

Abstract
Allosteric regulatory processes are implicated at all levels of biological function. Recent advances in our understanding of the diverse and functionally significant class of intrinsically disordered proteins have identified a multitude of ways in which disordered proteins function within the confines of the allosteric paradigm. Allostery within or mediated by intrinsically disordered proteins ensures robust and efficient signal integration through mechanisms that would be extremely unfavorable or even impossible for globular protein interaction partners. Here, we highlight recent examples that indicate the breadth of biological outcomes that can be achieved through allosteric regulation by intrinsically disordered proteins. Ongoing and future work in this rapidly evolving area of research will expand our appreciation of the central role of intrinsically disordered proteins in ensuring the fidelity and efficiency of cellular regulation.
Keywords: Allostery, protein disorder, protein-protein interaction, post-translational modification
Graphical Abstract
I. INTRODUCTION
Classical descriptions of allostery [1, 2] were based on observations of cooperativity between subunits of folded, oligomeric proteins. The concept of allostery has since been expanded to include regulatory mechanisms involving monomeric proteins, where binding of a ligand at one site can result in modulation of functional outcome at a distant site. Many experimental descriptions of allosteric regulation focused on the existence of discrete conformational states in the presence and absence of allosteric effector ligands. While these studies greatly advanced our understanding of how biological molecules can mediate a wide range of cellular signals, it became clear that changes in structure alone could not account for all observations of allosteric processes in biological systems [3–5]. This problem is addressed by the concept of dynamic allostery, which posits that allosteric signals can be transmitted through changes in the dynamic properties of amino acid networks connecting the ligand binding sites [6–9]. Protein dynamics is also central to ensemble models of allostery, in which binding of a ligand or other effector leads to a shift in the population of the various conformational states that are sampled through fluctuations of the protein structure [10]. There are numerous examples of biological systems that utilize both types of allostery and combinations thereof, providing exquisite insights into the functional importance of protein dynamics as well as structural changes in the context of signal transduction and enzyme catalysis.
Over the past decade, research on intrinsically disordered proteins (IDPs) and intrinsically disordered regions of proteins (IDRs) has prompted a re-evaluation of the definitions of allosteric phenomena. Approximately one-third of human proteins contain disordered regions that are 30 or more amino acids in length [11]. IDPs and IDRs are flexible and capable of rapidly sampling an ensemble of conformational states [12]. IDPs are common participants in multivalent interactions, frequently function as hubs in molecular interaction networks, and are highly enriched in sites for post-translational modification [13–17]. These inherent characteristics of IDPs make them ideally suited for cellular signaling and regulation [15, 18], and recent studies have indicated that the ability of disordered proteins to rapidly integrate and transmit a wide range of diverse cellular signals can be linked to mechanistic processes best described under the umbrella of allostery [12, 19–21]. In some cases, the allosteric phenomenon is observed within a single disordered polypeptide chain, in which a coupled folding-and-binding event or post-translational modification in one region of the protein influences subsequent interactions or modifications at a distant site within the same molecule. In other cases, the IDPs themselves act as extremely precise allosteric effectors, cooperatively modulating binding events on macromolecular surfaces and elegantly orchestrating responses to stimuli and decisions about cell fate.
In this review, we highlight the diversity of allosteric regulatory mechanisms that have been observed for molecular processes involving IDPs and the unique regulatory sensitivity that can be achieved through IDP-mediated allostery. Each example will be discussed with a focus on the biological and functional significance of the disordered proteins involved, as well as within the context of currently available theoretical descriptions and mechanistic models of allosteric regulation mediated by protein disorder. Finally, we will provide our perspectives on the future expansion of the concept of allostery to include the processes described here, and on the experimental methods and theoretical framework which will allow for further characterization of these complex yet highly important systems.
II. EXAMPLES OF ALLOSTERY INVOLVING DISORDERED PROTEINS
Thermodynamic coupling as a driver of allostery
The allosteric potential of intrinsically disordered proteins was first described theoretically in a series of papers by Hilser and colleagues [5, 10, 22–25]. Using thermodynamic models, the ensemble allosteric model (EAM) was developed to illustrate the energetic basis of allosteric coupling in multi-domain regulatory proteins. This framework is a natural and necessary extension of the concepts of structural and dynamic allostery: if each region of a protein is considered to exist as a distinct conformational ensemble, fluctuations within the conformational ensemble of one discrete protein domain (for example, upon ligand binding and/or folding) can play an important role in dictating functional output through energetic coupling (Δgint) to distal sites. By modeling energetic coupling in two- and three-domain proteins, it was demonstrated that there is a distinct thermodynamic advantage for disorder in optimizing allosteric coupling between domains (Figure 1) [10]. This formalism was further expanded to describe how intrinsically disordered protein ensembles can be tuned through a redistribution of conformational populations to enable a given protein region to function as both an allosteric agonist and antagonist [22].
Figure 1.
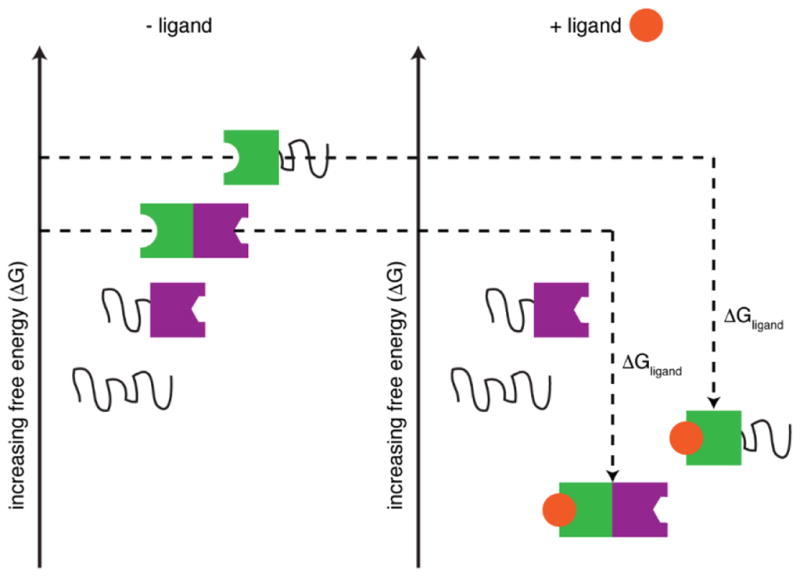
Schematic representation of the ensemble allosteric model for a two domain protein. Ligand binding stabilizes the active conformation of the effector domain (shown in green) to thermodynamically favor the population of the functional protein (with both green and purple domains in the folded state). Disordered domains are represented as black lines and the functional domain is shown in purple. ΔGligand represents the free energy change between apo and ligand bound states. (Adapted from refs. 10 and 23).
Application of the EAM to the human glucocorticoid receptor (GR), an allosterically regulated transcription factor from the steroid hormone receptor class of nuclear receptors, has enabled detailed, quantitative mechanistic studies of allostery involving disordered proteins. The human glucocorticoid receptor (GR) consists of three functional domains: an N-terminal intrinsically disordered (ID) domain, a central DNA binding domain (DBD), and a C-terminal ligand binding domain (LBD). All three domains of GR are inherently dynamic, and their conformational states can be altered by ligand or co-factor binding [26]. Studies of naturally occurring GR translational isoforms that differ only in the length of the N-terminal ID domain illustrate the functional importance of the disordered segments of GR, with the various isoforms displaying different physiological distribution and functional specificities [27, 28]. The changes in regulatory specificity for the N-terminal ID of GR can be observed even in studies of the ID domain in isolation [27]. The ID can be further broken down into two regions of distinct function: the regulatory (R) domain at the extreme N-terminus and the functional (F) domain that links the R domain to the DNA-binding domain and serves as a binding hub for cofactors and cellular activators required for transcriptional activation. Thermodynamic coupling between the R and F domains of the GR N-terminal ID varies with the length and overall stability of the R domain in the various isoforms, resulting in modulation of transcriptional activity [27]. Recently, these studies were extended to include the DBD of GR, allowing for characterization of a more complex biological system within the framework of the EAM [28]. In longer constructs of GR, functional output is modulated not only by thermodynamic coupling between the R and F domains of the ID, but also by thermodynamic coupling of the R and F domains to the DBD. Through careful analysis, the authors determined that only a small subset of the possible combinations of coupling energies allow for transcriptional activation or repression; the remaining combinations are functionally inactive due to “energetic frustration” caused by competing thermodynamic coupling between the domains.
