

Abstract
The molecular chaperone Hsp90 is one component of a highly complex and interactive cellular proteostasis network (PN) that participates in protein folding, directs misfolded and damaged proteins for destruction, and participates in regulating cellular transcriptional responses to environmental stress, thus promoting cell and organismal survival. Over the last 20 years, it has become clear that various disease states, including cancer, neurodegeneration, metabolic disorders, and infection by diverse microbes, impact the PN. Among PN components, Hsp90 was among the first to be pharmacologically targeted with small molecules. While the number of Hsp90 inhibitors described in the literature has dramatically increased since the first such small molecule was described in 1994, it has become increasingly apparent that not all of these agents have been sufficiently validated for specificity, mechanism of action, and lack of off-target effects. Given the less than expected activity of Hsp90 inhibitors in cancer-related human clinical trials, a re-evaluation of potentially confounding off-target effects, as well as confidence in target specificity and mechanism of action, is warranted. In this commentary, we provide feasible approaches to achieve these goals and we discuss additional considerations to improve the clinical efficacy of Hsp90 inhibitors in treating cancer and other diseases.
Keywords: Molecular chaperones, Hsp90, Inhibitors, Off-target effects, Secondary effects, Hsf1, Cancer, Clinical trials
Introduction
Maintenance of cellular protein homeostasis (also termed proteostasis) is now recognized to be of major importance for cell and organismal viability. In aging, or in various neurodegenerative diseases, proteostasis is generally compromised as a result of either reduced expression or inactivation of one or more components of the cellular proteostasis network (PN). These components (many of which serve in more than one capacity) include the proteasome, autophagy-related proteins, and molecular chaperones, notably Hsp90. Cancer cells, because of their genetic instability, frequent aneuploidy, and abundance of mutated/misfolded proteins, are highly dependent on the PN, components of which (e.g., Hsp90) are frequently upregulated compared to normal cells. Inhibition of one molecular component is akin to plucking a strand within a spider’s web, as the frequent result is upregulation of and potential compensation by other parts of the network. Thus, in developing and validating pharmacologic inhibitors of individual PN molecules, it is critically important to provide convincing evidence of target specificity and to document off-target effects where possible. This is particularly relevant in the case of inhibitors of Hsp90-family members, because these molecular chaperones play complex and multi-faceted roles in cellular proteostasis (Fig. 1). To further complicate matters, the Hsp90 family includes two cytoplasmic/nuclear isoforms, constitutively expressed Hsp90β and stress-induced Hsp90α, as well as isoforms restricted to the endoplasmic reticulum and mitochondria (Grp94 and Trap1, respectively). Thus, inhibiting “Hsp90” could reflect simultaneous inhibition of all or some of these family members, with potentially distinct cellular consequences. The Hsp90 family shares a highly conserved N-terminal domain containing an ATP-binding pocket that is the site targeted by all Hsp90 inhibitors that have been clinically evaluated in cancer patients at this writing. A second, C-terminal druggable site has also been identified. Although inhibitors targeted to this site have been developed, this C-terminal drug-binding site is less well characterized than is the N-terminal ATP-binding pocket (Fig. 2). However, one C-terminal inhibitor, RTA901 (Reata Pharmaceuticals) is currently in a phase 1 clinical trial (clinicaltrials.gov identifier: NCT02666963) for non-cancer indications. RTA901 is based on the novobiocin analog KU32 and has shown favorable activity in a range of preclinical models of neurodegeneration and neuroprotection, including diabetic neuropathy and neural inflammation (Ma et al. 2014).
Fig. 1.

Hsp90 impacts many components of the cellular PN, including autophagy and the unfolded protein response in the endoplasmic reticulum, as well as cytoplasmic protein quality control and the cytoplasmic stress response
Fig. 2.

General scheme of the domain structure of Hsp90 and of the target domains of the main classes of Hsp90 inhibitors. ADP and ATP illustrate the importance of the N-terminal domain for ATP binding and hydrolysis; M and C, middle and C-terminal domains; MEEVD, very C-terminal pentapeptide (hallmark of cytosolic Hsp90 isoforms) and binding site for most of the co-chaperones containing tetratricopeptide repeats
Although others have questioned the specificity of N-terminal domain inhibitors in comparison to compounds targeted to the C-terminal domain (Wang et al. 2017), one must be very careful to apply the same basic pharmacological principles of potency and specificity to the validation of all putative Hsp90 inhibitors. For example, little information is available concerning either the preference of inhibitors targeted to the C-terminal domain for the four Hsp90 family isoforms or a careful examination of their possible off-target effects. This commentary will discuss feasible experimental approaches for validating the binding specificity of inhibitors that target either of these sites, while also describing techniques to identify and to characterize possible off-target effects manifested by these agents. Finally, we will propose a new paradigm for more optimal use of Hsp90 inhibitors in cancer therapy—one that recognizes Hsp90 is but one node in the cellular PN of both a cancer and its host.
Authentication and validation of ATP-competitive inhibitors targeting the N-terminal domain
A survey of the current literature defines five distinct chemical scaffolds that have given rise to ATP-competitive inhibitors of Hsp90 and its paralogs Grp94 and Trap1 (Shi et al. 2012; Kitson and Moody 2013; Gewirth 2016). Figure 3 provides examples of each of these core scaffolds, which include purine-, benzoquinone ansamycin-, resorcinol-, indoline-, and tropane-based structures. From these core structures, many active analogs have been derived in structure-activity relationship (SAR) analysis campaigns designed to improve pharmacological properties in vivo. The structural diversity of these scaffolds is somewhat extraordinary within the context of ATP-competitive inhibitors targeting a single family of proteins such as Hsp90. Such structural diversity against a single conserved domain is rare. The extensively studied protein kinase family has yet to provide examples of multiple inhibitor classes that are as selective for an individual target as those targeting the Hsp90 family. Importantly, an extensive body of independently evaluated literature has established selectivity parameters and molecular insights on which one can build to define essential criteria for the validation of any putative new ATP-competitive agent as a bona fide inhibitor of the Hsp90 family. These criteria include a solved co-crystal structure, development of active and inactive analogs, affinity reagents, ATP-competition studies, and measurable effects on one or more Hsp90 client proteins (proteins that directly interact with and depend on Hsp90 for stability and function).
Fig. 3.
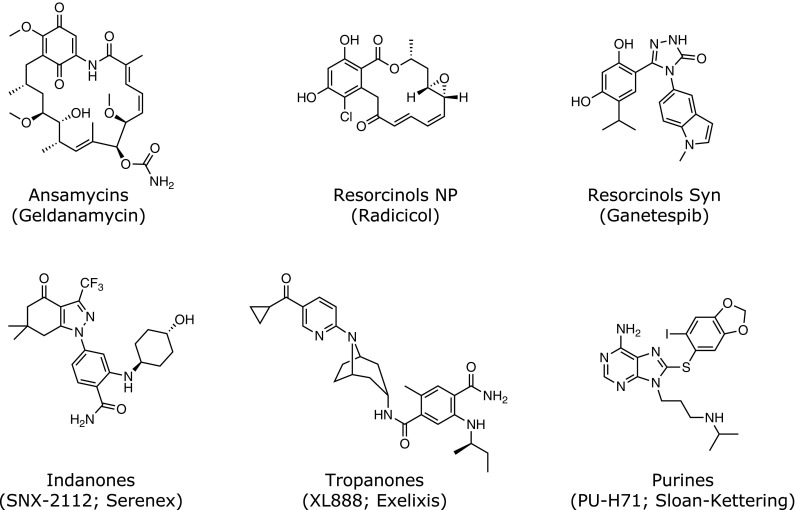
Five core scaffold structures common to selective inhibitors of Hsp90 and its paralogs. Specific examples and in some cases the institutional or industrial developer are indicated in parenthesis. Note that ganetespib is a totally synthetic resorcinol analog
Co-crystal structures
Two co-crystal structures of geldanamycin (GA) bound to Hsp90 appeared simultaneously in 1997 and revealed a drug binding pocket in the N-terminal domain that was highly conserved among Hsp90 family members across species (Prodromou et al. 1997; Stebbins et al. 1997). Based on the work of Toft and co-workers and the Pearl group, this pocket was quickly confirmed as an ATP-binding domain (Grenert et al. 1997; Prodromou et al. 1997). Shortly thereafter, the second natural product Hsp90 inhibitor, radicicol, was found to occupy the same nucleotide-binding pocket (Roe et al. 1999). A similar mode of nucleotide binding was observed in the complete structure of Grp94 by Gewirth and colleagues (Soldano et al. 2003). These structures showed the unique and unexpected orientation of the purine within the active site and they defined the essential contacts made with individual residues in the ATP-binding pocket. The orientation of the nucleotide is distinct from that of other classical ATPases and chaperones such as Heat shock protein 70 (Alderson et al. 2016). Figure 4 provides co-crystal structures for all five scaffolds shown in Fig. 3. Each of these co-crystal structures provides a molecular basis for the specificity of a particular scaffold for Hsp90 family members. With these structures, one can directly test the importance of various interactions by creating mutations of the protein or by chemically modifying the inhibitor. There are multiple examples of such studies in the literature (Chiosis et al. 2003; Wright et al. 2004; He et al. 2006; Immormino et al. 2006; Eccles et al. 2008; Fadden et al. 2010; Duerfeldt et al. 2012; Gewirth 2016). Other methods such as NMR or cryo-electron microscopy can serve as alternative biophysical approaches to interrogate and to validate small molecule—Hsp90 interactions (Verba et al. 2016).
Fig. 4.

Examples of the five core scaffolds of current Hsp90 inhibitors bound in the ATP-binding site of Hsp90. PDB designations of these structures are as follows: Ansamycins (GA) 4XDM; Natural Product Resorcinols NP (Radicicol) 4CE1; Synthetic Resorcinols (Ganetespib, Synta) 3TUH; Indanones (SNX-2112; Serenex) HSP902, 4NH7; Tropanones (XL888; Exelixis) 4AWO; Purines (PU-H71; Memorial Sloan-Kettering) 2FWZ
Analog development
If one reviews the history of all small molecule inhibitors recognized as selective for their targets, from natural products to fully synthetic molecules, invariably there is an accompanying literature of related active and inactive analogs. In this context, the five Hsp90-targeting scaffolds stack up well. From a pharmacologist’s or chemical biologist’s perspective, analogs of an active molecule are the equivalent (if not better) than mutation or knockout of the target itself. Unlike the genetic approach, active and inactive analogs can be tested alongside the original molecule across every relevant cell type and in any animal. Co-crystal structures provide a direct path for the development of analogs because they define the essential amino acid contacts between the inhibitor and binding site. Typically, one desires analogs without large substitutions because these could affect target specificity and bioavailability. For example, addition of a dimethyl group at the core amine of the SNX scaffold (see Fig. 3) reduces the affinity of the molecule for Hsp90 by 3 logs. The molecule no longer competes with ATP nor affects the stability of HER2, a highly dependent client of Hsp90 (Crowe et al. 2017). Hsp90 inhibitors that do not have a history of analog development should be viewed with caution, since it is likely that their mechanisms of action (and potential for promiscuous off-target effects) are poorly defined. As a general rule, second generation fully synthetic Hsp90 inhibitors seem to have fewer off target effects compared to natural products such as GA and radicicol (see below). This is likely due to the fact that second generation inhibitors have been designed expressly to inhibit Hsp90 and are smaller and structurally simpler than the natural products.
