

Abstract
Lipin-1 performs dual function during lipid metabolism, i.e., it functions as a transcriptional coactivator and as a phosphatidate phosphatase during triglyceride biosynthesis. We investigated whether exendin-4 prevented endoplasmic reticulum (ER) stress-induced hepatic steatosis and whether the protective effects of exendin-4 were associated with lipin-1 signaling. Tunicamycin and thapsigargin, ER stress inducers, increased triglycerides (TG) content and expression of genes encoding lipid droplet surface proteins. Exendin-4 decreased the expression of ER stress markers phosphorylated PKR like ER kinase (PERK), phosphorylated inositol-requiring enzyme 1 alpha (IRE1α), and glucose-regulated protein 78 kDa (GRP78) proteins and spliced X-box binding protein 1 (XBP-1s) mRNA and increased the expression of genes encoding lipolytic enzymes hormone-sensitive lipase (HSL) and monoacylglycerol lipase (MGL) and VLDL assembly-associated proteins microsomal triglyceride transfer protein (MTP) and apolipoprotein B (APOB) in tunicamycin-pretreated cells. Moreover, exendin-4 significantly decreased lipin-1β/α ratio by increasing SFRP10 and increased lipin-1 nuclear localization. The decrease in lipin-1β/α ratio was also observed in SIRT1 and AMPK agonist-treated cells. These data suggest that exendin-4 improves ER stress-induced hepatic lipid accumulation by increasing lipolysis and VLDL assembly, which is partially mediated by the regulation of lipin-1 signaling.
Electronic supplementary material
The online version of this article (10.1007/s12192-017-0872-z) contains supplementary material, which is available to authorized users.
Keywords: Exendin-4, ER stress, Lipid accumulation, Lipolysis, VLDL assembly, Lipin-1
Introduction
Excessive lipid accumulation in the liver induces nonalcoholic fatty liver disease (NAFLD), which is associated with various metabolic diseases such as obesity, insulin resistance, and dyslipidemia. Hepatic lipid accumulation is caused by an imbalance between energy intake and energy expenditure. Hepatic lipids, which are stored in the form of triglycerides (TGs) within lipid droplets, are broken down into free fatty acids (FFAs) by lipolytic enzymes, including adipose tissue triglyceride lipase (ATGL), hormone-sensitive lipase (HSL), and monoacylglycerol lipase (MGL). The fatty acids produced are oxidized through the regulation of peroxisome proliferator-activated receptor alpha (PPAR-alpha) (Sapiro et al. 2009) or are used for TG synthesis (Kawano and Cohen 2013). In addition, hepatic TG is released by very-low-density lipoprotein (VLDL) assembly pathway for lipid homeostasis (Mensenkamp et al. 2004). Interestingly, inhibition of hepatic TG-rich VLDL secretion leads to steatosis (Minehira et al. 2008).
The endoplasmic reticulum (ER) plays a central role in lipid and protein biosynthesis. Disruption of ER homeostasis leads to the accumulation of misfolded or unfolded proteins, which induces ER stress. ER stress is associated with obesity, insulin action, and type 2 diabetes (Gentile and Pagliassotti 2008; Ozcan et al. 2004; Li et al. 2011) and plays an important role in fatty liver disease development (Jo et al. 2013). At present, studies are in progress to determine the role of ER stress in fatty liver development. ER stress induces NAFLD and nonalcoholic steatohepatitis (NASH) through various related molecular mechanisms, including lipogenesis, inflammation, apoptosis, and autophagy (Malhi and Kaufman 2011; Lee et al. 2012b; Lee et al. 2012a). Interestingly, overexpression of chaperones that enhance the functional capacity of the ER or deletion of X-box binding protein 1 (XBP-1), a marker of ER stress, improves hepatic steatosis (Kammoun et al. 2009; Lee et al. 2008). Thus, improvement of ER function ameliorates NAFLD (Gentile and Pagliassotti 2008). However, the mechanism underlying ER stress-induced steatosis is unclear.
Lipin-1 plays a crucial role in lipid homeostasis. Lipin-1 is encoded by LPIN1 and is highly expressed in the key metabolic tissues, including the adipose tissue, skeletal muscle, and liver. Cytosolic lipin-1 functions as an Mg2+-dependent phosphatidate phosphatase type-1 in triglyceride synthesis pathway and nuclear lipin-1 functions as a transcriptional coactivator for the expression of fatty acid oxidation genes (Harris and Finck 2011; Ishimoto 2011). The liver and adipose tissue express two lipin-1 isoforms (lipin-1α and lipin-1β), which are generated by an alternative splicing factor serine-arginine-rich splicing factor 10 (SFRS10), that show different subcellular localization. Lipin-1α and lipin-1β are predominantly located in the nucleus and cytoplasm, respectively (Peterfy et al. 2005). Pihlajamäki et al. reported that lipin-1β/α ratio is significantly increased in the liver of obese humans (Pihlajamaki et al. 2011). Nuclear localization of lipin-1 represses the effects of sterol regulatory element-binding protein 1 (SREBP-1), which plays a major role in lipid and carbohydrate metabolism (Peterson et al. 2011). These results indicate the importance of lipin-1 signaling in regulating hepatic lipid metabolism.