By combining theoretical descriptions and experimental observations, these studies demonstrate that it is possible to obtain a great deal of information regarding complex allosteric regulatory mechanisms involving disordered proteins. In addition to the studies of GR highlighted here, recent work on the PPARγ-RXRα heterodimeric nuclear receptor has provided further insight into the mechanisms of allosteric signal transmission within multi-domain proteins in response to ligand and co-activator binding [29].
Conditional cooperativity
As suggested in the theoretical framework developed by Hilser and co-workers, intrinsic disorder may serve to optimize allosteric coupling between binding sites. This is indeed the case for the phd/doc toxin-antitoxin (TA) operon from bacteriophage P1. Early molecular biological studies of TA operons revealed a phenomenon termed “conditional cooperativity” whereby the toxin is able to function as both an enhancer and a repressor of transcription [30–32]. The “conditional cooperativity” of transcriptional regulation in the context of TA operons was found to be completely dependent on the ratio of available toxin (Doc) to antitoxin (Phd). Phd is comprised of a globular N-terminal domain required for dimerization and DNA binding, and a C-terminal intrinsically disordered region that adopts an α-helical structure in complex with Doc [33]. Transcriptional repression of the phd/doc operon is dependent on the N-terminal DNA binding domain of PhD.
To obtain a mechanistic description of the “conditional cooperativity” observed for regulation of the phd/doc operon, Loris and coworkers used structural methods to characterize the conformational ensemble of Phd in its free state and bound to Doc [34]. These studies revealed instability and conformational heterogeneity in the N-terminal DNA-binding domain of Phd. Remarkably, both the stability and DNA binding activity of the Phd N-terminus were dramatically enhanced by binding of Doc to the disordered C-terminal domain, indicating allosteric coupling between the N- and C-terminal domains of Phd. Further studies allowed for description of the molecular details behind the stoichiometry-dependent switch between transcriptional enhancement and repression (Figure 2), revealing that disordered regions within the antitoxin Phd function as entropic barriers that impair binding of a second Phd molecule to the operator DNA through negative cooperativity. Doc binding to the first Phd molecule lowers the entropic barrier and triggers an allosteric switch to positive cooperativity, which allows for strong repression. However, once Phd is fully saturated with Doc, the repressor complex becomes energetically unfavorable due to steric clashes between neighboring Doc molecules, enabling de-repression of transcription [35].
Figure 2.

Allosteric regulation of transcription of the phd/doc operon. The Phd dimer (blue and green) weakly binds to DNA through its partially ordered N-terminal DNA binding domains (left). The C-terminal domains of Phd fold upon binding Doc (purple), resulting in further stabilization of DNA binding (middle left). The stabilized Phd-Doc-Phd complex recruits additional Doc molecules to DNA (middle right), eventually resulting in steric clash between neighboring Doc molecules and dissociation of Phd and Doc from the DNA (right).
An important outcome of this study, which also pertains to many other systems, was the discovery that the degree of disorder in the C-terminal region of Phd is finely tuned to optimize allosteric coupling. Chimeric constructs in which all charged residues within the Phd C-terminal disordered region were replaced by polar or neutral amino acids, or in which of the entire C-terminal IDR was replaced with [SSSG]n further exacerbated negative cooperativity between binding sites, indicating that maintenance of a certain degree of disorder in the Phd C-terminal IDR is highly important for function [35].
Disordered proteins as allosteric effectors
The transcriptional co-activator CREB-binding protein (CBP) and its paralog p300 function as master regulators of cellular transcriptional programs by mediating interactions between a wide variety of transcription factors and the basal transcription machinery [36–38]. CBP is a large, multi-domain protein featuring seven stably folded domains, one molten globular domain, and many disordered regions of both known and unknown functionality [39, 40]. Both the structured and disordered regions of CBP interact with a multitude of disordered protein partners, and recent studies have implicated allosteric mechanisms as playing an important role in modulating the transcriptional co-activation function of CBP/p300.
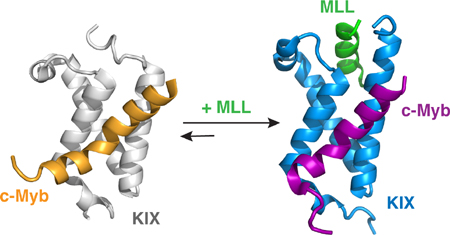
The globular KIX domain of CBP is an important binding site for a number of intrinsically disordered transcription factors involved in cell differentiation, leukemogenesis, and viral transformation [38, 41]. The kinase inducible activation domain (pKID) of the transcription factor CREB and the activation domain of the transcription factor c-Myb bind to a common site on KIX, whereas the mixed lineage leukemia protein (MLL) binds to a distinct surface on the opposite face of KIX (Figure 3) [42, 43]. Biochemical and structural studies show that MLL and pKID or MLL and c-Myb bind cooperatively to KIX to form ternary complexes; binding of MLL in its cognate site enhances the binding of both pKID and c-Myb and vice versa [42, 44, 45]. These studies indicated that KIX functions not only as a structured scaffold for binding of disordered ligands but also as an allosteric modulator of transcription.
Figure 3.

Structural basis of positive cooperativity for ligand binding by the KIX domain of CBP/p300. (A) The structures of the binary KIX:c-Myb complex (left, PDB ID: 1SB0) and the ternary MLL:KIX:c-Myb complex (right, PDB ID: 2AGH). For the binary complex, KIX is shown in gray and c-Myb is shown in orange. For the ternary complex, KIX is shown in blue, c-Myb is shown in purple, and MLL is shown in green. (B) Comparison of the structure of KIX in the binary (gray) and ternary (blue) complexes.
Numerous studies have sought to obtain a mechanistic explanation for the synergistic coupling between the distinct binding sites on KIX. ITC measurements of cooperativity in forming the c-Myb:KIX:MLL and pKID:KIX:MLL ternary complexes revealed differing thermodynamic contributions depending on the pair of ligands [42]. Cooperativity in binding of pKID to the KIX:MLL complex is driven by an increase in entropy, whereas enhanced binding of c-Myb to form the ternary complex is due to a favorable change in enthalpy. NMR studies by Bruschweiler, et al. [46, 47] identified dynamic amino acid networks linking the MLL binding site to the second ligand binding site on the other side of the KIX structure. Computational studies have provided additional insight into the mechanism of the cooperativity, although there are conflicting explanations for the origins of allostery within KIX arising from these studies. Molecular dynamics simulations of both the c-Myb:KIX:MLL [48] and pKID:KIX:MLL [49] ternary complexes suggested that cooperativity originates from a shift in the KIX conformational ensemble upon binding MLL, to populate a state that favors binding of c-Myb or pKID. However, the results of another computational study [50] suggested that binding of MLL lowers the entropic cost for binding of c-Myb, a result that is inconsistent with experiment [42]. Detailed kinetic studies using stopped-flow fluorescence methods suggested that allostery is mediated by changes in the binding kinetics of individual ligands in the presence or absence of a ligand in the opposing site, with the differences in kinetics observed primarily in the dissociation rate constants of the individual disordered ligands [51, 52].
Despite an abundance of data, a mechanistic explanation that can uniformly and reliably account for all of the observed allosteric processes involving KIX is lacking. The extreme diversity of ligands competing for the two binding sites on KIX (the MLL site and the pKID/c-Myb site) suggests that a general unifying mechanism might not be able to explain the role of cooperativity (either positive or negative) in function. By employing allostery to control occupancy of its two ligand binding sites, KIX could more effectively integrate cellular signals and ensure rapid transcriptional responses to the constantly changing environment in the cell.
