

Abstract
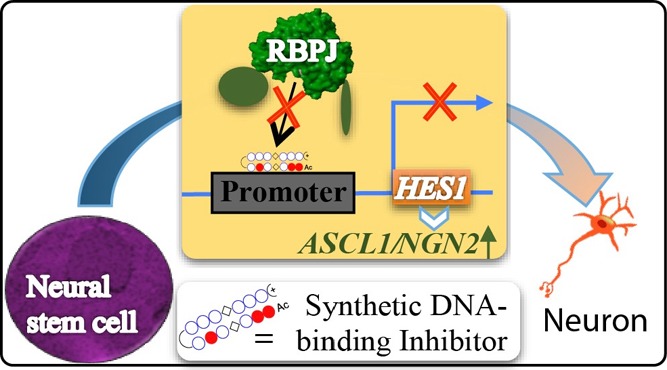
Synthetic DNA-binding inhibitors capable of gaining precise control over neurogenesis factors could obviate the current clinical barriers associated with the use of small molecules in regenerative medicine. Here, we report the design and bioefficacy of the synthetic ligand PIP-RBPJ-1, which caused promoter-specific suppression of neurogenesis-associated HES1 and its downstream genes. Furthermore, PIP-RBPJ-1 alone altered the neural-system-associated Notch-signaling factors and remarkably induced neurogenesis with an efficiency that was comparable to that of a conventional approach.
Introduction
Multipotent neural stem cells (NSCs) are responsible for neurogenesis and plasticity in regions of the mammalian adult brain.1,2 In neurodegenerative disorders such as Huntington’s and Alzheimer’s disease, neurogenesis (which involves the precise orchestration of diverse bioactive factors and intricate receptor signaling) is perturbed.1,3 Alterations in the complex transcription machinery of NSCs that depend on extrinsic and intrinsic factors decide their fate specification between a proliferation or a differentiation state into terminal neural cell types, such as neurons, astrocytes, and oligodendrocytes.4 In particular, the coordinated orchestration of activator and repressor markers in the promoter region of essential neurogenesis-regulating genes epigenetically controls cell fate specification.5 Notch signaling machinery is known to negatively regulate neurogenesis by promoting stem cell proliferation and gliogenesis.6,7 Synthetic inhibitors of EGF or γ-secretase receptors are known to regulate Notch signaling and were proclaimed to have clinical prospects in treating Alzheimer’s disease, but the chronic exposure of these Notch inhibitors may lead to toxicity.8,9 The DNA-binding protein RBPJ cooperates with four different Notch receptors in mature excitatory neurons, and a recent study in a mouse model showed that RBPJ inhibition did not affect the learning and memory.10 Therefore, the development of RBPJ-based Notch regulation is gaining attention, as it has a direct impact on key genes associated with neurogenesis. RBPJ is known to directly regulate the HES and HEY families of genes, which are negative regulators of neuronal differentiation.11
HES1 works as a HES1-Gro/TLE complex and is activated by the binding of the Notch intracellular domain (NICD).12 The HES1 expression oscillates through a negative response for a period of 2–3 h during proliferation and is known to be the master key factor of neurogenesis.13,14 The continued repression of HES1 and the ensuing expression of the downstream genes ASCL1/NGN2 shift the oscillation of neural progenitors in favor of neural differentiation.11 Therefore, direct regulation of HES1 is favored, as it is expected to have better control over neurogenesis than the Notch inhibitors that operate indirectly by targeting protein–protein interactions, and rely on the repression of the initial signaling processes. Consequently, several strategies have been developed to artificially regulate the HES1 gene expression. MicroRNAs are known to alter the HES1 expression and induce NSC differentiation into a specific neural subtype.15,16 Artificial knockdown of the HES1 expression in NSCs using small interfering RNA (siRNA) showed a significant reduction of Nestin+ neural progenitor cells and the consequent increase of Tuj1+ neuronal cells.17 However, the siRNA strategy encompasses handling difficulties and low chemical stability. Consequently, there is a need for a clinically friendly approach to directly control the HES1 expression and achieve directed differentiation of NSCs into neurons.
Among the several approaches that are known to regulate the HES1 expression, the use of small molecules is assured to have clinical potential because this approach is transgene-free and easily controllable. Accordingly, agalloside, an HES1 dimer inhibitor, was shown to accelerate the differentiation of mouse NSCs.18 The existing approaches for small molecule discovery mainly rely on high-throughput screening of large libraries of small molecules19,20 and are commonly time-consuming and sometimes unyielding. Our notion is that the promoter-specific repression of HES1 using a DNA-based synthetic inhibitor alone could trigger neural differentiation and induce neurogenesis, thereby obviating the need to screen a huge number of molecules. Dervan and colleagues discovered selective DNA-binding small molecules called pyrrole imidazole polyamides (PIPs) containing the N-methylpyrrole units (P) and N-methylimidazole units (I).21
Following an accepted binding rule, I/P and P/I in a hairpin structure bind to G, C and C, G, respectively, whereas P/P pairing binds to A, T or T, A. PIPs can be predesigned to recognize target DNA sequences and have been used extensively as a transcription repressor in living cells,22,23 PIPs could localize inside the nucleus of cells in a culture dish without any vehicle and have been proved to be functional in animal models.24,25 Consequently, we intended to explore the potential of PIPs to modulate DNA–protein interactions in the HES1 promoter region and induce promoter-specific transcription suppression. The recombining binding protein suppressor of hairless (RBPJ) associates with the promoter region of HES1 and regulates transcription in the Notch signaling pathway.26 By harnessing the sequence information, we have demonstrated for the first time that PIP could be designed as a DNA-binding inhibitor of HES1 to modulate key Notch signaling factors and shift the transcription program in hNSCs to one that favors neuronal differentiation. Furthermore, the designed PIP generated neurons with longer neurite outgrowth, thus validating the efficacy of our DNA-based synthetic strategy in targeted neuronal differentiation.
Results and Discussion
Previously, the transcription factor RBPJ was shown to bind the “(TG) TGGGAA” site.27,28 On the basis of this report, we interrogated the human HES1 genome sequence (gi|568815595) from NCBI and detected the binding sequence. By integrating the existing knowledge on the plausible binding site, the proximal sequence, and the mode of recognition by PIPs, we designed two PIPs termed PIP-RBPJ-1 and PIP-RBPJ-2 to target the RBPJ-binding motif “GGAAAGAA” and “TGGGAA”, respectively (Figure 1A). Also, a mismatched PIP-C was synthesized as the control (Figure S1A).
