

Abstract
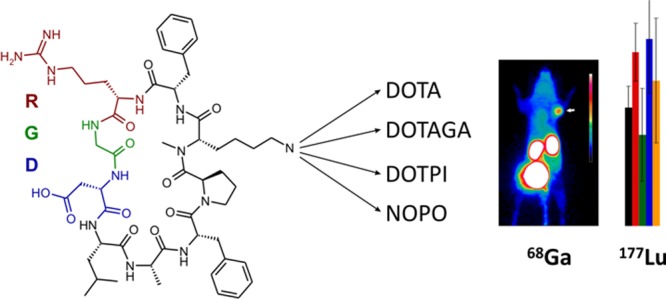
The epithelial integrin αvβ6 is expressed by many malignant carcinoma cell types, including pancreatic cancer, and thus represents a promising target for radionuclide therapy. The peptide cyclo(FRGDLAFp(NMe)K) was decorated with different chelators (DOTPI, DOTAGA, and DOTA). The Lu(III) complexes of these conjugates exhibited comparable αvβ6 integrin affinities (IC50 ranging from 0.3 to 0.8 nM) and good selectivities against other integrins (IC50 for αvβ8 >43 nM; for α5β1 >238 nM; and for αvβ3, αvβ5, and αIIbβ3 >1000 nM). Although different formal charges of the Lu(III) chelates (ranging from 0 to 4) resulted in strongly varying degrees of hydrophilicity (log D ranging from −3.0 to −4.1), biodistributions in murine H2009 xenografts of the Lu-177-labeled compounds (except the DOTPI derivative) were quite similar and comparable to our previously reported αvβ6 integrin positron emission tomography tracer Ga-68-avebehexin. Hence, combinations of existing Ga-68- and Lu-177-labeled c(FRGDLAFp(NMe)K) derivatives could be utilized for αvβ6 integrin-targeted theranostics, whereas our data nonetheless suggest that further improvement of pharmacokinetics might be necessary to ensure clinical success.
Introduction
Integrins,1 a family of 24 heterodimeric transmembrane receptors primarily mediating cell adhesion, have been in the focus of pharmaceutical research for a long time.2 Despite up-regulation of several integrin subtypes is related to pathological conditions,3 the development of specific inhibitors as well as molecular imaging agents has been predominantly focused on the subtype αvβ3 for decades because of its long-known key role in (tumor) angiogenesis4,5 However, more recently, other integrins have attracted considerable interest in view of their involvement in cancer pathogenesis.6 In particular, the subtype αvβ6 is poorly expressed by normal adult epithelia, but upregulated in a variety of carcinomas. It is also linked to poor prognosis and furthermore has been found to promote carcinoma invasion.7,8 Therefore, the αvβ6 integrin is an attractive target for molecular imaging and therapy of cancers.
Consequently, a number of radiolabeled αvβ6 integrin ligands for in vivo imaging of the αvβ6 integrin expression have been developed in recent years.9−15 Along these lines, we recently synthesized a small series of positron emission tomography (PET) imaging agents based on the metabolically stable and αvβ6 integrin-selective cyclic nonapeptide cyclo(FRGDLAFp(NMe)K).16 One to three peptide moieties were conjugated to the chelator TRAP (1,4,7-triazacyclononane-1,4,7-tris[methylene(2-carboxyethyl)phosphinic acid])17 and labeled with the positron emitter 68GaIII (t1/2 = 68 min). Because of its pronounced hydrophilicity promoting rapid renal clearance, the monomeric peptide conjugate 68Ga-avebehexin18 (see Chart 1) exhibited the most favorable image contrast (tumor-to-blood/muscle ratio) in μPET scans of subcutaneous murine H2009 (αvβ6 integrin-expressing human lung adenocarcinoma) xenografts. For this compound, we furthermore noticed a particularly low unspecific uptake in the pancreas, corresponding to the complete absence of αvβ6 integrin in the normal pancreatic tissue according to immunohistochemistry. On the other hand, the αvβ6 integrin is overexpressed by the tumor cells of almost 90% of all pancreatic ductal adenocarcinomas (PDACs)19−21 and thus represents a highly promising target for theranostics of PDAC. The fact that PDAC is one of the most lethal cancers with a five-year survival rate of only 8%,22 causing more annual deaths than prostate carcinoma,23 points at an unmet clinical need and the necessity to develop improved therapies particularly for this malignancy, whereas a substantial curative value for other cancers frequently expressing β6 integrin, such as head-and-neck squamous cell carcinoma,24 can be anticipated as well.
Chart 1. Chelator conjugates of the αvβ6 integrin-selective cyclic nonapeptide cyclo(FRGDLAFp(NMe)K).

However, although TRAP is suitable for elaboration of 64Cu25 and particularly of 68Ga radiopharmaceuticals26,27 because of its pronounced selectivity for small metal cations,28 it is not applicable for complexation of established therapeutic radionuclides,29 that is, β–- or α-emitting radiometal ions such as 177LuIII, 90YIII, 213BiIII, or 225AcIII, thus restricting the applicability of avebehexin to imaging. Hence, we synthesized a second series of conjugates with chelators for lanthanide-type ions shown in Chart 1, aiming at the corresponding therapeutic agents required for αvβ6 integrin addressing theranostics.
Results and Discussion
Because we previously found that conjugation of biomolecules to TRAP-type chelators can be done very conveniently by means of CuI-mediated azide–alkyne cycloaddition (CuAAC) followed by removal of TRAP-bound copper by competitive demetalation with 1,4,7-triazacyclononane-1,4,7-triacetic acid (NOTA),18,30 we again utilized this approach to decorate the alkyne-functionalized peptide c(FRGDLAFp(NMe)K(pentynoic acid)) with azide-equipped cyclen-based chelators DOTPI,31 DOTAGA, and DOTA (Chart 2).
Chart 2. Azide-decorated chelators for conjugation to alkyne-functionalized peptides by means of click chemistry (CuAAC).

We found that for all these chelators, transchelation indeed proceeds as expected.32 However, in contrast to Cu–TRAP and Cu–DOTPI complexes, Cu removal from DOTA and DOTAGA remained incomplete because the slightly higher thermodynamic stabilities of the respective CuII complexes prevented a sufficient shift of the equilibrium toward the Cu–NOTA side. Hence, the complete removal of Cu from the DOTA and DOTAGA conjugates ultimately afforded the use of sulfide, resulting in fairly low yields (7 and 18%) which are frequently observed for this method. Because of this situation and to elucidate the influence of the triazole moiety in the linkers, the latter two chelators were also conjugated directly via amide bonding (57 and 65% yield). Furthermore, an analogue of avebehexin featuring the TRAP-type chelator NOPO33 has been prepared to expand the set of conjugates amenable to 68Ga labeling for a preliminary, comparative 68Ga-PET study.
The selection of the chelators followed the rationale to equip the peptide with radiometal chelate moieties featuring different net charges, resulting in varying degrees of polarity and, therefore, distinctly different pharmacokinetics. As we previously observed that 68Ga-avebetrin, which contains a twofold negatively charged 68GaIII chelate, exhibited a lower background uptake compared to a radiolabeled c(FRGDLAFp(NMe)K)-NODAGA conjugate featuring an uncharged 68GaIII chelate,18 we expected that a similar beneficial effect of negatively charged chelates might be observed for the therapeutic 177LuIII complexes. Table 1 shows that indeed, the charge has the most pronounced influence on polarity (expressed as octanol–water distribution coefficients, log D, at pH 7.4), whereas the effect of the triazole is less substantial. A similar, systematic pattern is however not observed for the 68GaIII complexes, presumably owing to the fact that the respective hexacoordinate chelates are zwitterionic34 and the formal charge is less well-correlated to the actual number of charged atoms contained.