Exendin-4, a glucagon-like protein-1 (GLP-1) receptor agonist, is a representative drug for treating diabetes. Exendin-4 also improves hepatic steatosis and ER stress through silent mating-type information regulation 2 homolog 1 (SIRT1) and AMP-activated protein kinase (AMPK) signaling pathways, which regulate the increase in fatty acid oxidation and decrease in lipid synthesis (Xu et al. 2014). In the present study, we examined whether exendin-4 improved ER stress-induced steatosis and regulated lipin-1 expression under ER stress.
Materials and methods
Cell culture and pharmacological treatments
HepG2 cells were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) and were cultured in high glucose-containing Dulbecco’s modified Eagle’s medium (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37 °C in 5% CO2. The cells were pretreated with 3 μg/ml tunicamycin (Sigma-Aldrich Corp., St. Louis, MO, USA) for 24 h, followed by treatment with or without 100 nM exendin-4 (Sigma-Aldrich Corp.) for 24 h. To confirm the effect of SIRT1 and AMPK in hepatocytes, the cells were treated with 100 μM resveratrol (a SIRT1 agonist; Sigma-Aldrich Corp.), 2 mM AICAR (an AMPK agonist; Sigma-Aldrich Corp.), or 10 μM compound C (an AMPK inhibitor; Sigma-Aldrich Corp.) for 24 h.
Transient siRNA transfection
For siRNA-mediated gene silencing, HepG2 cells were transfected with an siRNA duplex against lipin-1 or a negative control siRNA duplex by using Lipofectamine RNAiMAX transfection reagent (Invitrogen, Carlsbad, CA, USA), according to the manufacturer’s instructions. Next, the cells were cultured under normal growth conditions (37 °C in 5% CO2) for 24 h and were harvested for further analysis.
Quantitative RT-PCR
Total RNA was extracted from the cells by using TRIzol reagent (Invitrogen), and 2 μg RNA was reverse transcribed using High-Capacity RNA-to-cDNA Kit (Applied Biosystems, Foster City, CA, USA), according to the manufacturer’s instructions. Quantitative RT-PCR was performed using SYBR Green (Roche, Lewis, UK) and Light-Cycler 480 (Roche). Relative gene expression levels were measured using comparative Ct method (2^delta delta Ct) and were normalized to the expression level of the β-actin gene (endogenous control). Primer pairs used for performing specific PCRs are listed in Online Resource 1 and Table S1 (Lindegaard et al. 2007; van Schadewijk et al. 2012).
Western blotting
Nuclear and cytoplasmic protein fractions were isolated from cultured cells by using a commercially available nuclear extraction kit (Cayman Chemical Co., Ann Arbor, MI, USA). Proteins present in each fraction (15 μg sample) were resolved on a 4–12% Bis-Tris NuPAGE gel (Invitrogen) and were transferred onto a polyvinylidene difluoride membrane by using iBlot2 PVDF stack (Invitrogen). The membrane was incubated with the following primary antibodies: anti-phosphorylated protein kinase A (PKA) (no. 5661; Cell Signaling Technology, Danvers, MA, USA), anti-PKA (no. 4782; Cell Signaling Technology), anti-lipin-1 (no. 5195; Cell Signaling Technology), anti-phosphorylated AMPK (no. 2531; Cell Signaling Technology), anti-AMPK (no. 2532; Cell Signaling Technology), anti-SIRT1 (sc-15,404; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), anti-glucose-regulated protein 78 kDa (GRP78) (sc-13968; Santa Cruz Biotechnology, Inc.), anti-phosphorylated PKR like ER kinase (PERK) (sc-3255; Santa Cruz Biotechnology, Inc.), anti-PERK (sc-13,073; Santa Cruz Biotechnology, Inc.), anti-phosphorylated inositol-requiring enzyme 1 alpha (IRE1α) (ab104157; Abcam, Cambridge, MA, USA), anti-IRE1α (no. 3294; Cell Signaling Technology), anti-β-actin (no. 4967; Cell Signaling Technology), and anti-lamin B1 (ab16048; Abcam). Next, the membranes were incubated with HRP-conjugated secondary antibodies, and immunoreactive bands were visualized using ECL detection kit (GE Healthcare, Piscataway, NJ, USA).
Measurement of intracellular triglyceride content
For the quantitative analysis of TGs, total TGs were extracted from whole HepG2 cells treated with 3 μg/ml tunicamycin or 0.5 μM thapsigargin (Sigma-Aldrich Corp.) for 24 h, and TG content was measured using a TG determination kit (TR0100, Sigma-Aldrich), according to the manufacturer’s instructions. Values were normalized to protein concentration measured using BCA protein assay kit (Thermo Scientific, Rockford, IL, USA).
Immunofluorescence analysis
HepG2 cells were cultured on coverslips, fixed with 4% paraformaldehyde for 5 min, permeabilized with pre-chilled 0.5% Triton X-100 for 5 min at − 20 °C, and incubated with a blocking buffer for 1 h at room temperature. Next, the cells were incubated overnight with the primary anti-lipin-1 antibody at 4 °C and were labeled with a secondary antibody for 1 h at room temperature. Tetramethylrhodamine isothiocyanate-conjugated anti-rabbit antibody (dilution, 1:100) was used as a fluorescent secondary antibody. Moreover, the nuclei were counterstained with DAPI (Sigma-Aldrich). Next, the cells were mounted using ProLong Gold antifade reagent (Invitrogen), and images were obtained using a fluorescence microscope (BX51; Olympus, Tokyo, Japan).