Figure 1.

Construction of PIPs to target the RBPJ-binding site in the promoter region of HES1. (A) Illustration of the reported14 binding site “(TG) TGGGAA” in the HES1 promoter region of RBPJ that is essential in governing the expression of the downstream gene HES1. (B) Chemical structures of the RBPJ-targeting PIPs termed PIP-RBPJ-1 and PIP-RBPJ-2.
According to previous reports,29 the PIPs were synthesized using an Fmoc-protected solid-phase synthesis system and were further purified by reverse-phase flash column chromatography (Figure S1B,C). The structures of the two PIPs are shown in Figure 1B. A Tm (melting temperature) assay was performed using two double-stranded DNA (ODN-1 and ODN-2) containing the RBPJ binding site and the PIP binding site (Figure S1D) to verify the binding affinity of the PIPs toward the predesigned target DNA sequence. As shown in Table 1, the addition of PIP-RBPJ1 resulted in a Tm shift of 26.1 °C for ODN-1 and 18.8 °C for ODN-2, which confirmed the binding to the target DNA. Similarly, the addition of PIP-RBPJ-2 caused a Tm shift of 5.1 and 9.6 °C, respectively, suggesting a relatively lower binding than that observed for PIP-RBPJ-1. The Tm assay performed using the mismatched PIP-C showed a Tm shift of only 2.2 °C, suggesting a weaker binding affinity. Taken together, the results of the Tm assay demonstrated that between the PIPs targeting RBPJ, the longer one (i.e., PIP-RBPJ-1) had a relatively better binding affinity to the target DNA. Furthermore, we verified the binding affinity of our designed PIPs using a luciferase reporter assay, which has been widely used to investigate promoter activity in eukaryotic cell lines.30 We designed a model for inserting the HES1 promoter region into the pMCS-Cypridina Luc vectors to clarify that the designer PIP could operate by inhibiting transcription factor interactions at the target DNA sequence. In a Notch-active environment, the binding of PIPs is expected to reduce luciferase activity (Figure 2A). Initially, two plasmids [pMCS-HES1-L (1009 bp) and pMCS-HES1-S (402 bp)] containing the key component of the HES1 promoter sequence were assessed using polymerase chain reaction (PCR) (Figure S2A). Before cell treatment, we characterized the NSC undifferentiated state by double staining of SOX2 and NESTIN (Figure S3). Upon transfection of the plasmid into hNSCs using liposome 3000 and coculturing for 2 days, the 1009 bp fragment plasmid showed a relatively better luciferase activity than did the 402 bp fragment plasmid (Figure S2B). Therefore, we chose to employ pMCS-HES1-L for the subsequent studies. After 2 h of transfection, PIPs at different concentrations (0.5, 2, and 5 μM) were added to the medium, and their effects were evaluated after 2 days of treatment. As shown in Figure 2B, PIP-RBPJ-1 significantly (P = 0.011 at 2 μM, P = 0.005 at 5 μM) repressed the promoter activity by reducing the luciferase emission in a concentration-dependent manner.
Table 1. Shift of Tm Value by RBPJ-Binding PIPsa.
| PIP-RBPJ-1 |
PIP-RBPJ-2 |
PIP-C |
|||||
|---|---|---|---|---|---|---|---|
| Tm/°C | Tm/°C | ΔTm/°C | Tm/°C | ΔTm/°C | Tm/°C | ΔTm/°C | |
| ODN-1 | 54.3(±0.1) | 80.4(±0.3) | 26.1 | 59.4(±0.2) | 5.1 | 56.5(±0.2) | 2.2 |
| ODN-2 | 37.9(±0.1) | 56.7(±0.6) | 18.8 | 47.4(±0.1) | 9.6 | 47.0(±0.6) | 2.2 |
Melting temperatures were calculated and analyzed, and each standard deviation is indicated in parentheses. Tm = Tm (DNA complex compound) – Tm (DNA).
Figure 2.

In vitro study using the designed PIPs. (A) Luciferase reporter assay model of the PIPs. In Notch-active models, the NICD translocates into the nucleus and operates with RBPJ to activate the downstream genes. PIP-RBPJ blocks the binding of RBPJ and results in the suppression of the gene expression. (B) Effect of PIPs on pHES1-L luciferase activity. PIP-RBPJ-1 decreases pHES1-L luciferase activity in a concentration-dependent manner. Three biological replicates were performed, and the mean ± SD are indicated, *P < 0.05, **P < 0.01. (C) Chromatin immunoprecipitation (ChIP) analysis using the RBPJ antibody in the promoter region of HES1 revealed a decrease in the amount of the promoter sequence in the PIP-RBPJ-1-treated hNSCs and not the dimethyl sulfoxide (DMSO)-treated hNSCs.
Contrastingly, PIP-RBPJ-2 yielded no reduction in the HES1 promoter luciferase activity. Thus, PIP-RBPJ-1 had a better binding affinity to the HES1 promoter region and notably altered the binding of the RBPJ protein to the target DNA sequence in hNSCs. Previously, the ChIP analysis demonstrated the ability of a designer PIP (PIP-S2) to recognize the cognate sequence in the promoter of SOX2 and alter the target gene expression by inhibiting the transcription factor.31 Likewise, here, we performed the ChIP analysis using the RBPJ antibody to evaluate the blocking effect of the designed PIP on RBPJ protein in the HES1 promoter region of hNSCs. As shown in Figure 2C, PIP-RBPJ-1 reduced the binding ability of RBPJ protein in the promoter region of HES1, as the amount of the promoter sequence in PIP-RBPJ-1-treated cells (3.86%) decreased when compared to that observed in DMSO-treated cells (10.68%). This result suggests that the PIP-RBPJ-1 could operate to block RBPJ inside a living cell.