Affinity reagents
A standard approach to defining the targets of a molecule is affinity purification. The experimental question is whether, if one attaches the molecule to a resin (e.g., Sepharose) or affinity tag (e.g., biotin), can one recover the expected target or its close paralogs—and nothing else—from a biological milieu. Affinity methods are valid means for defining selectivity of any Hsp90 inhibitor if one applies the following criteria. First, when immobilizing the molecule to a resin or coupling it to another handle such as biotin, the tether must be attached to a solvent-accessible region of the inhibitor based on the co-crystal structure; second, one must demonstrate competitive recovery of Hsp90 using either the free ligand, ATP, or another structurally distinct Hsp90 inhibitor sharing the same binding site. Understanding the point of attachment is vital to prevent steric hindrance by commonly used tethers (such as polyethylene based tethers). The choice of the tether is critical if one wants to minimize non-specific binding to the affinity matrix itself. It is widely known that all proteins tend to stick to all types of surfaces by ionic and non-ionic interactions. Even very stringent washes with high salt or detergents often do not overcome these interactions. Therefore, the choice of both resin and tether are crucial, as are adequate competitive-binding data to demonstrate that Hsp90 or other proteins bound to an affinity matrix are retained by a specific interaction (for an example, see Hughes et al. 2012).
Inhibition of ATP-binding and/or ATPase activity
All five scaffolds illustrated in Fig. 3 are known competitive inhibitors of ATP binding to Hsp90. This characteristic can be used to validate these agents as follows: measuring ATPase activity in the presence of increasing concentrations of inhibitor using purified protein; elution from γ-phosphate linked ATP resin in the presence of increasing inhibitor concentrations; assessing inhibitor impact on thermal stability of Hsp90 protein. Most cell-active Hsp90 inhibitors would be expected to have low nM potency in an ATPase inhibition assay. Purified native Hsp90 has extremely low ATPase activity and requires additional co-factors to stimulate this activity such as the co-chaperone Aha1. To avoid this issue, γ-phosphate linked ATP-Sepharose provides a relatively simple way to capture Hsp90 in native or recombinant form and to measure the affinity and selectivity of an Hsp90 inhibitor (see also Grenert et al. 1997; Fadden et al. 2010). Assessment of thermal stability is increasingly emerging as a preferred method for evaluation of drug binding to target proteins. Nucleotide binding pockets are large and generally, when they are occupied by either nucleotide or a potent inhibitor, the protein becomes more stable to thermal denaturation, shifting its melting temperature (Tm) by 10 °C or greater. For example, the SNX analog HS-10 shifts the Tm of native purified pig mammary Hsp90 and Grp94 by 11 °C from 45 to 56 °C (Crowe et al. 2017). This assay can be performed in a standard thermocycler, permitting many potential small molecule interactors to be assessed in parallel. Concentration-dependent shifts in Tm are directly proportional to the relative affinity of an inhibitor for its target.
An essential test for an Hsp90 inhibitor is to show potent intracellular activity as monitored by effects on established clients that are known to be highly sensitive to Hsp90 inhibition. One of the best characterized and most sensitive Hsp90 clients is HER2 (Trepel et al. 2010), an oncogenic receptor tyrosine kinase that is highly expressed in some breast cancer cell lines. The effects of an Hsp90 inhibitor on levels of this protein can simply be followed by immunoblotting. Typically, cells expressing a known client of interest are incubated with increasing concentrations of an active and, if available, inactive analog. Cells are lysed after 6–24 h (short exposure times are preferred as these are less likely to provide time for indirect or off-target effects) and immunoblotted with specific antibodies to the client protein. This method is adaptable to flow cytometry, especially if the client is expressed on the cell surface. Other highly sensitive Hsp90 kinase clients include mutated ALK, v-Src, and Cdk4. Since N-terminal Hsp90 inhibitors promote rapid ubiquitination of most client proteins that is detectable by western blot even before depletion of the client protein itself (within 15 min of drug exposure in the case of HER2) (Mimnaugh et al. 1996), immunoprecipitation of the client protein followed by western blotting for associated polyubiquitin chains is a valuable alternative or confirmatory approach to validate on-target activity.
Authentication and validation of competitive inhibitors targeting the C-terminal domain
Hsp90 C-terminal inhibitors impair Hsp90 molecular chaperone function by binding at or near its C-terminal dimerization domain and have been used as evidence for a putative second nucleotide-binding pocket within the protein (Marcu et al. 2000a, b; Allan et al. 2006; Matts et al. 2011b). Probably owing to the flexible nature of this binding pocket, a co-crystal structure of Hsp90 with a bound C-terminal inhibitor has yet to be solved. As a result, this criterion cannot be used as validation of Hsp90 C-terminal inhibitor binding. Instead, researchers have relied upon affinity chromatography and proteolytic fingerprinting to evaluate whether a compound binds the Hsp90 C-terminal domain (Marcu et al. 2000a, b; Matts and Manjarrez 2009; Yin et al. 2009; Matts et al. 2011a). As the antibiotic novobiocin is the prototypical C-terminal inhibitor, proposed inhibitors can be assayed for their ability to elute Hsp90 from immobilized novobiocin (Yin et al. 2009; Matts et al. 2011a). In addition, compounds can be similarly investigated for their ability to inhibit binding of the C-terminal fragment to immobilized ATP (Marcu et al. 2000a; Yin et al. 2009). Alternatively, once initial SARs for a new molecule have been defined, an affinity tag can be utilized to immobilize the proposed inhibitor, as has been done for novobiocin and inhibitors targeting the N-terminal domain (see above). Researchers can then confirm binding to a C-terminal fragment of Hsp90 and test whether the protein can be eluted by increasing concentrations of soluble inhibitor (Marcu et al. 2000b; Yin et al. 2009). Finally, a proteolytic footprint of Hsp90 produced upon incubation with the inhibitor should be used for final confirmation to determine whether it protects the same amino acids from trypsinolysis as that observed for novobiocin (Yun et al. 2004). Although, to our knowledge, thermal stability techniques have not yet been applied to study C-terminal inhibitors, these also may prove useful as a complement to proteolytic footprinting.
Generally speaking, once binding to the Hsp90 C-terminus has been established, C-terminal inhibitors can be assessed for cellular activity by (1) evaluating their ability to inhibit Hsp90 immunoprecipitation, (2) examining the degradation of Hsp90-dependent client proteins, (3) preventing C-terminal dimerization of two Hsp90 protomers, and (4) inhibiting Hsp90-dependent biochemical refolding activity. Specifically, C-terminal inhibitors inhibit the immunoprecipitation of Hsp90 by the mouse monoclonal antibody AC88, whose epitope is within the Hsp90 C-terminal domain (Yun et al. 2004). Finally, compounds can be evaluated for their ability to inhibit the Hsp90-dependent refolding of denatured luciferase or for their ability to prevent aggregation of thermally denatured citrate synthase (Buchner et al. 1998; Galam et al. 2007; Matts et al. 2011a; Sadikot et al. 2013). In the case of luciferase, care must be taken to control for possible direct (e.g., chaperone-independent) inhibition of luciferase by the compound in question.
Distinguishing off-target effects from secondary effects of Hsp90 inhibitors
Known off-target effects
There is a tendency to forget that small molecule inhibitors are chemicals. As chemicals, they may have multiple cellular targets, undergo metabolic conversion and generate toxic intermediates. In the case of Hsp90 inhibitors, this is a particularly important consideration, since the molecular chaperone affects many downstream signaling pathways, some of which may be more or less active in distinct cell systems. Clearly, on-target Hsp90 inhibition is by definition, functionally pleiotropic. Thus, for this discussion, the term “off-target effects” will be used for direct effects of Hsp90 inhibitors on unrelated molecular targets and non-specific chemical effects. The potentially unwanted inhibition of other Hsp90 isoforms, such as the mitochondrial homolog Trap1 or the endoplasmic reticulum isoform Grp94, by a particular inhibitor will not be considered an off-target effect for the purposes of this commentary. Indeed, although Hsp90 isoform-selective inhibitors are beginning to emerge (Chan et al. 2012; Duerfeldt et al. 2012; Hughes et al. 2012; Ye et al. 2015; Yim et al. 2016), the commonly used N-domain-directed agents target all four mammalian isoforms with little selectivity (when evaluated using purified proteins; the mitochondrial membrane proton gradient impedes cellular access of many Hsp90 inhibitors to Trap1). Of note, the recently developed inhibitor TAS116 targets only Hsp90α and Hsp90β (Ohkubo et al. 2015). Finally, Paracelsus’ tenet that “all things are poison, and nothing is without poison, the dosage alone makes it so a thing is not a poison" (Paracelsus 1538) also applies to off-target effects of the drugs discussed here.
The extent to which individual Hsp90 inhibitors have been tested for off-target effects varies enormously and is often not apparent from publications. To the best of our knowledge, few if any have been thoroughly tested with modern target profiling methods (Blagg and Workman 2014; see also below). The pioneering Hsp90 inhibitor GA and its derivatives have naturally been most closely scrutinized. Although these agents are no longer in clinical development, they are frequently used as a benchmark for confirming Hsp90 inhibition. GA and its derivatives, as the first small molecule Hsp90 inhibitors to be identified, have clearly been instrumental in demonstrating that Hsp90 is druggable, that its pharmacologic inhibition provides a significant means to unravel chaperone function and to identify its protein clientele, while also demonstrating that Hsp90 inhibition in vivo is not only feasible but is surprisingly well tolerated (Trepel et al. 2010). However, moving forward, we suggest that it is preferable to place a greater emphasis on use of second generation synthetic Hsp90 inhibitors in order to confirm Hsp90 dependence of a particular cellular activity, for the reasons mentioned above. For example, several off-target effects are unique to GA and its derivatives and these depend on their chemical nature as benzoquinones. At high concentrations (> 10 μM), GA induces the production of reactive oxygen species, dependent on the presence of the quinone moiety, whose cytotoxicity can be quenched by scavengers (Dikalov et al. 2002; Clark et al. 2009; Samuni et al. 2010). In vivo, the presence of the quinone moiety is responsible for the dose-limiting hepatotoxicity of the benzoquinone Hsp90 inhibitors (Samuni et al. 2010). The redox activity of the benzoquinone Hsp90 inhibitors also affects glutathione levels, which in turn could influence many cellular processes (McCollum et al. 2006; Lang et al. 2007; Mlejnek and Dolezel 2014). Further, GA and its clinical derivative 17-AAG directly bind to the voltage-dependent anion channel (VDAC) of the outer mitochondrial membrane. VDAC was discovered as a GA-binding protein by a pull-down with GA-conjugated beads. Because these natural products target VDAC at relatively low concentrations (0.1–1 μM), the benzoquinone class of Hsp90 inhibitors lead to mitochondrial membrane depolarization and increased intracellular Ca2+ levels (Xie et al. 2011).