Statistical analysis
Results are expressed as mean ± SEM. Statistical analysis was performed using SPSS 12.0 software (SPSS Inc., Chicago, IL, USA). Data were analyzed using one-way analysis of variance followed by Bonferroni/Dunn multiple range test to calculate significant differences between mean values obtained for the different experimental groups. Differences were considered significant at p < 0.05.
Results
ER stress induces lipid accumulation in hepatocytes
To determine whether ER stress induced hepatic steatosis, HepG2 cells were treated with 3 μg/ml tunicamycin or 0.5 μM thapsigargin. Tunicamycin and thapsigargin treatment increased intracellular TG levels (Fig. 1a, b). Next, we examined the effects of tunicamycin and thapsigargin on the expression of genes encoding lipid droplet surface proteins perilipin 1 (PLIN1), perilipin 2 (PLIN2), perilipin 3 (PLIN3), and cell death-inducing DFFA-like effector c (CIDEC; known as FSP27 or fat-specific protein 27 in rodents). Expression of PLIN1, PLIN2, and CIDEC but not of PLIN3 increased obviously in tunicamycin-treated cells compared with that in control cells (Fig. 1a). Thapsigargin increased the expression of PLIN2 and PLIN3 but not of PLIN1 and CIDEC compared with that in control cells (Fig. 1b). In contrast, exendin-4 treatment prevented tunicamycin-induced ER stress by decreasing the expression of ER stress markers, phosphorylated PKR like ER kinase (P-PERK), phosphorylated inositol-requiring enzyme 1 alpha (P-IRE1α), and GRP78 proteins and spliced XBP-1 (XBP-1s) mRNA (Fig. 1c, d).
Fig. 1.
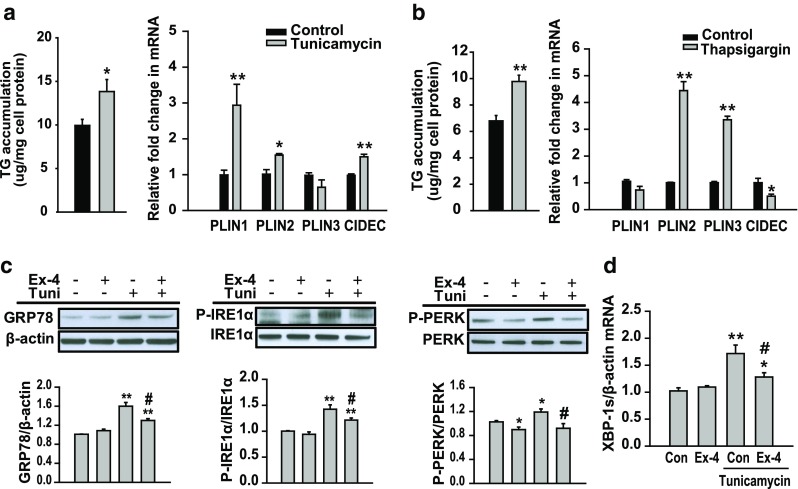
ER stress induces hepatic lipid accumulation. a, b HepG2 cells were treated with tunicamycin (3 μg/ml) or thapsigargin (0.5 μM) for 24 h. Lipid accumulation was measured by performing TG colorimetric assay. The mRNA expression levels of the genes encoding PLIN1, PLIN2, PLIN3, and CIDEC were measured by performing quantitative RT-PCR and were normalized to that of the β-actin gene. c, d HepG2 cells were pretreated with 3 μg/ml tunicamycin, followed by treatment with or without exendin-4 (Ex-4; 100 nM) for 24 h. Levels of phosphorylated PERK, phosphorylated IRE1α, and GRP78 were analyzed by performing western blotting and were normalized to the PERK, IRE1α, and β-actin of each sample, respectively. The expression of spliced XBP-1 (XBP-1s) mRNA was measured by performing quantitative RT-PCR and was normalized to that of the β-actin gene. All values are expressed as the mean ± SE (n = 5–6) *p < 0.05 and **p < 0.01 compared with control cells; #p < 0.05 compared with tunicamycin-treated cells
Exendin-4 increases lipolysis and VLDL assembly in tunicamycin-treated hepatocytes
PKA stimulates lipolysis by activating ATGL and HSL. To investigate whether exendin-4 affected the expression of genes involved in lipolysis, we examined the changes in the expression of PKA and lipolytic enzymes in exendin-4-treated cells. Exendin-4 treatment increased phosphorylated PKA level in a dose-dependent manner (Fig. 2a). The mRNA expression of the genes encoding ATGL, HSL, and MGL decreased in tunicamycin-treated cells compared with that in control cells; in contrast, the mRNA expression of the genes encoding HSL and MGL significantly increased in exendin-4-treated cells (Fig. 2b–d). The liver secretes TGs in the form of VLDL to maintain hepatic lipid homeostasis, and this process is initiated in the ER (Mensenkamp et al. 2004). Tunicamycin treatment decreased the mRNA expression of the genes encoding microsomal TG transfer protein (MTP) and apolipoprotein B (APOB) (Fig. 2e, f), which are required for VLDL assembly. However, exendin-4 treatment reversed the effect of tunicamycin on the expression of these genes. These data suggest that exendin-4 improves ER stress-induced steatosis by stimulating lipolysis and VLDL assembly.
Fig. 2.

