

Abstract
Insulin secretion is stimulated by glucose metabolism and inhibited by catecholamines through adrenergic receptor stimulation. We determined whether catecholamines suppress oxidative metabolism in β-cells through adrenergic receptors. In Min6 cells and isolated rat islets, epinephrine decreased oxygen consumption rates compared to vehicle control or co-administration of epinephrine with α2-adrenergic receptor antagonist yohimbine. Epinephrine also decreased forskolin-stimulated oxygen consumption rates, indicating cAMP dependent and independent actions. Furthermore, glucose oxidation rates were decreased with epinephrine, independent of the exocytosis of insulin, which was blocked with yohimbine. We evaluated metabolic targets through proteomic analysis after four hour epinephrine exposure that revealed 466 differentially expressed proteins that were significantly enriched for processes including oxidative metabolism, protein turnover, exocytosis, and cell proliferation. These results demonstrate that acute α2-adrenergic stimulation suppresses glucose oxidation in β-cells independent of nutrient availability and insulin exocytosis, while cAMP concentrations are elevated. Proteomics and immunoblots revealed changes in electron transport chain proteins that were correlated with lower metabolic reducing equivalents, intracellular ATP concentrations, and altered mitochondrial membrane potential implicating a new role for adrenergic control of mitochondrial function and ultimately insulin secretion.
Keywords: Pancreatic Islet, β-cell Metabolism, Proteomics, Adrenergic Receptor, Insulin Secretion, Oxidative Phosphorylation
1. Introduction
Pancreatic β-cells secrete insulin to regulate glucose homeostasis. The primary mechanism underlying glucose stimulated insulin secretion (GSIS) is the metabolism of glucose to increase the ratio of ATP/ADP, which closes ATP-sensitive K+ channels and depolarizes the plasma membrane (Giannaccini et al 1998, Seino 1999). This activates voltage dependent Ca2+ channels causing an influx of Ca2+ that stimulates insulin exocytosis (Malaisse et al 1978). β-cells optimize coupling between blood glucose concentration and insulin secretion through metabolic coupling factors and mitochondrial shuttles (Prentki et al 2013).
Norepinephrine and epinephrine are catecholamines (CA) released from synaptic nerves and the adrenal gland that mediate systemic responses to nutritional stressors (Cannon and De la Paz 1911). CA mobilize plasma glucose by suppressing insulin secretion from the pancreatic β-cell (Katada and Ui 1981, Laychock and Bilgin 1987) Nine isoforms of adrenergic receptors (ADR) mediate the actions of CA (Roth et al 1991). Predominantly the β isoforms (ADRβ1, ADRβ2, ADRβ3) couple to stimulatory G proteins (Gs). While, α isoforms (ADRα1A, ADRα1B, ADRα1C) couple to Gq and (ADRα2A, ADRα2B, ADRα2C) couple to inhibitory G proteins (Gi), however other G proteins, such as Gz and Go have recently been associated with ADR (Straub and Sharp 2012). Islets from all species examined express several isoforms of ADR but α2-ADR isoforms are responsible for CA inhibition of insulin secretion from β-cells (Kelly et al 2014, Lacey et al 1996, Urano et al 2004, Kelly et al 2017). Several mechanisms for α2-ADR inhibition of GSIS are described: 1) activation of ATP-sensitive K+ channels (KATP) and repolarization of the β-cells, 2) inhibition of L-type Ca2+ channels, 3) decreased activity of adenylyl cyclase (AC) and 4) inhibition of distal exocytosis through interactions with members of the SNARE protein family (Zhao et al 2010c, Zhao et al 2010b, Keahey et al 1989).
Although the mechanisms described for CA inhibition of insulin secretion have focused on the modulation of insulin secretion, the effects of CA through ADR to influence β-cell metabolism and trigger insulin secretion remained undefined. In this study we determined whether CA altered oxidative glucose metabolism in β-cells. CA actions on glycolytic metabolism in other metabolic tissues have been described, but to our knowledge there is no work aimed to elucidate the potential role for α2-ADR on oxidative phosphorylation in β-cells. Metabolic suppression in β-cells would expand CA-mediated mechanisms to inhibit insulin secretion given the tight coupling of cellular ATP production and insulin release. In addition to fully characterizing CA actions to suppress oxygen consumption rates in β-cells, proteomic approaches identify regulatory targets in oxidative metabolism that may explain suppressed glucose oxidation rates.
2. Material and Methods
2.1 Min6 Culture
Min6 cells (Miyazaki et al 1990) were cultured in RPMI 1640 (GIBCO 31800-022, Life Technologies, Carlsbad, CA) containing 11.1 mM glucose and 2 mM glutamine and fortified with 10 mM HEPES, 1 mM Sodium Pyruvate, 10% fetal bovine serum (Atlas Biologicals, Fort Collins, CO), and 1% penicillin-streptomycin (Sigma-Aldrich, St Louis, MO). Cells were cultured at 37°C with 5% CO2. When cells reached 80–90% confluency, cells were passaged (1:3) with 0.5% trypsin+EDTA (ThermoFisher, Rockford, IL). Experiments were conducted on Min6 cells between 35 and 45 passages. Before experiments, cells were cultured in 1 mM glucose RPMI 1640 supplemented with 10% FBS for 24 hours (h).
2.2 Islet Isolation & Culture
Sprague-Dawley male rats and B6 mice were humanely euthanized with the approval of and in accordance with guidelines established by the Institutional Animal Care and Use Committee at The University of Arizona standards. Islets were collected with a modified pancreatic digestion with Collagenase IV (Sigma-Aldrich) and subsequent Histopaque (Sigma-Aldrich) purification as described previously (Hiscox et al 2008). Rat and mouse islets were handpicked and cultured in RPMI 1640 supplemented with 11.1 mM glucose and 10% FBS for 24–48 h in an oxygen chamber supplemented with 95% O2 and 5% CO2 in a humidified incubator at 37°C.
2.3 Static Insulin Secretion Assay
Min6 were plated at a density of 100,000 cells per well in 24-well plates and grown to 70–80% confluency. Min6 cells were rinsed and incubated with 0 mM glucose Krebs Ringer Buffer (KRB; 115 mM NaCl, 5.9 mM KCl, 1.2 mM MgCl2, 1.2 mM NaH2PO4, 1.2 mM Na2SO4, 2.5 mM CaCl2, and 25 mM NaHCO3, pH 7.4) supplemented with 0.5% bovine serum albumin (BSA; Fisher Scientific, Waltham, MA) for 1 h. After preincubation the KRB was aspirated and Min6 cells were incubated in 1 ml of KRB supplemented with 0 mM glucose, 1 mM glucose or 20 mM glucose. To determine epinephrine dose response, epinephrine (Vedco Inc, St. Joseph, MO) was added (0–300 nM) for 1h, after which 500 l of the buffer was collected, and frozen at −80°C until insulin concentrations were measured. Total insulin content was collected from lysed cells by cold acid-ethanol extraction. The experiment was repeated three times (n=3; independent days) with four technical replicates per incubation condition. Insulin secretion assays were performed as described but replicated three additional times (n=3) with three technical replicates per incubation condition in the presence or absence of 100 M nifedipine (Sigma) and/or 250 M diazoxide (Sigma). Insulin concentrations were measured with mouse insulin ELISA (ALPCO Diagonstics, Windham, NH). The intra- and inter-assay variation was 5.4 ± 2.6 and 9.8 ± 2.1 respectively.
2.4 Expression of Adrenergic Receptor Transcripts
RNA was isolated from Min6, BTC3, INS-1, isolated mouse islets, and whole mouse fetus (positive control). Whole mouse fetus used for the positive control was obtained from all fetuses within a single pregnancy, snap frozen, and stored at −80°C until RNA extraction. Mouse islet RNA was extracted from two pooled isolations. All cell lines used for RNA extraction were cultured according to methods described for Min6 culture for at least 72 h. Cells were collected for RNA extraction from a single passage. RNA was extracted using the RNeasy Micro Kit (Qiagen, Valencia, CA) with DNAse 1 clean-up and eluted into RNAse free water. RNA quality and concentration was assessed using absorbance at 260 and 280 nm with a NanoDrop ND-1000 Spectrophotometer (NanoDrop, Wilmington, DE) before being reverse transcribed into cDNA with Superscript III (Invitrogen Life, Grand Island, NY). PCR was performed using primers designed against rodent sequences for the nine ADR isoforms (sequenced PCR products available upon request; Supplemental Table 1). The PCR products were separated by electrophoresis on a 1% agarose gel and visualized with ethidium bromide using ultraviolet light and a camera.
2.5 Oxygen Consumption
Using Instech’s MicroOxygen Uptake System (FO/SYSZ-P250 Plymouth Meeting, PA), oxygen consumption rates (OCR) were tested in cells and isolated islets for changes in response to glucose, epinephrine, adrenergic antagonists, and metabolic inhibitors (Papas et al 2007). Experiments in complete media refer to RPMI 1640 supplemented with 20 mM glucose. The disappearance of oxygen pressure (pO2) was plotted in real time to generate slopes (ΔpO2/Δt). An experimental batch of cells corresponds to one million Min6 cells or 100 isolated rat islets. On each experimental batch of cells test compounds were added and ambient ΔpO2 re-established, and repeat measurements made. Changes in OCR were recorded immediately after test compounds were added to cells and recorded for up to 20 minutes (min) following addition of the test compounds. A treatment period within an experiment was the average of two consecutive slopes, starting with a baseline period (initial OCR; Period 1) and then the addition of test compounds in Period 2 and Period 3. Initial experiments with Min6 cells and islets were performed with paired vehicle controls to determine the loss of OCR following repeated measurements. Repeated measurements of OCR in Min6 cells did not significantly decrease OCR (Supplemental Figure 1). However, vehicle controls were used to formulate correction factors of 8% for the treatment period (Period 2) and 15% for the subsequent treatment period (Period 3), which were applied only to experiments without paired vehicle controls. OCR was calculated via OCR = V×β×(ΔpO2/Δt). Where V is the volume of the chamber and β is Bunsen’s solubility coefficient for oxygen at 37°C, taken as 1.27 nmol/cm3×mmHg (Papas et al 2007). Each ΔpO2/Δt was verified with linear regression (R2 ≥ 0.88). A typical experimental set up was analyzed using the ratio of change from baseline period (initial OCR) to average of each treatment period after the appropriate correction factor.