This inhibition of RBPJ may induce chromatin remodeling in the HES1 promoter region and repress transcription. To evaluate the bioactivity of PIPs targeting the RBPJ binding site on the endogenous expression of HES1, we chose to study their effect on the endogenous expression of HES1. On the basis of our previous report, we chose to use PIP-RBPJ-1 and PIP-RBPJ-2 at 2 μM for 24 h. In accordance with the pattern observed in the luciferase assay, the quantitative real-time (qRT)-PCR studies showed that only PIP-RBPJ-1 significantly (P = 0.038) repressed the HES1 expression (PIP-RBPJ-2 did not have any notable effect on the HES1 expression) (Figure 3A).
Figure 3.

PIP-RBPJ-1 alters the expression of HES1 and its regulated downstream genes. (A) Effect of PIP-RBPJ-1 and PIP-RBPJ-2 on the HES1 expression. (B) Expression profile of HES1 in hNSCs treated with PIP-RBPJ-1 at different concentrations (0.5, 2, and 5 μM) for 24 h. (C) Effect of PIP-RBPJ-1 on the expression profile of HES1 at various time intervals (6, 12, 24, and 48 h). Expression profile of (D) ASCL1 and (E) NGN2 in PIP-RBPJ-1-treated hNSCs. The housekeeping gene β-actin is chosen to normalize the relative gene expression, as the expression of β-actin did not alter with both PIP-RBPJ-1 and PIP-RBPJ-2 treatment. Three biological replicates were performed, and the mean ± SD are indicated, *P < 0.05, **P < 0.01.
It is important to note here that the better binding affinity of PIP-RBPJ-1 toward its target DNA sequence and its gene-suppressing ability is consistent with the pattern observed in the Tm assay. Therefore, the shorter recognition capability of PIP-RBPJ2 and the resulting nonspecific binding to the target region are implied to hamper the bioactivity of PIP-RBPJ-2. The concentration dependency study performed using various concentrations (0.5, 2, and 5 μM) of PIP-RBPJ-1 for 24 h corroborated that PIP has an optimal gene-repressing ability at 2 μM (Figure 3B). Similarly, a time-dependency study performed at various time intervals (6, 12, 24, and 48 h) substantiated that 2 μM PIP-RBPJ-1 had significant activity at 12 h (P = 0.006) and 24 h (P = 0.020) (Figure 3C). Interestingly, PIP-RBPJ-1 treatment for just 6 h also yielded significant (P = 0.044) gene repression, suggesting that PIP-RBPJ-1 promptly localizes to the nuclei of hNSCs and binds the target sequence.
HES1 is a key antineurogenic protein that harbors a WRPW motif, a repressor peptide that recruits Groucho’s WD domain and a histone deacetylase enzyme to suppress downstream genes, such as ASCL1 and NGN2, which are associated with neural differentiation.32 The oscillatory expression of HES1 and ASCL1 is known to decide the cell fate specification between the proliferation of hNSCs and neural differentiation, respectively.10 Therefore, we performed qRT-PCR at 24 h to verify if the repression of HES1 could, in turn, induce the endogenous expression of ASCL1 and NGN2. As shown in Figure 3D,E, PIP-RBPJ-1 significantly induced the expression of both ASCL1 (P = 0.047) and NGN2 (P = 0.032) at 24 and 48 h. Therefore, PIP-RBPJ-1 remarkably resets the oscillatory state of HES1/ASCL1 into a stable state that favored neural differentiation. Subsequently, we carried out microarray analysis to evaluate the global expression profile of PIP-RBPJ-1-treated hNSCs using an Affymetrix Human Gene 2.1 ST Array Strip, which covers about 53 617 gene transcripts. To clarify the effect of PIP-RBPJ-1 both on HES1 and its regulated downstream genes, we chose to evaluate the hNSCs treated with 2 μM PIP-RBPJ-1 for 24 h. While keeping a 1.3-fold change as the cutoff value for a notable gene regulatory effect, PIP-RBPJ-1 was observed to downregulate 373 genes and upregulate 774 genes (t-test, P < 0.05) compared with DMSO (Figure S4). An ingenuity pathway analysis of the PIP-RBPJ-1-altered genes revealed Notch signaling as the top canonical pathway (Table 2). Also, the network analysis of PIP-RBPJ-1-modulated genes showed that gene networks were associated with neurological diseases and neural development and function (Figure S5). Because RBPJ is a well-known Notch effector, this result confirmed the regulatory role of PIP-RBPJ-1 in altering several Notch effectors, including lunatic fringe (LFNF), which is known to prevent individual cells from sending and receiving Notch signals at the same time.33 A qRT-PCR analysis validated the microarray data because the endogenous expression of NOTCH1 and the Notch ligand jagged1 (JAG1)34 was significantly suppressed by PIP-RBPJ-1 but not by PIP-RBPJ-2 in both 24 h- and 48 h-treated hNSCs (Figure 4). A functional annotation further suggested that PIP-RBPJ-1-induced genes favor neuronal development (Table S1). Interestingly, PIP-RBPJ-2 did not decrease the pHES1-L luciferase activity (Figure 2B) and slightly increased the endogenous expression of HES1 (Figure 3A). Also, PIP-RBPJ-2 increased the endogenous expression of NOTCH1 and JAG1 (Figure 4A,B). It is important to note here that PIP-RBPJ-1 and PIP-RBPJ-2 had distinctive bioactivity even when they both were designed to target the RBPJ protein-binding region in the HES1 promoter region. The shorter DNA sequence recognition ability of PIP-RBPJ-2 and the ensuing off-target effects are suggested to be the reason behind this altered bioactivity. Also, these data demonstrate the need for a combinatorial approach to design different PIPs targeting the core promoter region to identify and characterize the hit PIPs. Together, these data substantiate that in the Notch-active condition, PIP-RBPJ-1 altered Notch signaling and shifted hNSCs to differentiate into neural cells.
Table 2. Significantly Controlled Ingenuity Canonical Pathwaysa.
| top 5 ingenuity canonical pathways | –log(p-value) |
|---|---|
| Notch signaling | 2.15 |
| Nur77 signaling in T lymphocytes | 1.73 |
| mitochondrial dysfunction | 1.73 |
| threonine degradation II | 1.68 |
| calcium-induced T lymphocyte apoptosis | 1.54 |
Two biological replicates were performed in microarray data. Expression profile of PIP-RBPJ-1 versus DMSO with 1.3-fold change as the cutoff value for the notable gene regulatory effect.