Exendin-4 increases the expression of lipolysis- and VLDL assembly-associated genes in tunicamycin-treated hepatocytes. a Phosphorylated PKA levels in hepatocytes treated with different concentrations of exendin-4 were analyzed by performing western blotting. b–f HepG2 cells were pretreated with 3 μg/ml tunicamycin, followed by treatment with or without exendin-4 (Ex-4; 100 nM) for 24 h. The mRNA expression levels of the genes encoding ATGL, HSL, MGL, MTP, and APOB were normalized to that of the β-actin gene. All values are expressed as the mean ± SE (n = 5–6). *p < 0.05 and **p < 0.01 compared with control cells; #p < 0.05 and ##p < 0.01 compared with tunicamycin-treated cells
Exendin-4 decreases lipin-1β/α ratio and increases nuclear lipin-1 level in tunicamycin-treated hepatocytes
Lipin-1 is associated with lipid homeostasis, and lipin-1α and lipin-1β are involved in lipid oxidation and TG synthesis, respectively (Harris and Finck 2011; Ishimoto 2011). We examined whether exendin-4 regulated lipin-1 signaling. We observed that SFRS10 expression increased after exendin-4 treatment (Fig. 3a). Consistently, exendin-4 treatment increased lipin-1α mRNA expression compared with that in control cells but did not significantly alter lipin-1α mRNA expression in tunicamycin-pretreated cells (Fig. 3b). In contrast, exendin-4 treatment decreased tunicamycin-induced increase in lipin-1β mRNA expression (Fig. 3c). Consistently, lipin-1β/α ratio increased by twofold in tunicamycin-treated cells and decreased in exendin-4-treated cells (Fig. 3d). Exendin-4 also decreased lipin-1β/α ratio in palmitic acid (PA)-pretreated cells (Online Resource 1, Fig. S1a). Moreover, treatment with ER stress inhibitor TUDCA decreased lipin-1β/α ratio in tunicamycin-treated cells (Online Resource 1, Fig. S1b). These data suggest that increased lipin-1β/α ratio can serve as a biomarker of metabolic disruption and that exendin-4 regulates lipin-1 signaling in tunicamycin-treated hepatocytes.
Fig. 3.

Exendin-4 decreases lipin-1β/α ratio in tunicamycin-treated hepatocytes. a SFRS10 mRNA expression level in HepG2 cells treated with 100 nM exendin-4 (Ex-4) was analyzed by performing quantitative RT-PCR. b–d HepG2 cells were pretreated with 3 μg/ml tunicamycin, followed by treatment with or without exendin-4 (100 nM) for 24 h. The mRNA expression levels of the genes encoding lipin-1α and lipin-1β were normalized to that of the β-actin gene. All values are expressed as the mean ± SE (n = 4–6). *p < 0.05 compared with control cells; #p < 0.05 and ##p < 0.01 compared with tunicamycin-treated cells
Because lipin-1 isoforms show different subcellular distribution, we next examined lipin-1 localization by performing western blotting and immunofluorescence analysis. Tunicamycin treatment decreased nuclear lipin-1 levels and increased cytosolic lipin-1 levels. In contrast, exendin-4 treatment increased nuclear lipin-1 levels and decreased cytosolic lipin-1 levels compared with those in tunicamycin-treated cells (Fig. 4a). Consistent results were obtained by performing immunofluorescence analysis (Fig. 4b).
Fig. 4.

Exendin-4 increases nuclear lipin-1 level. Cytosolic and nuclear protein extracts were prepared using HepG2 cells pretreated with 3 μg/ml tunicamycin (Tuni) and treated with or without exendin-4 (Ex-4; 100 nM) for 24 h. a Nuclear and cytosolic lipin-1 levels were analyzed by performing western blotting and were normalized to the lamin B1 and β-actin of each sample, respectively. All values are expressed as the mean ± SE (n = 6). **p < 0.01 compared with control cells; #p < 0.05 and ##p < 0.01 compared with tunicamycin-treated cells. b Subcellular localization of lipin-1 was determined by performing immunofluorescence (IF) analysis, and the cells were photographed using a light microscope (magnification, ×400); scale bars = 100 μm
Exendin-4 regulates lipin-1 expression through SIRT–AMPK signaling
SIRT1 and phosphorylated AMPK levels decreased in tunicamycin-treated cells and increased in exendin-4-treated cells (Fig. 5a). To determine whether exendin-4-induced modulation in lipin-1 signaling increased SIRT1 and AMPK levels, HepG2 cells were treated with the SIRT1 agonist resveratrol or AMPK agonist AICAR. Resveratrol or AICAR treatment increased lipin-1 and lipin-1α expression and significantly decreased lipin-1β/α ratio compared with that in control cells (Fig. 5b, c). However, compound C (AMPK inhibitor) treatment decreased lipin-1 and lipin-1α expression and increased lipin-1β/α ratio compared with that in control cells (Online Resource 1, Fig. S2a). Exendin-4 did not stimulate lipin-1 expression in compound C-pretreated cells (Online Resource 1, Fig. S2b). Next, we investigated whether lipin-1 regulated SIRT1 and AMPK expression. However, no change in SIRT1 and AMPK expression was observed in lipin-1 siRNA-transfected cells (Fig. 5d). These data suggest that exendin-4-induced modulation of lipin-1 signaling in hepatocytes is mediated by SIRT1–AMPK signaling.
Fig. 5.