2.6 Glucose Oxidation Rate
Glucose oxidation rates were measured using [U-14C] D-glucose (PerkinElmer, Waltham, MA). Min6 cells (2×105 cells/reaction) were suspended in a 1 ml Nunc CryoTube affixed with epoxy inside a 20 ml glass scintillation vial (n > 5 per incubation condition) and sealed with a Suba-Seal Rubber Septa (Sigma-Aldrich). Cells were incubated in KRB + 0.05% BSA containing D-glucose (1.1, 7.5, or 20 mM) and [U-14C] D-glucose (8, 15, 20 µCi/ml, respectively), then pre-equilibrated in 95% O2/5% CO2 atmosphere at 37°C for 2 h (Ashcroft et al 1970) Blank reactions without cells were included to determine background levels. Reaction vials were cooled rapidly and the reaction stopped with 100 µl of 1N HCl. Solvable (0.5 ml; PerkinElmer) was added to the base of the scintillation vial to trap 14CO2. Vials were incubated at 37°C with gentle agitation for 90 min. The rubber stopper and reaction tube were removed, Ultima Gold scintillation fluid (2 ml; PerkinElmer) was added, and captured 14CO2 disintegrations per minute (dpm) determined with a LS5200 liquid scintillation analyzer (Beckman Coulter, Brea, CA). The specific activity of glucose in the KRB+0.05% BSA was determined. Glucose oxidation rate (moles/cell/h) was calculated by subtracting the blank dpm from the cell-incubated dpm, dividing by the specific activity for glucose, and dividing by the time of the incubation (Limesand et al 2006).
2.7 Cellular Redox
Min6 cells were seeded in black, clear bottom, tissue culture treated 96-well microplates (Corning Inc., Corning, NY) at a density of 10,000 cells/well. After overnight culture under standard conditions, cells were incubated in culture medium containing 100 nM epinephrine or vehicle control. Resazurin dye alamarBlue (Invitrogen) was added to each well and fluorescence was measured immediately (time zero) using a SpectraMax M2 with excitation and emission wavelength of 535 and 595 nm, respectively. For background subtraction, background fluorescence was determined for each experimental treatment from blank, cell-free wells containing incubation medium only. Plates were incubated for 4 h at 37°C and mea sured again. Values at 4 h were normalized to zero values.
2.8 ATP content
Confluent Min6 (n=8 wells/treatment from unique passages) were exposed to 100 nM epinephrine or vehicle control in standard culture media for 20 minutes. Media was aspirated and cells were rinsed with cold phosphate buffered saline (PBS). Cell lysate was collected and ATP content was determined using ATP Detection Assay Kit (Cayman Chemical, Ann Arbor, MI). Briefly, samples were run in duplicate, read with a Clarity Luminescence Microplate reader (BioTek, Winooski, VT), and sample content was calculated from a standard curve and normalized to cell number.
2.9 Mitochondrial Membrane Potential
Min6 cells were plated onto #1 glass coverslips and cultured as above for 4–5 days. Tetramethylrhodamine ethyl ester perchlorate (TMRE; Sigma), a membrane potential-sensitive probe, was used to monitor mitochondrial membrane potential (MMP). For excitation of fluorescence, white light was passed through a 10 nm band pass filter centered at 530 nm while the emitted light was passed through a 50 nm band pass filter centered at 580 nm. Cells were placed in stimulatory conditions in a 37°C chamber with Hank’s Balanced Salt Solution (HBSS) containing (in mM) 5 KCl, 0.3 KH2PO4, 138 NaCl, 0.2 NaHCO3, 0.3 Na2HPO4, 20 HEPES, 1.3 CaCl2, 0.4 MgSO4, 5.0 L-Glutamine and 5.6 glucose (pH 7.3). Cells were loaded with the TMRE by adding 100 nM to HBSS and incubating in chamber for 15 min. Baseline images were acquired every minute for a total of 5 min. Following epinephrine (100 nM) addition, images were acquired at 0.5, 1, 2, 3 and 5 min. On average, groups of 10–20 cells were imaged for each experiment. For quantification, a region of interest (ROI) was drawn around a mitochondrial dense region to obtain the average pixel intensity within that region. The maximum pixel intensity within the 5 min range of following addition of epinephrine was divided by the average baseline intensity to obtain the percent change of baseline. Pixel intensities were corrected by subtraction of a blank intensity prior to loading TMRE. Cyanide and FCCP (Sigma) were used to induce depolarization.
2.10 Protein Sample Preparation For Proteomic Analysis
Confluent Min6 were exposed to 4 h epinephrine or vehicle control and collected in three unique experiments. Each experiment included a control and treatment and was conducted on a different day and cell passage. Cell pellets were washed with PBS and lysed with 500 µl hexafluroisopropanol in a bath sonicator for 5 min. The lysate was transferred to 1.5 ml tubes and evaporated using vacuum centrifugation until dry. Reduction, alkylation, and trypsin digestion were performed as described previously (He et al 2016) Briefly, each tube of dried lysate was rehydrated using 10 µl of 100 mM ammonium bicarbonate (ABC). After 5 min, 10 µl of 100 mM dithiothreitol (Sigma 43815), 100 mM ABC was added to each tube and incubated to 30 min at 85°C. Once cooled to room temperature, 10 µl of 100 mM iodoacetamide (Sigma I1149), 100 mM ABC was added and samples were incubated for 30 min at room temperature in the dark. Two µg trypsin (Sigma T6567) dissolved in 170 µl 100 mM ABC was added to each tube (final volume 200 µl) and all were incubated overnight at 37°C using a ThermoMixer (Eppendorf, Hamburg, Germany) with continuous 600 RPM shaking.
Following digestion, each sample was desalted using a peptide trap (Optimize Technologies 10-04817-TN, Oregon City, OR) according to the manufacturer’s instructions. After desalting, samples were further cleaned using a strong cation exchange trap (Optimize Technologies 10-04817-TP) to remove hydrophobic compounds, which could interfere with the mass spectrometry. Samples were then desalted a second time using the same peptide trap as above. Samples were dried and resuspended in 10 µL of 2% acetonitrile (ACN), 0.1% formic acid (FA) and transferred to low retention vials in preparation for analysis using LC-MS/MS.
2.11 Proteomic Analysis by Mass Spectrometry
Peptides were analyzed using a Dionex UltiMate 3000 (ThermoFisher) high performance liquid chromatography machine (HPLC) coupled with an LTQ Velos Pro (ThermoFisher) tandem mass spectrometer. The HPLC was configured for reverse phase chromatography using an in-house prepared C18 column. The column, 75 µm by 10 cm, was packed with Halo 2.7 µm, 90Å C18 material (MAC-MOD Analytical Inc., Chadds Ford, PA), and located in the ion source just before a silica emitter tip. Peptides were separated for mass spectrometry analysis using a 4hr acetonitrile gradient starting at 2% ACN, 0.1% FA and reaching 50% ACN, 0.1% FA in 240 min, followed by a 15 min wash of 95% ACN, 0.1% FA. Flow was set at a constant 333 nl/min. Peptides were ionized using a Nanospray Flex ion source (ThermoFisher). High voltage was applied using a stainless steel liquid junction with the column located on the inlet side, and a silica emitter tip on the outlet side, minimizing dead volume between the column and emitter. Scan parameters for the LTQ Velos Pro were one MS scan followed by 10 MS/MS scans of the 5 most intense peaks. MS/MS scans were performed in pairs, a CID fragmentation scan followed a HCD fragmentation scan. All scans were performed in enhanced resolution mode. Dynamic exclusion was enabled with a mass exclusion time of 3 min and a repeat count of 1 within 30 s of initial m/z measurement.
Raw mass spectra were converted to MGF format for spectrum matching using the MSConvert program from the ProteoWizard software suite (Chambers et al 2012, Kessner et al 2008). The X! Tandem and OMSSA algorithms were used to match ms/ms spectra to the FASTA databases (Craig and Beavis 2004, Geer et al 2004) Precursor and fragment mass tolerances were set to 0.25 Da for both X! Tandem and OMSSA. Tryptic cleavage rules with ≤2 missed cleavages were used when calculating in-silico peptide precursor masses; a, b, and y ions were used for fragment m/z matching. Amino acid modifications that were included in the database searches were single and double oxidation of methionine, oxidation of proline, carbamidomethylation of cysteine, and deamidation of asparagine and glutamine. The FASTA database used for spectrum matching was the mouse protein set available from UniProt on January 28, 2015. A randomized version of the database was used calculate false discovery rates (FDR). Peptide-spectrum matches with e-values <0.01 were accepted for down-stream analysis. Peptide matches were organized by protein using Perl, at which time proteins identified by a single spectrum were removed. The mass spectrometry proteomics data has been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD006330 (Vizcaino et al 2014).