Figure 4.
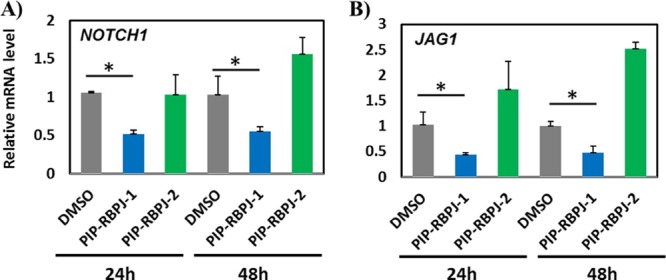
Effect of PIP-RBPJ-1 on Notch signaling factors. qRT-PCR of (A) NOTCH1 and (B) JAG1, which are critical Notch-signaling genes. The data are presented after normalization using β-actin. Mean ± SD from three wells. *P < 0.05, **P < 0.01.
Considering the remarkable bioactivity of PIP-RBPJ-1 to reset the global expression profile in hNSCs, we evaluated the efficacy of PIP-RBPJ-1 to induce neurogenesis in hNSCs. As shown in Figure 5A, hNSCs were treated with PIP-RBPJ-1 every 2 days with the medium exchange up to day 9, using PIP-RBPJ-2 as the control. A qRT-PCR analysis that was carried out to check the endogenous expression of the neuronal marker Tuj1 at regular time intervals (days 3, 6, and 9) revealed that until day 3, both PIPs exhibited no effect. However, at day 6, PIP-RBPJ-1 induced the endogenous expression of Tuj1, and at day 9, significant (P = 0.047) expression of Tuj1 was observed (Figure 5B). Immunostaining studies showed a similar pattern in the gene expression profile, and only PIP-RBPJ-1 (and not PIP-RBPJ-2) induced Tuj1+ cells (Figure 5C). A calculation of the neuronal induction efficiency by counting the number of Tuj1+ cells and examining the neurite length revealed that PIP-RBPJ-1 notably enhanced the generation of neurons and improved neurite growth by 42.2%/117.5 (efficiency/length) (Figure 5D, panel PIP-RBPJ-1 and Table S2). PIP-RBPJ-2 showed a relatively lower effect of 14.4%/59.1 (efficiency/length) (Figure 5D, panel PIP-RBPJ-1 and Table S2). To compare the efficacy of PIP-RBPJ-1 with the reported protocol, we chose to assess efficiency through immunostaining at day 9 using a previous protocol35 that employed cAMP/BDNF as a positive control. As shown in Figure 5E, PIP-RBPJ-1 induced neuronal differentiation in a fashion similar to that observed using the reported protocol.35 Similarly, the efficiency analysis clearly indicated that PIP-RBPJ-1 significantly (P < 0.001) induced neuronal differentiation with 68.4% efficiency, which is comparable to that observed for the positive control (70.3%) (Figure 5F). Interestingly, PIP-RBPJ-1 did not increase glial cells (Figure S6) and hence is suggested to favor neuronal differentiation. This proof-of-concept study indicates that our designed DNA-binding inhibitor of HES1 remarkably altered Notch signaling and induced neuronal differentiation with an efficiency that was comparable to that observed for the conventional approach targeting protein–protein interactions.15
Figure 5.

NSC differentiation using PIP-RBPJ-1. (A) Schematic representation of the neuronal differentiation protocol. After day 0, hNSCs were treated with the effectors (PIP-RBPJ-1 and PIP-RBPJ-2) every 2 days. (B) Gene expression profile of Tuj1 at various time intervals (days 3, 6, and 9) from three biological replicates. The mean ± SD are indicated. (C) Immunostaining of the neuronal marker Tuj1 analyzed at day 3 after treatment with effectors. (D) Efficiency of neuronal generation and neurite length at day 3 was analyzed using six different images for each sample. (E) Comparative analysis of neuronal differentiation using the effectors and the reported protocol using cAMP + BDNF as the positive control. (F) Neuronal efficiency of the effectors and the reported protocol calculated from 800 cells observed in five different images for each sample. Tuj1-positive cells were counted and normalized to total cell number. *P < 0.05, **P < 0.01, ***P < 0.001.
Conclusions
Directed differentiation of NSCs using gene-targeting small molecules alone presents potential clinical applications in regenerative medicine to treat neurodegenerative disorders. However, the identification of small molecules that are capable of targeting specific transcription machinery is not straightforward because of the involvement of diverse intrinsic and extrinsic signals. Consequently, the time gap between the identification of an essential neurogenesis factor and the development of synthetic ligands that can modulate them has widened in recent years. Despite the availability of small molecules that target signaling proteins associated with neurogenesis, there is a demand for developing DNA-based synthetic small molecules that can precisely alter the specific transcription machinery related to neurogenesis based on their potential in regenerative medicine. Moreover, there is a need to reduce the number of factors necessary to achieve neurogenesis because the current differentiation protocol requires two or more small molecules targeting distinct signaling neurogenesis factors. A designer molecule preprogrammed to target an essential neurogenesis factor could alone trigger neural differentiation and precisely alter the gene transcription program to induce neurogenesis by overcoming the clinical barriers associated with the use of small molecules. Therefore, we chose to construct a designer molecule using selective DNA-binding PIPs. Basic helix-loop-helix transcription factors, such as HES1, operate as negative regulators of neuronal differentiation and govern the transcription program associated with hNSCs.
To gain chemical control over neurogenesis, we first designed and characterized a PIP as the first-ever synthetic DNA-binding inhibitor of HES1 by harnessing the sequence information to target the binding site of a transcription factor called RBPJ. A Tm assay suggested the superior binding efficacy of the PIP termed PIP-RBPJ-1 over that termed PIP-RBPJ-2, and an in vitro luciferase reporter assay further verified the bioefficacy of PIP-RBPJ-1. In accordance with the pattern observed in the in vitro study, PIP-RBPJ-1 alone significantly (P = 0.038) suppressed the endogenous expression of HES1. The oscillation between HES1 and its downstream gene ASCL1 is known to decide the fate of hNSCs into either a proliferation-favoring state or a neural differentiation state. qRT-PCR of HES1-regulated downstream genes demonstrated that PIP-RBPJ-1 significantly (P < 0.04) induced the expression of ASCL1 and NGN2, which are critical neural system genes. Genome-wide gene expression studies revealed that PIP-RBPJ1 also regulated the Notch signaling pathway and shifted the transcription program to favor neuronal differentiation. This result is in accordance with a previous report that showed that alteration of Notch signaling induces neuronal migration.36 Moreover, PIP-RBPJ-1 successfully generated Tuj1 active neurons with longer neurite outgrowth with an efficiency that was comparable to that of the conventional protocol, suggesting the potential of this DNA-binding inhibitor to induce targeted differentiation.