Exendin-4 regulates lipin-1 signaling through SIRT1 and AMPK in HepG2 cells. a HepG2 cells were pretreated with 3 μg/ml tunicamycin (Tuni), followed by treatment with or without exendin-4 (Ex-4; 100 nM) for 24 h. SIRT1 and phosphorylated AMPK levels were analyzed by performing western blotting and were normalized to the lamin B1 and AMPK of each sample, respectively. b, c HepG2 cells were treated with 100 μM resveratrol (a SIRT1 agonist) or 2 mM AICAR (an AMPK agonist) for 24 h. d HepG2 cells transfected with 10 nM lipin-1 siRNA or control siRNA for 24 h. The mRNA expression levels of the genes encoding lipin-1, lipin-1α, lipin-1β, SIRT1, and AMPK were normalized to that of the β-actin gene. All values are expressed as the mean ± SE (n = 4–6). *p < 0.05 and **p < 0.01 compared with control cells; #p < 0.05 compared with tunicamycin-treated cells
Lipin-1 regulates VLDL assembly signaling
To determine whether lipin-1 regulated VLDL assembly for TG secretion, we examined APOB expression in lipin-1 siRNA-transfected cells by performing quantitative RT-PCR. The effect of exendin-4 on APOB expression was blunted in lipin-1 siRNA-transfected cells (Fig. 6a), suggesting that lipin-1 mediated the protective effect of exendin-4 against ER stress-associated lipid accumulation.
Fig. 6.

Lipin-1 increases APOB expression in exendin-4-treated cells. a HepG2 cells were transfected with 10 nM lipin-1 siRNA or control siRNA for 24 h and were pretreated with 3 μg/ml tunicamycin, followed by treatment with or without exendin-4 (Ex-4; 100 nM) for 24 h. APOB mRNA expression level was analyzed by performing quantitative RT-PCR. All values are expressed as the mean ± SE (n = 5–6). *p < 0.05 compared with control cells; ##p < 0.01 compared with tunicamycin-treated cells. b Schematic illustration of the signaling pathway discussed in this study
Discussion
In the present study, we found that exendin-4 improved ER stress-induced hepatic steatosis by stimulating lipolysis and VLDL assembly for TG secretion and that exendin-4-induced regulation of lipin-1 signaling was mediated by SIRT1–AMPK signaling (Fig. 6b). Tunicamycin and tunicamycin, ER stress inducers, increased the concentration of TGs and expression of genes encoding lipid droplet-associated proteins, indicating that ER stress induced hepatic steatosis. However, exendin-4 increased the expression of the genes encoding lipolytic enzymes HSL and MGL and VLDL assembly-associated proteins MTP and APOB in tunicamycin-treated cells. Exendin-4 dramatically decreased the ratio of lipin-1β/α by increasing SFRP10 expression and increased the nuclear localization of lipin-1 compared with that in tunicamycin-treated cells. SiRNA-induced inhibition of lipin-1 expression attenuated the effect of exendin-4 on APOB expression. These findings suggest that the protective effect of exendin-4 against ER stress-induced steatosis is at least partially mediated by increased lipolysis and VLDL secretion and that lipin-1 affects exendin-4-stimulated VLDL secretion.
High-fat diet-induced obesity causes ER stress (Castro et al. 2013; Kawasaki et al. 2012), and enhanced ER stress induces hepatic steatosis and insulin resistance (Jo et al. 2013; Zhang et al. 2013). Jo et al. reported that ER stress-associated hepatic steatosis is mediated by the increased expression of hepatic VLDL receptor (VLDLR) (Jo et al. 2013), which is partially consistent with our finding that VLDLR expression increased in tunicamycin-treated cells compared with that in control cells (Online Resource 1, Fig. S3). In the present study, expression of ER stress marker proteins phosphorylated PERK, phosphorylated IRE1α, and GRP78 increased in tunicamycin-treated cells and decreased in exendin-4-treated cells (Fig. 1). However, despite its protective effect against ER stress, exendin-4 did not affect VLDLR expression in tunicamycin-treated hepatocytes (Online Resource 1, Fig. S3). This result suggests that the protective effect of exendin-4 against ER stress-induced steatosis was mediated by another mechanism and not by the inhibition of lipoprotein uptake.
Exendin-4, a GLP-1 receptor agonist, exerts protective effect against hepatic steatosis in ob/ob mice and diet-induced obese mice by modulating lipid metabolism in the liver, muscle, and adipose tissue (Ding et al. 2006; Seo et al. 2016; Tanaka et al. 2014). PKA performs several functions, including regulation of glycogen, sugar, and lipid metabolism. PKA inhibits lipogenesis and stimulates lipolysis by phosphorylating important enzymes during metabolism (Dong et al. 2014). We determined the expression levels of lipid metabolism-associated genes in hepatocytes. We observed that exendin-4 increased the expression of phosphorylated PKA and lipolytic enzymes. In the liver, FFAs undergo β-oxidation or re-esterification into TGs that are then stored in lipid droplets or are exported from the liver as VLDLs (Dowman et al. 2010). We found that exendin-4 increased the expression of APOB and MTP, which are required for VLDL assembly, in tunicamycin-pretreated cells. These findings suggest that exendin-4 improves hepatic lipid metabolism by modulating lipolysis and lipid transport.