Differential expression of proteins between treatments was performed pairwise using peptide elution profiles as described by Wright et al (Wright et al 2016) Precursor mass spectra were extracted from the raw data in MS1 format using the MSConvert software from the ProteoWizard toolset (Chambers et al 2012, Kessner et al 2008). Peptide precursor m/z values were extracted from the previously compiled protein identifications using Perl. Peptide intensities were summed for each protein on a per-replicate basis. Data were normalized based on the mode of each replicate rather than the mean to minimize the effect of extreme values. A resampling analysis was performed for each pairwise comparison. Proteins were considered to be differentially expressed if the difference in means between conditions resulted in a P < 0.05 and the difference in means between one of the conditions and its baseline was P < 0.05. Differentially expressed proteins were filtered for redundancy and analyzed for functional annotations using online databases with KOBAS 3.0 for KEGG pathways and GO Terms (Xie et al 2011) Significance was determined if P<0.05 following Benjamini-Hochberg multiple test correction.
2.12 Immunoblot Analysis
Proteins were extracted from a confluent monolayer of Min6 cells (n=3 independent days) with Cell Lytic Reagent (Sigma-Aldrich), cell lysates were scraped, collected into tubes, and centrifuged at 13,000 g for 20 minutes. Supernatant protein concentration was measured with Pierce BCA assay (ThermoFisher). Immunoblots were performed as described previously (Limesand et al 2007, Camacho et al 2017). Briefly, protein lysates (20 or 30 µg) were separated by electrophoresis on a 10% or 12% Tris‐glycine gel and transferred onto polyvinylidene difluoride membranes (Bio‐Rad, Hercules, CA, USA). Transferred protein was stained using MemCode (ThermoFisher), photographed, and then removed. The membrane was blocked with 5% nonfat dry milk in TBS-T (20 mM Tris–HCl, 0.5 M NaCl, and 0.1% Tween‐20) and incubated with primary antibodies at 4°C for 24 h, followed by washing with TBS-T. Immunocomplexes were detected with horseradish peroxidase (HRP)‐conjugated secondary antibodies and enhanced chemiluminescence solution (32106; Thermo Scientific, Waltham, MA, USA). Primary antibodies used in this study included β-tubulin (Santa Cruz, sc66175), PSMB1 (ThermoFisher PA5-56219), ACSS2 (Santa Cruz, G1516), ATP6V0A1 (Abcam ab105937), and an OXPHOS cocktail (abcam ab110413) that included one each against CI subunit NDUFB8 (ab110242), CII-30kDa (ab14714), CIII-Core protein 2 (ab14745), CIV subunit I (ab14705) and CV alpha subunit (ab14748) as an optimized premixed cocktail. Protein levels were quantified using photographed images and densiometric analysis with ImageJ (Schneider et al 2012).
2.13 Statistical Analysis
Dose response curves and the IC50 for epinephrine were calculated with Prism (v7.0, Graphpad Software Inc., La Jolla, CA). Oxygen consumption rates were normalized as a percentage of baseline measurements (% of baseline) and statistical analysis was performed on the difference between baseline and treatment. Glucose oxidation rates were normalized as a percentage of control cells in stimulatory (20 mM) glucose. Protein quantification by immunoblot was normalized to β-tubulin or total protein where applicable. Differences in mean oxygen consumption rates, glucose oxidation rates, mitochondrial metabolic assays, and immunoblot protein quantification were analyzed by one-way ANOVA using general linear means procedure of SAS software (version 9.4; SAS Institute, Cary, NC), and differences were determined with a post hoc least significant difference test. Values were significant if P < 0.05.
3. Results
3.1 Min6 cells respond to epinephrine similar to islets
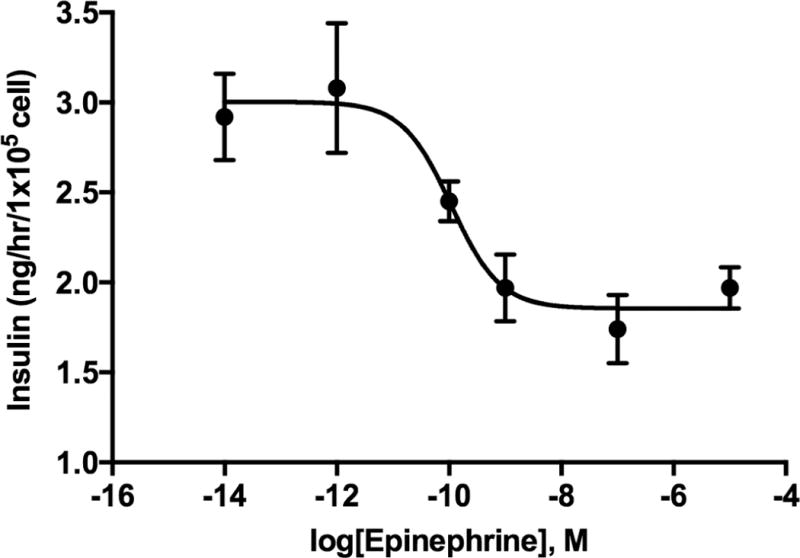
Adra1d and Adra2a, Adra2b, and Adra2c were expressed in Min6 cells and were similar to mouse islet expression of ADR mRNAs, with the exception of the presence of Adrb2 and Adra1b in mouse islets (Figure 1). The mRNA for α2A-ADR and α2C-ADR were present in mouse islets and rodent cell lines. The effective concentration of epinephrine to inhibit insulin secretion was determined in Min6 cells with a dose response curve (IC50 = 108 pM; Figure 2). Together, these data show the presence of functional, inhibitory ADRs in Min6 cells that reflect intact islets.
Figure 1.
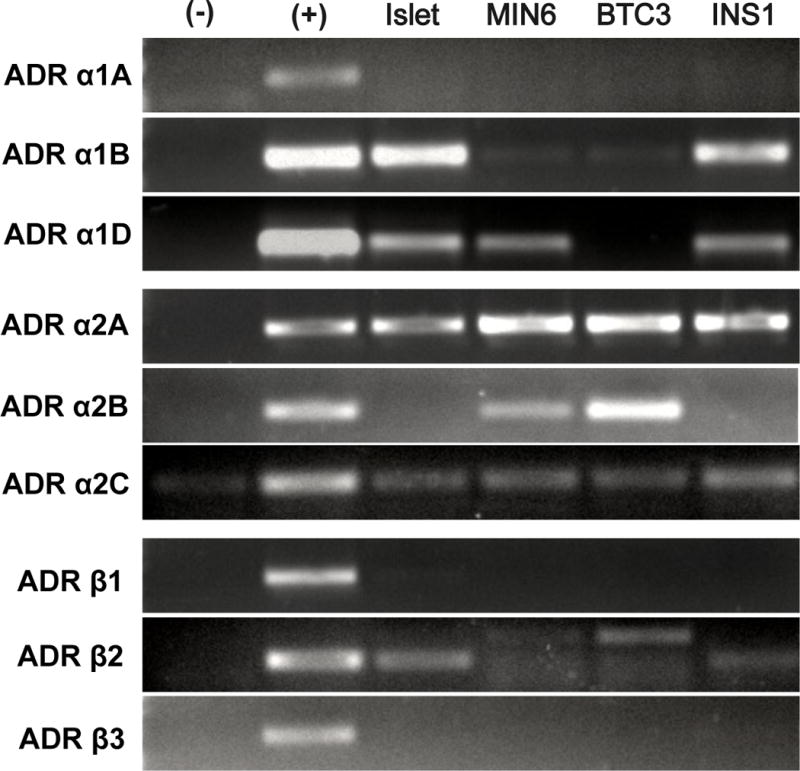
Figure 2.
3.2 Epinephrine suppresses oxygen consumption rates through α2-ADR in islets and Min6
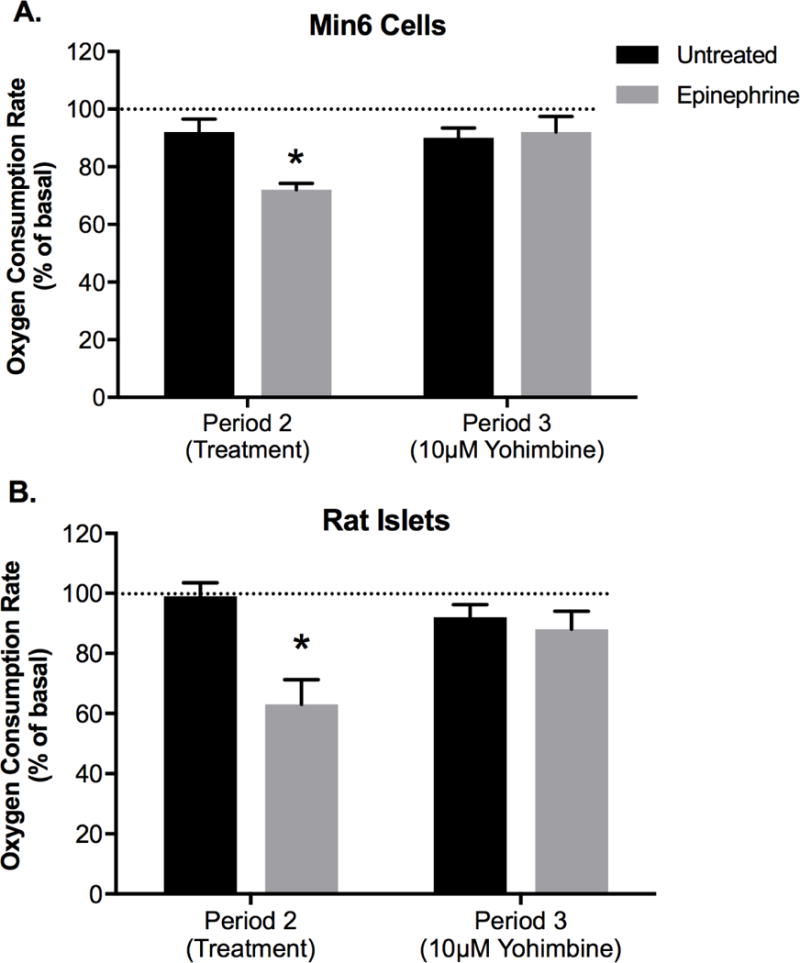
For Min6 cells in complete media, epinephrine reduced (P < 0.01) OCR to 72 ± 2.3% of baseline, whereas vehicle controls were 92 ± 4.5% of their baseline average and not different between experimental periods (Figure 3A). Addition of an α2-ADR antagonist, yohimbine (10 µM), reestablished OCR to baseline (Figure 3A). Epinephrine (100 nM) reduced (P < 0.01) OCR in rat islets to 63 ± 8.6% compared to vehicle OCR (99 ± 5.0%) and yohimbine reestablished OCR to baseline (Figure 3B). In Min6 cells with glucose-only KRB media, epinephrine decreased OCR 48 ± 6% (P < 0.05) compared to glucose-only vehicle OCR.