Table 1. n-Octanol-PBS Distribution Coefficients (log D7.4).
| radiometal | chelate chargea | 177Lu | 68Ga |
|---|---|---|---|
| c(avb6)-Trz-DOTPI | –4 | –4.13 ± 0.08 | no 68Ga labeling |
| c(avb6)-Trz-DOTAGA | –1 | –3.35 ± 0.03 | –3.32 ± 0.02 |
| c(avb6)-Trz-DOTA | 0 | –2.98 ± 0.08 | –3.10 ± 0.04 |
| c(avb6)-DOTAGA | –1 | –3.36 ± 0.07 | –3.49 ± 0.08 |
| c(avb6)-DOTA | 0 | –3.21 ± 0.24 | –3.37 ± 0.04 |
| c(avb6)-Ahx-NOPO | 0 | no 177Lu labeling | –3.06 ± 0.05 |
| c(avb6)-Trz-TRAP | |||
| (avebehexin) | –2 | no 177Lu labeling | –3.71 ± 0.03 |
Formal net charges for complexes with trivalent metal cations.
Notably, we observed that for c(avb6)-Trz-DOTA, the triazole linker actually affected radiolabeling, not only with 68Ga but also with 177Lu. Whereas the labeling of all other compounds under standard conditions (see Experimental Section) proceeded as expected and typically resulted in products with >98% purity according to radio-thin-layer chromatography (TLC), the triazole-DOTA precursor showed markedly inferior labeling yields and reduced radiochemical purity (90% and less). We hypothesize that close proximity of the additional triazole nitrogen donor thwarts the formation of the thermodynamically stable, kinetically inert “in-cage” chelate by stabilizing the initially formed, kinetically labile “out-of-cage” encounter complex.35 Hence, the usage of the CuAAC-conjugated DOTA-monoamide building block cannot be recommended for radiometalated compounds, at least not in the particular structural setup with a C3 alkyl chain between the chelator and azide/triazole functions.
Table 2 shows that the nature and the charge of the chelate have no substantial influence on the αvβ6 integrin affinities (expressed as 50% inhibition concentrations, IC50). Values in the similar sub-nanomolar range are invariantly observed for LuIII as well as for the GaIII complexes. The same applies to selectivities; despite somewhat larger variations, affinities to other integrins are low enough to rule out a significant degree of binding in an in vivo setting. An exception is observed for the NOPO conjugate, although an IC50 of approx. 2 nM is still to be considered absolutely sufficient for imaging.
Table 2. Binding Affinities Toward the Integrins αvβ6, αvβ8, α5β1, αvβ3, αvβ5, and αIIbβ3, Determined for the Nonradioactive Metal Complexes, Expressed as IC50 ± SD [nM].
| compound | αvβ6 | αvβ8 | α5β1 | αvβ3 | αvβ5 | αIIbβ3 |
|---|---|---|---|---|---|---|
| natLu-c(avb6)-Trz-DOTPI | 0.6 ± 0.1 | 116 ± 14 | 238 ± 59 | >1000 | >1000 | >1000 |
| natLu-c(avb6)-Trz-DOTAGA | 0.8 ± 0.2 | 113 ± 23 | 327 ± 3 | >1000 | >1000 | >1000 |
| natLu-c(avb6)-Trz-DOTA | 0.8 ± 0.1 | 158 ± 22 | 271 ± 83 | >1000 | >1000 | >1000 |
| natLu-c(avb6)-DOTAGA | 0.7 ± 0.1 | 123 ± 8 | 407 ± 96 | >1000 | >1000 | >1000 |
| natLu-c(avb6)-DOTA | 0.47 ± 0.03 | 43 ± 7 | 248 ± 24 | >1000 | >1000 | >1000 |
| natGa-c(avb6)-Trz-DOTAGA | 0.4 ± 0.2 | n.d. | n.d. | n.d. | n.d. | n.d. |
| natGa-c(avb6)-Trz-DOTA | 0.8 ± 0.3 | n.d. | n.d. | n.d. | n.d. | n.d. |
| natGa-c(avb6)-DOTAGA | 0.6 ± 0.1 | n.d. | n.d. | n.d. | n.d. | n.d. |
| natGa-c(avb6)-DOTA | 0.3 ± 0.03 | n.d. | n.d. | n.d. | n.d. | n.d. |
| natGa-c(avb6)-Ahx-NOPO | 1.98 ± 0.09 | 135 ± 26 | 440 ± 31 | >1000 | >1000 | >1000 |
| natGa-avebehexin18 | 0.26 ± 0.02 | n.d. | n.d. | n.d. | n.d. | n.d. |
Figure 1 shows that the results of ex vivo biodistribution studies of the 177Lu-labeled compounds in H2009 (αvβ6 integrin positive human lung adenocarcinoma) xenografted severe combined immunodeficiency (SCID) mice are remarkably similar, except for the heavily charged DOTPI derivative which showed markedly higher activity in the kidneys and strongly reduced organ/tissue uptakes. For the other compounds, tumor uptakes are also comparably low, which is however rooted in a fairly low αvβ6 integrin expression density of the tumor model (approx. 25% positive tumor cells in the solid tumor mass).18 Likewise, the substantial uptakes in several organs (lung, stomach, and intestines) are resulting from a low-level αvβ6 integrin expression in the respective murine organs and have been proven to be specific (i.e., fully blockable) in the context of preclinical evaluation of 68Ga-avebehexin.18
Figure 1.
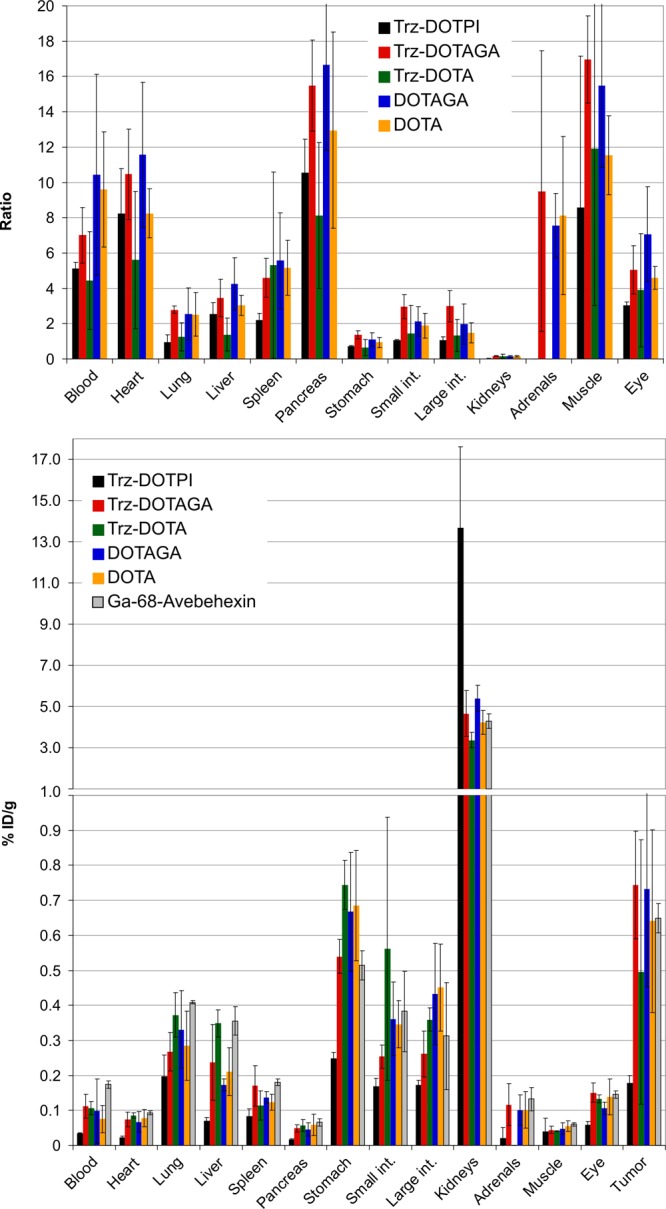
Tumor-to-organ/tissue ratios (top) and biodistribution (bottom, % injected dose per gram tissue) of approx. 2 MBq of the 177Lu-labeled c(avb6)-peptide conjugates in H2009 xenografted SCID mice, 90 min p.i. (mean ± SD, n = 3–5; for data in the numerical form, see Tables S1 and S2.). Literature data for 68Ga-avebehexin18 are plotted as a reference.