Development of artificial tools to regulate Notch signaling is in increasing demand owing to their clinical potential in treating neurodegenerative disorders.37 Consequently, several regulators of Notch signaling such as the inhibitors of the widely known therapeutic targets of Notch signaling such as EGF or γ-secretase receptors have been developed.38 However, the mode of action of the inhibitors of EGF and γ-secretase receptors depends on the repression of the initial signaling processes. Therefore, these two Notch inhibitors operate indirectly and hence, the ensuing potential off-target effect is a major concern. In this regard, our synthetic DNA-based inhibitor PIP-RBPJ-1 directly operates as it is designed to target the key region of the HES1 gene, a critical effector in neural proliferation and differentiation. However, it is important to note here that although PIP-RBPJ-1 significantly modulates Notch signaling and minimizes the probability of side effects, this synthetic DNA-binding inhibitor is not entirely specific to Notch signaling. To this end, the future studies will aim to improve the recognition capability of PIP-RBPJ-1 and obviate any potential off-target effects that limit the clinical application. Because PIPs can be preprogrammed to target specific DNA sequences of interest, this strategy may be expanded to differentiate stem cells into a desired neural subtype through the rational design of distinct PIPs targeting a specific regulatory sequence motif. The advancement of our DNA-based approach by harnessing the accumulated sequence information may offer versatile utility to gain chemical control over neurogenesis, which in turn may have a broad range of clinical applications.
Experimental Section
PIP Synthesis
PIPs were synthesized using a PSSM-8 (Shimadzu) system with computer-assisted operation by using Fmoc chemistry as described before.18 HPLC analysis were performed with a JASCO PU-2080 plus HPLC pump and an 807-IT UV/vis detector. A Chemcobond 5-ODS-H reversed-phase column (4.6 × 150 mm) in 0.1% TFA in water with acetonitrile as the eluent at a flow rate of 2.0 mL min–1 with detection at 254 nm. Electrospray ionization time-of-flight mass spectrometry and matrix-assisted laser desorption/ionization mass spectrometry were performed by using a positive ionization mode. All the PIPs were obtained as a white powder.
PIP-RBPJ-1 MS (ESI-TOF) m/z: calcd for C86H105N32O163+ [M + 3H]3+, 613.95; found, 613.75; m/z calcd for C86H104N32O162+ [M + 2H]2+, 920.41; found, 920.13; HPLC: tR = 17.933 min (0.1% TFA/MeCN, linear gradient 0–50%, 0–20 min).
PIP-RBPJ-2 MS (ESI-TOF) m/z: calcd for C56H70N22O102+ [M + 2H]2+, 605.66; found, 605.16; HPLC: tR = 16.867 min (0.1% TFA/MeCN, linear gradient 0–50%, 0–20 min).
PIP-C MS (MALDI-TOF) m/z: calcd for C84H95N31NaO18+ [M + Na]+, 1849.87; found, 1849.47; m/z calcd for C84H95KN31O18+ [M + K]+, 1865.98; found, 1865.49; HPLC: tR = 18.633 min (0.1% TFA/MeCN, linear gradient 0–50%, 0–20 min).
Human NSC Culture and Differentiation
Human H9 hESC-derived neural stem cell (hNSC) were acquired from Invitrogen and cultured by the recommended method. The StemPro NSC SFM complete medium component: KnockOut D-MEM/F-12 medium (Invitrogen), GlutaMAX-I Supplement (Invitrogen) 2 mM, βFGF 20 ng/mL (Invitrogen), EGF 20 ng/mL (Invitrogen), and Stempro Neural Supplement (Invitrogen) 2%. Cells were cultured on the poly-l-ornithine 20 μg/mL (SIGMA) and laminin 2 μg/mL (Invitrogen) matrix-coated plate. For the spontaneous differentiation, the hNSCs were seeded in the coated plate at 2.5 × 104 cells/cm2. After 2 days, the medium exchange was done with StemPro NSC SFM without the growth factors. For the neuron differentiation, we used the neuronal differentiation medium: Neurobasal (GIBCO), B27 supplement 2% (Invitrogen), GlutaMAX-I Supplement 2 mM, β-mercaptoethanol 0.1 Mm (SIGMA), BDNF 10 ng/mL (PeproTech), and cAMP 100 μM.35 All the cells were used between passage 3 and passage 5. Before the treatment of effectors (designed PIPs and DMSO), we carried out immunostaining experiments to verify the expression of NESTIN and SOX2 using the antibodies bought from Abcam (Figure S3). Consistent with our previous report,23 our initial optimization studies showed that until 5 μM, no significant changes in cell viability were observed.
Construction of Luciferase Plasmid
The 402 bp and 1009 bp HES1 genes in the promoter region were cloned from the hNSC genome with an EcoR1 enzyme site in the 5′ prime and a Hind3 enzyme site at the 3′ prime. The promoter segment and the valid pCMCS-Cypridina-Luc plasmid were double-digested by EcoR1 and Hind3 enzymes in M buffer for 1 h (TOYOBO), and then the products were purified by Wizard SV gel and the PCR Clean-up system (Promega Corporation). We then ligated the vector and the segment by T4 DNA ligase (TOYOBO) at 4 centigrade overnight and then transfected into the Escherichia coli JM109 competent cells (TAKARA) following the given protocol. After attaining the target vectors pMCS-HES1 1K-Luc and pMCS-HES1 0.4K-Luc, we confirmed it using PCR gel (Figure S2).