Figure 3.
3.3 Epinephrine suppresses forskolin-stimulated OCR
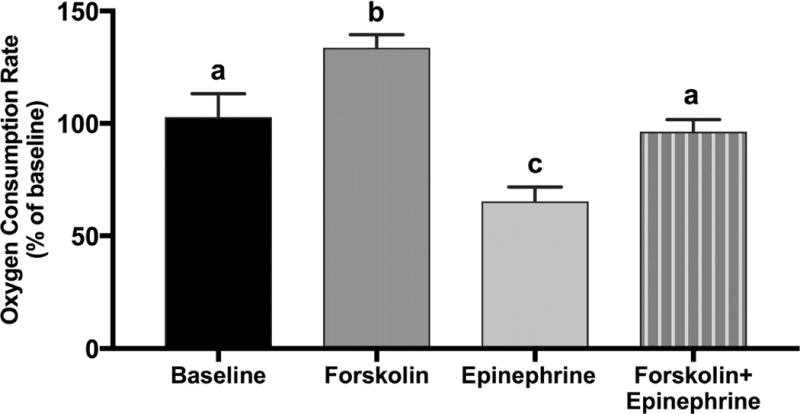
In complete media forskolin (100 µM) increased OCR in Min6 cells 31.7 ± 5.3% compared to baseline (P < 0.05; Figure 4). When forskolin (100 µM) and epinephrine (100 nM) were both present in complete media, OCR was reduced (P < 0.05) 22.3 ± 2.9% when compared to forskolin-stimulated OCR (Figure 4) but was not different than baseline OCR. Whether epinephrine or forskolin was added first within an experiment did not affect the OCR of the combined epinephrine and forskolin condition (data not shown).
Figure 4.
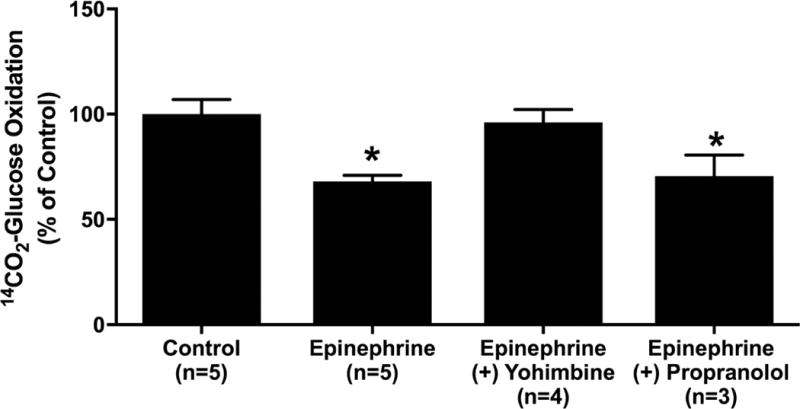
3.4 Epinephrine suppresses glucose oxidation rates independent of insulin secretion
Epinephrine decreased (P < 0.05) the rate of glucose oxidation in Min6 cells incubated with 20 mM glucose (Figure 5). The addition of yohimbine (10 µM) to incubations removed the reduction caused by epinephrine. Incubation with the β-ADR antagonist propranolol (10 µM) did not abate epinephrine suppression of glucose oxidation (Figure 5), indicating α2-ADRs are responsible for reduction.
Figure 5.
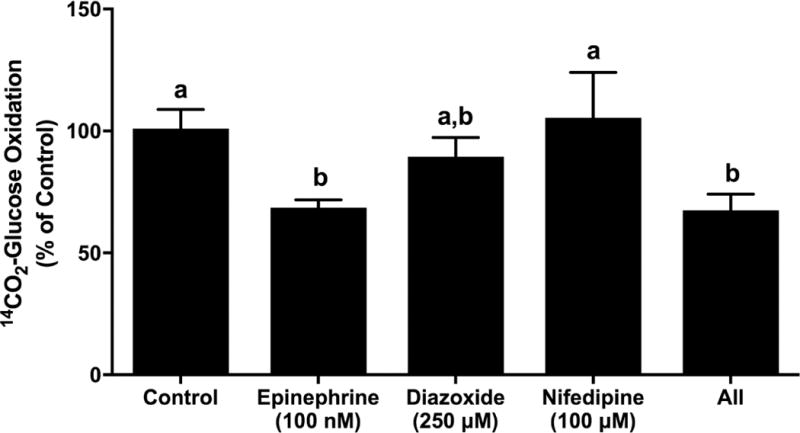
To determine whether the decreased glucose oxidation rates reflect reduced energy demands due to epinephrine’s inhibition of insulin secretion, we measured glucose oxidation rate in the presence of pharmacologic blockade of insulin secretion in glucose-only KRB. Diazoxide (250 µM), a potent activator of the KATP channels, and nifedipine (100 µM), a Ca2+-channel blocker, inhibited GSIS in Min6 cells. In KRB media with 20 mM glucose, Min6 cells secreted 2.7 ± 0.3 ng/cell of insulin but the addition of diazoxide and nifedipine reduced insulin secretion to 0.7 ± 0.04 ng/cell and 0.5 ± 0.03 ng/cell, respectively (P < 0.05, n=4).
The concentrations of nifedipine and diazoxide that effectively blocked insulin secretion in Min6 cells did not reduce glucose oxidation rates compared to control cell rates (Figure 6). Combination of diazoxide, nifedipine, and epinephrine lowered (P < 0.05; 67.6 ± 6.5%) glucose oxidation rates compared to control glucose oxidation rates, similar to the decrease seen from epinephrine alone (68.6 ± 3.2% of control; Figure 6). However, glucose oxidation rates in the presence of diazoxide were also not different from rates in the presence of epinephrine. These results indicate epinephrine decreases glucose oxidation rates independent from insulin secretion.
Figure 6.
3.5 Proteomic analysis reveals protein targets that link α2-ADR stimulation to oxidative metabolism
Min6 cells exposed to epinephrine for 4 h had 466 differentially expressed proteins compared to vehicle controls (P < 0.05; Supplemental Data 1). Proteins were enriched for pathways of oxidative metabolism, proteasome, ribosome (protein metabolism), and DNA replication/mitosis (Table 1). A comprehensive list of metabolic proteins differentially expressed following epinephrine exposure is in Table 2. Glycolytic proteins include decreased glyceraldehyde-3-phosphate dehydrogenase as well as increased lactate dehydrogenase and fructose-bisphosphate aldolase, consistent with inhibitory adrenergic stimulation. Yet, the majority of proteins in metabolic pathways were involved in oxidative phosphorylation and at least one subunit of every complex in the electron transport chain (ETC) was affected by the epinephrine exposure. Protein subunits in NADH dehydrogenase (complex I), cytochrome bc1 complex (complex III), and cytochrome c oxidase (complex IV) were more abundant in epinephrine-exposed cells. However, protein subunits for succinate dehydrogenase (complex II) and ATP synthase (Complex V) were lower (Table 2). In addition, acute epinephrine exposure altered concentrations of ribosomal subunits and increased 18 unique subunits of the proteasome complex, indicating epinephrine exposure alters protein traffic in Min6 cells.
Table 1.
Pathways associated with proteins affected by acute adrenergic stimulation in Min6 cells
| Pathway | P-Value | Number of Proteins |
|---|---|---|
| Metabolic Pathways | 4.9 × 10−11 | 73 |
| Oxidative Phosphorylation | 2.6 × 10−11 | 23 |
| Ribosome | 1.7 × 10−11 | 24 |
| Proteasome | 2.4 × 10−10 | 15 |
| Synaptic Vesicle Cycle | 1.5 × 10−6 | 12 |
| DNA Replication | 8.8 × 10−5 | 8 |
| Cell Cycle | 1.5 × 10−5 | 10 |
Non-redundant pathways over-represented (Corrected P < 2×10−5) in differentially expressed proteins. The number of proteins is the total number of proteins found differentially expressed in Min6 cells and annotated to each pathway in the online KEGG database.
Table 2.