Because of similar biodistributions, none of the investigated compounds clearly stands out in terms of tumor-to-organ ratios, although the two DOTAGA derivatives appear to be somewhat favored. The actual extent is however low, and it remains questionable whether these results in murine models would translate to the human setting. Data for natLu-c(avb6)-Trz-DOTA are reported for the sake of completeness but should be considered less relevant because of the above-mentioned irregularities in terms of radiolabeling.
Although not being the primary focus of this study, we also performed a preliminary comparison of PET imaging properties of the corresponding 68Ga-labeled compounds. Figure 2 shows that the image contrast is quite similar for all tracers, however slightly favoring our previous lead candidate 68Ga-avebehexin18 which features a particularly low background uptake. As an exception, 68Ga-c(avb6)-Trz-DOTA shows high blood pool activity (indicated by hotspots in the regions of heart and carotid), which is most likely caused by radiometal dissociation, consistent with our hypothesis that this chelate is actually present as a kinetically labile “out-of-cage” complex (see above). Furthermore, substantial variations of absolute tumor uptakes are observed in contrast to the 177Lu-labeled species, which however cannot be fully explained on the basis of our limited data set.
Figure 2.

PET images (maximum intensity projections) for 68Ga-labeled c(avb6)-peptide conjugates in H2009 tumor-bearing SCID mice (approx. 12 MBq, 60 min p.i.). Images are scaled for similar visual intensity in the tumor (positions indicated by white arrows), resulting in different upper limits (% injected dose per milliliter) of scale bars.
Discussion
As inferred from previous studies,16,18 cyclo(FRGDLAFp(NMe)K) was found to tolerate a variety of functionalizations without substantial impact on the integrin affinity profile. Contrary to our expectations, however, the fourfold negatively charged 177Lu-DOTPI derivative actually showed inferior in vivo properties, namely, a comparatively low uptake in tumor and αvβ6 integrin expressing organs but a much higher activity accumulation in the kidneys. The first observation is explained by the fact that an increased polarity accelerates blood clearance and owing to fewer tissue passages, reduces target accumulation of a tracer. However, it is surprising that the presence of several negative charges multiplies kidney retention because one would expect a more rapid and complete excretion instead. Presumably, the specific configuration of the monoconjugated DOTPI complex is subjected to a specific reabsorption mechanism. Irrespective of different chelators and formal charges of radiometal chelates (0 and −1), biodistribution of the other 177Lu-labeled species is remarkably similar and furthermore matches the in vivo properties of the PET imaging agent 68Ga-avebehexin. Hence, combinations of these 68Ga and 177Lu radiopharmaceuticals are, by principle, applicable as αvβ6 integrin-targeted theranostic pairs.
However, in contrast to the imaging agents which should primarily exhibit a high target-to-background ratio to ensure a good image contrast, clinical applicability of therapeutic radiopharmaceuticals ultimately depends on the delivered radiation dose per tissue volume, which should be as high as possible. In this context, we consider it problematic that the tumor uptake of the 177Lu-labeled c(FRGDLAFp(NMe)K) derivatives is low and by far does not match the usual values known from clinically successful radiopeptides and theranostics, such as sst2 ligands or prostate-specific membrane antigen inhibitors. In principle, this does not implicate a general unsuitability for therapeutic purposes, particularly in view of the above-mentioned, comparatively low target expression density in the subcutaneous tumor models, which might not truly reflect the clinical situation.
We nevertheless hold the opinion that even our preliminary data set clearly indicates a yet unsatisfactory performance and a lot of room for improvement, suggesting that the properties of these simple peptide–chelate conjugates do not warrant clinical transfer. This applies even more because past experience has shown that the premature clinical transfer of nonoptimal compounds sometimes hampered or even inhibited subsequent clinical implementation of more advanced agents, ultimately compromising patient welfare. A plethora of strategies for the optimization of pharmacokinetics and tuning of in vivo properties have been developed over the decades, for example, the introduction of pharmacokinetic modifiers into the molecular frameworks. In the present case, such approaches should be systematically exploited to obtain compounds with more adequate properties before taking the next steps.
Against this background, we also decided to refrain from a more extended in vivo evaluation of the 177Lu labeled and particularly of the 68Ga-PET agents. Despite the low animal numbers and the absence of control experiments limit the significance of our study and may render it methodically weak, it appears to be the most appropriate decision with respect to the 3R-rule (reduce, refine, and replace). In the context of an increasingly critical public opinion towards animal experiments and a growing societal awareness for animal welfare, the number of animals necessary for a given experiment should probably not be characterized by strict adherence to statistics. In view of the logical proposition that a theory cannot be proven by any finite data set but be refuted by a single experiment, even a small trial may already supply enough information to experienced researchers to enable them to predict a long-term failure and to adjust a study plan accordingly. We nonetheless believe that examples of such data (e.g., Figure 2) deserve to be disseminated to help others to avoid the same pitfall, ultimately resulting in a more responsible use of resources and animals.
Conclusions
In summary, the αvβ6 integrin-selective peptide cyclo(FRGDLAFp(NMe)K) can be equipped with different chelators and subsequently labeled with the therapeutic radionuclide 177Lu, without substantial loss of target affinity and selectivity. Notwithstanding this, the in vivo properties of the resulting therapeutic radiopharmaceuticals are essentially not satisfactory, particularly in terms of absolute tumor uptake, not warranting immediate clinical translation. Further modification of the structures, aiming at the optimization of tumor uptake and retention, is required before the potential of c(FRGDLAFp(NMe)K)-based αvβ6 integrin-targeted radiopharmaceuticals that can be harnessed for cancer therapy in a clinical setting.
Experimental Section
General
Unless otherwise noted, all commercially available reagents and solvents were of analytical grade and were used without further purification. 3-Azido-1-propylamine, Cu(OAc)2·H2O, 4-pentynoic acid, diisopropylamine (DIPEA), and sodium ascorbate were purchased from Sigma-Aldrich (Darmstadt, Germany). NOTA was purchased from Macrocyclics, Inc. (Plano, Texas, USA). DOTA-tris(tBu)ester and DOTAGA anhydride were obtained from CheMatech (Dijon, France). HATU was obtained from Bachem Holding AG (Bubendorf, Switzerland). The chlorotrityl chloride resin was purchased from Iris Biotech GmbH (Marktredwitz, Germany). N-terminal Fmoc-protected amino acids were purchased from Iris Biotech GmbH, Alfa Aesar (Karlsruhe, Germany) and Carbolution Chemicals GmbH (St. Ingbert, Germany). The Solid-phase peptide synthesis was performed in 20 mL syringes by B. Braun (Melsungen, Germany) equipped with an additional frit. NOPO,36 DOTPI,37 as well as cyclo(F-R-G-D-L-A-F-p-(NMe)K(pentynoic acid)), TRAP(azide)1, and avebehexin18 were synthesized as described previously.