A more recent example of allostery driven by binding of disordered proteins involves the TAZ1 domain of CBP/p300, which forms the binding site for numerous regulatory proteins [53]. The cellular transcriptional response to oxygen deficiency is mediated by the transcription factor HIF-1 [54–56], which interacts with the TAZ1 domain of CBP/p300 through the disordered C-terminal transactivation domain of its oxygen-sensitive α subunit to activate transcription of critical adaptive genes [57, 58]. Upon restoration of normal oxygen levels, the transcriptional program and overall stability of HIF-1α is downregulated by negative feedback loops [59, 60]. These feedback loops are essential for HIF-1α regulation, as dysregulation or uncontrolled HIF-1α transcriptional activity is associated with metabolic disorders and abnormal angiogenesis, promoting a number of undesirable outcomes, including tumor growth, cardiovascular disease, and prolonged inflammatory response [54].
HIF-1α transcriptional activity is downregulated by the intrinsically disordered protein CITED2, which is under direct transcriptional control of HIF-1α and competes with HIF-1α for binding to TAZ1 [61]. Solution structures of TAZ1 bound to the disordered C-terminal transactivation domains of HIF-1α [62, 63] and CITED2 [64, 65] reveal that the proteins bind to a partially overlapping site on TAZ1, with a conserved four residue LP(Q/E)L motif occupying an identical binding groove on the TAZ1 surface. The HIF-1α and CITED2 activation domains bind tightly to TAZ1 with equal affinity (~10 nM) [64, 66], yet fluorescence and NMR studies of HIF-1α and CITED2 competition for TAZ1 binding yielded a surprising result [66]. In mixtures containing equimolar amounts of HIF-1α, CITED2, and TAZ1, only CITED2 is bound to TAZ1, implying that CITED2 binding disfavors the HIF-1α bound state. Detailed analysis of the biophysical and structural data revealed that CITED2 displaces the bound HIF-1α from its complex with TAZ1 by a complex allosteric mechanism which relies heavily on the disorder of both HIF-1α and CITED2, as well as structural plasticity of the TAZ1 domain (Figure 4). In its TAZ1-bound state, HIF-1α retains flexibility throughout its N-terminus and the LPQL motif to facilitate access to the TAZ1 surface by CITED2. Once CITED2 binds to TAZ1 via its N-terminal α helix, coupling between the CITED2 α helix and the LPEL motif facilitates displacement of the HIF-1α LPQL motif from the common binding site. These initial binding events trigger a conformational change in TAZ1, from a conformation that is more favorable for HIF-1α binding to a conformation that strictly favors CITED2 binding, effectively skewing the equilibrium such that the only observable state is the TAZ1:CITED2 complex. Importantly, this process is only weakly reversible—while equimolar amounts of CITED2 are sufficient to fully displace HIF-1α from its complex with TAZ1, very large amounts of HIF-1α are required to displace CITED2 from its complex with TAZ1. The ability of CITED2 to fully displace bound HIF-1α from its complex with TAZ1 at equimolar concentrations implies that regulatory efficiency can be achieved without requiring the accumulation of high concentrations of CITED2 to shut off HIF-1α transcriptional activation. Thus, HIF-1α, CITED2, and TAZ1 function as a highly efficient, unidirectional molecular switch capable of responding to environmental signals within the cell [66].
Figure 4.

Unidirectional allosteric regulation of the hypoxic response by CITED2. (Left) HIF-1α (orange) binds to the TAZ1 domain of CBP/p300 (gray) using three motifs (αA, LPQL-αB, and αC). (Middle left) The CITED2 αA helix (blue) binds to the TAZ1:HIF-1α complex, displacing the flexible HIF-1α αA motif. (Middle right) CITED2 further destabilizes the TAZ1:HIF-1α complex by competing for a shared binding site with its LPEL motif and triggering a conformational change in TAZ1. (Right) CITED2 successfully displaces HIF-1α to form a stable complex with TAZ1. All helices are represented as cylinders, with the α2 helix of TAZ1 omitted for clarity. ΔgC represents the thermodynamic coupling between the CITED2 αA and LPEL motifs. ΔgH represents the thermodynamic coupling between the LPQL-αB and αC motifs of HIF-1α. First published in Nature, vol. 543, p. 450, 2017 by Springer Nature.
The allosteric behavior observed for competition between HIF-1α and CITED2 for TAZ1 binding is largely dependent on the flexible and disordered nature of the HIF-1α and CITED2 transactivation domains. CITED2 exploits the nascent flexibility of the HIF-1α N-terminus to use its own N-terminal α helix as an anchor to TAZ1, bringing the CITED2 LPEL motif into close proximity to its binding site and driving efficient displacement of HIF-1α. The allosteric mechanism here shares many features with the “anchor and driver” model of allostery proposed by Nussinov and coworkers [67, 68] in which the “anchor” of the allosteric effector (CITED2 α helix) dictates the potency of the allosteric enhancement while the “driver” (CITED2 LPEL motif) influences the efficiency of the allosteric process.
Conformational changes within the TAZ1 domain itself also play an important role in allosteric regulation. Plasticity in the TAZ1 domain allows for subtle conformational rearrangement in its complexes with HIF-1α and CITED2, and structural differences in the TAZ1:HIF-1α and TAZ1:CITED2 complexes dictate whether a competing ligand can bind or not. Parallels can be drawn to the example of cooperative binding interactions of the KIX domain of CBP discussed above and summarized in Figure 3. In both cases, structural changes in globular domains that bind multiple ligands at distinct binding sites can be instrumental in determining the bound (and functional) state of the protein at equilibrium.
The molecular switch between HIF-1α and CITED2 also shares similarities with examples from the literature that have been described as “unidirectional allostery” [69, 70], “competitive allostery” [71], and “facilitated dissociation” mechanisms [72–74]. In all of these mechanisms, competition for a shared binding site accelerates dissociation of the bound ligand. These types of regulatory mechanisms would be highly advantageous in cellular signaling processes where it might be detrimental to wait for bound ligands or partner proteins to spontaneously dissociate.
Disorder within molecular hub proteins can modulate functional output
A key property of intrinsically disordered proteins is that they often contain multiple interaction motifs (binding sites) for molecular partners. As such, they can potentially bind to multiple cellular partners in a variety of combinations, allowing them to function as molecular hubs in interaction networks [13, 75]. Molecular hub proteins display varying degrees of binding promiscuity, with some binding modules favoring interactions with both high affinity and specificity, while other interactions can feature low affinity and low specificity, and all possible combinations in between. One such intrinsically disordered molecular hub protein is the adenovirus oncoprotein early region 1A (E1A), which interacts with the TAZ2 domain of CBP/p300 and the retinoblastoma protein (pRb) to epigenetically reprogram transcriptional processes in the host cell [76, 77]. Intriguingly, E1A interactions with partner proteins can be modulated by both positive and negative cooperativity, depending on the occupancy or availability of various binding sites [78, 79]. For instance, an E1A construct containing only conserved region 1 (CR1), which is capable of binding both pRb and TAZ2 in isolation, displays negative cooperativity for formation of an E1A-TAZ2-pRb ternary complex when the E1A CR1 region is already bound to the other partner protein. Extension of the E1A construct to include the N-terminal domain results in formation of ternary E1A-TAZ2-pRb complexes with positive cooperativity (Figure 5). This switch from negative to positive cooperativity is driven by the incorporation of another TAZ2 interaction site, implying an allosteric regulatory mechanism in which occupancy of the N-terminal domain modulates pRb binding in the CR1 region. Notably, binding promiscuity of E1A and its complexes with partner proteins have important implications for the functional output. Binary interactions between E1A and pRb regulate the cell cycle, while interactions between E1A and TAZ2 facilitate acetylation of E1A by CBP/p300 and subsequent epigenetic regulation of transcription. The allosterically-enhanced ternary complex formed between E1A, CBP/p300, and pRb allows for effective disruption of the cell cycle by promoting pRb acetylation by CBP/p300, thus targeting pRb for degradation and allowing the virus to hijack the cellular transcriptional machinery, forcing cell cycle progression and uncontrolled cellular proliferation [78].
Figure 5.

A cooperativity switch in the adenoviral oncoprotein E1A. Phase diagrams for pRb and TAZ2 binding illustrate how formation of pRb:E1A:TAZ2 ternary complexes can be described by either negative (left) or positive cooperativity (right), depending on the availability of binding sites. Kd values for formation of various complexes with the E1A constructs shown above the phase diagrams are indicated at the phase boundaries. First published in Nature, vol. 498, p. 392, 2013 by Springer Nature.