Hsp90 is a member of the GHKL superfamily of ATPases/kinases that utilize a common Bergerat fold to bind ATP. In addition to Hsp90, these proteins also include DNA topoisomerase II, the DNA-mismatch-repair enzyme MutL and histidine kinases (Dutta and Inouye 2000). It is therefore possible that some N-domain ATP-competitive Hsp90 inhibitors could target one or more of these important enzymes, and this should be evaluated on a case by case basis. For example, the resorcinol radicicol, but not the benzoquinone GA, inhibits the archeal DNA topoisomerase type IIB (Gadelle et al. 2005) and binds its ATPase domain in a manner equivalent to its binding to Hsp90 (Corbett and Berger 2006). Ironically, novobiocin, another natural product, was initially examined as a potential Hsp90 inhibitor because it was known to interact with the ATP-binding pocket of the bacterial GHKL protein DNA gyrase B (Maxwell 1993). However, novobiocin proved to bind to the C-terminal region of Hsp90 (Marcu et al. 2000a). Nonetheless, novobiocin is not only a potent inhibitor of bacterial DNA gyrase B, it inhibits mammalian DNA polymerase α and topoisomerases I and II (Burke et al. 1979; Edenberg 1980; Hussy et al. 1986).
Additional off-targets have been reported for radicicol. It inhibits several eukaryotic members of the GHKL family in addition to the archeal DNA topoisomerase IIB. At high concentrations, radicicol also inhibits the yeast Sln1 histidine kinase (Besant et al. 2002). Further, radicicol inhibits mammalian citrate lyase (Ki et al. 2000) as well as mammalian branched-chain α-keto acid dehydrogenase kinase and mammalian pyruvate dehydrogenase kinase (PDHK) (Tuganova et al. 2001; Kato et al. 2007). Indeed, based on this pharmacophore, novel dual inhibitors have been developed that target both Hsp90 and PDHK with remarkable affinities (Meng et al. 2014).
The resorcinolic Hsp90 inhibitor AUY922 (Brough et al. 2008) has received extensive clinical evaluation (although it is no longer in clinical trials). However, like radicicol, AUY922 inhibits PDHK in the low μM range (Meng et al. 2014). While other off-targets have not been reported for this drug, lack of such information in the published record for this and other clinically evaluated Hsp90 inhibitors is a critical concern.
Considering the list of known off-target interaction reported, it seems clear that, at the very least, all members of the GHKL superfamily of proteins should be directly tested for inhibition with any new compound targeting the ATP-binding pocket of Hsp90.
Experimental approaches for the characterization of off-target effects
The most straightforward approach to determine whether a drug exhibits off-target effects is to eliminate its molecular target or to replace the molecular target with a mutant version that is unable to bind the drug. Any remaining drug effects would have to be off-target. An early example of this approach is the deletion of the genes for immunophilins and calcineurin in budding yeast to assess the genome-wide off-target effects of FK506 and cyclosporin A by gene expression profiling (Marton et al. 1998). This was further developed into chemogenomic profiling using yeast deletion libraries, which yields a list of interactions, both on- and off-target, of a given drug with each gene based on a growth assay (Giaever et al. 2004). Indeed, this was done only a few years later for the Hsp90 inhibitor macbecin (McClellan et al. 2007). With the CRISPR/Cas9 system, single target gene/protein tests or genome-wide screens are now also possible in human cells (Shalem et al. 2014; Shalem et al. 2015).
However, this approach is difficult in the case of Hsp90, since its multiple isoforms are encoded by different genes. While the two organelle-restricted isoforms, Trap1 and Grp94, are not essential for cell viability (Randow and Seed 2001; Yoshida et al. 2013), the cytosolic isoforms are (Borkovich et al. 1989; Voss et al. 2000; Zou et al. 2017). Maintaining at least some expression of one cytosolic isoform is essential in eukaryotes (Borkovich et al. 1989; Picard et al. 1990; Picard 2012), and the deletion of one may in some cases be compensated by increased expression of the other (Zou et al. 2017). This complexity is reduced somewhat for isoform-selective drugs, but most of these have yet to undergo the rigorous testing and validation for specificity described above. In the case of the two cytosolic Hsp90 isoforms, the fundamental problem remains. Hence, it has not been possible so far to generate a eukaryotic biological system, be it yeast, a mammalian cell line or a multicellular organism, without any cytosolic Hsp90. Mammalian cell lines may tolerate transient shRNA- or siRNA-mediated knockdowns of Hsp90, but a residual amount must remain to ensure cell viability and might account for persistent (or even enhanced) drug sensitivity. Obviously, if one is interested in inhibitors of cytosolic Hsp90, both Hsp90α and Hsp90β have to be considered, i.e., specifically eliminated or reduced to reveal off-target effects. Swapping the endogenous Hsp90 for an inhibitor-resistant point mutant would be a very elegant approach, but our understanding of the ATP-binding pocket is insufficient to design a mutant that loses inhibitor binding but remains otherwise fully functional. It has been argued that Hsp90 of the nematode Caenorhabditis elegans does not bind GA (David et al. 2003), but it is not clear whether this also holds true for other Hsp90 inhibitors. Humicola fuscoatra, the fungus that produces radicicol, has an amino acid substitution in the ATP-binding pocket that makes its Hsp90 a very poor binder for radicicol without affecting GA binding (Prodromou et al. 2009). Intriguingly, several Hsp90 point mutants that confer resistance to 17-AAG and radicicol have been isolated by selection in yeast and then transferred into human Hsp90 and human cells; however, they do not seem to map to sites of interactions with the inhibitors and their molecular mechanisms remain incompletely understood (Zurawska et al. 2010).
Hence, there is no perfect experiment to identify off-target effects with genetic tools. For human cells, depending on the specific experimental question and isoform selectivity of the drug, comparing the effects of a partial knockdown of both Hsp90α and Hsp90β with the effects of the pharmacological inhibitor may be an acceptable compromise. This type of experimental argument has been used quite often for single readouts such as the impact on one particular Hsp90 client (see https://www.picard.ch/downloads/Hsp90facts). To exclude off-target effects, a broader panel of readouts would have to show the same or very similar results. And that may never happen because the absence of the target (e.g., Hsp90α) or even multiple targets cannot be equated with the presence of the drug-inhibited target. The latter may still have some residual or even altered functions. With the terminology commonly applied to hormones, one would say that a drug can be both a partial antagonist and a partial agonist. Differentiating between off-target and partial agonistic effects of a drug may be experimentally challenging.
There is yet another complication that precludes equating the genetically induced long-term absence of the target (e.g., Hsp90) with the more acute presence of an inhibitor. The literature is replete with evidence indicating that the effects of Hsp90 inhibitors are extensively influenced by post-translational modifications of both Hsp90 and co-chaperones, by the levels of Hsp90 and co-chaperones, and by the cell state-specific nature of the Hsp90 complex (Kamal et al. 2003; Forafonov et al. 2008; Holmes et al. 2008; Mollapour et al. 2010; Mollapour and Neckers 2012; Neckers and Workman 2012; Beebe et al. 2013; Mollapour et al. 2014; Dunn et al. 2015; Zong et al. 2015; Oberoi et al. 2016; Rodina et al. 2016; Woodford et al. 2016a; Woodford et al. 2016b; Zuehlke et al. 2017). This diversity of cellular states cannot be accurately captured by genetic disruption.
Unbiased screens, typically involving gene expression profiling, proteomics or yeast 3-hybrid schemes (Terstappen et al. 2007; Rix and Superti-Furga 2009; Chan et al. 2010; Chidley et al. 2011; Lee and Bogyo 2013), can help in target de-convolution. A particularly exciting recent development exploits label-free proteomics to monitor the impact of a given drug on the thermal stability of all detectable proteins (Savitski et al. 2014; Martinez Molina and Nordlund 2016; Leuenberger et al. 2017). This approach could potentially even identify indirect effects of a drug acting on non-proteinaceous targets, as long as these ultimately affect proteins.
Whatever the approach taken, a remaining issue is that extrapolating from a given experimental test system to other cell types, tissues, or the whole organism may be problematic. Applied to a clinical setting, this is where the molecular concept of “off-target” gives way to the medical concept of “side-effects.” The latter are all unwanted effects of a drug and its metabolites that include both target-related and non-target-related effects. In the case of Hsp90, some of these undesirable effects may include the consequences of inhibiting other Hsp90 isoforms, and the results of inhibiting Hsp90 in normal cells and unrelated tissues.
Activation of Hsf1 by Hsp90 inhibitors: is it on-target or off-target and should it be used as a pharmacodynamic surrogate of Hsp90 inhibition?
The molecular mechanisms by which Hsp90 alters the transcriptional activity of Hsf1 are far more complex than the relatively simplistic titration model that remains commonly accepted today. Two decades of work have revealed the inadequacies of this model and argue for a revised, more systems-level conceptualization of how Hsp90 function alters Hsf1 activation state, both directly and indirectly. Activation of the Hsf1-regulated heat-shock response can be driven by compounds acting directly on Hsp90 or by compounds that alter Hsp90 function indirectly by perturbing protein homeostasis in a variety of ways. Conversely, a variety of allosteric inhibitors and modulators of Hsp90/co-chaperone engagement have now been reported to alter Hsp90 function in more selective ways than compounds which bind the N-terminal ATP-binding pocket of Hsp90 (Garg et al. 2016). These inhibitors are less prone to perturb overall protein homeostasis and many do not activate Hsf1. As a corollary, the observation that a compound does or does not activate Hsf1 provides little or no evidence that it acts as a direct inhibitor of Hsp90, especially since some inhibitors targeted to the C-terminal domain do and some do not activate Hsf1 (Conde et al. 2009; Neef et al. 2010; Eskew et al. 2011). In the case of novobiocin analogs, this characteristic appears to depend on the size of the amide side chain (Ghosh et al. 2016). Lastly, the effects of genetic reduction of Hsp90 levels are not phenocopied by most Hsp90 inhibitors (see above). In particular, N-terminal inhibitors confer a positive gain of function in driving the degradation of most client proteins through recruitment of CHIP (Cyr et al. 2002; Xu et al. 2002). In contrast, Hsp90 levels can be significantly reduced with genetic techniques without depleting clients or activating Hsf1 (Whitesell et al. 2014). Much remains to be learned about the dynamic interplay between the chaperone machinery, exemplified by Hsp90 and the major transcriptional regulator of inducible chaperone expression, Hsf1. What has been learned, however, makes clear that development of new, more realistic mechanistic models to clarify how Hsp90 affects Hsf1 are long overdue.
Lessons learned
Largely to explain the activation of Hsf1 by Hsp90 inhibitors, a model was proposed decades ago that invokes the existence of a repressive association between Hsf1 and an Hsp90-containing multi-chaperone complex under basal conditions (Zou et al. 1998). Upon exposure to proteotoxic stressors, including Hsp90 inhibitors, these chaperone associations are “titered away” by an increased demand for chaperone assistance in combating protein misfolding. As a result, Hsf1 is released to trimerize and undergo the extensive post-translational modifications associated with acquisition of transcriptional transactivating activity (Anckar and Sistonen 2011). While conceptually satisfying, this simple titration model is no longer adequate to accommodate an ever-expanding body of experimental observations. The most prominent of these are summarized below with admittedly author-biased, far from comprehensive reference to the available literature.