Metabolic Related Proteins Differentially Expressed Following Acute Adrenergic Stimulation in Min6.
| Entry | Protein names | Symbol | Ratio | P-value | |
|---|---|---|---|---|---|
| Glycolytic Metabolism | |||||
| Q3UDU4 | L-lactate dehydrogenase | Ldh | 8.0 | 1.10E-03 | up |
| Q9WUA3 | ATP-dependent 6-phosphofructokinase | Pfkp | 157.5 | 1.24E-02 | up |
| A2CEK3 | Phosphoglucomutase-2 | Pgm1 | 131.8 | 2.40E-03 | up |
| S4R1N5 | Glyceraldehyde-3-phosphate dehydrogenase | Gapdh | 0.005 | 5.80E-05 | down |
| Redox | |||||
| Q8C355 | Superoxide dismutase [Cu-Zn] | Sod1 | 0.001 | 2.00E-02 | down |
| P09671 | Superoxide dismutase [Mn], mitochondrial | Sod2 | 0.040 | 5.00E-02 | down |
| Q9QUH0 | Glutaredoxin-1 (Thioltransferase-1) (TTase-1) | Glrx | 0.010 | 2.44E-03 | down |
| Oxidative Metabolism | |||||
| A7MCU9 | Pyruvate kinase | Pklr | 110.5 | 9.40E-04 | up |
| Complex I | |||||
| Q99LC3 | NADH dehydrogenase 1α subcomplex 10 | Ndufa10 | 56.9 | 5.00E-02 | up |
| Q9CQZ6 | NADH dehydrogenase 1β subcomplex subunit 3 | Ndufb3 | 57.6 | 1.20E-03 | up |
| Q9DCS9 | NADH dehydrogenase 1β subcomplex subunit 10 | Ndufb10 | 0.01 | 1.20E-03 | down |
| Q8K3J1 | NADH dehydrogenase 1β subcomplex subunit 8 | Ndufb8 | 57.5 | 1.29E-03 | up |
| Complex II | |||||
| Q8K2B3 | Succinate dehydrogenase (ubiquinone) subunit | Sdha | 0.01 | 2.00E-03 | down |
| Complex III | |||||
| Q9DB77 | Cytochrome b-c1 complex subunit 2 | Uqcrc2 | 4.10 | 2.00E-02 | up |
| Q9CZ13 | Cytochrome b-c1 complex subunit 1 | Uqcrc1 | 160.8 | 2.00E-02 | up |
| Q9DCN2 | NADH-cytochrome b5 reductase 3 | Cyb5r3 | 3.30 | 2.60E-02 | up |
| Complex IV | |||||
| P00405 | Cytochrome c oxidase subunit 2 | Cox2 | 108.2 | 7.00E-04 | up |
| P12787 | Cytochrome c oxidase subunit 5A | Cox5a | 141.7 | 9.00E-06 | up |
| P19783 | Cytochrome c oxidase subunit 4 isoform 1 | Cox4i1 | 155.6 | 1.00E-05 | up |
| Complex V | |||||
| Q9DB20 | ATP synthase subunit O, mitochondrial | Atp5o | 0.26 | 2.00E-02 | down |
| P56135 | ATP synthase subunit f, mitochondrial | Atp5j2 | 0.02 | 1.00E-03 | down |
| Q9DCX2 | ATP synthase subunit d, mitochondrial | Atp5h | 0.48 | 5.00E-02 | down |
| ATPases involved in Ca2+, ion transport, and novel | |||||
| Q8R3A9 | Sodium/Potassium transporting ATPase subunit α2 | Atp1a2 | 79.7 | 6.75E-03 | up |
| Q8R0B0 | Sodium/Potassium transporting ATPase subunit α3 | Atp1a3 | 78.7 | 6.82E-03 | up |
| Q64436 | Potassium transporting ATPase α chain 1 | Atp4a | 0.02 | 7.09E-03 | down |
| Q8R429 | Sarco/endo-plasmic reticulum Ca2+ATPase 1 | Atp2a1 | 0.02 | 6.69E-03 | down |
| O54994 | Sarco/endo-plasmic reticulum Ca2+ ATPase 2 | Atp2a2 | 0.26 | 1.83E-02 | down |
| P57746 | V-Type proton ATPase subunit D | Atp6v1d | 81.46 | 9.07E-03 | up |
| Q8BVE3 | ATPase H+ transporting lysosomal V1 subunit H | Atp6v1h | 78.29 | 9.46E-03 | up |
| Q9D1K2 | ATPase H+ transporting lysosomal V1 subunit F | Atp6v1f | 0.02 | 1.48E-03 | down |
| Q99M55 | ATPase H+ transporting lysosomal V0 subunit A1 | Atp6v0a1 | 155.49 | 3.87E-03 | up |
| P51863 | ATPase H+ transporting lysosomal V0 subunit D1 | Atp6v0d1 | 0.02 | 4.99E-02 | down |
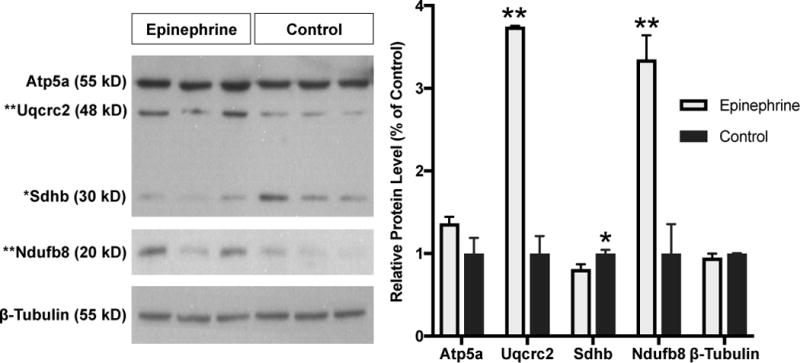
3.6 Confirmation of Proteomic Analysis by Immunoblot
To confirm the aberrant oxidative phosphorylation identified in the proteomic analysis, immunoblots for mitochondrial proteins confirmed defects in ETC proteins (Figure 7). Succinate dehydrogenase [ubiquinone] iron-sulfur subunit (Sdhb) was lower (P < 0.05) in cells exposed to 4 h epinephrine compared to vehicle control, which is consistent with lower ratios of succinate dehydrogenase [ubiquinone] flavoprotein subunits identified by proteomics. Furthermore, subunits of mitochondrial complex I and cytochrome bc1 complex III subunits (Uqcrc2 and Uqcrc1) were higher in proteomics and were confirmed by immunoblots. Uqcrc2 and NADH:Ubiquinone oxidoreductase subunit B8 (Ndufb8) were higher (P < 0.05) in cells exposed to 4 h epinephrine compared to vehicle controls (Figure 7). Mitochondrial ATP synthase subunit alpha (Atp5a) was not different in immunoblot or proteomics. Mitochondrial encoded cytochrome c oxidase (Mtco1 also known as Cox1) was not detected on immunoblots or in proteomic analysis. To improve confidence in proteomic analysis, we also investigated non-oxidative candidates. The proteasome was over represented in proteomics and 8 subunits were higher in cells exposed to epinephrine compared to vehicle. Epinephrine exposure did not change the abundance of subunit A1 (Psma1; 1.06 ± 0.40 relative to vehicle control) or subunit B1 (Psmb1; 0.96 ± 0.10 relative to vehicle control) with immunoblot analysis. Acetyl CoA synthetase 2 (Acss2) was lower in cells exposed to epinephrine compared to vehicle in proteomics (P < 0.0001) and by immunoblot (P < 0.01; 0.43 ± 0.06 relative to control).
Figure 7.
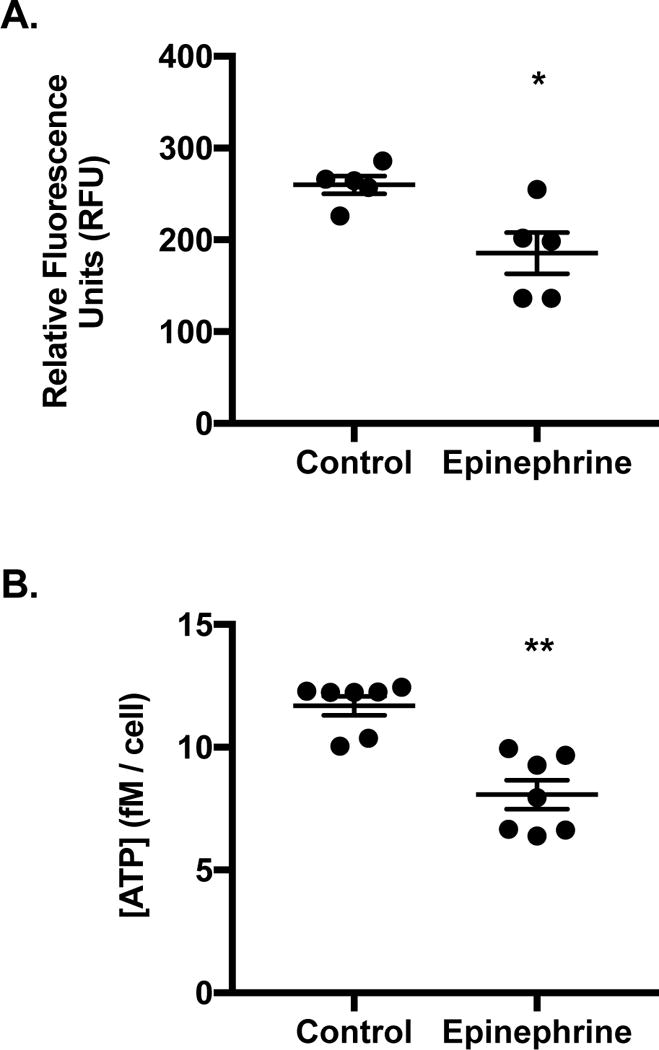
3.7 Mitochondrial Metabolism in Min6 After Epinephrine Exposure
To determine whether protein changes in the ETC would reflect physiological changes in mitochondrial metabolism of Min6 cells, redox, ATP content, and MMP were measured. Min6 were incubated under optimal conditions with alamarBlue for 4 h. When mitochondrial electron carriers such as NADH, FADH2, or cytochromes become reduced alamarBlue fluoresces. At 4 h, control cells exhibited higher (P < 0.05) relative fluorescent intensity than epinephrine exposed cells (Figure 8A; 260 ± 9.7 versus 186 ± 22.4 respectively), indicating epinephrine lowered the overall level of cellular reducing equivalents. Intracellular ATP content of Min6 exposed to epinephrine was lower compared to controls (Figure 8B; P < 0.01; 8.1 ± 1.6 and 11.7 ± 1.0 fM/cell respectively). Despite these findings, the MMP following acute epinephrine exposure was increased (115 ± 3.2% of baseline, P < 0.01) in Min6 cells in stimulatory media.