Post-translational modifications as allosteric modulators
Another functional advantage of protein disorder is to maintain accessibility of binding sites for modifying enzymes and partner proteins. Experimental data and computational predictions indicate that IDPs are highly enriched in sites for post-translational modifications [80–82]. These post-translational modifications can play important roles in transmitting signals between structurally distinct regions of disordered proteins or enhancing or abrogating interactions with biological partners [83]. In some cases, accumulation of post-translational modifications within a disordered protein leads to a threshold response. In these cases, allostery is inferred from the non-linear biological response as a function of an increasing number of modifications. In other cases, post-translational modifications can drive interactions at remote sites, triggering transmission of an allosteric signal across the molecule even in the absence of changes in the overall conformational ensemble. Examples of both types of allostery driven by post-translational modifications are discussed below.
An early example of how post-translational modifications can mediate allosteric signal transmission in disordered proteins involves p27Kip1, which regulates cell cycle progression through inhibitory interactions with Cdk2/CyclinA and Cdk2/CyclinE complexes [84]. Relief of cell cycle inhibition is achieved through phosphorylation events in both the kinase inducible domain near the p27Kip1 N-terminus and within the C-terminal disordered domain. Inherent flexibility of p27Kip1 in its inhibitory complex with Cdk2/CyclinA preserves accessibility of tyrosine residues in its kinase inducible domain to modifying enzymes like the tyrosine kinases Abl and Src [85]. Phosphorylation of Tyr88 in the kinase inducible domain triggers further phosphorylation events within the p27Kip1 polypeptide chain, namely phosphorylation of Thr187 located 100 residues away in the C-terminal disordered domain. Thr187 phosphorylation is entirely dependent on the flexibility of p27Kip1 both in its free state and in its complex with Cdk2/CyclinA [85, 86]. It has been proposed that this flexibility allows for threonine phosphorylation by a unimolecular mechanism in which full dissociation of p27Kip1 is not required for conformational remodeling to allow the C-terminal threonine residue to access the Cdk2 active site.
Multi-site phosphorylation of the intrinsically disordered protein Sic1 provides a prototypical example of a threshold response. Sic1 functions as an inhibitor of Cdk1, blocking cell cycle progression [87]. Sic1 possesses nine distinct phosphorylation sites, and phosphorylation of at least six of these sites is required to target Sic1 for degradation through interaction with the Cdc4 subunit of the SCF ubiquitin ligase [88], thus allowing successful entry into S phase of the cell cycle. Surprisingly, this molecular switch is not driven by changes in the conformational ensemble, as phosphorylated forms of Sic1 retain high levels of flexibility and conformational disorder [89, 90]. Instead, Sic1 relies on the cumulative electrostatic contributions of the phosphoryl groups to produce a switch-like response once the phosphorylation threshold has been met [87, 88]. Individual phosphodegron motifs of Sic1 participate in weak, transient interactions with Cdc4 that are insufficient to target Sic1 for degradation, but six or more phosphorylation sites function cooperatively to facilitate robust interactions with Cdc4 to initiate cell cycle progression and DNA replication. Further studies identified that Sic1 phosphodegron motifs can recognize a negative allosteric site on Cdc4 in addition to the primary phosphodegron binding pocket. Negative allostery between the primary and allosteric sites on Cdc4 facilitates exchange of phosphodegron motifs that bind to the allosteric site, enabling conformational remodeling and increasing the probability of additional phosphodegron binding events at the primary interaction site [91].
A similar allosteric threshold response to phosphorylation was recently described in the context of T-cell receptor (TCR) activation [92]. The early stages of TCR signaling are driven by tyrosine phosphorylation events in the linker for activation of T-cells (LAT) protein by the kinase ZAP-70 [93, 94]. LAT functions as a signaling scaffold and possesses multiple tyrosine residues that can be phosphorylated to recruit downstream signaling partners such as the SH2-domain containing adaptor protein Grb2 [94]. Using a cleverly designed kinetic assay, Groves and coworkers were able to show that there is a nonlinear relationship between the binding kinetics of LAT:Grb2 and the number of phosphorylated tyrosine residues on LAT [92]. The nonlinearity of the response implies that binding of individual phosphorylated tyrosine residues is coupled, such that single phosphorylation events are not sufficient for Grb2 recruitment and activation of downstream signaling events, but that the signal for Grb2 binding must be amplified by additional phosphorylation events. The kinetics of the process suggest that ZAP-70 phosphorylates LAT in a weakly processive manner, indicating that phosphorylation of a single tyrosine residue in LAT accelerates phosphorylation of successive tyrosines. This processivity enables a sensitive response to upstream signals without unnecessarily triggering downstream signal amplification through single site phosphorylation events [92]. While the structural origins of this allosteric process are still unknown, it is intriguing to speculate that multi-site phosphorylation of LAT leads to remodeling of the conformational ensemble to favor interactions with Grb2 and other cellular partners [92].
In other systems, accumulation of post-translational modifications does not result in an abrupt molecular switch; instead, additional modifications function as a molecular rheostat to control signal output. In the case of the transcription factor Ets-1, transcriptional activity is regulated by a partially disordered auto-inhibitory domain that functions by reducing the affinity of Ets-1 for DNA [95, 96]. Cooperative binding interactions of Ets-1 with its binding partner RUNX1 and DNA allow for relief of auto-inhibition [97] and enable transcription to proceed. Transcriptional inhibition is restored by a series of calcium-dependent phosphorylation events in a serine-rich disordered region, in which successive phosphorylation of serine residues yields a graded increase in transcriptional auto-inhibition [98]. Serine phosphorylation alters the conformational ensemble of Ets-1 by favoring coupled folding and binding of the Ets-1 H1 helix to its own auto-inhibitory module such that DNA binding becomes less favorable and the auto-inhibited state becomes the dominant state in solution. Phosphorylation-induced changes in the conformational ensemble are transmitted through an allosteric network connecting the auto-inhibitory module to the DNA binding site [98, 99].
Allostery in phase separation
There is currently great interest in the role played by intrinsically disordered proteins in liquid-liquid phase separation processes, which mediate compartmentalization and spatial organization of proteins and nucleic acids within the cell through formation of structures commonly referred to as membraneless organelles or, more recently, biomolecular condensates [100–102]. IDPs rely upon weak, multivalent interactions to form these macromolecular assemblies that can function as reserves of critical signaling components [15, 103]. The protein components of biomolecular condensates associate through multivalent interactions that rely on synergy between tandem repeats of modular domains as well as the disordered linkers between them. The neuronal Wiskott-Aldrich syndrome protein (N-WASP) integrates a wide range of cellular inputs to control formation of actin filaments via interactions with the Arp2/3 complex [104]. Interactions between N-WASP and its biological partners Nck and nephrin were shown to promote phase separation in vitro, with the propensity for phase transition depending on the valency of the ligands [103]. The signaling adaptor protein Nck utilizes its three SH3 domains to interact with the six proline-rich motifs of the neuronal Wiskott-Aldrich syndrome protein (N-WASP) [105]. Nck also binds to nephrin through its SH2 domain, which can recognize three tyrosine phosphorylation sites on the cytoplasmic tail of nephrin [106, 107]. Phase separation of Nck, N-WASP, and nephrin is also dependent on a disordered linker between the first two SH3 domains of Nck [108]. Intriguingly, this disordered linker enables Nck to function not only as a driver of phase separation, but also as an allosteric activator of N-WASP—the Nck linker region binds to the N-WASP GTPase binding domain and competes with binding of the N-WASP VCA segment to relieve autoinhibition and promote interactions with the Arp2/3 complex [109]. The multiple functions of the disordered linker between the Nck SH3 domains illustrates the complexity of cellular regulatory processes and suggests that allosteric regulation involving disordered regions may play a major role in phase separation processes.