Alternate, chaperone-independent modes of Hsf1 activation, operating in response to specific stressors are now well described (Anckar and Sistonen 2011).
Hsp90 is present in great excess of basal requirements making it a relatively poor initial sensor of proteotoxic stress. Under non-stress conditions, its cellular level and chaperoning function can be reduced substantially in many organisms and cell types (both normal and neoplastic) without impairing overall protein homeostasis or activating Hsf1 (Jarosz et al. 2010; Karras et al. 2017).
Hsf1 is not localized to the cytoplasm, but rather the nucleus in the majority of human tumors and cancer cell lines that have been examined (Mendillo et al. 2012).
In Xenopus oocytes, Hsf1 is predominantly localized to the nucleus under basal conditions. Treatment with the classical Hsp90 inhibitor GA under non-stress conditions does not activate Hsf1, but rather impairs activation of a heat-shock reporter in these cells (Ali et al. 1998; Bharadwaj et al. 1999).
Hsp90 association with Hsf1 has been demonstrated primarily by introducing recombinant protein into reticulocyte lysate or by cross-linking in intact cells (Zou et al. 1998). While the association may have functional significance, it is quite weak. Furthermore, evidence for in vitro reconstitution of Hsf1:: Hsp90 interaction is remarkably limited. In contrast, robust association of Hsf1 with Hsp70 is readily detected without resort to recombinant proteins or cross-linkers (Shi et al. 1998; Taipale et al. 2014; Zheng et al. 2016).
In addition to repressing activation-associated Hsf1 oligomerization, a role for Hsp90-containing complexes has been reported for removing Hsf1 trimers from their association with DNA and attenuating Hsf1 transactivating activity (Guo et al. 2001; Conde et al. 2009).
Biochemical evidence indicates that Hsp90β can actually potentiate Hsf1 activation (Hentze et al. 2016).
Unlike most clients that are conformationally stabilized by Hsp90 and depleted by N-terminal Hsp90 inhibitors, Hsf1 is not (Anckar and Sistonen 2011).
Treatment of cells with Hsp90 inhibitors results in modest activation of Hsf1 relative to the extent they compromise Hsp90 function. A number of explanations have been suggested including the destabilization of Hsp90 client proteins (kinases and co-regulators) that are required for robust activation of Hsf1 (Whitesell and Lindquist 2009).
Hsp90 has far reaching effects on gene expression, including that of heat-shock genes only some of which are mediated by Hsf1. Non-Hsf1 dependent effects may be mediated by other sequence-specific transcription factors, chromatin remodeling factors and elements of the basal transcriptional machinery (Calderwood and Neckers 2016).
The transcriptional regulation of most heat shock protein genes is complex, often involving input from not just Hsf1 but other transcription factors as well, such as NRF2, NFκB, AP1, and YY1 in a stress- and cell-type-specific manner (Mendillo et al. 2012). Given this reality, the ability of a compound under investigation to increase the level of one or more heat shock protein levels is not sufficient evidence to conclude that the increase is indeed mediated via Hsf1.
Many thiol-reactive electrophilic compounds have been reported that exert significant oxidative stress in a concentration-dependent manner that can independently alter both Hsp90 and Hsf1 function (Santagata et al. 2012).
Remodeling considerations
The observations described above highlight the complex relationship between Hsp90 function and Hsf1 activation state. The biology is much more complicated than originally conceived. Indeed, Hsf1 has emerged as a highly networked sensor of protein homeostasis that integrates diverse inputs by multiple mechanisms. Some of these may involve direct or indirect interaction with Hsp90 while others may have little to do with Hsp90 or its chaperone function. As an additional layer of complexity, the Hsf1 regulatory network is context dependent with potential for variation across different organisms, cell types and tissues. As a starting point for discussion, the cartoon presented in Fig. 5 lays out the most prominent factors that need to be considered in developing new, more realistic models for the regulation of Hsf1 activity. In the realm of chaperone-targeted drug development efforts, more realistic models are unlikely to diminish the value of the heat-shock response as a biomarker for heat shock-active drugs of known mechanism (even though such an effect may be unwanted in the context of cancer, see below). In the realm of drug discovery, however, the complexity of Hsf1 activation mechanisms precludes any value to use of the heat shock-response in establishing the proximal target of action for putative inhibitors of Hsp90 or other chaperones.
Fig. 5.

Network-based model for the regulation of Hsf1 by Hsp90. Sentinel references for the interactions depicted are indicated in parentheses (1, Guo et al. 2001; 2, Anckar and Sistonen 2011; 3a, Boyault et al. 2007; 3b, Raychaudhuri et al. 2014; 4, Whitesell and Lindquist 2009; 5, Calderwood and Neckers 2016; 6, Fritah et al. 2009)
Should the clinical development of Hsp90 inhibitors for cancer treatment be re-focused?
Design flaws of Hsp90-targeted clinical trials
Eighteen Hsp90 inhibitors have entered clinical development as anti-cancer agents during the last 20 years, but only a few remain under active clinical investigation. There are several possible reasons for failure to obtain the robust preclinical activity observed in mouse xenograft models. Some factors that may have contributed to the inability thus far to bring an Hsp90 inhibitor to regulatory approval relate to the pharmacodynamic markers of target engagement used in most Hsp90 clinical trials. The most common cells analyzed for biomarkers have been peripheral blood mononuclear cells (PBMCs). It is understandable why this tumor surrogate cell population has been favored; in patients with locally advanced or metastatic cancer it is very difficult to obtain biopsies pre- and post-therapy, while obtaining peripheral blood samples pre- and post-therapy is almost always feasible. There are, however, several reasons why PBMCs are not an optimal surrogate tissue for Hsp90 inhibitor clinical trials. First, the pharmacokinetics of Hsp90 inhibitors differs significantly between tumor and normal tissue (Xu et al. 2003; Chiosis and Neckers 2006). In addition, certain tumor cells appear to be more sensitive to N-terminal Hsp90 inhibitors than are normal cells (Kamal et al. 2003; Woodford et al. 2016b). As reported by Kamal et al. (Kamal et al. 2003), the Hsp90 complex in cancer cells is biochemically distinct from non-transformed cells, contributing to a high affinity binding state for certain Hsp90 inhibitors. More recently, Chiosis and colleagues have reported that in approximately 50% of cancers, especially in MYC-driven tumors, Hsp90 and Hsc70 can act as scaffolds for functionally integrated complexes they term the epichaperome, which can confer sensitivity to Hsp90 inhibitors (Moulick et al. 2011; Rodina et al. 2016).
An additional reason why PBMCs are not optimal as a PD surrogate tissue is that many of the most sensitive Hsp90 client proteins are putative tumor drivers that are not expressed in PBMCs, including ALK fusion proteins and HER2. Indeed, the most frequently monitored PD endpoint in PBMCs is Hsp70 induction, which is mediated at the transcriptional level by Hsf1. Unfortunately, and as discussed above, Hsf1 activation is a poor surrogate for on-target in vivo activity of an Hsp90 inhibitor. Further, Hsf1 in tumors is thought to contribute to malignancy (Dai et al. 2007; Mendillo et al. 2012), and its stimulation by high dose Hsp90 inhibitors is likely to contribute to drug resistance. In addition, inhibition of a significant portion of Hsp90 is likely to impact protein homeostasis markedly, engendering upregulation of other components of the PN in the tumor, including Hsf1. Thus, treating to the maximum tolerated dose, a common feature of all Hsp90 inhibitor clinical trials to date, is very likely to be counterproductive with respect to anti-tumor activity. In contrast, preclinical evidence supports administration of a chronic low level, non-heat shock response-inducing concentration of Hsp90 inhibitor combined with other cytotoxic or targeted therapy in order to provide an effective strategy to inhibit the emergence of resistance (Whitesell et al. 2014). This strategy may also take advantage of the recognized property of Hsp90 inhibitors to accumulate in tumors and to persist for a significantly greater time compared to normal tissues (Eiseman et al. 2005; Daozhen et al. 2007).
Nuclear functions of Hsp90
Another factor that may contribute to the less than expected clinical activity of Hsp90 inhibitors is the many roles played by Hsp90 in mediating numerous cellular processes beyond stabilization of oncogenic kinases and the pathways they drive. For example, there is an ever expanding number of critical nuclear events affected by Hsp90 (Trepel et al. 2010), including transcriptional regulation, RNA polymerase II pausing (Sawarkar et al. 2012), mRNA splicing (Lu et al. 2015; Ferraldeschi et al. 2016), and induction of apoptosis (Solier et al. 2012). As recently proposed by Sawarkar and Paro, the depth and breadth of the Hsp90 interactome, which is frequently updated in a database of interactors maintained by Didier Picard at the University of Geneva (https://www.picard.ch/Hsp90Int/index.php), suggests that in addition to direct involvement in chromatin, there exists a cohort of Hsp90 interactors among RNA processing/spicing proteins and DNA replication/damage-response proteins. Thus, Hsp90 may functionally coordinate processes such as the DNA damage response, mRNA splicing, DNA replication, transcription, and nuclear architecture (Sawarkar and Paro 2013). Little to no consideration of possible impact of Hsp90 inhibitors on these processes is evident in the design of many preclinical studies, as well as past and current clinical trials evaluating these agents.
Hsp90 and host immunity
Finally, the vast majority of preclinical models used to evaluate Hsp90 inhibitors rely on mice with compromised immune systems. Thus, the impact of these agents on host immunity and the consequences of that impact on anti-tumor activity have not been rigorously evaluated. In fact, in vitro data suggest that Hsp90 inhibition achieved by conventional dosing is immunosuppressive (Graner 2016). It is surprising therefore that little has been published on the impact of Hsp90 inhibitors on either the tumor microenvironment or on systemic immunity (Proia and Kaufmann 2015). As of this writing, a single trial sponsored by Emory University that is not yet open for recruitment proposes to combine a checkpoint inhibitory antibody with the Hsp90 inhibitor XL-888 (https://clinicaltrials.gov/ct2/show/NCT03095781). An additional Hsp90-related consideration with relevance to anti-cancer immunity involves the concept of immunogenic cell death (ICD). It has become clear that tumor cells can die in a manner that limits the development of anti-tumor immunity by inducing tolerance. In contrast, cell death under circumstances that give rise to increased tumor cell surface expression of Hsp90, Hsp70 and calreticulin, as well as secretion of ATP and HMGB1, can lead to induction of adaptive immunity and immunologic memory (Galluzzi et al. 2017). Many years ago, heat stress was shown to stimulate the later form of cell death (Feng et al. 2001). Since heat stress and Hsp90 inhibition share significant molecular consequences, appropriately dosed Hsp90 inhibitors may also promote ICD, but this remains to be examined. It is critical to understand how to make therapy less toleragenic and to enhance anti-cancer immunity, with the goal of inducing a sustained systemic anti-tumor response. It is likely that knowledge gained of the role of Hsp90 in, and the impact of Hsp90 inhibitors on the ICD process will greatly enhance the utility of targeting Hsp90 as a component of a multipronged therapeutic regimen.