Figure 8.
4. Discussion
We show that epinephrine exposure suppresses oxidative metabolism in β-cells and islets under optimal (nutrient rich) and glucose-only substrate conditions through α2-ADR, which expands previously defined mechanisms for ADR inhibition of insulin secretion as well as designates novel downstream effects of the α2-ADRs. Epinephrine stimulated Gi-coupled α2-ADRs to decrease β-cell oxidative metabolism through glycolytic and mitochondrial respiration. We establish that the decrease in oxidative metabolism occurred in the presence of pharmacologically stimulated AC and occurred independent from insulin secretion. Thus we proposed novel metabolic targets downstream of Gi-coupled ADR signaling that we evaluated with proteomics on Min6 cells exposed to epinephrine for four hours. The proteomic analysis for epinephrine exposure implicated metabolic rearrangements of proteins involved in the ETC and the citric acid cycle. These differentially expressed proteins were confirmed with immunoblots and correlated with metabolic assays that show decreased cellular reducing agents, lower intracellular ATP concentrations, and higher MMP. These results indicate that epinephrine causes an interruption in mitochondrial respiratory chain reactions and ATP production by β-cells.
These data reaffirm that stimulation through α2-ADR inhibits insulin secretion but also indicate a previously undefined mechanism that involves the oxidative metabolism of glucose (Aarnio et al 2001, Katada and Ui 1981). The significance of α2-ADR regulation of β-cell function has been demonstrated through studies manipulating expression of the predominant isoform Adrα2a. Mice that lack Adrα2a are hyperinsulinemic and hypoglycemic, indicating loss of inhibitory control in β-cells (Savontaus et al 2008). Overexpression of Adrα2a decreased β-cell function and contributed to the development of diabetes in adulthood (Rosengren et al 2010, Liggett 2009, Ahren 2009). The ADR expression profile of Min6 in concert with physiological findings using α2-ADR antagonist yohimbine demonstrate α2-ADR are responsible for decreased oxidative phosphorylation of islets and Min6, which expands the mechanisms underlying adrenergic signaling in β-cells.
In rat islets glucose utilization rates were inhibited with epinephrine and the α2-ADR agonist clonidine (Laychock and Bilgin 1987). However, others have demonstrated epinephrine suppresses insulin secretion but does not affect glucose utilization rates (Hermann and Deckert 1977). We showed epinephrine through α2-ADR decreases glucose oxidation rate and oxygen consumption rates in complete nutrient (amino acid and glucose supplemented) as well as nutrient-deprived (glucose-only) situations in isolated rat islets and Min6 cells. Furthermore, pharmacologic activation of AC to elevate intracellular cAMP increased OCR for Min6 cells but this enhancement was suppressed with epinephrine. Therefore, mechanisms independent of cAMP may be partially responsible for epinephrine inhibition of OCR. Lower AC activity to reduce cAMP is a prominent mechanism underlying Gi-coupled inhibition of insulin secretion (Howell and Montague 1973, Liggett 2009). Thus we hypothesized that α2-ADR activation would suppress OCR through lowering cAMP production. Here, we found epinephrine suppressed forskolin-stimulated OCR but not to the magnitude seen in control media. However, these results are reflected by studies that show epinephrine decreased insulin secretion when cAMP concentrations were maintained and studies that demonstrate α2-ADR has cAMP independent mechanisms to suppress insulin secretion (Jones et al 1987, Bradley et al 2005, Gloerich and Bos 2010, Straub and Sharp 2012). Further studies are required to determine the exact role of cAMP in coordinating oxidative metabolism and α2-ADR activation.
In the present study we also found α2-ADR stimulation decreased glucose oxidation rates during a pharmacological blockade of insulin exocytosis. This demonstrates that although epinephrine suppresses insulin secretion, the observed effects on oxidative metabolism do not merely reflect lower energy requirements from less insulin exocytosis. In fact, we found blockade of insulin secretion alone did not affect glucose oxidation rates in Min6 cells. Therefore exocytosis of insulin does not have a measurable cost on glucose oxidation or that stimulated β-cells still have the ability to generate ATP despite its utilization by exocytosis.
A clear link between α2-ADR stimulation and decreased respiration in β-cells is established and consistent with proteomics data. Current discoveries in islets have identified physical interactions between ADR-coupled G proteins and cellular components such as insulin vesicles and ion channels (Zhao et al 2010c, Zhao et al 2010b, Straub and Sharp 2012). For example, Gαo reduces insulin vesicle docking and in our data, is increased following ADR stimulation (Zhao et al 2010a). Moreover, studies in other cell types have demonstrated that G proteins, including Gαi, are present in mitochondria providing framework for changes observed in oxidative phosphorylation (Lyssand and Bajjalieh 2007, Beninca et al 2014). We believe these studies support activation of α2-ADR may cause changes in protein abundance through novel downstream targets of G protein signaling.
Rearrangements in metabolic proteins after epinephrine exposure in Min6 cells support a direct role in β-cell oxidative metabolism. Given the evidence that G proteins can act directly on mitochondria and that many G protein functions are unknown, we hypothesize that proteins involved in oxidative phosphorylation are downstream targets for α2-ADR signaling. We found epinephrine exposure altered the abundance of every complex in the ETC. Complex I had increased expression in an essential subunit and mixed expression changes in the accessory catalytic subunits (Su et al 2012). Cytochrome c subunits of complex III and complex IV were also increased, inconsistent with physiological assessments. However, succinate dehydrogenase (complex II) and several essential subunits to ATP synthase were decreased with epinephrine. Change in complex II abundance was confirmed by immunoblot. While complex II does not promote ATP synthesis directly through contribution to the proton gradient, it is the only ETC enzyme involved in both respiration and the citric acid cycle (Rutter et al 2010). When we evaluated the mitochondrial function of Min6 cells after epinephrine exposure there was a decrease in cellular reducing capacity and ATP concentration, consistent with lower levels of succinate dehydrogenase (complex II). However, MMP was increased in cells following acute epinephrine exposure, which might reflect a decrease in proton carriers coincident with reduced cytochrome c oxidase activity (electron transfer to molecular oxygen) even though some Complex IV subunits are upregulated. Although ATP synthase subunit Atp5a was not different by either measure, downregulation of Atp5o, Atp5jp, and Atp5h in the proteomics analysis indicate reductions in the synthesis of ATP consistent with the decreased intracellular ATP concentration following epinephrine exposure. Additional mechanisms not tested in the present study may include decreased production/mobilization and translocation of ADP into the mitochondrial matrix (Chance and Williams 1955, Gnaiger 2001). Together, the upregulation of subunits for Complexes I and III and decrease in ATP concentration through inhibition in ATP synthase can explain the increase in MMP, but additional work is required to determine the overall regulation of mitochondrial respiration by adrenergic stimulation.
Differentially expressed proteins expanded beyond oxidative phosphorylation and included protein turnover and DNA replication. We anticipated the acute response to stress (CA) would consist of signals that suppress cell growth, which reflects a balance between the production of new proteins and protein breakdown. Proteins annotated to ribosomal pathways reflected increased and decreased expression, yet the 18 subunits of the proteasome were increased, however subunits Psma1 and Psmb1 were not different when evaluated by immunoblot. The proteomic results suggest increased protein breakdown through up-regulation of the proteasome and changes in normal protein translation (ribosomes). Although we could not confirm changes in Psma1 and Psmb1 by immunoblot, the proteasome was previously implicated in islets following chronic ADR stimulation (Kelly et al 2017) and in diabetic islets (Bugliani et al 2013).
In vivo CA mediate hypoxic, nutritional, and neurologic stress responses. Stimulation of ADRα2A by these hormones to reduce β-cell metabolism has an obvious role in suppressing insulin secretion (Arun 2004). Since β-cell metabolism and insulin secretion are linked, mechanisms that inhibit metabolism also reduce insulin secretion. Suppression of β-cell metabolism may protect against longer bouts of hypoxia or stress. β-cells are susceptible to hypoxia because they rely on quantitative production of ATP by oxidative metabolism to secrete insulin (Cantley et al 2010). Signals that suppress oxidative metabolism independent of nutrient sensing may benefit β-cells during hypoxia, in order to prevent depletion of oxidative pathways and increase redox status. Especially given prolonged elevations of CA are seen in pathologies such as fetal growth restriction, which is characterized by low plasma oxygen, as well as in individuals undergoing intense exercise training or frequent hypoxia bouts (Macko et al 2016, Davy et al 1995, Silverman and Mazzeo 1996, Barnholt et al 2006). Whether this acute mechanism to suppress metabolism ultimately prevents damage to β-cells under chronic stress (high CA) conditions long term has yet to be determined, but the co-occurrence of hypoxia and CA in vivo provide a compelling basis for CA regulation of islet and β-cell metabolism.