Analytical and preparative high-performance liquid chromatography (HPLC) were performed on Shimadzu gradient systems, each equipped with a SPD-20A dual wavelength UV–vis detector (λ1 = 220 nm, λ2 = 254 nm). For analytical purposes, a flow rate of 1.0 mL/min with a Nucleosil 100-5 C18 column (125 × 4.6 mm, 5 μm particle size) was used for linear gradients in 15 or 20 min. A mixture of acetonitrile (J.T.Baker Ultra Gradient HPLC grade, supplemented with 5% H2O) and purified water (from Millipore system) was used as an eluent, containing 0.1% trifluoroacetic acid (TFA); gradient A: 15–65% MeCN in 15 min, gradient B: 0–40% MeCN in 20 min. Preparative HPLC purification was performed with a Multospher 100 RP 18-5μ column (250 × 10 mm, 5 μm particle size) at a flow rate of 5.0 mL/min in 15 min or with a Multospher 100 RP 18-5μ column (150 × 10 mm, 5 μm particle size) at a flow rate of 3.0 mL/min in 20 min. Mass spectra were acquired on an expressionL CMS mass spectrometer with electrospray ionization and a quadrupole analyzer (Advion Inc. Ithaca, USA). NMR spectra were measured on an AVHD 300 (Bruker, Billerica, USA) device at 300 K. Determination of pH values was done using a calibrated SevenEasy pH meter from Mettler Toledo (Gießen, Germany). For lyophilization, an Alpha 1-2 lyophilization apparatus (Christ, Osterode, Germany) with a rotary vane pump RZ-2 (Vacuubrand, Wertheim, Germany) was used. Centrifugation was done using a Heraeus Megafuge 16R centrifuge (Thermo Fisher Scientific Inc., Waltham, USA). Quantification of tissue activities was done using a 2480 WIZARD2 automatic γ-counter (PerkinElmer, Waltham, USA).
Integrin Binding Assays
IC50 values were determined as described previously,37,38 using the following compounds as internal standards: cilengitide39 (αvß3—0.54 nM, αvß5—8 nM, and α5ß1—15.4 nM), the linear RTD-peptide RTDLDSLRT40 (αvß6—33 nM and αvß8—100 nM), and tirofiban41 (αIIbß3—1.2 nM). Briefly, the flat-bottom 96-well enzyme-linked immunosorbent assay plates (BRAND, Wertheim, Germany) were coated with the respective extracellular matrix protein in a carbonate buffer (15 mM Na2CO3, 35 mM NaHCO3, pH 9.6) at 4 °C overnight. After washing the plates with phosphate-buffered saline (PBS)-T-buffer (PBS/Tween-20), free binding sites were blocked by incubation with TS-B-buffer (Tris-saline/bovine serum albumin). Dilution series of the respective inactive LuIII and GaIII compounds (20 μM to 6.4 nM) were prepared and incubated in 1:1 mixtures with the respective integrin. Surface-bound integrin was detected by subsequent incubation with a specific primary antibody and a secondary peroxidase-labeled antibody (antimouse IgG-POD, Sigma-Aldrich). After the addition of the dye SeramunBlau (Seramun Diagnostic, Heidesee, Germany) and quenching of the reaction by the addition of 3 M H2SO4, the absorbance at λ = 405 nm was measured with a microplate reader (Genius, Tecan, Männedorf, Switzerland). The IC50 value for each compound was determined in duplicate, and the inhibition curves were analyzed using OriginPro 9.0 software. All determined IC50 values were referenced to the activity of the respective internal standards.
Metal Complexation and Radiochemistry
For the synthesis of the nonradioactive GaIII and LuIII complexes, 50 μL of 4 mM aq solution of Ga(NO3)3 or LuCl3 were added to 100 μL of 2 mM aq solution of the respective chelator conjugate. The reaction mixture was stirred at room temperature (rt) (GaIII, 5 min) or 90 °C (LuIII) for 5 min (DOTPI) or 30 min (DOTAGA and DOTA conjugates). The purity of all synthesized chelates was determined by analytical reversed-phase (RP)-HPLC and mass spectrometry (MS) and was always ≥95%.
Radiometal incorporation and radiochemical purity of labeled compounds was determined by radio-TLC on the instant TLC silica-impregnated chromatography paper (Agilent, Santa Clara, USA; eluents: 0.1 M trisodium citrate or a 1:1 (v/v) mixture of 1 M ammonium acetate and methanol) and analyzed using a scan-RAM radio-TLC detector by LabLogic systems Inc. (Brandon, USA). 177Lu-labeling experiments were performed by mixing a 1 mM solution of the precursor (0.5–4 nmol) and 10 μL of an aqueous NH4OAc solution (1 M) to adjust pH to 5.9. Subsequently, 10–24 MBq of [177Lu]LuCl3 (0.8 GBq/mL, 0.04 M HCl(aq), as obtained from ITG, Garching, Germany) was added, and the solution was diluted with tracepure water to attain a final volume of 100 μL. The mixture was heated for 30 min at 95 °C. The labeling efficiency was determined by radio-TLC and was always ≥99%. 68Ga-labeling experiments were performed using a fully-automated on-site system (GallElut+ by Scintomics, Lindach, Germany) as described previously.42 Briefly, the eluate of a 68Ge/68Ga generator with a SnO2 matrix (by IThemba LABS, SA; 1.25 mL, eluent: 1 M aq. HCl, containing approx. 500 MBq 68Ga) was adjusted to pH 2 by the addition of aq. N-(2-hydroxyethyl)piperazine-N′-ethanesulfonic acid (HEPES) buffer (450 μL, 2.7 M) and applied for labeling of 2 nmol of the NOPO or TRAP conjugate, respectively, for 2 min at 95 °C. In the case of the DOTA and DOTAGA compounds, the generator eluate was adjusted to pH 3 by the addition of aq. HEPES buffer (900 μL, 2.7 M) and applied for labeling of 5 nmol of the respective ligand for 5 min at 100 °C. The radiolabeled peptides were trapped on Sep-Pak C8 light solid-phase extraction cartridges, which were purged with water (10 mL). The product was eluted with 2 mL of aq. EtOH (50%). After evaporation of the ethanol, the labeling efficiency was determined by radio-TLC and was always found to be ≥96%, except for 68Ga-c(avb6)-Trz-DOTA (<90%).
Radio-HPLC of the 177Lu-labeled compounds was performed on a Nucleosil 100-5 C18 column (125 × 4.6 mm, 5 μm particle size). A mixture of acetonitrile (J. T. Baker Ultra Gradient HPLC grade, supplemented with 5% H2O) and purified water (from Millipore system) was used as an eluent, containing 0.1% TFA. For linear gradients in 15 min, a Sykam S2100 HPLC gradient system (Sykam GmbH, Eresing, Germany) was utilized with a linear UV–vis detector (λ = 220 nm) by Linear Instruments Corp. (Reno, USA) and by an ACE Mate 925-SCINT scintillation counter with a multichannel analyzer by EG&G Ortec Inc. (Oak Ridge, USA). Radiochemical purity was always ≥98%.
Determination of log D Values
For the determination of n-octanol-PBS distribution coefficients (log D7.4), 500 μL of 1-octanol and 500 μL of PBS were combined in a 1.5 mL Eppendorf tube. Approx. 0.5 MBq of the 177Lu-labeled compound or 1 MBq of the 68Ga-labeled compound was added and vortexed vigorously for 3 min. The samples were centrifuged (9.000 rpm, 5 min), and the activities in 200 μL of the organic phase and 20 μL of the aqueous phase were quantified in a γ-counter. Each experiment was repeated eight times.
Animal Studies
All animal studies have been performed in accordance with general animal welfare regulations in Germany and the institutional guidelines for the care and use of animals. To establish xenograft models, three- to four-month-old female CB17 SCID mice (Charles River, Sulzfeld, Germany) were injected subcutaneously with 0.8 × 106 H2009 human lung adenocarcinoma cells (ATCC CRL-5911; American Type Culture Collection, Manassas, VA, USA) in Matrigel (CultrexBME, Type 3 PathClear, Trevigen, GENTAUR GmbH, Aachen, Germany). Mice were used for biodistribution and PET studies, when tumors had grown to a diameter of 4–9 mm (8–30 d after inoculation).