Mutations in LPIN1 were identified in fatty liver dystrophic (fld) mice, which induces lipodystrophy by impairing adipocyte differentiation (Chen et al. 2012; Reue and Dwyer 2009). Donkor et al. examined the adipose tissue of young healthy men and found that lipin-1 expression in the adipose tissue was correlated with adiposity, insulin sensitivity, and oxidative, lipogenic, and lipolytic gene expression (Donkor et al. 2008). In the liver, lipin-1 performs dual functions during lipid metabolism, i.e., it functions as a transcriptional coactivator and as a phosphatidate phosphatase during TG biosynthesis (Reue and Zhang 2008). Yin et al. reported that liver-specific deletion of SIRT1 aggravated alcoholic fatty liver by altering lipin-1 signaling (Yin et al. 2014). Increased lipin-1β expression and hepatic lipin-1β/α ratio and decreased total lipin-1 and lipin-1α expression have been observed in the livers of high-fat diet-fed mice (Zhang et al. 2014). The present study is the first to show that exendin-4 improved lipin-1 signaling in tunicamycin-treated hepatocytes. Furthermore, we found that SIRT1 and AMPK promoted the alternative splicing of hepatic lipin-1 mRNA and decreased lipin-1β/α ratio.
Lipin-1 expression is associated with hepatic VLDL assembly and secretion (Bou Khalil et al. 2009). Lipin-1 interacts with HNF4-alpha and PPARs, which function as nuclear hormone receptors, to enhance fatty acid oxidation (Chen et al. 2012; Schweitzer et al. 2015). Interestingly, inhibition of HNF4-alpha decreases APOB expression, thus increasing TG content (Xu et al. 2015). Treatment with Wy-14,643, a PPAR-alpha agonist, increases APOB-100 secretion by increasing MTP expression (Linden et al. 2002; Ameen et al. 2005). We found that lipin-1 inhibition attenuated exendin-4-induced APOB expression, indicating that lipin-1 can be used as a therapeutic target of exendin-4 to prevent ER stress-induced steatosis. However, further studies are needed to determine whether lipin-1α or lipin-1β regulate APOB expression.
In conclusion, the present study showed that exendin-4 improved ER stress-induced hepatic steatosis by stimulating lipolysis and lipid transport and regulated lipin-1β/α ratio through SIRT1–AMPK signaling. We also found that lipin-1 affected lipid transport in hepatocytes. Thus, exendin-4-induced activation of SIRT1–AMPK–lipin-1 signaling could be a potential therapeutic approach for treating fatty liver diseases and could help in explaining the relationship between ER stress and steatosis.
Electronic supplementary material
(PDF 199 kb)
Acknowledgments
This study was supported by Samsung Biomedical Research Institute (SBRI) (http://www.sbri.or.kr), the Medical Research Funds from Kangbuk Samsung Hospital, and the National Research Foundation (NRF), which is funded by the Korean government (NRF-2016R1A6A3A11930792) (http://www.nrf.re.kr). The funders had no role in the study design, data collection, and analysis, the decision to publish, or preparation of the manuscript.
Abbreviations
- AMPK
AMP-activated protein kinase
- APOB
Apolipoprotein B
- ATGL
Adipose tissue triglyceride lipase
- CIDEC
Cell death-inducing DFFA-like effector c
- ER
Endoplasmic reticulum
- GLP-1
Glucagon-like protein-1
- GRP78
Glucose-regulated protein 78 kDa
- HSL
Hormone-sensitive lipase
- IRE1α
Inositol-requiring enzyme 1 alpha
- MGL
Monoacylglycerol lipase
- MTP
Microsomal triglyceride transfer protein
- NAFLD
Nonalcoholic fatty liver disease
- NASH
Nonalcoholic steatohepatitis
- PA
Palmitic acid
- PERK
PKR like ER kinase
- PKA
Protein kinase A
- PLIN
Perilipin
- SFRS10
Serine-arginine-rich splicing factor 10
- SIRT1
Silent mating-type information regulation 2 homolog 1
- SREBP-1
Sterol regulatory element-binding protein 1
- TG
Triglycerides
- VLDL
Very-low-density lipoprotein
- XBP-1
X-box binding protein 1
Footnotes
Electronic supplementary material
The online version of this article (10.1007/s12192-017-0872-z) contains supplementary material, which is available to authorized users.