5. Conclusion
These data establish a direct, but still evolving, link between β-cell oxidative metabolism and acute α2-ADR stimulation. Epinephrine suppression of oxidative metabolism occurred independent of nutrient availability, insulin exocytosis and cAMP concentrations. Proteomic analysis supported metabolic rearrangements and proposed mitochondrial protein targets down-stream of α2-ADR signaling that were confirmed by additional protein quantification methods and metabolic assays. This work provides a framework for identifying ADR-regulated targets in order to explore interventions that alleviate effects of chronic ADR stimulation.
Supplementary Material
Epinephrine decreases oxidative metabolism in β-cells via α2-adrenergic receptors.
Oxidative metabolism was reduced despite interruptions in cAMP and insulin exocytosis.
Proteomics identified novel mitochondrial, ribosomal, and proteasomal protein targets.
Epinephrine altered expression of electron transport chain proteins in β-cells.
Epinephrine increased mitochondrial membrane potential and decreased ATP content.
Acknowledgments
We would like to acknowledge Jenna Miner for her technical support on conducting oxygen consumption measurements. We would also like to thank Miranda Anderson and Dustin Yates for their laboratory support and guidance.
Funding: This work was supported by National Institutes of Health R01 DK084842 (S.W.L. Principal Investigator).
Abbreviations
- GSIS
glucose stimulated insulin secretion
- CA
catecholamine
- ADR
adrenergic receptor
- GO
gene ontology
- GPCR
G protein coupled receptor
- KRB
Krebs Ringer Buffer
- OCR
oxygen consumption rate
- AC
adenylyl cyclase
- ETC
electron transport chain
- TMRE
tetramethylrhodamine ethyl ester perchlorate
- HBSS
Hank’s Phosphate Buffered Saline
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure: The authors have nothing to disclose.
References
- Aarnio P, Lauritsen T, Dela F. Insulin secretion and glucose kinetics during exercise with and without pharmacological alpha(1)- and alpha(2)-receptor blockade. Diabetes. 2001;50:1834–1843. doi: 10.2337/diabetes.50.8.1834. [DOI] [PubMed] [Google Scholar]
- Ahren B. Islet G protein-coupled receptors as potential targets for treatment of type 2 diabetes. Nat Rev Drug Discov. 2009;8:369–385. doi: 10.1038/nrd2782. [DOI] [PubMed] [Google Scholar]
- Arun CP. Fight or flight, forbearance and fortitude: the spectrum of actions of the catecholamines and their cousins. Ann N Y Acad Sci. 2004;1018:137–140. doi: 10.1196/annals.1296.016. [DOI] [PubMed] [Google Scholar]
- Ashcroft SJ, Hedeskov CJ, Randle PJ. Glucose metabolism in mouse pancreatic islets. Biochem J. 1970;118:143–154. doi: 10.1042/bj1180143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnholt KE, Hoffman AR, Rock PB, Muza SR, Fulco CS, Braun B, Holloway L, Mazzeo RS, Cymerman A, Friedlander AL. Endocrine responses to acute and chronic high-altitude exposure (4,300 meters): modulating effects of caloric restriction. Am J Physiol Endocrinol Metab. 2006;290:E1078–88. doi: 10.1152/ajpendo.00449.2005. [DOI] [PubMed] [Google Scholar]
- Beninca C, Planaguma J, de Freitas Shuck A, Acin-Perez R, Munoz JP, de Almeida MM, Brown JH, Murphy AN, Zorzano A, Enriquez JA, Aragay AM. A new non-canonical pathway of Galpha(q) protein regulating mitochondrial dynamics and bioenergetics. Cell Signal. 2014;26:1135–1146. doi: 10.1016/j.cellsig.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley J, Reisert J, Frings S. Regulation of cyclic nucleotide-gated channels. Curr Opin Neurobiol. 2005;15:343–349. doi: 10.1016/j.conb.2005.05.014. [DOI] [PubMed] [Google Scholar]
- Bugliani M, Liechti R, Cheon H, Suleiman M, Marselli L, Kirkpatrick C, Filipponi F, Boggi U, Xenarios I, Syed F, Ladriere L, Wollheim C, Lee MS, Marchetti P. Microarray analysis of isolated human islet transcriptome in type 2 diabetes and the role of the ubiquitin-proteasome system in pancreatic beta cell dysfunction. Mol Cell Endocrinol. 2013;367:1–10. doi: 10.1016/j.mce.2012.12.001. [DOI] [PubMed] [Google Scholar]
- Camacho LE, Chen X, Hay WW, Jr, Limesand SW. Enhanced insulin secretion and insulin sensitivity in young lambs with placental insufficiency-induced intrauterine growth restriction. Am J Physiol Regul Integr Comp Physiol. 2017;313:R101–R109. doi: 10.1152/ajpregu.00068.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon W, De la Paz D. Emotional stimulation of adrenal secretion. Am J Physiol. 1911;28:64–70. [Google Scholar]
- Cantley J, Grey ST, Maxwell PH, Withers DJ. The hypoxia response pathway and beta-cell function. Diabetes Obes Metab. 2010;12(Suppl 2):159–167. doi: 10.1111/j.1463-1326.2010.01276.x. [DOI] [PubMed] [Google Scholar]
- Chambers MC, Maclean B, Burke R, Amodei D, Ruderman DL, Neumann S, Gatto L, Fischer B, Pratt B, Egertson J, Hoff K, Kessner D, Tasman N, Shulman N, Frewen B, Baker TA, Brusniak MY, Paulse C, Creasy D, Flashner L, Kani K, Moulding C, Seymour SL, Nuwaysir LM, Lefebvre B, Kuhlmann F, Roark J, Rainer P, Detlev S, Hemenway T, Huhmer A, Langridge J, Connolly B, Chadick T, Holly K, Eckels J, Deutsch EW, Moritz RL, Katz JE, Agus DB, MacCoss M, Tabb DL, Mallick P. A cross-platform toolkit for mass spectrometry and proteomics. Nat Biotechnol. 2012;30:918–920. doi: 10.1038/nbt.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chance B, Williams GR. Respiratory enzymes in oxidative phosphorylation. I. Kinetics of oxygen utilization. J Biol Chem. 1955;217:383–393. [PubMed] [Google Scholar]
- Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinformatics. 2004;20:1466–1467. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- Davy KP, Johnson DG, Seals DR. Cardiovascular, plasma norepinephrine, and thermal adjustments to prolonged exercise in young and older healthy humans. Clin Physiol. 1995;15:169–181. doi: 10.1111/j.1475-097x.1995.tb00441.x. [DOI] [PubMed] [Google Scholar]
- Geer LY, Markey SP, Kowalak JA, Wagner L, Xu M, Maynard DM, Yang X, Shi W, Bryant SH. Open mass spectrometry search algorithm. J Proteome Res. 2004;3:958–964. doi: 10.1021/pr0499491. [DOI] [PubMed] [Google Scholar]
- Giannaccini G, Lupi R, Trincavelli ML, Navalesi R, Betti L, Marchetti P, Lucacchini A, Del Guerra S, Martini C. Characterization of sulfonylurea receptors in isolated human pancreatic islets. J Cell Biochem. 1998;71:182–188. doi: 10.1002/(sici)1097-4644(19981101)71:2<182::aid-jcb4>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Gloerich M, Bos JL. Epac: defining a new mechanism for cAMP action. Annu Rev Pharmacol Toxicol. 2010;50:355–375. doi: 10.1146/annurev.pharmtox.010909.105714. [DOI] [PubMed] [Google Scholar]
- Gnaiger E. Bioenergetics at low oxygen: dependence of respiration and phosphorylation on oxygen and adenosine diphosphate supply. Respir Physiol. 2001;128:277–297. doi: 10.1016/s0034-5687(01)00307-3. [DOI] [PubMed] [Google Scholar]
- He Y, Schmidt MA, Erwin C, Guo J, Sun R, Pendarvis K, Warner BW, Herman EM. Transgenic Soybean Production of Bioactive Human Epidermal Growth Factor (EGF) PLoS One. 2016;11:e0157034. doi: 10.1371/journal.pone.0157034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann LS, Deckert T. The effect of epinephrine and isoproterenol on insulin secretion and glucose utilization in isolated islets of Langerhans from mice. Acta Endocrinol (Copenh) 1977;84:105–114. doi: 10.1530/acta.0.0840105. [DOI] [PubMed] [Google Scholar]
- Hiscox AM, Stone AL, Limesand S, Hoying JB, Williams SK. An islet-stabilizing implant constructed using a preformed vasculature. Tissue Eng Part A. 2008;14:433–440. doi: 10.1089/tea.2007.0099. [DOI] [PubMed] [Google Scholar]
- Howell SL, Montague W. Adenylate cyclase activity in isolated rat islets of Langerhans. Effects of agents which alter rates of insulin secretion. Biochim Biophys Acta. 1973;320:44–52. doi: 10.1016/0304-4165(73)90163-3. [DOI] [PubMed] [Google Scholar]
- Jones PM, Fyles JM, Persaud SJ, Howell SL. Catecholamine inhibition of Ca2+-induced insulin secretion from electrically permeabilised islets of Langerhans. FEBS Lett. 1987;219:139–144. doi: 10.1016/0014-5793(87)81206-1. [DOI] [PubMed] [Google Scholar]
- Katada T, Ui M. Islet-activating protein. A modifier of receptor-mediated regulation of rat islet adenylate cyclase. J Biol Chem. 1981;256:8310–8317. [PubMed] [Google Scholar]