Luciferase Reporter Assay
Experiments were performed by a Piece Renilla Luciferase Glow assay kit (Thermo Fisher Scientific). The hNSCs were seeded at 1 × 104 cells per well at 96-well plates and incubated overnight. The lipofectamine 3000 reagent (Thermo Fisher Scientific) was used to transfect the constructed plasmids into the hNSC following the suggested protocol and was incubated for 48 h. After 48 h, luciferase activity was checked to select the better plasmid for screening the PIPs. For testing the PIP activity, 1 × 104 cells were seeded in 96-well plates and after 1 day seeding, the medium exchange was done to the StemPro NSC SFM without the growth factors. The plasmid pMCS-HES1-L was then transfected, and PIPs were added after 2 h transfection. The hNSCs were treated with different concentrations of the PIPs (0.5, 2, 5 μM) for another 48 h. We then added 10 μL of cell medium supernatant to the opaque 96-well plate followed by mixing 50 μL working solution (100× Coelenterazine and Renilla Glow Assay) to each well. After 10 min for signal stabilization, we programmed the luminometer by SpectraMax M2/M2e (Molecular Devices) using a 463 nm light to detect the light output.
Chromatin Immunoprecipitation-PCR
ChIP-PCR was done following the protocol. hNSCs were cultured and treated with PIPs (2.0 μM) for 2 days and then fixed by 1% formaldehyde solution using the nuclear lysis buffer [Tris-HCl (pH 7.5) 10 mM, NaCl 200 mM, ethylenediaminetetraacetic acid 10 mM, sodium dodecyl sulfate (SDS) 1% (v/v)] on the cell for 10 min on ice. Sonication was then used to shear the chromatin to ∼200–1000 bp. Dynabeads Protein G (Invitrogen) was used for IP after pretreating the sample, and with the first antibody RBPJ (Abcam), samples were rotated for 3 h at 4 °C by reverse cross-linking with 10% SDS and purified DNA with QIAGEN quick PCR purification kits. After samples were prepared, real-time PCR was performed using the following primers: HES1-ChIP-F: 5′-ATTGGCCGCCAGACCTTGTG-3′ and HES1-ChIP-R: 5′-CGGATCCTGTGTGATCCCTAGG-3’.
Tm Assay
DNA duplex (2.5 μM) was mixed with PIPs (5.0 μM) in a 100 μL mixture containing sodium cacodylate (10 mM, pH 7.0), sodium chloride (10 mM), and 2.5% (v/v) of DMSO. The sequence of ODN-1 5′-CTGTGGGAAAGAAAGTTTGGGAAGTTTCA-3′ and ODN-2 5′-TGTGGGAAAGAAAGT-3′ was duplex DNA. Annealing was performed by heating the mixture at 95 °C for 5 min and then decreasing to room temperature for about 3 h. DNA melting was assessed through the absorbance of the mixture at λ = 260 nm from 25 to 90 °C (1 °C/min, 1 measurement/°C) on a V-650 UV–Vis spectrophotometer (JASCO).
Quantitative Real-Time PCR
RNA was extracted using an RNeasy Mini Kit (QIAGEN), and then the cDNA was reverse-transcribed from 200 ng total RNA by ReverTra Ace qPCR RT Master Mix with a gDNA Remover (Toyobo). SYBR green real-time PCR amplifications were carried out with THUNDERBIRD SYBR qPCR Mix (Toyobo) and analyzed by using the ABI 7300 Real-time PCR System (Applied Biosystems, USA). The primer information is in the Supporting Information, Table S3.
Microarray
The hNSCs were treated with 2 μM of the compound for 1 day, and then the total RNA was isolated by the RNeasy Mini Kit (QIAGEN). RNA quality was examined using the Agilent RNA 6000 Pico Kit (Agilent), and the Gene chip WT PLUS Reagent Kit (Agilent) was used to amplify RNA into cRNA. After the cRNA purification and quantitation, ss-cDNA was synthesized. Using the Gene chip WT Terminal Labeling Kit, we fragmented and labeled the ss-cDNA, and hybridization was performed in human gene 2.1 ST array strip (Agilent).
Ingenuity Pathway Analysis
The raw data were tabled with the genes upregulated more than 1.3 fold and the P-value less than 0.05. PIP versus DMSO samples were then subjected to the network analysis by QIAGEN’s Ingenuity Pathway Analysis (QIAGEN, www.qiagen.com/ingenuity) software.
Immunostaining
For the hNSCs, immunostaining of Tuj1 (SYSY) was performed in 3 and 9 days after the 2 μM of compound treatment, while 0.1% DMSO was used as the negative control. For differentiation, the medium we used is the StemPro NSC SFM complete medium without the NSC maintaining factors β-FGF and EGF, and we changed the medium every 2 days. After 3 days of culture, we fixed the cells using 4% paraformaldehyde (Sigma) in phosphate-buffered saline (PBS) for 30 min at room temperature and then permeabilized the cells by 0.5% Triton X-100 (Nacalai Tesque) in PBS for 10 min. Bovine serum albumin (BSA, 5%, Nacalai Tesque) was used for blocking for 1 h at room temperature. The cells were then incubated with first antibody Tuj1 rabbit anti-human in the ratio of 1:200 in 1% BSA solutions at 4 °C overnight. The next day, the second antibody alex488 goat anti-rabbit (Invitrogen) in the ratio of 1:500 was incubated for 1 h at room temperature. Nuclear staining with 1 μg/mL DAPI/PBS solution was done for 15 min at room temperature. To check the glial cell differentiation rates, a 9 day-treated cell was used for immunostaining by the GFAP (Abcam) antibody using the same protocol. The immunostaining result was checked using a confocal microscope.
Calculation of Neuron Efficiency and Counting of the Neurite Length
The hNSCs treated with PIPs and the control effectors were fixed and stained with Tuj1 and DAPI to calculate the total neuron number (Tuj1+) and the total nuclei number (DAPI), respectively. For each sample, six pictures were taken using confocal (20×) microscopy, and calculation was done. To calculate the neuronal generation efficiency, hNSCs treated for 3 and 9 days were calculated. Total nuclei were counted using imagine J plugin by counting the DAPI, while total neurons were counted manually. Neuron efficiency was calculated by the formula below. Neuron efficiency = total neuron number (Tuj1+)/total nuclei number (DAPI). In three days of hNSC treatment, the neurite length was calculated using the image J plugin Neurite Tracer39 as the manual. Neurite length = total neurite length/total neuron number.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b00220.