Future clinical prospects
In summary, while several N-terminal Hsp90 inhibitors remain in clinical development, a re-imagining of how to achieve their optimal benefit in cancer patients is urgently needed. To date, emphasis has been placed on impacting client protein levels (with a primary focus on oncogenic kinases), while other critical but more complex activities of Hsp90 that may be associated with the malignant phenotype have not been taken into account in understanding clinical response. Perhaps consideration should be given to employing Hsp90 inhibitors as modulators of central signaling hubs including transcriptional control, epigenetic state and maintenance of DNA integrity. Such a re-orientation of Hsp90 inhibitor usage would likely lead to investigating different types of tumors and ultimately selecting different agents for combination. Further, dosing strategies must be reconsidered to avoid global disruption of cellular proteostasis and activation of compensatory components of the PN, including Hsf1, while providing enough sustained low-level Hsp90 inhibitor exposure to limit development of resistance to other co-administered anti-cancer drugs. Lastly, a better understanding of the dose-dependent impact of Hsp90 inhibitors on host systemic immunity and the tumor microenvironment may facilitate productive combination of Hsp90 inhibitors with immunotherapies.
Conclusions
From the preceding discussion, it is clear that target validation and determination of off-target effects are both critical components of Hsp90 inhibitor development, whether the inhibitor is destined for use as a bioprobe of Hsp90 function or for clinical use in cancer or other diseases. We have assembled a rich toolbox of approaches that can be used for this purpose (see Box 1). Although some may not be applicable in individual cases, we strongly recommend that several orthogonal techniques be used for on-target validation and for identifying promiscuous off-target effects. The later sections of this commentary have focused on re-imagining the way that Hsp90 inhibitors may be used clinically. While most of our focus has been on cancer, it should be clear from our remarks that Hsp90 inhibition may have a useful role in targeting other diseases as well. In each case, one must be cognizant of compensatory responses of the PN to Hsp90 inhibition and of the concept that moderate inhibition in vivo may be preferable to maximally tolerated inhibition. In going forward, we must understand that Hsp90 inhibitors are likely to behave differently at the organismal level in humans compared to immunocompromised mice. Failure to do so is part of a more general failure so far to investigate in depth the impact of Hsp90 inhibitors on non-oncogene-driven processes, whether the host immune responses or the relatively unexplored roles of Hsp90 in the nucleus. As Hsp90 inhibitors are increasingly applied to non-cancer indications, these considerations will take on added importance. Finally, without validation of on-target specificity in cells and animals, and absent careful consideration of possibly confounding contributions of inevitable off-target effects, Hsp90 inhibitors will not achieve their full potential, either as probes of chaperone function in complex systems or in their successful application in the clinic.
Box 1.
Criteria for selecting, authenticating and validating Hsp90 inhibitors
| • Direct biochemical or biophysical binding assays. • Competition against ATP for binding (for inhibitors that bind the same site(s) as ATP). • Specific binding of Hsp90 to immobilized inhibitor (“affinity purification”) and release by the same compound, ATP and/or other Hsp90 inhibitors targeting the same or an allosteric site. • Stringent affinity purification followed by mass spectrometry to determine specificity of binding. • Inhibition of Hsp90 functions in vitro (prevention of aggregation of citrate synthase, refolding of luciferase, etc.). • Co-crystal structure or equivalent experimental structure (still unresolved for inhibitors targeting the C-terminal domain). • Chemical synthesis and evaluation of structure-activity relationships for active and inactive analogs. • Demonstrated effect on validated Hsp90 client proteins in cells and/or organisms. |
Contributor Information
Len Neckers, Email: len@helix.nih.gov.
Didier Picard, Email: didier.picard@unige.ch.
References
- Alderson TR, Kim JH, Markley JL. Dynamical structures of Hsp70 and Hsp70-Hsp40 complexes. Structure. 2016;24(7):1014–1030. doi: 10.1016/j.str.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali A, Bharadwaj S, O'Carroll R, Ovsenek N. HSP90 interacts with and regulates the activity of heat shock factor 1 in Xenopus oocytes. Mol Cell Biol. 1998;18(9):4949–4960. doi: 10.1128/mcb.18.9.4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan RK, Mok D, Ward BK, Ratajczak T. Modulation of chaperone function and cochaperone interaction by novobiocin in the C-terminal domain of Hsp90: evidence that coumarin antibiotics disrupt Hsp90 dimerization. J Biol Chem. 2006;281(11):7161–7171. doi: 10.1074/jbc.M512406200. [DOI] [PubMed] [Google Scholar]
- Anckar J, Sistonen L. Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu Rev Biochem. 2011;80(1):1089–1115. doi: 10.1146/annurev-biochem-060809-095203. [DOI] [PubMed] [Google Scholar]
- Beebe K, Mollapour M, Scroggins B, Prodromou C, Xu W, Tokita M, Taldone T, Pullen L, Zierer BK, Lee MJ, Trepel J, Buchner J, Bolon D, Chiosis G, Neckers L. Posttranslational modification and conformational state of heat shock protein 90 differentially affect binding of chemically diverse small molecule inhibitors. Oncotarget. 2013;4(7):1065–1074. doi: 10.18632/oncotarget.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besant PG, Lasker MV, Bui CD, Turck CW. Inhibition of branched-chain α-keto acid dehydrogenase kinase and Sln1 yeast histidine kinase by the antifungal antibiotic radicicol. Mol Pharmacol. 2002;62(2):289–296. doi: 10.1124/mol.62.2.289. [DOI] [PubMed] [Google Scholar]
- Bharadwaj S, Ali A, Ovsenek N. Multiple components of the HSP90 chaperone complex function in regulation of heat shock factor 1 in vivo. Mol Cell Biol. 1999;19:8033–8041. doi: 10.1128/mcb.19.12.8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagg J, Workman P. Chemical biology approaches to target validation in cancer. Curr Opin Pharmacol. 2014;17:87–100. doi: 10.1016/j.coph.2014.07.007. [DOI] [PubMed] [Google Scholar]
- Borkovich KA, Farrelly FW, Finkelstein DB, Taulien J, Lindquist S. Hsp82 is an essential protein that is required in higher concentrations for growth of cells at higher temperatures. Mol Cell Biol. 1989;9(9):3919–3930. doi: 10.1128/mcb.9.9.3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyault C, Zhang Y, Fritah S, et al. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev. 2007;21:2172–2181. doi: 10.1101/gad.436407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brough PA, Aherne W, Barril X, et al. 4,5-diarylisoxazole Hsp90 chaperone inhibitors: potential therapeutic agents for the treatment of cancer. J Med Chem. 2008;51:196–218. doi: 10.1021/jm701018h. [DOI] [PubMed] [Google Scholar]
- Buchner J, Grallert H, Jakob U. Analysis of chaperone function using citrate synthase as nonnative substrate protein. Methods Enzymol. 1998;290:323–338. doi: 10.1016/s0076-6879(98)90029-5. [DOI] [PubMed] [Google Scholar]
- Burke JF, Duff PM, Pearson CK. Effect of drugs on deoxyribonucleic acid synthesis in isolated mammalian cell nuclei. Comparison with partially purified deoxyribonucleic acid polymerases. Biochem J. 1979;178(3):621–626. doi: 10.1042/bj1780621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderwood SK, Neckers L. Hsp90 in cancer: transcriptional roles in the nucleus. Adv Cancer Res. 2016;129:89–106. doi: 10.1016/bs.acr.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CT, Reeves RE, Geller R, Yaghoubi SS, Hoehne A, Solow-Cordero DE, Chiosis G, Massoud TF, Paulmurugan R, Gambhir SS. Discovery and validation of small-molecule heat-shock protein 90 inhibitors through multimodality molecular imaging in living subjects. Proc Natl Acad Sci U S A. 2012;109(37):E2476–E2485. doi: 10.1073/pnas.1205459109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JN, Nislow C, Emili A. Recent advances and method development for drug target identification. Trends Pharmacol Sci. 2010;31:82–88. doi: 10.1016/j.tips.2009.11.002. [DOI] [PubMed] [Google Scholar]
- Chidley C, Haruki H, Pedersen MG, Muller E, Johnsson K. A yeast-based screen reveals that sulfasalazine inhibits tetrahydrobiopterin biosynthesis. Nat Chem Biol. 2011;7:375–383. doi: 10.1038/nchembio.557. [DOI] [PubMed] [Google Scholar]
- Chiosis G, Lucas B, Huezo H, Solit D, Basso A, Rosen N. Development of purine-scaffold small molecule inhibitors of Hsp90. Curr Cancer Drug Targets. 2003;3(5):371–376. doi: 10.2174/1568009033481778. [DOI] [PubMed] [Google Scholar]
- Chiosis G, Neckers L. Tumor selectivity of Hsp90 inhibitors: the explanation remains elusive. ACS Chem Biol. 2006;1(5):279–284. doi: 10.1021/cb600224w. [DOI] [PubMed] [Google Scholar]
- Clark CB, Rane MJ, El Mehdi D, Miller CJ, Sachleben LR, Jr, Gozal E. Role of oxidative stress in geldanamycin-induced cytotoxicity and disruption of Hsp90 signaling complex. Free Radic Biol Med. 2009;47(10):1440–1449. doi: 10.1016/j.freeradbiomed.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conde R, Belak ZR, Nair M, O'Carroll RF, Ovsenek N. Modulation of Hsf1 activity by novobiocin and geldanamycin. Biochem Cell Biol. 2009;87(6):845–851. doi: 10.1139/o09-049. [DOI] [PubMed] [Google Scholar]
- Corbett KD, Berger JM. Structural basis for topoisomerase VI inhibition by the anti-Hsp90 drug radicicol. Nucleic Acids Res. 2006;34(15):4269–4277. doi: 10.1093/nar/gkl567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe LB, Hughes PF, Alcorta DA, Osada T, Smith AP, Totzke J, Loiselle DR, Lutz ID, Gargesha M, Roy D, Roques J, Darr D, Lyerly HK, Spector NL, Haystead TAJ. A fluorescent Hsp90 probe demonstrates the unique association between extracellular Hsp90 and malignancy in vivo. ACS Chem Biol. 2017;12(4):1047–1055. doi: 10.1021/acschembio.7b00006. [DOI] [PubMed] [Google Scholar]