III. FUTURE PERSPECTIVES
The examples discussed in this review constitute only a first glimpse into the functions of intrinsically disordered proteins in allosteric regulation of cellular signaling. Given the abundance of IDPs and their central role in regulation of the cell, these examples undoubtedly represent only the tip of the iceberg. With growing knowledge of the myriad biological functions of intrinsically disordered proteins, we can anticipate rapid advances in our understanding of the mechanisms by which IDPs exert allosteric control over key cellular processes. Identifying novel allosteric regulatory processes and providing quantitative descriptions of their molecular mechanisms will undoubtedly be challenging. Integrated approaches bridging biological, physical, and chemical disciplines, as already applied in a number of detailed functional studies of disordered proteins [110–115] (including those that participate in allosteric regulatory mechanisms [116, 117]) will be instrumental in expanding our views of how IDPs function within the confines of pre-existing paradigms regarding protein structure and function, including allostery.
Given their role in key cellular regulatory pathways and their involvement in numerous debilitating diseases, intrinsically disordered proteins and their binding partners are of great interest as potential therapeutic targets. The realization that many IDPs function allosterically makes them even more attractive targets for drug design. Indeed, initial success has been reported in development of an allosteric inhibitor that effectively targets the disordered C-terminal region of a protein tyrosine phosphatase [118]. Given the abundance of proteins in the human proteome that contain both ordered and disordered regions that function synergistically to regulate biological activity, this could be a valuable strategy for development of allosteric inhibitors of challenging drug targets. Further studies of allosteric mechanisms involving IDPs will undoubtedly enhance our understanding of this functionally significant class of proteins, shedding new light on their role in regulation and organization of the cell and providing new opportunities for therapeutic intervention.
HIGHLIGHTS.
Intrinsically disordered proteins (IDPs) are important for cellular signaling and regulation
Recent studies highlight the many roles of IDPs in allosteric processes
Acknowledgments
The authors gratefully acknowledge support from grants CA96865 (P.E.W.), CA214054 (P.E.W.), GM113251 (H.J.D.) from the National Institutes of Health and from the Skaggs Institute for Chemical Biology (P.E.W.). R.B.B. was supported by a postdoctoral fellowship from the American Cancer Society (125343-PF-13-202-01-DMC).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Koshland DE, Jr, Nemethy G, Filmer D. Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry. 1966;5:365–85. doi: 10.1021/bi00865a047. [DOI] [PubMed] [Google Scholar]
- 2.Monod J, Wyman J, Changeux JP. On the Nature of Allosteric Transitions: A Plausible Model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 3.Cooper A, Dryden DT. Allostery without conformational change. A plausible model. Eur Biophys J. 1984;11:103–9. doi: 10.1007/BF00276625. [DOI] [PubMed] [Google Scholar]
- 4.Nussinov R, Tsai CJ. Allostery without a conformational change? Revisiting the paradigm. Current opinion in structural biology. 2015;30:17–24. doi: 10.1016/j.sbi.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 5.Hilser VJ, Wrabl JO, Motlagh HN. Structural and energetic basis of allostery. Annu Rev Biophys. 2012;41:585–609. doi: 10.1146/annurev-biophys-050511-102319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kern D, Zuiderweg ER. The role of dynamics in allosteric regulation. Current opinion in structural biology. 2003;13:748–57. doi: 10.1016/j.sbi.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 7.Smock RG, Gierasch LM. Sending signals dynamically. Science. 2009;324:198–203. doi: 10.1126/science.1169377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Popovych N, Sun S, Ebright RH, Kalodimos CG. Dynamically driven protein allostery. Nature structural & molecular biology. 2006;13:831–8. doi: 10.1038/nsmb1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oyen D, Fenwick RB, Stanfield RL, Dyson HJ, Wright PE. Cofactor-Mediated Conformational Dynamics Promote Product Release From Escherichia coli Dihydrofolate Reductase via an Allosteric Pathway. J Am Chem Soc. 2015;137:9459–68. doi: 10.1021/jacs.5b05707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hilser VJ, Thompson EB. Intrinsic disorder as a mechanism to optimize allosteric coupling in proteins. Proc Natl Acad Sci U S A. 2007;104:8311–5. doi: 10.1073/pnas.0700329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van der Lee R, Buljan M, Lang B, Weatheritt RJ, Daughdrill GW, Dunker AK, et al. Classification of intrinsically disordered regions and proteins. Chemical reviews. 2014;114:6589–631. doi: 10.1021/cr400525m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Csizmok V, Follis AV, Kriwacki RW, Forman-Kay JD. Dynamic Protein Interaction Networks and New Structural Paradigms in Signaling. Chemical reviews. 2016;116:6424–62. doi: 10.1021/acs.chemrev.5b00548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haynes C, Oldfield CJ, Ji F, Klitgord N, Cusick ME, Radivojac P, et al. Intrinsic disorder is a common feature of hub proteins from four eukaryotic interactomes. PLoS computational biology. 2006;2:e100. doi: 10.1371/journal.pcbi.0020100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dosztanyi Z, Chen J, Dunker AK, Simon I, Tompa P. Disorder and sequence repeats in hub proteins and their implications for network evolution. J Proteome Res. 2006;5:2985–95. doi: 10.1021/pr060171o. [DOI] [PubMed] [Google Scholar]
- 15.Wright PE, Dyson HJ. Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Biol. 2015;16:18–29. doi: 10.1038/nrm3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bah A, Forman-Kay JD. Modulation of Intrinsically Disordered Protein Function by Post-translational Modifications. J Biol Chem. 2016;291:6696–705. doi: 10.1074/jbc.R115.695056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berlow RB, Dyson HJ, Wright PE. Functional advantages of dynamic protein disorder. FEBS letters. 2015;589:2433–40. doi: 10.1016/j.febslet.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iakoucheva LM, Brown CJ, Lawson JD, Obradovic Z, Dunker AK. Intrinsic disorder in cell-signaling and cancer-associated proteins. J Mol Biol. 2002;323:573–84. doi: 10.1016/s0022-2836(02)00969-5. [DOI] [PubMed] [Google Scholar]
- 19.Tompa P. Multisteric regulation by structural disorder in modular signaling proteins: an extension of the concept of allostery. Chemical reviews. 2014;114:6715–32. doi: 10.1021/cr4005082. [DOI] [PubMed] [Google Scholar]
- 20.Flock T, Weatheritt RJ, Latysheva NS, Babu MM. Controlling entropy to tune the functions of intrinsically disordered regions. Current opinion in structural biology. 2014;26:62–72. doi: 10.1016/j.sbi.2014.05.007. [DOI] [PubMed] [Google Scholar]
- 21.Papaleo E, Saladino G, Lambrughi M, Lindorff-Larsen K, Gervasio FL, Nussinov R. The Role of Protein Loops and Linkers in Conformational Dynamics and Allostery. Chemical reviews. 2016;116:6391–423. doi: 10.1021/acs.chemrev.5b00623. [DOI] [PubMed] [Google Scholar]