- Keahey HH, Boyd AE, 3rd, Kunze DL. Catecholamine modulation of calcium currents in clonal pancreatic beta-cells. Am J Physiol. 1989;257:C1171–6. doi: 10.1152/ajpcell.1989.257.6.C1171. [DOI] [PubMed] [Google Scholar]
- Kelly AC, Bidwell CA, McCarthy FM, Taska DJ, Anderson MJ, Camacho LE, Limesand SW. RNA sequencing exposes novel adaptive and immune responses to intrauterine growth restriction in fetal sheep islets. Endocrinology. 2017 doi: 10.1210/en.2016-1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly AC, Steyn LV, Kitzmann JP, Anderson MJ, Mueller KR, Hart NJ, Lynch RM, Papas KK, Limesand SW. Function and expression of sulfonylurea, adrenergic, and glucagon-like peptide 1 receptors in isolated porcine islets. Xenotransplantation. 2014 doi: 10.1111/xen.12101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessner D, Chambers M, Burke R, Agus D, Mallick P. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics. 2008;24:2534–2536. doi: 10.1093/bioinformatics/btn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey RJ, Chan SL, Cable HC, James RF, Perrett CW, Scarpello JH, Morgan NG. Expression of alpha 2- and beta-adrenoceptor subtypes in human islets of Langerhans. J Endocrinol. 1996;148:531–543. doi: 10.1677/joe.0.1480531. [DOI] [PubMed] [Google Scholar]
- Laychock SG, Bilgin S. Alpha 2-adrenergic inhibition of pancreatic islet glucose utilization is mediated by an inhibitory guanine nucleotide regulatory protein. FEBS Lett. 1987;218:7–10. doi: 10.1016/0014-5793(87)81007-4. [DOI] [PubMed] [Google Scholar]
- Liggett SB. alpha2A-adrenergic receptors in the genetics, pathogenesis, and treatment of type 2 diabetes. Sci Transl Med. 2009;1:12ps15. doi: 10.1126/scitranslmed.3000606. [DOI] [PubMed] [Google Scholar]
- Limesand SW, Rozance PJ, Smith D, Hay WW., Jr Increased insulin sensitivity and maintenance of glucose utilization rates in fetal sheep with placental insufficiency and intrauterine growth restriction. Am J Physiol Endocrinol Metab. 2007;293:E1716–25. doi: 10.1152/ajpendo.00459.2007. [DOI] [PubMed] [Google Scholar]
- Limesand SW, Rozance PJ, Zerbe GO, Hutton JC, Hay WW., Jr Attenuated insulin release and storage in fetal sheep pancreatic islets with intrauterine growth restriction. Endocrinology. 2006;147:1488–1497. doi: 10.1210/en.2005-0900. [DOI] [PubMed] [Google Scholar]
- Lyssand JS, Bajjalieh SM. The heterotrimeric [corrected] G protein subunit G alpha i is present on mitochondria. FEBS Lett. 2007;581:5765–5768. doi: 10.1016/j.febslet.2007.11.044. [DOI] [PubMed] [Google Scholar]
- Macko AR, Yates DT, Chen X, Shelton LA, Kelly AC, Davis MA, Camacho LE, Anderson MJ, Limesand SW. Adrenal Demedullation and Oxygen Supplementation Independently Increase Glucose-Stimulated Insulin Concentrations in Fetal Sheep With Intrauterine Growth Restriction. Endocrinology. 2016;157:2104–2115. doi: 10.1210/en.2015-1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaisse WJ, Herchuelz A, Devis G, Somers G, Boschero AC, Hutton JC, Kawazu S, Sener A, Atwater IJ, Duncan G, Ribalet B, Rojas E. Regulation of calcium fluxes and their regulatory roles in pancreatic islets. Ann N Y Acad Sci. 1978;307:562–582. doi: 10.1111/j.1749-6632.1978.tb41982.x. [DOI] [PubMed] [Google Scholar]
- Miyazaki J, Araki K, Yamato E, Ikegami H, Asano T, Shibasaki Y, Oka Y, Yamamura K. Establishment of a pancreatic beta cell line that retains glucose-inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology. 1990;127:126–132. doi: 10.1210/endo-127-1-126. [DOI] [PubMed] [Google Scholar]
- Papas KK, Pisania A, Wu H, Weir GC, Colton CK. A stirred microchamber for oxygen consumption rate measurements with pancreatic islets. Biotechnol Bioeng. 2007;98:1071–1082. doi: 10.1002/bit.21486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prentki M, Matschinsky FM, Madiraju SR. Metabolic signaling in fuel-induced insulin secretion. Cell Metab. 2013;18:162–185. doi: 10.1016/j.cmet.2013.05.018. [DOI] [PubMed] [Google Scholar]
- Rosengren AH, Jokubka R, Tojjar D, Granhall C, Hansson O, Li DQ, Nagaraj V, Reinbothe TM, Tuncel J, Eliasson L, Groop L, Rorsman P, Salehi A, Lyssenko V, Luthman H, Renstrom E. Overexpression of alpha2A-adrenergic receptors contributes to type 2 diabetes. Science. 2010;327:217–220. doi: 10.1126/science.1176827. [DOI] [PubMed] [Google Scholar]
- Roth NS, Lefkowitz RJ, Caron MG. Structure and function of the adrenergic receptor family. Adv Exp Med Biol. 1991;308:223–238. doi: 10.1007/978-1-4684-6015-5_20. [DOI] [PubMed] [Google Scholar]
- Rutter J, Winge DR, Schiffman JD. Succinate dehydrogenase - Assembly, regulation and role in human disease. Mitochondrion. 2010;10:393–401. doi: 10.1016/j.mito.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savontaus E, Fagerholm V, Rahkonen O, Scheinin M. Reduced blood glucose levels, increased insulin levels and improved glucose tolerance in alpha2A-adrenoceptor knockout mice. Eur J Pharmacol. 2008;578:359–364. doi: 10.1016/j.ejphar.2007.09.015. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seino S. ATP-sensitive potassium channels: a model of heteromultimeric potassium channel/receptor assemblies. Annu Rev Physiol. 1999;61:337–362. doi: 10.1146/annurev.physiol.61.1.337. [DOI] [PubMed] [Google Scholar]
- Silverman HG, Mazzeo RS. Hormonal responses to maximal and submaximal exercise in trained and untrained men of various ages. J Gerontol A Biol Sci Med Sci. 1996;51:B30–7. doi: 10.1093/gerona/51a.1.b30. [DOI] [PubMed] [Google Scholar]
- Straub SG, Sharp GW. Evolving insights regarding mechanisms for the inhibition of insulin release by norepinephrine and heterotrimeric G proteins. Am J Physiol Cell Physiol. 2012;302:C1687–98. doi: 10.1152/ajpcell.00282.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su T, Esser L, Xia D, Yu CA, Yu L. Generation, characterization and crystallization of a cytochrome c(1)-subunit IV fused cytochrome bc(1) complex from Rhodobacter sphaeroides. Biochim Biophys Acta. 2012;1817:298–305. doi: 10.1016/j.bbabio.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urano Y, Sakurai T, Ueda H, Ogasawara J, Sakurai T, Takei M, Izawa T. Desensitization of the inhibitory effect of norepinephrine on insulin secretion from pancreatic islets of exercise-trained rats. Metabolism. 2004;53:1424–1432. doi: 10.1016/j.metabol.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Vizcaino JA, Deutsch EW, Wang R, Csordas A, Reisinger F, Rios D, Dianes JA, Sun Z, Farrah T, Bandeira N, Binz PA, Xenarios I, Eisenacher M, Mayer G, Gatto L, Campos A, Chalkley RJ, Kraus HJ, Albar JP, Martinez-Bartolome S, Apweiler R, Omenn GS, Martens L, Jones AR, Hermjakob H. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat Biotechnol. 2014;32:223–226. doi: 10.1038/nbt.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright ML, Pendarvis K, Nanduri B, Edelmann MJ, Jenkins HN, Reddy JS, Wilson JG, Ding X, Broadway PR, Ammari MG, Paul O, Roberts B, Donaldson JR. The Effect of Oxygen on Bile Resistance in Listeria monocytogenes. J Proteomics Bioinform. 2016;9:107–119. doi: 10.4172/jpb.1000396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie C, Mao X, Huang J, Ding Y, Wu J, Dong S, Kong L, Gao G, Li CY, Wei L. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011;39:W316–22. doi: 10.1093/nar/gkr483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao A, Ohara-Imaizumi M, Brissova M, Benninger RK, Xu Y, Hao Y, Abramowitz J, Boulay G, Powers AC, Piston D, Jiang M, Nagamatsu S, Birnbaumer L, Gu G. Galphao represses insulin secretion by reducing vesicular docking in pancreatic beta-cells. Diabetes. 2010a;59:2522–2529. doi: 10.2337/db09-1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Fang Q, Straub SG, Lindau M, Sharp GW. Hormonal inhibition of endocytosis: novel roles for noradrenaline and G protein G(z) J Physiol. 2010b;588:3499–3509. doi: 10.1113/jphysiol.2010.190116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Fang Q, Straub SG, Lindau M, Sharp GW. Noradrenaline inhibits exocytosis via the G protein betagamma subunit and refilling of the readily releasable granule pool via the alpha(i1/2) subunit. J Physiol. 2010c;588:3485–3498. doi: 10.1113/jphysiol.2010.190090. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.