Funding for this work was provided by JSPS [16H06356 to H.S., 16K12896 to G.N.P.]
The authors declare no competing financial interest.
Notes
All the experiments were conducted in the Kyoto University Graduate School of Science, Department of Chemistry, Kitashirakawa-Oiwakecho, 606-8502 and Kyoto University Institute for Integrated Cell-Material Sciences (WPI-iCeMS), Yoshida-Ushinomiyacho, 606-8501. One of the co-author J.D. was involved in this research during his internship at Kyoto University and now has the affiliation with Indian Institute of Technology-Delhi, India. Department of Biochemical Engineering and Biotechnology, Hauz Khas, 110-016, New Delhi, India.
Supplementary Material
References
- Gonzalez-Perez O. Neural stem cells in the adult human brain. Biol. Biomed. Rep. 2012, 2, 59–69. [PMC free article] [PubMed] [Google Scholar]
- Ma D. K.; Bonaguidi M. A.; Ming G.-l.; Song H. Adult neural stem cells in the mammalian central nervous system. Cell Res. 2009, 19, 672–682. 10.1038/cr.2009.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T.; Ninomiya M.; Acosta P. H.; Garcia-Verdugo J. M.; Sunabori T.; Sakaguchi M.; Adachi K.; Kojima T.; Hirota Y.; Kawase T.; Araki N.; Abe K.; Okano H.; Sawamoto K. Subventricular zone-derived neuroblasts migrate and differentiate into mature neurons in the post-stroke adult striatum. J. Neurosci. 2006, 26, 6627–6636. 10.1523/jneurosci.0149-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massirer K. B.; Carromeu C.; Griesi-Oliveira K.; Muotri A. R. Maintenance and differentiation of neural stem cells. Wiley Interdiscip. Rev.: Syst. Biol. Med. 2011, 3, 107–114. 10.1002/wsbm.100. [DOI] [PubMed] [Google Scholar]
- Juliandi B.; Abematsu M.; Nakashima K. Epigenetic regulation in neural stem cell differentiation. Dev., Growth Differ. 2010, 52, 493–504. 10.1111/j.1440-169x.2010.01175.x. [DOI] [PubMed] [Google Scholar]
- Pierfelice T.; Alberi L.; Gaiano N. Notch in the vertebrate nervous system: an old dog with new tricks. Neuron 2011, 69, 840–855. 10.1016/j.neuron.2011.02.031. [DOI] [PubMed] [Google Scholar]
- Shimojo H.; Ohtsuka T.; Kageyama R. Dynamic expression of notch signaling genes in neural stem/progenitor cells. Front. Neurosci. 2011, 5, 78–85. 10.3389/fnins.2011.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe D. J. Presenilin, Notch, and the genesis and treatment of Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 11039–11041. 10.1073/pnas.211352598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe M. S. γ-Secretase inhibitors and modulators for Alzheimer’s disease. J. Neurochem. 2012, 120, 89–98. 10.1111/j.1471-4159.2011.07501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato C.; Turkoz M.; Dearborn J. T.; Wozniak D. F.; Kopan R.; Hass M. R. Loss of RBPj in Postnatal Excitatory Neurons Does Not Cause Neurodegeneration or Memory Impairments in Aged Mice. PLoS One 2012, 7, e48180 10.1371/journal.pone.0048180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homem C. C. F.; Repic M.; Knoblich J. A. Proliferation control in neural stem and progenitor cells. Nat. Rev. Neurosci. 2015, 16, 647–659. 10.1038/nrn4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuthall H. N.; Husain J.; McLarren K. W.; Stifani S. Role for Hes1-Induced Phosphorylation in Groucho-Mediated Transcriptional Repression. Mol. Cell. Biol. 2002, 22, 389–399. 10.1128/mcb.22.2.389-399.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kageyama R.; Shimojo H.; Imayoshi I. Dynamic expression and roles of Hes factors in neural development. Cell Tissue Res. 2015, 359, 125–133. 10.1007/s00441-014-1888-7. [DOI] [PubMed] [Google Scholar]
- Imayoshi I.; Isomura A.; Harima Y.; Kawaguchi K.; Kori H.; Miyachi H.; Fujiwara T.; Ishidate F.; Kageyama R. Oscillatory Control of Factors Determining Multipotency and Fate in Mouse Neural Progenitors. Science 2013, 342, 1203–1208. 10.1126/science.1242366. [DOI] [PubMed] [Google Scholar]
- Tan S.-L.; Ohtsuka T.; González A.; Kageyama R. MicroRNA9 regulates neural stem cell differentiation by controlling Hes1 expression dynamics in the developing brain. Genes Cells 2012, 17, 952–961. 10.1111/gtc.12009. [DOI] [PubMed] [Google Scholar]
- Shi X.; Yan C.; Liu B.; Yang C.; Nie X.; Wang X.; Zheng J.; Wang Y.; Zhu Y. miR-381 Regulates Neural Stem Cell Proliferation and Differentiation via Regulating Hes1 Expression. PLoS One 2015, 10, e0138973 10.1371/journal.pone.0138973. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Young F. I.; Keruzore M.; Nan X.; Gennet N.; Bellefroid E. J.; Li M. The doublesex-related Dmrta2 safeguards neural progenitor maintenance involving transcriptional regulation of Hes1. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, e5599–e5607. 10.1073/pnas.1705186114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai M. A.; Ishikawa N.; Tanaka M.; Uemura K.; Sugimitsu N.; Suganami A.; Tamura Y.; Koyano T.; Kowithayakorn T.; Ishibashi M. Hes1 inhibitor isolated by target protein oriented natural products isolation (TPO-NAPI) of differentiation activators of neural stem cells. Chem. Sci. 2016, 7, 1514–1520. 10.1039/c5sc03540f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wawer M. J.; Li K.; Gustafsdottir S. M.; Ljosa V.; Bodycombe N. E.; Marton M. A.; Sokolnicki K. L.; Bray M.-A.; Kemp M. M.; Winchester E.; Taylor B.; Grant G. B.; Hon C. S.-Y.; Duvall J. R.; Wilson J. A.; Bittker J. A.; Dancik V.; Narayan R.; Subramanian A.; Winckler W.; Golub T. R.; Carpenter A. E.; Shamji A. F.; Schreiber S. L.; Clemons P. A. Toward performance-diverse small-molecule libraries for cell-based phenotypic screening using multiplexed high-dimensional profiling. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 10911–10916. 10.1073/pnas.1410933111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M.; Peh J.; Hergenrother P. J.; Cunningham B. T. Detection of protein-small molecule binding using a self-referencing external cavity laser biosensor. J. Am. Chem. Soc. 2014, 136, 5840–5843. 10.1021/ja500636p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White S.; Baird E. E.; Dervan P. B. On the pairing rules for recognition in the minor groove of DNA by pyrrole-imidazole polyamides. Chem. Biol. 1997, 4, 569–578. 10.1016/s1074-5521(97)90243-x. [DOI] [PubMed] [Google Scholar]