PET Imaging
H2009 tumor-bearing SCID mice were anesthesized with isoflurane for intravenous administration of the 68Ga-labeled compound. The administered activity per mouse ranged between 11 and 16 MBq (50–340 pmol, depending on variations in timing of production and administration). PET imaging was performed on a Siemens Inveon small-animal PET system, 60 min p.i. with an acquisition time of 20 min. Data were reconstructed using Siemens Inveon Research Workspace software, employing a three-dimensional ordered subset expectation maximum algorithm without scatter and attenuation correction.
Biodistribution
For biodistribution studies, 0.8–2.2 MBq (approx. 100–350 pmol) of the 177Lu-labeled compounds was injected into the tail vein of H2009 tumor-bearing SCID mice. The mice were sacrificed 90 min after injection, a blood sample was taken, and the organs of interest [heart, lung, liver, spleen, pancreas, stomach (empty), small intestine (empty), large intestine (empty), kidneys, adrenals, muscle, eye, tumor, and tail] were dissected. Quantification of the activity in weighted tissue samples was done using a γ-counter. Injected dose per gram tissue (% ID/g) was calculated from the organ weights and counted activities.
Syntheses
DOTPI(Azide)1
DOTPI dihydrate (100 mg, 124 μmol, 1.0 equiv) was dissolved in dry dimethyl sulfoxide (DMSO, 400 μL), and DIPEA (303 mg, 399 μL, 2.35 mmol, 19 equiv) was added. 3-Azido-1-propylamine (13.7 mg, 13.7 μL, 136 μmol, 1.1 equiv) was added quickly in one portion together with HATU (141 mg, 371 μmol, 3.0 equiv) under stirring at rt. After 60 min, the reaction mixture was quenched with water, adjusted to pH 9, and extracted with CHCl3 (8×). Aqueous phases were combined and evaporated in vacuo. The crude product was dissolved in water (1 mL) and purified by size-exclusion chromatography (Sephadex G-10; mobile phase: water, adjusted to pH 2–3 with 1 M aq. HCl). Fractions containing the product were identified by MS. The solvent was removed in vacuo, and the crude product was dissolved in a mixture of MeCN and water (1:1 by volumes, 1 mL) and subjected to preparative RP-HPLC purification (7–14% MeCN containing 0.1% TFA, 15 min, λ = 220 nm, tR = 10.9 min). After the removal of the organic solvent, the solution was lyophilized to yield DOTPI(azide)1 (21.0 mg, 24.6 μmol, 20%) as a colorless, hygroscopic solid. MS (ESI, positive): m/z = 854.6 [M + H]+, 427.9 [M + 2H]2+. HPLC (gradient: 0–25% MeCN containing 0.1% TFA, in 20 min): tR = 9.98 min. 1H NMR (300 MHz, D2O): δ 1.26 (t, 3JHH = 7.0 Hz, 2H, N3–CH2), 1.71 (p, 3JHH = 6.7 Hz, 2H, N3–CH2CH2), 1.99 (dt, 2JPH = 13.5 Hz,3JHH = 7.8 Hz, 8H, P–CH2CH2), 2.39–2.48 (m, 2H, NH–CH2CH2), 2.58 (dt, 3JPH = 12.9 Hz, 3JHH = 7.7 Hz, 6H, CH2–COOH), 3.20 (t, 3JHH = 6.7 Hz, 2H, CH2–CONH), 3.26–3.53 (m, broad, 24H, ring-CH2, N–CH2–P) ppm. 13C NMR (75 MHz, D2O): δ 23.9 (PBCC), 25.2 (PACC), 26.3 (PBCC), 27.5 (CCC), 27.6 (PACC), 36.8 (N(H)CH2), 48.6 (N3–CH2), 51.0 (m, ring-C), 52.1 (NCPA), 54.3 (NCPB), 162.6 (d, 3JPC = 36.1 Hz, N(H)C=O), 176.5 (d, 3JPC = 13.5 Hz, COOH) ppm. 31P NMR (122 MHz, D2O) δ 39.9 (broad, PA), 55.0 (PB) ppm. Superscript indices A and B indicate P atoms belonging to the undecoratedA or decoratedB side arm, respectively.
DOTAGA-Azide
DOTAGA anhydride (30.0 mg, 60.7 μmol, 1.0 equiv) was dissolved in dry DMSO (1.5 mL) under nitrogen atmosphere, and DIPEA (70.5 mg, 92.7 μL, 546 μmol, 9.0 equiv) was added. 3-Azido-1-propylamine (7.90 mg, 7.90 μL, 78.9 μmol, 1.3 equiv) was added under stirring at rt. After 4 h, the reaction mixture was evaporated in vacuo. Purification was performed by preparative RP-HPLC (0–35% MeCN containing 0.1% TFA, 20 min, λ = 220 nm, tR = 11.5 min). After the removal of the organic solvent, the aqueous eluate was lyophilized to yield DOTAGA-azide (21.7 mg, 38.9 μmol, 64%) as a colorless oil. MS (ESI, positive): m/z = 559.0 [M + H]+, 1117.4 [2M + H]+. HPLC (gradient B): tR = 6.73 min.
DOTAGA-Azide
DOTA-tris(tBu)ester (50.0 mg, 87.3 μmol, 1.0 equiv) was dissolved in dry dimethylformamide (DMF) (400 μL), and DIPEA (214 mg, 282 μL, 1.7 mmol, 19 equiv) was added. 3-Azido-1-propylamine (10.5 mg, 10.5 μL, 105 μmol, 1.2 equiv) was added quickly in one portion together with HATU (99.5 mg, 262 μmol, 3.0 equiv) under stirring at rt. After 2 h, the mixture was quenched with water and the solvent was evaporated in vacuo. Cleavage of tert-butyl groups was performed by the addition of TFA (1 mL) and stirring for 1 h at rt. After the removal of TFA, the crude product was dissolved in water and purified by size-exclusion chromatography (Sephadex G-10; mobile phase: water, adjusted to pH 2–3 with 1 M aq. HCl). Fractions containing the product were identified by MS. The solvent was removed in vacuo, and the crude product was dissolved in a mixture of MeCN and water (1:1 by volume, 1 mL) and purified by preparative HPLC (9–12% MeCN containing 0.1% TFA, 15 min, λ = 220 nm, tR = 7.0 min). After evaporation of the organic component in vacuo, the aqueous eluate was lyophilized to yield DOTAGA-azide (8.0 mg, 16.5 μmol, 19%) as a pale-yellow oil. MS (ESI, positive): m/z = 487.3 [M + H]+.
Cyclo-[F-R(Pbf)-G-D(tBu)-L-A-F-p-(NMe)K(aminohexanoic acid)]
Fmoc-6-aminohexanoic acid (3.1 mg, 8.9 μmol, 1.2 equiv), HATU (8.4 mg, 22 μmol, 3.0 equiv), and DIPEA (5.7 mg, 7.5 μL, 44 μmol, 6.0 equiv) were stirred in anhydrous DMF (1 mL) for 15 min. Cyclo-[F-R(Pbf)-G-D(tBu)-L-A-F-p-(NMe)K] (10.0 mg, 7.4 μmol, 1.0 equiv) was dissolved in anhydrous DMF (1 mL) and was added dropwise. After 2 h, piperidine (400 μL) was added and the mixture was stirred at rt for 30 min. All volatiles were evaporated in vacuo. The crude product was purified by preparative HPLC (55% MeCN containing 0.1% TFA, 20 min, λ = 220 nm, tR = 8.53 min). After the removal of the organic solvent, the product was lyophilized, yielding a colorless solid (7.1 mg, 4.8 μmol, 65%). MS (ESI, positive): m/z = 1468.0 [M + H]+, 734.5 [M + 2H]2+, 706.3 [M – tBu + 2H]2+. HPLC (gradient: 20–80% MeCN containing 0.1% TFA, in 15 min): tR = 12.6 min.
