 10.1016/j.jmb.2005.07.075. [DOI] [PubMed] [Google Scholar]
- Carter D. C.; Ho J. X. Structure of serum albumin. Adv. Protein Chem. 1994, 45, 153–203. 10.1016/s0065-3233(08)60640-3. [DOI] [PubMed] [Google Scholar]
- Tajmir-Riahi H. A. An overview of drug binding to human serum albumin: protein folding and unfolding. Sci. Iran. 2007, 14, 87–95. [Google Scholar]
- Caterina P.; Antonello D. P.; Chiara G.; Chiara C.; Giacomo L.; Antonio S.; Giovambattista D. S.; Luca G. Pharmacokinetic drug-drug interaction and their implication in clinical management. J. Res. Med. Sci. 2013, 18, 600–609. [PMC free article] [PubMed] [Google Scholar]
- Nuin E.; Jiménez M. C.; Sastre G.; Andreu I.; Miranda M. A. Drug–Drug Interactions within Protein Cavities Probed by Triplet–Triplet Energy Transfer. J. Phys. Chem. Lett. 2013, 4, 1603–1607. 10.1021/jz400640s. [DOI] [PubMed] [Google Scholar]
- Yamasaki K.; Chuang V. T. G.; Maruyama T.; Otagiri M. Albumin–drug interaction and its clinical implication. Biochim. Biophys. Acta 2013, 1830, 5435–5443. 10.1016/j.bbagen.2013.05.005. [DOI] [PubMed] [Google Scholar]
- Maciążek-Jurczyk M.; Sułkowska A.; Bojko B.; Równicka J.; Sułkowski W. W. Fluorescence analysis of competition of phenylbutazone and methotrexate in binding to serum albumin in combination treatment in rheumatology. J. Mol. Struct. 2009, 924, 378–384. 10.1016/j.molstruc.2008.12.023. [DOI] [Google Scholar]
- Jha N. S.; Kishore N. Thermodynamic studies on the interaction of folic acid with bovine serum albumin. J. Chem. Thermodyn. 2011, 43, 814–821. 10.1016/j.jct.2010.12.024. [DOI] [Google Scholar]
- Whittle S. L.; Hughes R. A. Folate supplementation and methotrexate treatment in rheumatoid arthritis: a review. Rheumatology 2004, 43, 267–271. 10.1093/rheumatology/keh088. [DOI] [PubMed] [Google Scholar]
- Poe M. Acidic dissociation constants of folic acid, dihydrofolic acid, and methotrexate. J. Biol. Chem. 1977, 252, 3724–3728. [PubMed] [Google Scholar]
- Lakowicz J. R.Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: New York, 2006. [Google Scholar]
- Facchiano A.; Ragone R. Modification of Job’s method for determining the stoichiometry of protein–protein complexes. Anal. Biochem. 2003, 313, 170–172. 10.1016/s0003-2697(02)00562-6. [DOI] [PubMed] [Google Scholar]
- Hiratsuka T. Conformational-changes in the 23-kilodalton NH2-terminal peptide segment of myosin ATpase associated with ATP hydrolysis. J. Biol. Chem. 1990, 265, 18786–18790. [PubMed] [Google Scholar]
- Datta S.; Halder M. Effect of encapsulation in the anion receptor pocket of sub-domain IIA of human serum albumin on the modulation of pKa of warfarin and structurally similar acidic guests: A possible implication on biological activity. J. Photochem. Photobiol., B 2014, 130, 76–85. 10.1016/j.jphotobiol.2013.10.013. [DOI] [PubMed] [Google Scholar]
- Bolel P.; Mahapatra N.; Halder M. Optical spectroscopic exploration of binding of cochineal red A with two homologous serum albumins. J. Agric. Food Chem. 2012, 60, 3727–3734. 10.1021/jf205219w. [DOI] [PubMed] [Google Scholar]
- Lahti J. L.; Tang G. W.; Capriotti E.; Liu T.; Altman R. B.. Bioinformatics and variability in drug response: A protein structural perspective. J. R.Thereforec., Interface 2012, 9, 1409–1437. 10.1098/rsif.2011.0843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas L. H.; Price K. E.; Larive C. K.. Epitope mapping and competitive binding of HSA drug site II ligands by NMR diffusion measurements. J. Am. Chem.Thereforec. 2004, 126, 14258–14266. 10.1021/ja0479538 [DOI] [PubMed] [Google Scholar]
- Sekula B.; Bujacz A. Structural insights into the competitive binding of diclofenac and naproxen by equine serum albumin. J. Med. Chem. 2016, 59, 82–89. 10.1021/acs.jmedchem.5b00909. [DOI] [PubMed] [Google Scholar]
- Shea B.; Swinden M. V.; Ghogomu E. T.; Ortiz Z.; Katchamart W.; Rader T.; Bombardier C.; Wells G. A.; Tugwell P. Folic acid and folinic acid for reducing side effects in patients receiving methotrexate for rheumatoid arthritis. J. Rheumatol. 2014, 41, 1049–1060. 10.3899/jrheum.130738. [DOI] [PubMed] [Google Scholar]
- Cronstein B. N. Low-dose methotrexate: A mainstay in the treatment of rheumatoid arthritis. Pharmacol. Rev. 2005, 57, 163–172. 10.1124/pr.57.2.3. [DOI] [PubMed] [Google Scholar]
- Morgan S. L.; Baggott J. E.; Vaughn W. H.; Austin J. S.; Veitch T. A.; Lee J. Y.; Koopman W. J.; Krumdiek C. L.; Alarcon G. S. Supplementation with folic acid during methotrexate therapy for rheumatoid arthritis: A double-blind, placebo-controlled trial. Ann. Intern. Med. 1994, 121, 833–841. 10.7326/0003-4819-121-11-199412010-00002. [DOI] [PubMed] [Google Scholar]
- Khanna D.; Park G. S.; Paulus H. E.; Simpson K. M.; Elashoff D.; Cohen S. B.; Emery P.; Dorrier C.; Furst D. E. Reduction of the efficacy of methotrexate by the use of folic acid: Post hoc analysis from two randomized controlled studies. Arthritis Rheumatol. 2005, 52, 3030–3038. 10.1002/art.21295. [DOI] [PubMed] [Google Scholar]
- Lugo M. R.; Sharom F. J. Interaction of LDS-751 and rhodamine 123 with P-glycoprotein: Evidence for simultaneous binding of both drugs. Biochemistry 2005, 44, 14020–14029. 10.1021/bi0511179. [DOI] [PubMed] [Google Scholar]
- Fletcher A. N. Relative Fluorescence Quantum Yields of Quinine Sulfate and 2-Aminopurine. J. Mol. Spectrosc. 1967, 23, 221–224. 10.1016/0022-2852(67)90012-4. [DOI] [Google Scholar]
- Sugio S.; Kashima A.; Mochizuki S.; Noda M.; Kobayashi K. Crystal structure of human serum albumin at 2.5 Å resolution. Protein Eng., Des. Sel. 1999, 12, 439–446. 10.1093/protein/12.6.439. [DOI] [PubMed] [Google Scholar]
- Yang H.; Yang S.; Kong J.; Dong A.; Yu S. Obtaining information about protein secondary structures in aqueous solution using Fourier transform IR spectroscopy. Nat. Protoc. 2015, 10, 382–396. 10.1038/nprot.2015.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.