- Muzikar K. A.; Nickols N. G.; Dervan P. B. Repression of DNA-binding dependent glucocorticoid receptor-mediated gene expression. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 16598–16603. 10.1073/pnas.0909192106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed J.; Pandian G. N.; Sato S.; Taniguchi J.; Chandran A.; Hashiya K.; Bando T.; Sugiyama H. Targeted suppression of EVI1 oncogene expression by sequence-specific pyrrole-imidazole polyamide. Chem. Biol. 2014, 21, 1370–1380. 10.1016/j.chembiol.2014.07.019. [DOI] [PubMed] [Google Scholar]
- Yang F.; Nickols N. G.; Li B. C.; Marinov G. K.; Said J. W.; Dervan P. B. Antitumor activity of a pyrrole-imidazole polyamide. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 1863–1868. 10.1073/pnas.1222035110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao E.-H.; Fukuda N.; Ueno T.; Matsuda H.; Nagase H.; Matsumoto Y.; Sugiyama H.; Matsumoto K. A pyrrole-imidazole polyamide targeting transforming growth factor-beta1 inhibits restenosis and preserves endothelialization in the injured artery. Cardiovasc. Res. 2009, 81, 797–804. 10.1093/cvr/cvn355. [DOI] [PubMed] [Google Scholar]
- Lake R. J.; Tsai P.-F.; Choi I.; Won K.-J.; Fan H.-Y. RBPJ, the major transcriptional effector of Notch signaling, remains associated with chromatin throughout mitosis, suggesting a role in mitotic bookmarking. PLoS Genet. 2014, 10, e1004204 10.1371/journal.pgen.1004204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Zou J.; Zhao B.; Johannsen E.; Ashworth T.; Wong H.; Pear W. S.; Schug J.; Blacklow S. C.; Arnett K. L.; Bernstein B. E.; Kieff E.; Aster J. C. Genome-wide analysis reveals conserved and divergent features of Notch1/RBPJ binding in human and murine T-lymphoblastic leukemia cells. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 14908–14913. 10.1073/pnas.1109023108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake R. J.; Tsai P.-F.; Choi I.; Won K.-J.; Fan H.-Y. RBPJ, the major transcriptional effector of Notch signaling, remains associated with chromatin throughout mitosis, suggesting a role in mitotic bookmarking. PLoS Genet. 2014, 10, e1004204 10.1371/journal.pgen.1004204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y.; Pandian G. N.; Zou T.; Taniguchi J.; Sato S.; Kashiwazaki G.; Vaijayanthi T.; Hidaka T.; Bando T.; Sugiyama H. A Multi-target Small Molecule for Targeted Transcriptional Activation of Therapeutically Significant Nervous System Genes. ChemistryOpen 2016, 5, 517–521. 10.1002/open.201600125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould S. J.; Subramani S. Firefly luciferase as a tool in molecular and cell biology. Anal. Biochem. 1988, 175, 5–13. 10.1016/0003-2697(88)90353-3. [DOI] [PubMed] [Google Scholar]
- Taniguchi J.; Pandian G. N.; Hidaka T.; Hashiya K.; Bando T.; Kim K. K.; Sugiyama H. A synthetic DNA-binding inhibitor of SOX2 guides human induced pluripotent stem cells to differentiate into mesoderm. Nucleic Acids Res. 2017, 45, 9219–9228. 10.1093/nar/gkx693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buscarlet M.; Perin A.; Laing A.; Brickman J. M.; Stifani S. Inhibition of cortical neuron differentiation by Groucho/TLE1 requires interaction with WRPW, but not Eh1, repressor peptides. J. Biol. Chem. 2008, 283, 24881–24888. 10.1074/jbc.m800722200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBon L.; Lee T. V.; Sprinzak D.; Jafar-Nejad H.; Elowitz M. B. Fringe proteins modulate Notch-ligand cis and trans interactions to specify signaling states. eLife 2014, 3, e02950 10.7554/elife.02950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandbarbe L. Delta-Notch signaling controls the generation of neurons/glia from neural stem cells in a stepwise process. Development 2003, 130, 1391–1402. 10.1242/dev.00374. [DOI] [PubMed] [Google Scholar]
- Shin S.; Vemuri M.. Culture and Differentiation of Human Neural Stem Cells. In Protocols for Neural Cell Culture; Doering L., Ed.; Springer Protocols Handbooks; Humana Press, 2010; pp 51–73. [Google Scholar]
- Hashimoto-Torii K.; Torii M.; Sarkisian M. R.; Bartley C. M.; Shen J.; Radtke F.; Gridley T.; Šestan N.; Rakic P. Interaction between Reelin and Notch signaling regulates neuronal migration in the cerebral cortex. Neuron 2008, 60, 273–284. 10.1016/j.neuron.2008.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathia J. D.; Mattson M. P.; Cheng A. Notch: from neural development to neurological disorders. J. Neurochem. 2008, 107, 1471–1481. 10.1111/j.1471-4159.2008.05715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X.; Wu H.; Xu H.; Xiong H.; Chu Q.; Yu S.; Wu G. S.; Wu K. Notch signaling: an emerging therapeutic target for cancer treatment. Cancer Lett. 2015, 369, 20–27. 10.1016/j.canlet.2015.07.048. [DOI] [PubMed] [Google Scholar]
- Frangi A. F.; Niessen W. J.; Vincken K. L.; Viergever M. A.. Multiscale vessel enhancement filtering. International Conference on Medical Image Computing and Computer-Assisted Intervention; Springer, 1998; pp 130–137.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.