- 22.Motlagh HN, Hilser VJ. Agonism/antagonism switching in allosteric ensembles. Proc Natl Acad Sci U S A. 2012;109:4134–9. doi: 10.1073/pnas.1120519109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Motlagh HN, Li J, Thompson EB, Hilser VJ. Interplay between allostery and intrinsic disorder in an ensemble. Biochemical Society transactions. 2012;40:975–80. doi: 10.1042/BST20120163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Motlagh HN, Wrabl JO, Li J, Hilser VJ. The ensemble nature of allostery. Nature. 2014;508:331–9. doi: 10.1038/nature13001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wrabl JO, Gu J, Liu T, Schrank TP, Whitten ST, Hilser VJ. The role of protein conformational fluctuations in allostery, function, and evolution. Biophysical chemistry. 2011;159:129–41. doi: 10.1016/j.bpc.2011.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hilser VJ, Thompson EB. Structural dynamics, intrinsic disorder, and allostery in nuclear receptors as transcription factors. J Biol Chem. 2011;286:39675–82. doi: 10.1074/jbc.R111.278929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li J, Motlagh HN, Chakuroff C, Thompson EB, Hilser VJ. Thermodynamic dissection of the intrinsically disordered N-terminal domain of human glucocorticoid receptor. J Biol Chem. 2012;287:26777–87. doi: 10.1074/jbc.M112.355651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li J, White JT, Saavedra H, Wrabl JO, Motlagh HN, Liu K, et al. Genetically tunable frustration controls allostery in an intrinsically disordered transcription factor. Elife. 2017:6. doi: 10.7554/eLife.30688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Vera IMS, Zheng J, Novick S, Shang J, Hughes TS, Brust R, et al. Synergistic Regulation of Coregulator/Nuclear Receptor Interaction by Ligand and DNA. Structure. 2017;25:1506–18e4. doi: 10.1016/j.str.2017.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Afif H, Allali N, Couturier M, Van Melderen L. The ratio between CcdA and CcdB modulates the transcriptional repression of the ccd poison-antidote system. Mol Microbiol. 2001;41:73–82. doi: 10.1046/j.1365-2958.2001.02492.x. [DOI] [PubMed] [Google Scholar]
- 31.Magnuson R, Yarmolinsky MB. Corepression of the P1 addiction operon by Phd and Doc. J Bacteriol. 1998;180:6342–51. doi: 10.1128/jb.180.23.6342-6351.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Overgaard M, Borch J, Jorgensen MG, Gerdes K. Messenger RNA interferase RelE controls relBE transcription by conditional cooperativity. Mol Microbiol. 2008;69:841–57. doi: 10.1111/j.1365-2958.2008.06313.x. [DOI] [PubMed] [Google Scholar]
- 33.Garcia-Pino A, Christensen-Dalsgaard M, Wyns L, Yarmolinsky M, Magnuson RD, Gerdes K, et al. Doc of prophage P1 is inhibited by its antitoxin partner Phd through fold complementation. J Biol Chem. 2008;283:30821–7. doi: 10.1074/jbc.M805654200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garcia-Pino A, Balasubramanian S, Wyns L, Gazit E, De Greve H, Magnuson RD, et al. Allostery and intrinsic disorder mediate transcription regulation by conditional cooperativity. Cell. 2010;142:101–11. doi: 10.1016/j.cell.2010.05.039. [DOI] [PubMed] [Google Scholar]
- 35.Garcia-Pino A, De Gieter S, Talavera A, De Greve H, Efremov RG, Loris R. An intrinsically disordered entropic switch determines allostery in Phd-Doc regulation. Nat Chem Biol. 2016;12:490–6. doi: 10.1038/nchembio.2078. [DOI] [PubMed] [Google Scholar]
- 36.Blobel GA. CREB-binding protein and p300: molecular integrators of hematopoietic transcription. Blood. 2000;95:745–55. [PubMed] [Google Scholar]
- 37.Giordano A, Avantaggiati ML. p300 and CBP: partners for life and death. J Cell Physiol. 1999;181:218–30. doi: 10.1002/(SICI)1097-4652(199911)181:2<218::AID-JCP4>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 38.Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000;14:1553–77. [PubMed] [Google Scholar]
- 39.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 40.Dyson HJ, Wright PE. Role of Intrinsic Protein Disorder in the Function and Interactions of the Transcriptional Coactivators CREB-binding Protein (CBP) and p300. J Biol Chem. 2016;291:6714–22. doi: 10.1074/jbc.R115.692020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thakur JK, Yadav A, Yadav G. Molecular recognition by the KIX domain and its role in gene regulation. Nucleic acids research. 2014;42:2112–25. doi: 10.1093/nar/gkt1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goto NK, Zor T, Martinez-Yamout M, Dyson HJ, Wright PE. Cooperativity in transcription factor binding to the coactivator CREB-binding protein (CBP). The mixed lineage leukemia protein (MLL) activation domain binds to an allosteric site on the KIX domain. J Biol Chem. 2002;277:43168–74. doi: 10.1074/jbc.M207660200. [DOI] [PubMed] [Google Scholar]
- 43.Radhakrishnan I, Perez-Alvarado GC, Parker D, Dyson HJ, Montminy MR, Wright PE. Solution structure of the KIX domain of CBP bound to the transactivation domain of CREB: a model for activator:coactivator interactions. Cell. 1997;91:741–52. doi: 10.1016/s0092-8674(00)80463-8. [DOI] [PubMed] [Google Scholar]
- 44.De Guzman RN, Goto NK, Dyson HJ, Wright PE. Structural basis for cooperative transcription factor binding to the CBP coactivator. J Mol Biol. 2006;355:1005–13. doi: 10.1016/j.jmb.2005.09.059. [DOI] [PubMed] [Google Scholar]
- 45.Ernst P, Wang J, Huang M, Goodman RH, Korsmeyer SJ. MLL and CREB bind cooperatively to the nuclear coactivator CREB-binding protein. Mol Cell Biol. 2001;21:2249–58. doi: 10.1128/MCB.21.7.2249-2258.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bruschweiler S, Konrat R, Tollinger M. Allosteric communication in the KIX domain proceeds through dynamic repacking of the hydrophobic core. ACS chemical biology. 2013;8:1600–10. doi: 10.1021/cb4002188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bruschweiler S, Schanda P, Kloiber K, Brutscher B, Kontaxis G, Konrat R, et al. Direct observation of the dynamic process underlying allosteric signal transmission. J Am Chem Soc. 2009;131:3063–8. doi: 10.1021/ja809947w. [DOI] [PubMed] [Google Scholar]
- 48.Korkmaz EN, Nussinov R, Haliloglu T. Conformational control of the binding of the transactivation domain of the MLL protein and c-Myb to the KIX domain of CREB. PLoS computational biology. 2012;8:e1002420. doi: 10.1371/journal.pcbi.1002420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Palazzesi F, Barducci A, Tollinger M, Parrinello M. The allosteric communication pathways in KIX domain of CBP. Proc Natl Acad Sci U S A. 2013;110:14237–42. doi: 10.1073/pnas.1313548110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Law SM, Gagnon JK, Mapp AK, Brooks CL., 3rd Prepaying the entropic cost for allosteric regulation in KIX. Proc Natl Acad Sci U S A. 2014;111:12067–72. doi: 10.1073/pnas.1405831111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shammas SL, Travis AJ, Clarke J. Allostery within a transcription coactivator is predominantly mediated through dissociation rate constants. Proc Natl Acad Sci U S A. 2014;111:12055–60. doi: 10.1073/pnas.1405815111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang N, Lodge JM, Fierke CA, Mapp AK. Dissecting allosteric effects of activator-coactivator complexes using a covalent small molecule ligand. Proc Natl Acad Sci U S A. 2014;111:12061–6. doi: 10.1073/pnas.1406033111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Devel. 2000;14:1553–77. [PubMed] [Google Scholar]
- 54.Semenza GL. HIF-1 and human disease: one highly involved factor. Genes Dev. 2000;14:1983–91. [PubMed] [Google Scholar]
- 55.Semenza GL. Hypoxia-inducible factor 1: oxygen homeostasis and disease pathophysiology. Trends Mol Med. 2001;7:345–50. doi: 10.1016/s1471-4914(01)02090-1. [DOI] [PubMed] [Google Scholar]
- 56.Semenza GL. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu Rev Pathol. 2014;9:47–71. doi: 10.1146/annurev-pathol-012513-104720. [DOI] [PubMed] [Google Scholar]