- Cyr DM, Hohfeld J, Patterson C. Protein quality control: U-box-containing E3 ubiquitin ligases join the fold. Trends Biochem Sci. 2002;27:368–375. doi: 10.1016/s0968-0004(02)02125-4. [DOI] [PubMed] [Google Scholar]
- Dai C, Whitesell L, Rogers AB, Lindquist S. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell. 2007;130(6):1005–1018. doi: 10.1016/j.cell.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daozhen C, Lu L, Min Y, Xinyu J, Ying H. Synthesis of (131)I-labeled-[(131)I]iodo-17-allylamino-17-demethoxy geldanamycin ([(131)I]iodo-17-AAG) and its biodistribution in mice. Cancer Biother Radiopharm. 2007;22(5):607–612. doi: 10.1089/cbr.2006.363. [DOI] [PubMed] [Google Scholar]
- David CL, Smith HE, Raynes DA, Pulcini EJ, Whitesell L. Expression of a unique drug-resistant Hsp90 ortholog by the nematode Caenorhabditis elegans. Cell Stress Chaperones. 2003;8(1):93–104. doi: 10.1379/1466-1268(2003)8<93:eoaudh>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikalov S, Landmesser U, Harrison DG. Geldanamycin leads to superoxide formation by enzymatic and non-enzymatic redox cycling. J Biol Chem. 2002;277:25480–25485. doi: 10.1074/jbc.M203271200. [DOI] [PubMed] [Google Scholar]
- Duerfeldt AS, Peterson LB, Maynard JC, Ng CL, Eletto D, Ostrovsky O, Shinogle HE, Moore DS, Argon Y, Nicchitta CV, Blagg BSJ. Development of a Grp94 inhibitor. J Am Chem Soc. 2012;134(23):9796–9804. doi: 10.1021/ja303477g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn DM, Woodford MR, Truman AW, Jensen SM, Schulman J, Caza T, Remillard TC, Loiselle D, Wolfgeher D, Blagg BSJ, Franco L, Haystead TA, Daturpalli S, Mayer MP, Trepel JB, Morgan RML, Prodromou C, Kron SJ, Panaretou B, Stetler-Stevenson WG, Landas SK, Neckers L, Bratslavsky G, Bourboulia D, Mollapour M. c-Abl mediated tyrosine phosphorylation of Aha1 activates its co-chaperone function in cancer cells. Cell Rep. 2015;12(6):1006–1018. doi: 10.1016/j.celrep.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta R, Inouye M. GHKL, an emergent ATPase/kinase superfamily. Trends Biochem Sci. 2000;25(1):24–28. doi: 10.1016/s0968-0004(99)01503-0. [DOI] [PubMed] [Google Scholar]
- Eccles SA, Massey A, Raynaud FI, Sharp SY, Box G, Valenti M, Patterson L, de Haven Brandon A, Gowan S, Boxall F, Aherne W, Rowlands M, Hayes A, Martins V, Urban F, Boxall K, Prodromou C, Pearl L, James K, Matthews TP, Cheung KM, Kalusa A, Jones K, McDonald E, Barril X, Brough PA, Cansfield JE, Dymock B, Drysdale MJ, Finch H, Howes R, Hubbard RE, Surgenor A, Webb P, Wood M, Wright L, Workman P. NVP-AUY922: a novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer Res. 2008;68(8):2850–2860. doi: 10.1158/0008-5472.CAN-07-5256. [DOI] [PubMed] [Google Scholar]
- Edenberg HJ. Novobiocin inhibition of simian virus 40 DNA replication. Nature. 1980;286(5772):529–531. doi: 10.1038/286529a0. [DOI] [PubMed] [Google Scholar]
- Eiseman JL, Lan J, Lagattuta TF, Hamburger DR, Joseph E, Covey JM, Egorin MJ. Pharmacokinetics and pharmacodynamics of 17-demethoxy 17-[[(2-dimethylamino)ethyl]amino]geldanamycin (17DMAG, NSC 707545) in C.B-17 SCID mice bearing MDA-MB-231 human breast cancer xenografts. Cancer Chemother Pharmacol. 2005;55(1):21–32. doi: 10.1007/s00280-004-0865-3. [DOI] [PubMed] [Google Scholar]
- Eskew JD, Sadikot T, Morales P, et al. Development and characterization of a novel C-terminal inhibitor of Hsp90 in androgen dependent and independent prostate cancer cells. BMC Cancer. 2011;11:468. doi: 10.1186/1471-2407-11-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadden P, Huang KH, Veal JM, Steed PM, Barabasz AF, Foley B, Hu M, Partridge JM, Rice J, Scott A, Dubois LG, Freed TA, Silinski MAR, Barta TE, Hughes PF, Ommen A, Ma W, Smith ED, Spangenberg AW, Eaves J, Hanson GJ, Hinkley L, Jenks M, Lewis M, Otto J, Pronk GJ, Verleysen K, Haystead TA, Hall SE. Application of chemoproteomics to drug discovery: identification of a clinical candidate targeting hsp90. Chem Biol. 2010;17(7):686–694. doi: 10.1016/j.chembiol.2010.04.015. [DOI] [PubMed] [Google Scholar]
- Feng H, Zeng Y, Whitesell L, Katsanis E. Stressed apoptotic tumor cells express heat shock proteins and elicit tumor-specific immunity. Blood. 2001;97(11):3505–3512. doi: 10.1182/blood.v97.11.3505. [DOI] [PubMed] [Google Scholar]
- Ferraldeschi R, Welti J, Powers MV, et al. Second-generation Hsp90 inhibitor onalespib blocks mRNA splicing of androgen receptor variant 7 in prostate cancer cells. Cancer Res. 2016;76:2731–2742. doi: 10.1158/0008-5472.CAN-15-2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forafonov F, Toogun OA, Grad I, Suslova E, Freeman BC, Picard D. p23/Sba1p protects against Hsp90 inhibitors independently of its intrinsic chaperone activity. Mol Cell Biol. 2008;28(10):3446–3456. doi: 10.1128/MCB.02246-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritah S, Col E, Boyault C, et al. Heat-shock factor 1 controls genome-wide acetylation in heat-shocked cells. Mol Biol Cell. 2009;20:4976–4984. doi: 10.1091/mbc.E09-04-0295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadelle D, Bocs C, Graille M, Forterre P. Inhibition of archaeal growth and DNA topoisomerase VI activities by the Hsp90 inhibitor radicicol. Nucleic Acids Res. 2005;33(7):2310–2317. doi: 10.1093/nar/gki526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galam L, Hadden MK, Ma Z, Ye Q-Z, Yun B-G, Blagg BSJ, Matts RL. High-throughput assay for the identification of hsp90 inhibitors based on Hsp90-dependent refolding of firefly luciferase. Bioorg Med Chem. 2007;15(5):1939–1946. doi: 10.1016/j.bmc.2007.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Reply: the complement system is also important in immunogenic cell death. Nat Rev Immunol. 2017;17:143. doi: 10.1038/nri.2016.143. [DOI] [PubMed] [Google Scholar]
- Garg G, Khandelwal A, Blagg BS. Anticancer inhibitors of Hsp90 function: beyond the usual suspects. Adv Cancer Res. 2016;129:51–88. doi: 10.1016/bs.acr.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gewirth DT. Paralog specific Hsp90 inhibitors - a brief history and a bright future. Curr Top Med Chem. 2016;16(25):2779–2791. doi: 10.2174/1568026616666160413141154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Liu Y, Garg G, Anyika M, McPherson NT, Ma J, Dobrowsky RT, Blagg BS. Diverging novobiocin anti-cancer activity from neuroprotective activity through modification of the amide tail. ACS Med Chem Lett. 2016;7(8):813–818. doi: 10.1021/acsmedchemlett.6b00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaever G, Flaherty P, Kumm J, et al. Chemogenomic profiling: identifying the functional interactions of small molecules in yeast. Proc Natl Acad Sci U S A. 2004;101:793–798. doi: 10.1073/pnas.0307490100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graner MW. HSP90 and immune modulation in cancer. Adv Cancer Res. 2016;129:191–224. doi: 10.1016/bs.acr.2015.10.001. [DOI] [PubMed] [Google Scholar]
- Grenert JP, Sullivan WP, Fadden P, Haystead TAJ, Clark J, Mimnaugh E, Krutzsch H, Ochel HJ, Schulte TW, Sausville E, Neckers LM, Toft DO. The amino-terminal domain of heat shock protein 90 (hsp90) that binds geldanamycin is an ATP/ADP switch domain that regulates hsp90 conformation. J Biol Chem. 1997;272(38):23843–23850. doi: 10.1074/jbc.272.38.23843. [DOI] [PubMed] [Google Scholar]
- Guo Y, Guettouche T, Fenna M, Boellmann F, Pratt WB, Toft DO, Smith DF, Voellmy R. Evidence for a mechanism of repression of heat shock factor 1 transcriptional activity by a multichaperone complex. J Biol Chem. 2001;276(49):45791–45799. doi: 10.1074/jbc.M105931200. [DOI] [PubMed] [Google Scholar]
- He H, Zatorska D, Kim J, Aguirre J, Llauger L, She Y, Wu N, Immormino RM, Gewirth DT, Chiosis G. Identification of potent water soluble purine-scaffold inhibitors of the heat shock protein 90. J Med Chem. 2006;49(1):381–390. doi: 10.1021/jm0508078. [DOI] [PubMed] [Google Scholar]
- Hentze N, Le Breton L, Wiesner J, Kempf G, Mayer MP. Molecular mechanism of thermosensory function of human heat shock transcription factor Hsf1. elife. 2016;5:e11576. doi: 10.7554/eLife.11576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes JL, Sharp SY, Hobbs S, Workman P. Silencing of HSP90 cochaperone AHA1 expression decreases client protein activation and increases cellular sensitivity to the HSP90 inhibitor 17-allylamino-17-demethoxygeldanamycin. Cancer Res. 2008;68(4):1188–1197. doi: 10.1158/0008-5472.CAN-07-3268. [DOI] [PubMed] [Google Scholar]
- Hughes PF, Barrott JJ, Carlson DA, Loiselle DR, Speer BL, Bodoor K, Rund LA, Haystead TA. A highly selective Hsp90 affinity chromatography resin with a cleavable linker. Bioorg Med Chem. 2012;20(10):3298–3305. doi: 10.1016/j.bmc.2012.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussy P, Maass G, Tümmler B, Grosse F, Schomburg U. Effect of 4-quinolones and novobiocin on calf thymus DNA polymerase α primase complex, topoisomerases I and II, and growth of mammalian lymphoblasts. Antimicrob Agents Chemother. 1986;29:1073–1078. doi: 10.1128/aac.29.6.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Immormino RM, Kang Y, Chiosis G, Gewirth DT. Structural and quantum chemical studies of 8-aryl-sulfanyl adenine class Hsp90 inhibitors. J Med Chem. 2006;49(16):4953–4960. doi: 10.1021/jm060297x. [DOI] [PubMed] [Google Scholar]
- Jarosz DF, Taipale M, Lindquist S. Protein homeostasis and the phenotypic manifestation of genetic diversity: principles and mechanisms. Annu Rev Genet. 2010;44:189–216. doi: 10.1146/annurev.genet.40.110405.090412. [DOI] [PubMed] [Google Scholar]
- Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, Burrows FJ. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425(6956):407–410. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- Karras GI, Yi S, Sahni N, et al. HSP90 shapes the consequences of human genetic variation. Cell. 2017;168(856–866):e812. doi: 10.1016/j.cell.2017.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Li J, Chuang JL, Chuang DT. Distinct structural mechanisms for inhibition of pyruvate dehydrogenase kinase isoforms by AZD7545, dichloroacetate, and radicicol. Structure. 2007;15(8):992–1004. doi: 10.1016/j.str.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ki SW, Ishigami K, Kitahara T, Kasahara K, Yoshida M, Horinouchi S. Radicicol binds and inhibits mammalian ATP citrate lyase. J Biol Chem. 2000;275(50):39231–39236. doi: 10.1074/jbc.M006192200. [DOI] [PubMed] [Google Scholar]
- Kitson RR, Moody CJ. Learning from nature: advances in geldanamycin- and radicicol-based inhibitors of Hsp90. J Org Chem. 2013;78(11):5117–5141. doi: 10.1021/jo4002849. [DOI] [PubMed] [Google Scholar]
- Lang W, Caldwell GW, Li J, Leo GC, Jones WJ, Masucci JA. Biotransformation of geldanamycin and 17-allylamino-17-demethoxygeldanamycin by human liver microsomes: reductive versus oxidative metabolism and implications. Drug Metab Dispos. 2007;35(1):21–29. doi: 10.1124/dmd.106.009639. [DOI] [PubMed] [Google Scholar]
- Lee J, Bogyo M. Target deconvolution techniques in modern phenotypic profiling. Curr Opin Chem Biol. 2013;17(1):118–126. doi: 10.1016/j.cbpa.2012.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuenberger P, Ganscha S, Kahraman A, Cappelletti V, Boersema PJ, von Mering C, Claassen M, Picotti P. Cell-wide analysis of protein thermal unfolding reveals determinants of thermostability. Science. 2017;355:812. doi: 10.1126/science.aai7825. [DOI] [PubMed] [Google Scholar]
- Lu Y, Xu W, Ji J, Feng D, Sourbier C, Yang Y, Qu J, Zeng Z, Wang C, Chang X, Chen Y, Mishra A, Xu M, Lee MJ, Lee S, Trepel J, Linehan WM, Wang X, Yang Y, Neckers L. Alternative splicing of the cell fate determinant Numb in hepatocellular carcinoma. Hepatology. 2015;62(4):1122–1131. doi: 10.1002/hep.27923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Farmer KL, Pan P, Urban MJ, Zhao H, Blagg BS, Dobrowsky RT. Heat shock protein 70 is necessary to improve mitochondrial bioenergetics and reverse diabetic sensory neuropathy following KU-32 therapy. J Pharmacol Exp Ther. 2014;348(2):281–292. doi: 10.1124/jpet.113.210435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcu MG, Chadli A, Bouhouche I, Catelli M, Neckers LM. The heat shock protein 90 antagonist novobiocin interacts with a previously unrecognized ATP-binding domain in the carboxyl terminus of the chaperone. J Biol Chem. 2000;275(47):37181–37186. doi: 10.1074/jbc.M003701200. [DOI] [PubMed] [Google Scholar]
- Marcu MG, Schulte TW, Neckers L. Novobiocin and related coumarins and depletion of heat shock protein 90-dependent signaling proteins. J Natl Cancer Inst. 2000;92(3):242–248. doi: 10.1093/jnci/92.3.242. [DOI] [PubMed] [Google Scholar]
- Martinez Molina D, Nordlund P. The cellular thermal shift assay: a novel biophysical assay for in situ drug target engagement and mechanistic biomarker studies. Annu Rev Pharmacol Toxicol. 2016;56(1):141–161. doi: 10.1146/annurev-pharmtox-010715-103715. [DOI] [PubMed] [Google Scholar]
- Marton MJ, DeRisi JL, Bennett HA, Iyer VR, Meyer MR, Roberts CJ, Stoughton R, Burchard J, Slade D, Dai H, Bassett DE, Hartwell LH, Brown PO, Friend SH. Drug target validation and identification of secondary drug target effects using DNA microarrays. Nat Med. 1998;4(11):1293–1301. doi: 10.1038/3282. [DOI] [PubMed] [Google Scholar]
- Matts RL, Brandt GEL, Lu Y, Dixit A, Mollapour M, Wang S, Donnelly AC, Neckers L, Verkhivker G, Blagg BSJ. A systematic protocol for the characterization of Hsp90 modulators. Bioorg Med Chem. 2011;19(1):684–692. doi: 10.1016/j.bmc.2010.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matts RL, Dixit A, Peterson LB, et al. Elucidation of the Hsp90 C-terminal inhibitor binding site. ACS Chem Biol. 2011;6:800–807. doi: 10.1021/cb200052x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matts RL, Manjarrez JR. Assays for identification of Hsp90 inhibitors and biochemical methods for discriminating their mechanism of action. Curr Top Med Chem. 2009;9(15):1462–1478. doi: 10.2174/156802609789895692. [DOI] [PubMed] [Google Scholar]
- Maxwell A. The interaction between coumarin drugs and DNA gyrase. Mol Microbiol. 1993;9(4):681–686. doi: 10.1111/j.1365-2958.1993.tb01728.x. [DOI] [PubMed] [Google Scholar]
- McClellan AJ, Xia Y, Deutschbauer AM, Davis RW, Gerstein M, Frydman J. Diverse cellular functions of the Hsp90 molecular chaperone uncovered using systems approaches. Cell. 2007;131(1):121–135. doi: 10.1016/j.cell.2007.07.036. [DOI] [PubMed] [Google Scholar]
- McCollum AK, Teneyck CJ, Sauer BM, Toft DO, Erlichman C. Up-regulation of heat shock protein 27 induces resistance to 17-allylamino-demethoxygeldanamycin through a glutathione-mediated mechanism. Cancer Res. 2006;66(22):10967–10975. doi: 10.1158/0008-5472.CAN-06-1629. [DOI] [PubMed] [Google Scholar]
- Mendillo ML, Santagata S, Koeva M, et al. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell. 2012;150:549–562. doi: 10.1016/j.cell.2012.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng T, Zhang D, Xie Z, Yu T, Wu S, Wyder L, Regenass U, Hilpert K, Huang M, Geng M, Shen J. Discovery and optimization of 4,5-diarylisoxazoles as potent dual inhibitors of pyruvate dehydrogenase kinase and heat shock protein 90. J Med Chem. 2014;57(23):9832–9843. doi: 10.1021/jm5010144. [DOI] [PubMed] [Google Scholar]
- Mimnaugh EG, Chavany C, Neckers L. Polyubiquitination and proteasomal degradation of the p185c-erbB-2 receptor protein-tyrosine kinase induced by geldanamycin. J Biol Chem. 1996;271:22796–22801. doi: 10.1074/jbc.271.37.22796. [DOI] [PubMed] [Google Scholar]
- Mlejnek P, Dolezel P. N-acetylcysteine prevents the geldanamycin cytotoxicity by forming geldanamycin-N-acetylcysteine adduct. Chem Biol Interact. 2014;220:248–254. doi: 10.1016/j.cbi.2014.06.025. [DOI] [PubMed] [Google Scholar]
- Mollapour M, Bourboulia D, Beebe K, et al. Asymmetric Hsp90 N domain SUMOylation recruits Aha1 and ATP-competitive inhibitors. Mol Cell. 2014;53:317–329. doi: 10.1016/j.molcel.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollapour M, Neckers L. Post-translational modifications of Hsp90 and their contributions to chaperone regulation. Biochim Biophys Acta. 2012;1823(3):648–655. doi: 10.1016/j.bbamcr.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollapour M, Tsutsumi S, Donnelly AC, et al. Swe1(Wee1)-dependent tyrosine phosphorylation of Hsp90 regulates distinct facets of chaperone function. Mol Cell. 2010;37:333–343. doi: 10.1016/j.molcel.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulick K, Ahn JH, Zong H, Rodina A, Cerchietti L, Gomes DaGama EM, Caldas-Lopes E, Beebe K, Perna F, Hatzi K, Vu LP, Zhao X, Zatorska D, Taldone T, Smith-Jones P, Alpaugh M, Gross SS, Pillarsetty N, Ku T, Lewis JS, Larson SM, Levine R, Erdjument-Bromage H, Guzman ML, Nimer SD, Melnick A, Neckers L, Chiosis G. Affinity-based proteomics reveal cancer-specific networks coordinated by Hsp90. Nat Chem Biol. 2011;7(11):818–826. doi: 10.1038/nchembio.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neckers L, Workman P. Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res. 2012;18(1):64–76. doi: 10.1158/1078-0432.CCR-11-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neef DW, Turski ML, Thiele DJ. Modulation of heat shock transcription factor 1 as a therapeutic target for small molecule intervention in neurodegenerative disease. PLoS Biol. 2010;8(1):e1000291. doi: 10.1371/journal.pbio.1000291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberoi J, Dunn DM, Woodford MR, Mariotti L, Schulman J, Bourboulia D, Mollapour M, Vaughan CK. Structural and functional basis of protein phosphatase 5 substrate specificity. Proc Natl Acad Sci U S A. 2016;113(32):9009–9014. doi: 10.1073/pnas.1603059113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkubo S, Kodama Y, Muraoka H, Hitotsumachi H, Yoshimura C, Kitade M, Hashimoto A, Ito K, Gomori A, Takahashi K, Shibata Y, Kanoh A, Yonekura K. TAS-116, a highly selective inhibitor of heat shock protein 90α and β, demonstrates potent antitumor activity and minimal ocular toxicity in preclinical models. Mol Cancer Ther. 2015;14(1):14–22. doi: 10.1158/1535-7163.MCT-14-0219. [DOI] [PubMed] [Google Scholar]
- Picard D. Preface to Hsp90. Biochim Biophys Acta. 2012;1823(3):605–606. doi: 10.1016/j.bbamcr.2012.02.004. [DOI] [PubMed] [Google Scholar]
- Picard D, Khursheed B, Garabedian MJ, Fortin MG, Lindquist S, Yamamoto KR. Reduced levels of hsp90 compromise steroid receptor action in vivo. Nature. 1990;348(6297):166–168. doi: 10.1038/348166a0. [DOI] [PubMed] [Google Scholar]
- Prodromou C, Nuttall JM, Millson SH, Roe SM, Sim TS, Tan D, Workman P, Pearl LH, Piper PW. Structural basis of the radicicol resistance displayed by a fungal hsp90. ACS Chem Biol. 2009;4(4):289–297. doi: 10.1021/cb9000316. [DOI] [PubMed] [Google Scholar]
- Prodromou C, Roe SM, O'Brien R, Ladbury JE, Piper PW, Pearl LH. Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell. 1997;90(1):65–75. doi: 10.1016/s0092-8674(00)80314-1. [DOI] [PubMed] [Google Scholar]
- Proia DA, Kaufmann GF. Targeting heat-shock protein 90 (HSP90) as a complementary strategy to immune checkpoint blockade for cancer therapy. Cancer Immunol Res. 2015;3(6):583–589. doi: 10.1158/2326-6066.CIR-15-0057. [DOI] [PubMed] [Google Scholar]
- Randow F, Seed B. Endoplasmic reticulum chaperone gp96 is required for innate immunity but not cell viability. Nat Cell Biol. 2001;3(10):891–896. doi: 10.1038/ncb1001-891. [DOI] [PubMed] [Google Scholar]
- Raychaudhuri S, Loew C, Korner R, Pinkert S, Theis M, Hayer-Hartl M, Buchholz F, Hartl FU. Interplay of acetyltransferase EP300 and the proteasome system in regulating heat shock transcription factor 1. Cell. 2014;156(5):975–985. doi: 10.1016/j.cell.2014.01.055. [DOI] [PubMed] [Google Scholar]
- Rix U, Superti-Furga G. Target profiling of small molecules by chemical proteomics. Nat Chem Biol. 2009;5(9):616–624. doi: 10.1038/nchembio.216. [DOI] [PubMed] [Google Scholar]