c(avb6)-Trz-DOTPI
DOTPI(azide)1 (2.9 mg, 3.4 μmol, 1.1 equiv) was dissolved in a mixture of tert-butanol and water (1:1 by volume, 10 mM) and combined with a solution of cyclo-[F-R-G-D-L-A-F-p-(NMe)K(pentynoic acid)] (3.5 mg, 3.1 μmol, 1.0 equiv) in a mixture of tert-butanol and water (1:1 by volume, 300 μL). Sodium ascorbate (30.6 mg, 154 μmol, 50 equiv) was dissolved in water (400 μL) and added to the mixture as well as Cu(OAc)2·H2O (0.74 mg, 3.70 μmol, 1.2 equiv) in 100 μL water. Vortexing resulted in a brown precipitate, turning later into a blue clear reaction mixture. After 1 h, the reaction mixture was diluted with water (2 mL). The product was demetalated by the addition of NOTA (18.7 mg, 62 μmol, 20 equiv), adjusted to pH 2.2 using 1 M aq. HCl, and left to react for 1 h at 60 °C, followed by stirring overnight at rt. The mixture was purified by preparative HPLC (30% MeCN containing 0.1% TFA, 20 min, λ = 220 nm, tR = 18.6 min). After the removal of the organic solvent, the aqueous solution was lyophilized, yielding c(avb6)-Trz-DOTPI (2.6 mg, 1.3 μmol, 42%) as a colorless solid. MS (ESI, positive): m/z = 1980.8 [M + H]+, 990.9 [M + 2H]2+, 661.0 [M + 3H]3+. HPLC (gradient A): tR = 8.25 min.
c(avb6)-Trz-DOTAGA
DOTAGA-azide (4.0 mg, 7.1 μmol, 2.0 equiv) was dissolved in a mixture of tert-butanol and water (1:1 by volume, 200 μL) and combined with a solution of cyclo-[F-R-G-D-L-A-F-p-(NMe)K(pentynoic acid)] (4.0 mg, 3.6 μmol, 1.0 equiv) in a mixture of tert-butanol and water (1:1 by volume, 400 μL). Sodium ascorbate (35 mg, 178 μmol, 50 equiv) was dissolved in water (400 μL) and added to the mixture, followed by Cu(OAc)2·H2O (0.85 mg, 4.3 μmol, 1.2 equiv) in 100 μL water. Vortexing resulted in a brown precipitate, which was turning into a clear, green reaction mixture. After 1 h, the reaction mixture was diluted with water (2 mL). The product was demetalated by the addition of excess Na2S·9H2O at pH 7.5. The precipitate was separated by centrifugation, and the clear reaction mixture was purified by preparative HPLC purification (32–33% MeCN containing 0.1% TFA, 20 min, λ = 220 nm, tR = 10.5 min). After the removal of the organic solvent, the aqueous solution was lyophilized, yielding c(avb6)-Trz-DOTAGA (1.1 mg, 0.65 μmol, 18%) as a colorless solid. MS (ESI, positive): m/z = 1685.3 [M + H]+, 843.2 [M + 2H]2+, 562.5 [M + 3H]3+. HPLC (gradient A): tR = 9.60 min.
c(avb6)-Trz-DOTA
DOTAGA-azide (9.6 mg, 20 μmol, 2.0 equiv) was dissolved in a mixture of tert-butanol and water (1:1 by volume, 600 μL) and combined with a solution of cyclo-[F-R-G-D-L-A-F-p-(NMe)K(pentynoic acid)] (11.1 mg, 9.86 μmol, 1.0 equiv) in a mixture of tert-butanol and water (1:1 by volume, 800 μL). Sodium ascorbate (97.7 mg, 493 μmol, 50 equiv) was dissolved in water (600 μL) and added to the mixture, followed by Cu(OAc)2·H2O (2.4 mg, 12 μmol, 1.2 equiv) in 100 μL of water. Vortexing resulted in a brown precipitate, which was turning into a clear, green reaction mixture. After 1 h, the reaction mixture was diluted with water (2 mL). The product was demetalated by the addition of excess Na2S·9H2O at pH 7.5. The precipitate was separated by centrifugation, and the clear reaction mixture was purified by preparative HPLC purification (34% MeCN containing 0.1% TFA, 15 min, λ = 220 nm, tR = 12.1 min). After the removal of the organic solvent, the aqueous eluate was lyophilized, yielding c(avb6)-Trz-DOTA (1.0 mg, 0.64 μmol, 7%) as a colorless solid. MS (ESI, positive): m/z = 1613.4 [M + H]+, 807.1 [M + 2H]2+, 538.5 [M + 3H]3+. HPLC (gradient A): tR = 9.55 min.
c(avb6)-DOTAGA
A solution of DOTAGA anhydride (5.1 mg, 10 μmol, 2.0 equiv) in anhydrous DMF (1 mL) was combined with DIPEA (4.0 mg, 5.3 μL, 31 μmol, 6.0 equiv) under nitrogen atmosphere, and the reaction mixture was stirred for 15 min. Cyclo-[F-R(Pbf)-G-D(tBu)-L-A-F-p-(NMe)K] (7.0 mg, 5.2 μmol, 1.0 equiv) was dissolved in anhydrous DMF (1 mL) and was added dropwise. After 2 h, the solvent was evaporated in vacuo. The residue was stirred in a mixture of TFA/TIPS/H2O (90/5/5 by volume, 1 mL) for 1.5 h at rt. After the removal of TFA by flushing with nitrogen, the crude product was dissolved in DMF and purified by preparative HPLC (25–50% MeCN containing 0.1% TFA, 15 min, λ = 220 nm, tR = 12.3 min). The organic solvent was removed, and the aqueous residue was lyophilized, yielding c(avb6)-DOTAGA (5.1 mg, 3.4 μmol, 65%) as a colorless solid. MS (ESI, positive): m/z = 1505.5 [M + H]+, 753.2 [M + 2H]2+, 502.5 [M + 3H]3+. HPLC (gradient A): tR = 9.51 min.
c(avb6)-DOTA
A solution of DOTA-tris-tert-butyl ester (5.9 mg, 10 μmol, 2.0 equiv) in anhydrous DMF (1 mL) was combined with DIPEA (4.0 mg, 5.3 μL, 31 μmol, 6.0 equiv) and HATU (5.9 mg, 16 μmol, 3.0 equiv), and the yellow reaction mixture was stirred for 15 min. Cyclo-[F-R(Pbf)-G-D(tBu)-L-A-F-p-(NMe)K] (7.0 mg, 5.2 μmol, 1.0 equiv) was dissolved in anhydrous DMF (1 mL) and was added dropwise. After 2 h, the solvent was removed in vacuo. The residue was stirred in a mixture of TFA/TIPS/H2O (90/5/5 by volume, 1 mL) for 2 h at rt. After the removal of TFA by flushing with nitrogen, the crude product was dissolved in DMF and purified by preparative HPLC (33–45% MeCN containing 0.1% TFA, 15 min, λ = 220 nm, tR = 10.7 min). The organic solvent was removed, and the aqueous residue was lyophilized, yielding c(avb6)-DOTA (4.3 mg, 3.0 μmol, 57%) as a colorless solid. MS (ESI, positive): m/z = 1433.7 [M + H]+, 717.3 [M + 2H]2+, 487.5 [M + 3H]3+. HPLC (gradient A): tR = 9.46 min.
c(avb6)-Ahx-NOPO
Cyclo-[F-R(Pbf)-G-D(tBu)-L-A-F-p-(NMe)K(aminohexanoic acid)] (5.0 mg, 3.4 μmol, 1.0 equiv) was dissolved in dry DMF (800 μL) and dry DMSO (200 μL). NOPO·0.6H2O (5.2 mg, 10.2 μmol, 3.0 equiv) and DIPEA (4.4 mg, 5.8 μL, 34 μmol, 10 equiv) were added, and subsequently HATU (7.7 mg, 21 μmol, 6.0 equiv) was added in portions. The yellow solution was stirred for 1.5 h at rt. After quenching the reaction with water (1 mL), the solvent was removed in vacuo. The residue was stirred in a mixture of TFA/TIPS/H2O (90/5/5 by volume, 1 mL) for 1.5 h. Precipitation of the peptide was performed by the addition of the reaction mixture to ice-cold diethyl ether (15 mL). After centrifugation, the crude product was dried overnight in a desiccator. The residue was purified by preparative HPLC (35–40% MeCN containing 0.1% TFA, 15 min, λ = 220 nm, tR = 10.1 min). The organic solvent was removed, and the aqueous residue was lyophilized, yielding c(avb6)-Ahx-NOPO (3.9 mg, 2.4 μmol, 70%) as a colorless solid. MS (ESI, positive): m/z = 1635.8 [M + H]+, 818.6 [M + 2H]2+.