- 57.Arany Z, Huang LE, Eckner R, Bhattacharya S, Jiang C, Goldberg MA, et al. An essential role for p300/CBP in the cellular response to hypoxia. Proc Natl Acad Sci U S A. 1996;93:12969–73. doi: 10.1073/pnas.93.23.12969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gu J, Milligan J, Huang LE. Molecular mechanism of hypoxia-inducible factor 1alpha -p300 interaction. A leucine-rich interface regulated by a single cysteine. J Biol Chem. 2001;276:3550–4. doi: 10.1074/jbc.M009522200. [DOI] [PubMed] [Google Scholar]
- 59.Henze AT, Acker T. Feedback regulators of hypoxia-inducible factors and their role in cancer biology. Cell cycle. 2010;9:2749–63. doi: 10.4161/cc.9.14.12591. [DOI] [PubMed] [Google Scholar]
- 60.Ivan M, Kaelin WG., Jr The EGLN-HIF O2-Sensing System: Multiple Inputs and Feedbacks. Molecular cell. 2017;66:772–9. doi: 10.1016/j.molcel.2017.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bhattacharya S, Michels CL, Leung MK, Arany ZP, Kung AL, Livingston DM. Functional role of p35srj, a novel p300/CBP binding protein, during transactivation by HIF-1. Genes Dev. 1999;13:64–75. doi: 10.1101/gad.13.1.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dames SA, Martinez-Yamout M, De Guzman RN, Dyson HJ, Wright PE. Structural basis for Hif-1 alpha/CBP recognition in the cellular hypoxic response. Proc Natl Acad Sci U S A. 2002;99:5271–6. doi: 10.1073/pnas.082121399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Freedman SJ, Sun ZY, Poy F, Kung AL, Livingston DM, Wagner G, et al. Structural basis for recruitment of CBP/p300 by hypoxia-inducible factor-1 alpha. Proc Natl Acad Sci U S A. 2002;99:5367–72. doi: 10.1073/pnas.082117899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.De Guzman RN, Martinez-Yamout MA, Dyson HJ, Wright PE. Interaction of the TAZ1 domain of the CREB-binding protein with the activation domain of CITED2: regulation by competition between intrinsically unstructured ligands for non-identical binding sites. J Biol Chem. 2004;279:3042–9. doi: 10.1074/jbc.M310348200. [DOI] [PubMed] [Google Scholar]
- 65.Freedman SJ, Sun ZY, Kung AL, France DS, Wagner G, Eck MJ. Structural basis for negative regulation of hypoxia-inducible factor-1alpha by CITED2. Nat Struct Biol. 2003;10:504–12. doi: 10.1038/nsb936. [DOI] [PubMed] [Google Scholar]
- 66.Berlow RB, Dyson HJ, Wright PE. Hypersensitive termination of the hypoxic response by a disordered protein switch. Nature. 2017;543:447–51. doi: 10.1038/nature21705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nussinov R, Tsai CJ. Unraveling structural mechanisms of allosteric drug action. Trends Pharmacol Sci. 2014;35:256–64. doi: 10.1016/j.tips.2014.03.006. [DOI] [PubMed] [Google Scholar]
- 68.Nussinov R, Tsai CJ, Liu J. Principles of allosteric interactions in cell signaling. J Am Chem Soc. 2014;136:17692–701. doi: 10.1021/ja510028c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Byeon IJ, Dao KK, Jung J, Keen J, Leiros I, Doskeland SO, et al. Allosteric communication between cAMP binding sites in the RI subunit of protein kinase A revealed by NMR. J Biol Chem. 2010;285:14062–70. doi: 10.1074/jbc.M110.106666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Guo C, Zhou HX. Unidirectional allostery in the regulatory subunit RIalpha facilitates efficient deactivation of protein kinase A. Proc Natl Acad Sci U S A. 2016;113:E6776–E85. doi: 10.1073/pnas.1610142113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Singh AK, Ekka MK, Kaushik A, Pandya V, Singh RP, Banerjee S, et al. Substrate-Induced Facilitated Dissociation of the Competitive Inhibitor from the Active Site of O-Acetyl Serine Sulfhydrylase Reveals a Competitive-Allostery Mechanism. Biochemistry. 2017;56:5011–25. doi: 10.1021/acs.biochem.7b00500. [DOI] [PubMed] [Google Scholar]
- 72.Bergqvist S, Alverdi V, Mengel B, Hoffmann A, Ghosh G, Komives EA. Kinetic enhancement of NF-kappaBxDNA dissociation by IkappaBalpha. Proc Natl Acad Sci U S A. 2009;106:19328–33. doi: 10.1073/pnas.0908797106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kamar RI, Banigan EJ, Erbas A, Giuntoli RD, Olvera de la Cruz M, Johnson RC, et al. Facilitated dissociation of transcription factors from single DNA binding sites. Proc Natl Acad Sci U S A. 2017;114:E3251–E7. doi: 10.1073/pnas.1701884114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tsai MY, Zhang B, Zheng W, Wolynes PG. Molecular Mechanism of Facilitated Dissociation of Fis Protein from DNA. J Am Chem Soc. 2016;138:13497–500. doi: 10.1021/jacs.6b08416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kim PM, Sboner A, Xia Y, Gerstein M. The role of disorder in interaction networks: a structural analysis. Mol Syst Biol. 2008;4:179. doi: 10.1038/msb.2008.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ferrari R, Pellegrini M, Horwitz GA, Xie W, Berk AJ, Kurdistani SK. Epigenetic reprogramming by adenovirus e1a. Science. 2008;321:1086–8. doi: 10.1126/science.1155546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.White E. Regulation of the cell cycle and apoptosis by the oncogenes of adenovirus. Oncogene. 2001;20:7836–46. doi: 10.1038/sj.onc.1204861. [DOI] [PubMed] [Google Scholar]
- 78.Ferreon AC, Ferreon JC, Wright PE, Deniz AA. Modulation of allostery by protein intrinsic disorder. Nature. 2013;498:390–4. doi: 10.1038/nature12294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ferreon JC, Martinez-Yamout MA, Dyson HJ, Wright PE. Structural basis for subversion of cellular control mechanisms by the adenoviral E1A oncoprotein. Proc Natl Acad Sci U S A. 2009;106:13260–5. doi: 10.1073/pnas.0906770106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Iakoucheva LM, Radivojac P, Brown CJ, O’Connor TR, Sikes JG, Obradovic Z, et al. The importance of intrinsic disorder for protein phosphorylation. Nucleic acids research. 2004;32:1037–49. doi: 10.1093/nar/gkh253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pejaver V, Hsu WL, Xin F, Dunker AK, Uversky VN, Radivojac P. The structural and functional signatures of proteins that undergo multiple events of post-translational modification. Protein Sci. 2014;23:1077–93. doi: 10.1002/pro.2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tompa P, Davey NE, Gibson TJ, Babu MM. A million peptide motifs for the molecular biologist. Molecular cell. 2014;55:161–9. doi: 10.1016/j.molcel.2014.05.032. [DOI] [PubMed] [Google Scholar]
- 83.Csizmok V, Forman-Kay JD. Complex regulatory mechanisms mediated by the interplay of multiple post-translational modifications. Curr Opin Struct Biol. 2018;48:58–67. doi: 10.1016/j.sbi.2017.10.013. [DOI] [PubMed] [Google Scholar]
- 84.Galea CA, Wang Y, Sivakolundu SG, Kriwacki RW. Regulation of cell division by intrinsically unstructured proteins: intrinsic flexibility, modularity, and signaling conduits. Biochemistry. 2008;47:7598–609. doi: 10.1021/bi8006803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Grimmler M, Wang Y, Mund T, Cilensek Z, Keidel EM, Waddell MB, et al. Cdk-inhibitory activity and stability of p27Kip1 are directly regulated by oncogenic tyrosine kinases. Cell. 2007;128:269–80. doi: 10.1016/j.cell.2006.11.047. [DOI] [PubMed] [Google Scholar]
- 86.Galea CA, Nourse A, Wang Y, Sivakolundu SG, Heller WT, Kriwacki RW. Role of intrinsic flexibility in signal transduction mediated by the cell cycle regulator, p27 Kip1. J Mol Biol. 2008;376:827–38. doi: 10.1016/j.jmb.2007.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nash P, Tang X, Orlicky S, Chen Q, Gertler FB, Mendenhall MD, et al. Multisite phosphorylation of a CDK inhibitor sets a threshold for the onset of DNA replication. Nature. 2001;414:514–21. doi: 10.1038/35107009. [DOI] [PubMed] [Google Scholar]