- Rodina A, Wang T, Yan P, et al. The epichaperome is an integrated chaperome network that facilitates tumour survival. Nature. 2016;538:397–401. doi: 10.1038/nature19807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe SM, Prodromou C, O'Brien R, Ladbury JE, Piper PW, Pearl LH. Structural basis for inhibition of the hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J Med Chem. 1999;42(2):260–266. doi: 10.1021/jm980403y. [DOI] [PubMed] [Google Scholar]
- Sadikot T, Swink M, Eskew JD, et al. Development of a high-throughput screening cancer cell-based luciferase refolding assay for identifying Hsp90 inhibitors. Assay Drug Dev Technol. 2013;11:478–488. doi: 10.1089/adt.2012.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuni Y, Ishii H, Hyodo F, Samuni U, Krishna MC, Goldstein S, Mitchell JB. Reactive oxygen species mediate hepatotoxicity induced by the Hsp90 inhibitor geldanamycin and its analogs. Free Radic Biol Med. 2010;48:1559–1563. doi: 10.1016/j.freeradbiomed.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santagata S, Xu YM, Wijeratne EM, et al. Using the heat-shock response to discover anticancer compounds that target protein homeostasis. ACS Chem Biol. 2012;7(2):340–349. doi: 10.1021/cb200353m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitski MM, Reinhard FB, Franken H, et al. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science. 2014;346(6205):1255784. doi: 10.1126/science.1255784. [DOI] [PubMed] [Google Scholar]
- Sawarkar R, Paro R. Hsp90@chromatin.nucleus: an emerging hub of a networker. Trends Cell Biol. 2013;23(4):193–201. doi: 10.1016/j.tcb.2012.11.007. [DOI] [PubMed] [Google Scholar]
- Sawarkar R, Sievers C, Paro R. Hsp90 globally targets paused RNA polymerase to regulate gene expression in response to environmental stimuli. Cell. 2012;149:807–818. doi: 10.1016/j.cell.2012.02.061. [DOI] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343(6166):84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Zhang F. High-throughput functional genomics using CRISPR-Cas9. Nat Rev Genet. 2015;16(5):299–311. doi: 10.1038/nrg3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Van de Water R, Hong K, et al. EC144 is a potent inhibitor of the heat shock protein 90. J Med Chem. 2012;55:7786–7795. doi: 10.1021/jm300810x. [DOI] [PubMed] [Google Scholar]
- Shi Y, Mosser DD, Morimoto RI. Molecular chaperones as HSF1-specific transcriptional repressors. Genes Dev. 1998;12(5):654–666. doi: 10.1101/gad.12.5.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldano KL, Jivan A, Nicchitta CV, Gewirth DT. Structure of the N-terminal domain of GRP94. Basis for ligand specificity and regulation. J Biol Chem. 2003;278(48):48330–48338. doi: 10.1074/jbc.M308661200. [DOI] [PubMed] [Google Scholar]
- Solier S, Kohn KW, Scroggins B, Xu W, Trepel J, Neckers L, Pommier Y. Heat shock protein 90α (HSP90α), a substrate and chaperone of DNA-PK necessary for the apoptotic response. Proc Natl Acad Sci U S A. 2012;109(32):12866–12872. doi: 10.1073/pnas.1203617109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stebbins CE, Russo AA, Schneider C, Rosen N, Hartl FU, Pavletich NP. Crystal structure of an Hsp90-geldanamycin complex: targeting of a protein chaperone by an antitumor agent. Cell. 1997;89(2):239–250. doi: 10.1016/s0092-8674(00)80203-2. [DOI] [PubMed] [Google Scholar]
- Taipale M, Tucker G, Peng J, et al. A quantitative chaperone interaction network reveals the architecture of cellular protein homeostasis pathways. Cell. 2014;158:434–448. doi: 10.1016/j.cell.2014.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terstappen GC, Schlupen C, Raggiaschi R, Gaviraghi G. Target deconvolution strategies in drug discovery. Nat Rev Drug Discov. 2007;6(11):891–903. doi: 10.1038/nrd2410. [DOI] [PubMed] [Google Scholar]
- Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer. 2010;10(8):537–549. doi: 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuganova A, Yoder MD, Popov KM. An essential role of Glu-243 and His-239 in the phosphotransfer reaction catalyzed by pyruvate dehydrogenase kinase. J Biol Chem. 2001;276(21):17994–17999. doi: 10.1074/jbc.M009327200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verba KA, Wang RY, Arakawa A, Liu Y, Shirouzu M, Yokoyama S, Agard DA. Atomic structure of Hsp90-Cdc37-Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science. 2016;352(6293):1542–1547. doi: 10.1126/science.aaf5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss AK, Thomas T, Gruss P. Mice lacking HSP90b fail to develop a placental labyrinth. Development. 2000;127:1–11. doi: 10.1242/dev.127.1.1. [DOI] [PubMed] [Google Scholar]
- Wang Y, Koay YC, McAlpine SR. Redefining the phenotype of Heat Shock Protein 90 (Hsp90) inhibitors. Chemistry. 2017;23(9):2010–2013. doi: 10.1002/chem.201604807. [DOI] [PubMed] [Google Scholar]
- Whitesell L, Lindquist S. Inhibiting the transcription factor HSF1 as an anticancer strategy. Expert Opin Ther Targets. 2009;13(4):469–478. doi: 10.1517/14728220902832697. [DOI] [PubMed] [Google Scholar]
- Whitesell L, Santagata S, Mendillo ML, Lin NU, Proia DA, Lindquist S. HSP90 empowers evolution of resistance to hormonal therapy in human breast cancer models. Proc Natl Acad Sci U S A. 2014;111(51):18297–18302. doi: 10.1073/pnas.1421323111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodford MR, Dunn DM, Blanden AR, Capriotti D, Loiselle D, Prodromou C, Panaretou B, Hughes PF, Smith A, Ackerman W, Haystead TA, Loh SN, Bourboulia D, Schmidt LS, Marston Linehan W, Bratslavsky G, Mollapour M. The FNIP co-chaperones decelerate the Hsp90 chaperone cycle and enhance drug binding. Nat Commun. 2016;7:12037. doi: 10.1038/ncomms12037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodford MR, Truman AW, Dunn DM, Jensen SM, Cotran R, Bullard R, Abouelleil M, Beebe K, Wolfgeher D, Wierzbicki S, Post DE, Caza T, Tsutsumi S, Panaretou B, Kron SJ, Trepel JB, Landas S, Prodromou C, Shapiro O, Stetler-Stevenson WG, Bourboulia D, Neckers L, Bratslavsky G, Mollapour M. Mps1 mediated phosphorylation of Hsp90 confers renal cell carcinoma sensitivity and selectivity to Hsp90 inhibitors. Cell Rep. 2016;14(4):872–884. doi: 10.1016/j.celrep.2015.12.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright L, Barril X, Dymock B, et al. Structure-activity relationships in purine-based inhibitor binding to HSP90 isoforms. Chem Biol. 2004;11:775–785. doi: 10.1016/j.chembiol.2004.03.033. [DOI] [PubMed] [Google Scholar]
- Xie Q, Wondergem R, Shen Y, et al. Benzoquinone ansamycin 17AAG binds to mitochondrial voltage-dependent anion channel and inhibits cell invasion. Proc Natl Acad Sci U S A. 2011;108:4105–4110. doi: 10.1073/pnas.1015181108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Eiseman JL, Egorin MJ, D'Argenio DZ. Physiologically-based pharmacokinetics and molecular pharmacodynamics of 17-(allylamino)-17-demethoxygeldanamycin and its active metabolite in tumor-bearing mice. J Pharmacokinet Pharmacodyn. 2003;30(3):185–219. doi: 10.1023/a:1025542026488. [DOI] [PubMed] [Google Scholar]
- Xu W, Marcu M, Yuan X, Mimnaugh E, Patterson C, Neckers L. Chaperone-dependent E3 ubiquitin ligase CHIP mediates a degradative pathway for c-ErbB2/Neu. Proc Natl Acad Sci U S A. 2002;99(20):12847–12852. doi: 10.1073/pnas.202365899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye BX, Deng X, Shao LD, et al. Vibsanin B preferentially targets Hsp90β, inhibits interstitial leukocyte migration, and ameliorates experimental autoimmune encephalomyelitis. J Immunol. 2015;194:4489–4497. doi: 10.4049/jimmunol.1402798. [DOI] [PubMed] [Google Scholar]
- Yim KH, Prince TL, Qu S, Bai F, Jennings PA, Onuchic JN, Theodorakis EA, Neckers L. Gambogic acid identifies an isoform-specific druggable pocket in the middle domain of Hsp90β. Proc Natl Acad Sci U S A. 2016;113(33):E4801–E4809. doi: 10.1073/pnas.1606655113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Z, Henry EC, Gasiewicz TA. (-)-Epigallocatechin-3-gallate is a novel Hsp90 inhibitor. Biochemist. 2009;48(2):336–345. doi: 10.1021/bi801637q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida S, Tsutsumi S, Muhlebach G, et al. Molecular chaperone TRAP1 regulates a metabolic switch between mitochondrial respiration and aerobic glycolysis. Proc Natl Acad Sci U S A. 2013;110:E1604–E1612. doi: 10.1073/pnas.1220659110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun BG, Huang W, Leach N, Hartson SD, Matts RL. Novobiocin induces a distinct conformation of Hsp90 and alters Hsp90-cochaperone-client interactions. Biochemist. 2004;43(25):8217–8229. doi: 10.1021/bi0497998. [DOI] [PubMed] [Google Scholar]
- Zheng X, Krakowiak J, Patel N, Beyzavi A, Ezike J, Khalil AS, Pincus D. Dynamic control of Hsf1 during heat shock by a chaperone switch and phosphorylation. elife. 2016;5:e18638. doi: 10.7554/eLife.18638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong H, Gozman A, Caldas-Lopes E, et al. A hyperactive signalosome in acute myeloid leukemia drives addiction to a tumor-specific Hsp90 species. Cell Rep. 2015;13:2159–2173. doi: 10.1016/j.celrep.2015.10.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J, Guo Y, Guettouche T, Smith DF, Voellmy R. Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell. 1998;94(4):471–480. doi: 10.1016/s0092-8674(00)81588-3. [DOI] [PubMed] [Google Scholar]
- Zou M, Bhatia A, Dong H, et al. Evolutionarily conserved dual lysine motif determines the non-chaperone function of secreted Hsp90α in tumour progression. Oncogene. 2017;36:2160–2171. doi: 10.1038/onc.2016.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuehlke AD, Reidy M, Lin C, et al. An Hsp90 co-chaperone protein in yeast is functionally replaced by site-specific posttranslational modification in humans. Nat Commun. 2017;8:15328. doi: 10.1038/ncomms15328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurawska A, Urbanski J, Matuliene J, Baraniak J, Klejman MP, Filipek S, Matulis D, Bieganowski P. Mutations that increase both Hsp90 ATPase activity in vitro and Hsp90 drug resistance in vivo. Biochim Biophys Acta. 2010;1803(5):575–583. doi: 10.1016/j.bbamcr.2010.03.002. [DOI] [PubMed] [Google Scholar]