Acknowledgments
The authors thank Prof. Markus Schwaiger (Department of Nuclear Medicine, TUM) for providing laboratory space and granting access to imaging devices and Sybille Reder, Markus Mittelhäuser for assistance with animal PET.
Glossary
Abbreviations
- PET
positron emission tomography
- PDAC
pancreatic ductal adenocarcinoma
- PBS
phosphate-buffered saline
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b00035.
Biodistribution data in numerical form and miscellaneous information (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Deutsche Forschungsgemeinschaft, #NO822/4-1 and SFB 824.
The authors declare no competing financial interest.
Supplementary Material
References
- Hynes R. O. Integrins. Cell 2002, 110, 673–687. 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- Kapp T. G.; Rechenmacher F.; Sobahi T. R.; Kessler H. Integrin modulators: a patent review. Expert Opin. Ther. Pat. 2013, 23, 1273–1295. 10.1517/13543776.2013.818133. [DOI] [PubMed] [Google Scholar]
- Niu G.; Chen X. Why integrin as a primary target for imaging and therapy. Theranostics 2011, 1, 30–45. 10.7150/thno/v01p0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks P. C.; Clark R.; Cheresh D. A. Requirement of vascular integrin alpha vbeta 3 for angiogenesis. Science 1994, 264, 569–571. 10.1126/science.7512751. [DOI] [PubMed] [Google Scholar]
- Avraamides C. J.; Garmy-Susini B.; Varner J. A. Integrins in angiogenesis and lymphangiogenesis. Nat. Rev. Cancer 2008, 8, 604–617. 10.1038/nrc2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieberler M.; Reuning U.; Reichart F.; Notni J.; Wester H.-J.; Schwaiger M.; Weinmüller M.; Räder A.; Steiger K.; Kessler H. Exploring the Role of RGD-Recognizing Integrins in Cancer. Cancers 2017, 9, 116. 10.3390/cancers9090116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay A.; Raghavan S. Defining the Role of Integrin αvβ6 in Cancer. Curr. Drug Targets 2009, 10, 645–652. 10.2174/138945009788680374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu J.; Li Z. The roles of integrin αvβ6 in cancer. Canc. Lett. 2017, 403, 128–137. 10.1016/j.canlet.2017.06.012. [DOI] [PubMed] [Google Scholar]
- Li S.; McGuire M. J.; Lin M.; Liu Y.-H.; Oyama T.; Sun X.; Brown K. C. Synthesis and characterization of a high-affinity αvβ6-specific ligand for in vitro and in vivo applications. Mol. Cancer Ther. 2009, 8, 1239–1249. 10.1158/1535-7163.mct-08-1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.; Wu Y.; Wang F.; Liu Z. Molecular imaging of integrin αvβ6 expression in living subjects. Am. J. Nucl. Med. Mol. Imaging 2014, 4, 333–345. [PMC free article] [PubMed] [Google Scholar]
- John A. E.; Luckett J. C.; Tatler A. L.; Awais R. O.; Desai A.; Habgood A.; Ludbrook S.; Blanchard A. D.; Perkins A. C.; Jenkins R. G.; Marshall J. F. Preclinical SPECT/CT imaging of αvβ6 integrins for molecular stratification of idiopathic pulmonary fibrosis. J. Nucl. Med. 2013, 54, 2146–2152. 10.2967/jnumed.113.120592. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Liu H.; Ma T.; Sun X.; Shi J.; Jia B.; Sun Y.; Zhan J.; Zhang H.; Zhu Z.; Wang F. Integrin αvβ6-Targeted SPECT Imaging for Pancreatic Cancer Detection. J. Nucl. Med. 2014, 55, 989–994. 10.2967/jnumed.113.132969. [DOI] [PubMed] [Google Scholar]
- Zhu X.; Li J.; Hong Y.; Kimura R. H.; Ma X.; Liu H.; Qin C.; Hu X.; Hayes T. R.; Benny P.; Gambhir S. S.; Cheng Z. 99mTc-labeled cystine knot peptide targeting integrin αvβ6 for tumor SPECT imaging. Mol. Pharm. 2014, 11, 1208–1217. 10.1021/mp400683q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausner S. H.; Abbey C. K.; Bold R. J.; Gagnon M. K.; Marik J.; Marshall J. F.; Stanecki C. E.; Sutcliffe J. L. Targeted in vivo imaging of integrin αvβ6 with an improved radiotracer and its relevance in a pancreatic tumor model. Cancer Res. 2009, 69, 5843–5850. 10.1158/0008-5472.can-08-4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A. N.; McGuire M. J.; Li S.; Hao G.; Kumar A.; Sun X.; Brown K. C. Dimerization of a phage-display selected peptide for imaging of αvβ6-integrin: two approaches to the multivalent effect. Theranostics 2014, 4, 745–760. 10.7150/thno.7811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltsev O. V.; Marelli U. K.; Kapp T. G.; Di Leva F. S.; Di Maro S.; Nieberler M.; Reuning U.; Schwaiger M.; Novellino E.; Marinelli L.; Kessler H. Stable Peptides Instead of Stapled Peptides: Highly Potent αvβ6-Selective Integrin Ligands. Angew. Chem., Int. Ed. 2016, 55, 1535–1539. 10.1002/anie.201508709. [DOI] [PubMed] [Google Scholar]
- Notni J.; Hermann P.; Havlíčková J.; Kotek J.; Kubíček V.; Plutnar J.; Loktionova N.; Riss P. J.; Rösch F.; Lukeš I. A triazacyclononane-based bifunctional phosphinate ligand for the preparation of multimeric 68Ga tracers for positron emission tomography. Chem.—Eur. J. 2010, 16, 7174–7185. 10.1002/chem.200903281. [DOI] [PubMed] [Google Scholar]
- Notni J.; Reich D.; Maltsev O. V.; Kapp T. G.; Steiger K.; Hoffmann F.; Esposito I.; Weichert W.; Kessler H.; Wester H.-J. In Vivo PET Imaging of the Cancer Integrin αvβ6 Using 68Ga-Labeled Cyclic RGD Nonapeptides. J. Nucl. Med. 2017, 58, 671–677. 10.2967/jnumed.116.182824. [DOI] [PubMed] [Google Scholar]
- Sipos B.; Hahn D.; Carceller A.; Piulats J.; Hedderich J.; Kalthoff H.; Goodman S. L.; Kosmahl M.; Kloppel G. Immunohistochemical screening for β6-integrin subunit expression in adenocarcinomas using a novel monoclonal antibody reveals strong up-regulation in pancreatic ductal adenocarcinomas in vivo and in vitro. Histopathol. 2004, 45, 226–236. 10.1111/j.1365-2559.2004.01919.x. [DOI] [PubMed] [Google Scholar]