- 88.Borg M, Mittag T, Pawson T, Tyers M, Forman-Kay JD, Chan HS. Polyelectrostatic interactions of disordered ligands suggest a physical basis for ultrasensitivity. Proc Natl Acad Sci U S A. 2007;104:9650–5. doi: 10.1073/pnas.0702580104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mittag T, Marsh J, Grishaev A, Orlicky S, Lin H, Sicheri F, et al. Structure/function implications in a dynamic complex of the intrinsically disordered Sic1 with the Cdc4 subunit of an SCF ubiquitin ligase. Structure. 2010;18:494–506. doi: 10.1016/j.str.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mittag T, Orlicky S, Choy WY, Tang X, Lin H, Sicheri F, et al. Dynamic equilibrium engagement of a polyvalent ligand with a single-site receptor. Proc Natl Acad Sci U S A. 2008;105:17772–7. doi: 10.1073/pnas.0809222105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Csizmok V, Orlicky S, Cheng J, Song J, Bah A, Delgoshaie N, et al. An allosteric conduit facilitates dynamic multisite substrate recognition by the SCF(Cdc4) ubiquitin ligase. Nat Commun. 2017;8:13943. doi: 10.1038/ncomms13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Huang WYC, Ditlev JA, Chiang HK, Rosen MK, Groves JT. Allosteric Modulation of Grb2 Recruitment to the Intrinsically Disordered Scaffold Protein, LAT, by Remote Site Phosphorylation. J Am Chem Soc. 2017;139:18009–15. doi: 10.1021/jacs.7b09387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Balagopalan L, Coussens NP, Sherman E, Samelson LE, Sommers CL. The LAT story: a tale of cooperativity, coordination, and choreography. Cold Spring Harb Perspect Biol. 2010;2:a005512. doi: 10.1101/cshperspect.a005512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Samelson LE. Signal transduction mediated by the T cell antigen receptor: the role of adapter proteins. Annu Rev Immunol. 2002;20:371–94. doi: 10.1146/annurev.immunol.20.092601.111357. [DOI] [PubMed] [Google Scholar]
- 95.Petersen JM, Skalicky JJ, Donaldson LW, McIntosh LP, Alber T, Graves BJ. Modulation of transcription factor Ets-1 DNA binding: DNA-induced unfolding of an alpha helix. Science. 1995;269:1866–9. doi: 10.1126/science.7569926. [DOI] [PubMed] [Google Scholar]
- 96.Wang H, McIntosh LP, Graves BJ. Inhibitory module of Ets-1 allosterically regulates DNA binding through a dipole-facilitated phosphate contact. J Biol Chem. 2002;277:2225–33. doi: 10.1074/jbc.M109430200. [DOI] [PubMed] [Google Scholar]
- 97.Goetz TL, Gu TL, Speck NA, Graves BJ. Auto-inhibition of Ets-1 is counteracted by DNA binding cooperativity with core-binding factor alpha2. Mol Cell Biol. 2000;20:81–90. doi: 10.1128/mcb.20.1.81-90.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pufall MA, Lee GM, Nelson ML, Kang HS, Velyvis A, Kay LE, et al. Variable control of Ets-1 DNA binding by multiple phosphates in an unstructured region. Science. 2005;309:142–5. doi: 10.1126/science.1111915. [DOI] [PubMed] [Google Scholar]
- 99.Lee GM, Donaldson LW, Pufall MA, Kang HS, Pot I, Graves BJ, et al. The structural and dynamic basis of Ets-1 DNA binding autoinhibition. J Biol Chem. 2005;280:7088–99. doi: 10.1074/jbc.M410722200. [DOI] [PubMed] [Google Scholar]
- 100.Shin Y, Brangwynne CP. Liquid phase condensation in cell physiology and disease. Science. 2017:357. doi: 10.1126/science.aaf4382. [DOI] [PubMed] [Google Scholar]
- 101.Mitrea DM, Kriwacki RW. Phase separation in biology; functional organization of a higher order. Cell Commun Signal. 2016;14:1. doi: 10.1186/s12964-015-0125-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Banani SF, Lee HO, Hyman AA, Rosen MK. Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol. 2017;18:285–98. doi: 10.1038/nrm.2017.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Li P, Banjade S, Cheng HC, Kim S, Chen B, Guo L, et al. Phase transitions in the assembly of multivalent signalling proteins. Nature. 2012;483:336–40. doi: 10.1038/nature10879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Padrick SB, Rosen MK. Physical mechanisms of signal integration by WASP family proteins. Annu Rev Biochem. 2010;79:707–35. doi: 10.1146/annurev.biochem.77.060407.135452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rohatgi R, Nollau P, Ho HY, Kirschner MW, Mayer BJ. Nck and phosphatidylinositol 4,5-bisphosphate synergistically activate actin polymerization through the N-WASP-Arp2/3 pathway. J Biol Chem. 2001;276:26448–52. doi: 10.1074/jbc.M103856200. [DOI] [PubMed] [Google Scholar]
- 106.Jones N, Blasutig IM, Eremina V, Ruston JM, Bladt F, Li H, et al. Nck adaptor proteins link nephrin to the actin cytoskeleton of kidney podocytes. Nature. 2006;440:818–23. doi: 10.1038/nature04662. [DOI] [PubMed] [Google Scholar]
- 107.Blasutig IM, New LA, Thanabalasuriar A, Dayarathna TK, Goudreault M, Quaggin SE, et al. Phosphorylated YDXV motifs and Nck SH2/SH3 adaptors act cooperatively to induce actin reorganization. Mol Cell Biol. 2008;28:2035–46. doi: 10.1128/MCB.01770-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Banjade S, Wu Q, Mittal A, Peeples WB, Pappu RV, Rosen MK. Conserved interdomain linker promotes phase separation of the multivalent adaptor protein Nck. Proc Natl Acad Sci U S A. 2015;112:E6426–35. doi: 10.1073/pnas.1508778112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Okrut J, Prakash S, Wu Q, Kelly MJ, Taunton J. Allosteric N-WASP activation by an inter-SH3 domain linker in Nck. Proc Natl Acad Sci U S A. 2015;112:E6436–45. doi: 10.1073/pnas.1510876112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Theillet FX, Binolfi A, Bekei B, Martorana A, Rose HM, Stuiver M, et al. Structural disorder of monomeric alpha-synuclein persists in mammalian cells. Nature. 2016;530:45–50. doi: 10.1038/nature16531. [DOI] [PubMed] [Google Scholar]
- 111.Milles S, Mercadante D, Aramburu IV, Jensen MR, Banterle N, Koehler C, et al. Plasticity of an Ultrafast Interaction between Nucleoporins and Nuclear Transport Receptors. Cell. 2015;163:734–45. doi: 10.1016/j.cell.2015.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Borcherds W, Theillet FX, Katzer A, Finzel A, Mishall KM, Powell AT, et al. Disorder and residual helicity alter p53-Mdm2 binding affinity and signaling in cells. Nat Chem Biol. 2014;10:1000–2. doi: 10.1038/nchembio.1668. [DOI] [PubMed] [Google Scholar]
- 113.Boeynaems S, Bogaert E, Kovacs D, Konijnenberg A, Timmerman E, Volkov A, et al. Phase Separation of C9orf72 Dipeptide Repeats Perturbs Stress Granule Dynamics. Molecular cell. 2017;65:1044–55e5. doi: 10.1016/j.molcel.2017.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gustafson CL, Parsley NC, Asimgil H, Lee HW, Ahlbach C, Michael AK, et al. A Slow Conformational Switch in the BMAL1 Transactivation Domain Modulates Circadian Rhythms. Molecular cell. 2017;66:447–57e7. doi: 10.1016/j.molcel.2017.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Monahan Z, Ryan VH, Janke AM, Burke KA, Rhoads SN, Zerze GH, et al. Phosphorylation of the FUS low-complexity domain disrupts phase separation, aggregation, and toxicity. EMBO J. 2017;36:2951–67. doi: 10.15252/embj.201696394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Follis AV, Chipuk JE, Fisher JC, Yun MK, Grace CR, Nourse A, et al. PUMA binding induces partial unfolding within BCL-xL to disrupt p53 binding and promote apoptosis. Nat Chem Biol. 2013;9:163–8. doi: 10.1038/nchembio.1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Follis AV, Llambi F, Kalkavan H, Yao Y, Phillips AH, Park CG, et al. Regulation of apoptosis by an intrinsically disordered region of Bcl-xL. Nat Chem Biol. 2018 doi: 10.1038/s41589-018-0011-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Krishnan N, Koveal D, Miller DH, Xue B, Akshinthala SD, Kragelj J, et al. Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nat Chem Biol. 2014;10:558–66. doi: 10.1038/nchembio.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]