References
- Ameen C, Edvardsson U, Ljungberg A, Asp L, Akerblad P, Tuneld A, Olofsson SO, Linden D, Oscarsson J. Activation of peroxisome proliferator-activated receptor alpha increases the expression and activity of microsomal triglyceride transfer protein in the liver. J Biol Chem. 2005;280(2):1224–1229. doi: 10.1074/jbc.M412107200. [DOI] [PubMed] [Google Scholar]
- Bou Khalil M, Sundaram M, Zhang HY, Links PH, Raven JF, Manmontri B, Sariahmetoglu M, Tran K, Reue K, Brindley DN, Yao Z. The level and compartmentalization of phosphatidate phosphatase-1 (lipin-1) control the assembly and secretion of hepatic VLDL. J Lipid Res. 2009;50(1):47–58. doi: 10.1194/jlr.M800204-JLR200. [DOI] [PubMed] [Google Scholar]
- Castro G, MF CA, Weissmann L, Quaresma PG, Katashima CK, Saad MJ, Prada PO. Diet-induced obesity induces endoplasmic reticulum stress and insulin resistance in the amygdala of rats. FEBS Open Bio. 2013;3(1):443–449. doi: 10.1016/j.fob.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Gropler MC, Mitra MS, Finck BN. Complex interplay between the lipin 1 and the hepatocyte nuclear factor 4 alpha (HNF4alpha) pathways to regulate liver lipid metabolism. PLoS One. 2012;7(12):e51320. doi: 10.1371/journal.pone.0051320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding X, Saxena NK, Lin S, Gupta NA, Anania FA. Exendin-4, a glucagon-like protein-1 (GLP-1) receptor agonist, reverses hepatic steatosis in ob/ob mice. Hepatology. 2006;43(1):173–181. doi: 10.1002/hep.21006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Q, Giorgianni F, Deng X, Beranova-Giorgianni S, Bridges D, Park EA, Raghow R, Elam MB. Phosphorylation of sterol regulatory element binding protein-1a by protein kinase A (PKA) regulates transcriptional activity. Biochem Biophys Res Commun. 2014;449(4):449–454. doi: 10.1016/j.bbrc.2014.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donkor J, Sparks LM, Xie H, Smith SR, Reue K. Adipose tissue lipin-1 expression is correlated with peroxisome proliferator-activated receptor alpha gene expression and insulin sensitivity in healthy young men. J Clin Endocrinol Metab. 2008;93(1):233–239. doi: 10.1210/jc.2007-1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowman JK, Tomlinson JW, Newsome PN. Pathogenesis of non-alcoholic fatty liver disease. QJM: Month J Assoc Phys. 2010;103(2):71–83. doi: 10.1093/qjmed/hcp158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentile CL, Pagliassotti MJ. The endoplasmic reticulum as a potential therapeutic target in nonalcoholic fatty liver disease. Curr Opin Investig Drugs. 2008;9(10):1084–1088. [PMC free article] [PubMed] [Google Scholar]
- Harris TE, Finck BN. Dual function lipin proteins and glycerolipid metabolism. Trends Endocrinol Metab. 2011;22(6):226–233. doi: 10.1016/j.tem.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimoto K. Lipin 1 in lipid metabolism. Yakugaku zasshi J Pharm Soc Jpn. 2011;131(8):1189–1194. doi: 10.1248/yakushi.131.1189. [DOI] [PubMed] [Google Scholar]
- Jo H, Choe SS, Shin KC, Jang H, Lee JH, Seong JK, Back SH, Kim JB. Endoplasmic reticulum stress induces hepatic steatosis via increased expression of the hepatic very low-density lipoprotein receptor. Hepatology. 2013;57(4):1366–1377. doi: 10.1002/hep.26126. [DOI] [PubMed] [Google Scholar]
- Kammoun HL, Chabanon H, Hainault I, Luquet S, Magnan C, Koike T, Ferre P, Foufelle F. GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J Clin Invest. 2009;119(5):1201–1215. doi: 10.1172/JCI37007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano Y, Cohen DE. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J Gastroenterol. 2013;48(4):434–441. doi: 10.1007/s00535-013-0758-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki N, Asada R, Saito A, Kanemoto S, Imaizumi K. Obesity-induced endoplasmic reticulum stress causes chronic inflammation in adipose tissue. Sci Rep. 2012;2(1):799. doi: 10.1038/srep00799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AH, Scapa EF, Cohen DE, Glimcher LH. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science. 2008;320(5882):1492–1496. doi: 10.1126/science.1158042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Mendez R, Heng HH, Yang ZQ, Zhang K. Pharmacological ER stress promotes hepatic lipogenesis and lipid droplet formation. Am J Transl Res. 2012;4(1):102–113. [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Zheng Z, Mendez R, Ha SW, Xie Y, Zhang K. Pharmacologic ER stress induces non-alcoholic steatohepatitis in an animal model. Toxicol Lett. 2012;211(1):29–38. doi: 10.1016/j.toxlet.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Xu S, Giles A, Nakamura K, Lee JW, Hou X, Donmez G, Li J, Luo Z, Walsh K, Guarente L, Zang M. Hepatic overexpression of SIRT1 in mice attenuates endoplasmic reticulum stress and insulin resistance in the liver. FASEB J. 2011;25(5):1664–1679. doi: 10.1096/fj.10-173492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindegaard B, Larsen LF, Hansen AB, Gerstoft J, Pedersen BK, Reue K. Adipose tissue lipin expression levels distinguish HIV patients with and without lipodystrophy. Int J Obes. 2007;31(3):449–456. doi: 10.1038/sj.ijo.0803434. [DOI] [PubMed] [Google Scholar]
- Linden D, Lindberg K, Oscarsson J, Claesson C, Asp L, Li L, Gustafsson M, Boren J, Olofsson SO. Influence of peroxisome proliferator-activated receptor alpha agonists on the intracellular turnover and secretion of apolipoprotein (Apo) B-100 and ApoB-48. J Biol Chem. 2002;277(25):23044–23053. doi: 10.1074/jbc.M110416200. [DOI] [PubMed] [Google Scholar]
- Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease. J Hepatol. 2011;54(4):795–809. doi: 10.1016/j.jhep.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mensenkamp AR, Van Luyn MJ, Havinga R, Teusink B, Waterman IJ, Mann CJ, Elzinga BM, Verkade HJ, Zammit VA, Havekes LM, Shoulders CC, Kuipers F. The transport of triglycerides through the secretory pathway of hepatocytes is impaired in apolipoprotein E deficient mice. J Hepatol. 2004;40(4):599–606. doi: 10.1016/j.jhep.2003.12.011. [DOI] [PubMed] [Google Scholar]
- Minehira K, Young SG, Villanueva CJ, Yetukuri L, Oresic M, Hellerstein MK, Farese RV, Jr, Horton JD, Preitner F, Thorens B, Tappy L. Blocking VLDL secretion causes hepatic steatosis but does not affect peripheral lipid stores or insulin sensitivity in mice. J Lipid Res. 2008;49(9):2038–2044. doi: 10.1194/jlr.M800248-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306(5695):457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- Peterfy M, Phan J, Reue K. Alternatively spliced lipin isoforms exhibit distinct expression pattern, subcellular localization, and role in adipogenesis. J Biol Chem. 2005;280(38):32883–32889. doi: 10.1074/jbc.M503885200. [DOI] [PubMed] [Google Scholar]
- Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, Balderas E, Guertin DA, Madden KL, Carpenter AE, Finck BN, Sabatini DM. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. 2011;146(3):408–420. doi: 10.1016/j.cell.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pihlajamaki J, Lerin C, Itkonen P, Boes T, Floss T, Schroeder J, Dearie F, Crunkhorn S, Burak F, Jimenez-Chillaron JC, Kuulasmaa T, Miettinen P, Park PJ, Nasser I, Zhao Z, Zhang Z, Xu Y, Wurst W, Ren H, Morris AJ, Stamm S, Goldfine AB, Laakso M, Patti ME. Expression of the splicing factor gene SFRS10 is reduced in human obesity and contributes to enhanced lipogenesis. Cell Metab. 2011;14(2):208–218. doi: 10.1016/j.cmet.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reue K, Dwyer JR. Lipin proteins and metabolic homeostasis. J Lipid Res. 2009;50(Suppl):S109–S114. doi: 10.1194/jlr.R800052-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reue K, Zhang P. The lipin protein family: dual roles in lipid biosynthesis and gene expression. FEBS Lett. 2008;582(1):90–96. doi: 10.1016/j.febslet.2007.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapiro JM, Mashek MT, Greenberg AS, Mashek DG. Hepatic triacylglycerol hydrolysis regulates peroxisome proliferator-activated receptor alpha activity. J Lipid Res. 2009;50(8):1621–1629. doi: 10.1194/jlr.M800614-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitzer GG, Chen Z, Gan C, McCommis KS, Soufi N, Chrast R, Mitra MS, Yang K, Gross RW, Finck BN. Liver-specific loss of lipin-1-mediated phosphatidic acid phosphatase activity does not mitigate intrahepatic TG accumulation in mice. J Lipid Res. 2015;56(4):848–858. doi: 10.1194/jlr.M055962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo MH, Lee J, Hong SW, Rhee EJ, Park SE, Park CY, Oh KW, Park SW, Lee WY. Exendin-4 inhibits hepatic lipogenesis by increasing beta-catenin signaling. PLoS One. 2016;11(12):e0166913. doi: 10.1371/journal.pone.0166913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Masaki Y, Tanaka M, Miyazaki M, Enjoji M, Nakamuta M, Kato M, Nomura M, Inoguchi T, Kotoh K, Takayanagi R. Exenatide improves hepatic steatosis by enhancing lipid use in adipose tissue in nondiabetic rats. World J Gastroenterol: WJG. 2014;20(10):2653–2663. doi: 10.3748/wjg.v20.i10.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Schadewijk A, van't Wout EF, Stolk J, Hiemstra PS. A quantitative method for detection of spliced X-box binding protein-1 (XBP1) mRNA as a measure of endoplasmic reticulum (ER) stress. Cell Stress Chaperones. 2012;17(2):275–279. doi: 10.1007/s12192-011-0306-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F, Li Z, Zheng X, Liu H, Liang H, Xu H, Chen Z, Zeng K, Weng J. SIRT1 mediates the effect of GLP-1 receptor agonist exenatide on ameliorating hepatic steatosis. Diabetes. 2014;63(11):3637–3646. doi: 10.2337/db14-0263. [DOI] [PubMed] [Google Scholar]
- Xu Y, Zalzala M, Xu J, Li Y, Yin L, Zhang Y. A metabolic stress-inducible miR-34a-HNF4alpha pathway regulates lipid and lipoprotein metabolism. Nat Commun. 2015;6:7466. doi: 10.1038/ncomms8466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Hu M, Liang X, Ajmo JM, Li X, Bataller R, Odena G, Stevens SM, Jr, You M. Deletion of SIRT1 from hepatocytes in mice disrupts lipin-1 signaling and aggravates alcoholic fatty liver. Gastroenterology. 2014;146(3):801–811. doi: 10.1053/j.gastro.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Hietakangas V, Wee S, Lim SC, Gunaratne J, Cohen SM. ER stress potentiates insulin resistance through PERK-mediated FOXO phosphorylation. Genes Dev. 2013;27(4):441–449. doi: 10.1101/gad.201731.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZF, Fan SH, Zheng YL, Lu J, Wu DM, Shan Q, Hu B. Troxerutin improves hepatic lipid homeostasis by restoring NAD(+)-depletion-mediated dysfunction of lipin 1 signaling in high-fat diet-treated mice. Biochem Pharmacol. 2014;91(1):74–86. doi: 10.1016/j.bcp.2014.07.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 199 kb)