- Steiger K.; Schlitter A.-M.; Weichert W.; Esposito I.; Wester H.-J.; Notni J. Perspective of αvβ6-Integrin Imaging for Clinical Management of Pancreatic Carcinoma and Its Precursor Lesions. Mol. Imaging 2017, 16, 153601211770938. 10.1177/1536012117709384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.; Lin P.; Gao C.; Peng C.; Liu S.; Gao H.; Wang B.; Wang J.; Niu J.; Niu W. Integrin β6 acts as an unfavorable prognostic indicator and promotes cellular malignant behaviors via ERK-ETS1 pathway in pancreatic ductal adenocarcinoma (PDAC). Tumor Biol. 2016, 37, 5117–5131. 10.1007/s13277-015-4353-7. [DOI] [PubMed] [Google Scholar]
- Siegel R. L.; Miller K. D.; Jemal A. Cancer Statistics, 2017. Ca-Cancer J. Clin. 2017, 67, 7–30. 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- Krebs in Deutschland 2011/2012, 10. Ausgabe. Editors: Robert Koch-Institut and Gesellschaft der epidemiologischen Krebsregister in Deutschland e.V.: Berlin, 2015. [Google Scholar]
- Ahmedah H.; Patterson L.; Shnyder S.; Sheldrake H. RGD-binding integrins in head and neck cancers. Cancers 2017, 9, 56. 10.3390/cancers9060056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šimeček J.; Wester H.-J.; Notni J. Copper-64 labelling of triazacyclononane-triphosphinate chelators. Dalton Trans. 2012, 41, 13803–13806. 10.1039/c2dt31880f. [DOI] [PubMed] [Google Scholar]
- Notni J.; Šimeček J.; Wester H.-J. Phosphinic acid functionalized polyazacycloalkane chelators for radiodiagnostics and radiotherapeutics: unique characteristics and applications. ChemMedChem 2014, 9, 1107–1115. 10.1002/cmdc.201400055. [DOI] [PubMed] [Google Scholar]
- Notni J.; Pohle K.; Wester H.-J. Comparative gallium-68 labeling of TRAP-, NOTA-, and DOTA-peptides: practical consequences for the future of gallium-68-PET. EJNMMI Res. 2012, 2, 28. 10.1186/2191-219x-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šimeček J.; Hermann P.; Wester H.-J.; Notni J. How is 68Ga Labeling of Macrocyclic Chelators Influenced by Metal Ion Contaminants in 68Ge/68Ga Generator Eluates?. ChemMedChem 2013, 8, 95–103. 10.1002/cmdc.201200471. [DOI] [PubMed] [Google Scholar]
- Wurzer A.; Seidl C.; Morgenstern A.; Bruchertseifer F.; Schwaiger M.; Wester H.-J.; Notni J. Dual-Nuclide Radiopharmaceuticals for Positron Emission Tomography Based Dosimetry in Radiotherapy. Chem.—Eur. J. 2017, 24, 547. 10.1002/chem.201702335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranyai Z.; Reich D.; Vágner A.; Weineisen M.; Tóth I.; Wester H.-J.; Notni J. A shortcut to high-affinity Ga-68 and Cu-64 radiopharmaceuticals: one-pot click chemistry trimerisation on the TRAP platform. Dalton Trans. 2015, 44, 11137–11146. 10.1039/c5dt00576k. [DOI] [PubMed] [Google Scholar]
- Šimeček J.; Hermann P.; Havlíčková J.; Herdtweck E.; Kapp T. G.; Engelbogen N.; Kessler H.; Wester H.-J.; Notni J. A Cyclen-Based Tetraphosphinate Chelator for the Preparation of Radiolabeled Tetrameric Bioconjugates. Chem.—Eur. J. 2013, 19, 7748–7757. 10.1002/chem.201300338. [DOI] [PubMed] [Google Scholar]
- Notni J.; Wester H.-J. A Practical Guide on the Synthesis of Metal Chelates for Molecular Imaging and Therapy by Means of Click Chemistry. Chem.—Eur. J. 2016, 22, 11500–11508. 10.1002/chem.201600928. [DOI] [PubMed] [Google Scholar]
- Šimeček J.; Zemek O.; Hermann P.; Wester H.-J.; Notni J. A Monoreactive Bifunctional Triazacyclononane Phosphinate Chelator with High Selectivity for Gallium-68. ChemMedChem 2012, 7, 1375–1378. 10.1002/cmdc.201200261. [DOI] [PubMed] [Google Scholar]
- Heppeler A.; Froidevaux S.; Mäcke H. R.; Jermann M.; Béhé M.; Powell P.; Hennig M. Radiometal-Labelled Macrocyclic Chelator-Derivatised Somatostatin Analogue with Superb Tumour-Targeting Properties and Potential for Receptor-Mediated Internal Radiotherapy. Chem.—Eur. J. 1999, 5, 1974–1981. . [DOI] [Google Scholar]
- Toth E.; Brucher E.; Lazar I.; Toth I. Kinetics of Formation and Dissociation of Lanthanide(III)-DOTA Complexes. Inorg. Chem. 1994, 33, 4070–4076. 10.1021/ic00096a036. [DOI] [Google Scholar]
- Šimeček J.; Zemek O.; Hermann P.; Notni J.; Wester H.-J. Tailored Gallium(III) Chelator NOPO: Synthesis, Characterization, Bioconjugation, and Application in Preclinical Ga-68-PET Imaging. Mol. Pharm. 2014, 11, 3893–3903. 10.1021/mp400642s. [DOI] [PubMed] [Google Scholar]
- Frank A. O.; Otto E.; Mas-Moruno C.; Schiller H. B.; Marinelli L.; Cosconati S.; Bochen A.; Vossmeyer D.; Zahn G.; Stragies R.; Novellino E.; Kessler H. Conformational control of integrin-subtype selectivity in isoDGR peptide motifs: a biological switch. Angew. Chem., Int. Ed. 2010, 49, 9278–9281. 10.1002/anie.201004363. [DOI] [PubMed] [Google Scholar]
- Kapp T. G.; Rechenmacher F.; Neubauer S.; Maltsev O. V.; Cavalcanti-Adam A. E.; Zarka R.; Reuning U.; Notni J.; Wester H.-J.; Mas-Moruno C.; Spatz J.; Geiger B.; Kessler H. A Comprehensive Evaluation of the Activity and Selectivity Profile of Ligands for RGD-binding Integrins. Sci. Rep. 2017, 7, 39805. 10.1038/srep39805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mas-Moruno C.; Rechenmacher F.; Kessler H. Cilengitide: the first anti-angiogenic small molecule drug candidate. Design, synthesis and clinical evaluation. Adv. Anticancer Agents Med. Chem. 2010, 10, 753–768. 10.2174/187152010794728639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft S.; Diefenbach B.; Mehta R.; Jonczyk A.; Luckenbach G. A.; Goodman S. L. Definition of an Unexpected Ligand Recognition Motif for αvβ6 Integrin. J. Biol. Chem. 1999, 274, 1979–1985. 10.1074/jbc.274.4.1979. [DOI] [PubMed] [Google Scholar]
- Hartman G. D.; Egbertson M. S.; Halczenko W.; Laswell W. L.; Duggan M. E.; Smith R. L.; Naylor A. M.; Manno P. D.; Lynch R. J. Non-peptide fibrinogen receptor antago-nists. 1. Discovery and design of exosite inhibitors. J. Med. Chem. 1992, 35, 4640–4642. 10.1021/jm00102a020. [DOI] [PubMed] [Google Scholar]
- Notni J.; Šimeček J.; Hermann P.; Wester H.-J. TRAP, a powerful and versatile framework for gallium-68 radiopharmaceuticals. Chem.—Eur. J. 2011, 17, 14718–14722. 10.1002/chem.201103503. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.