

Summary
Lysosome membrane recycling occurs at the end of the autophagic pathway and requires proteins that are mostly encoded by genes mutated in neurodegenerative diseases. However, its implication in neuronal death is still unclear. Here, we show that spatacsin, which is required for lysosome recycling and whose loss of function leads to hereditary spastic paraplegia 11 (SPG11), promotes clearance of gangliosides from lysosomes in mouse and human SPG11 models. We demonstrate that spatacsin acts downstream of clathrin and recruits dynamin to allow lysosome membrane recycling and clearance of gangliosides from lysosomes. Gangliosides contributed to the accumulation of autophagy markers in lysosomes and to neuronal death. In contrast, decreasing ganglioside synthesis prevented neurodegeneration and improved motor phenotype in a SPG11 zebrafish model. Our work reveals how inhibition of lysosome membrane recycling leads to the deleterious accumulation of gangliosides, linking lysosome recycling to neurodegeneration.
Keywords: lipid metabolism, lysosomes, membrane trafficking, neurodegenerative disease, glycosphingolipids, autophagic lysosome recovery, autophagy, induced pluripotent stem cells, organoids, knockout
Graphical Abstract
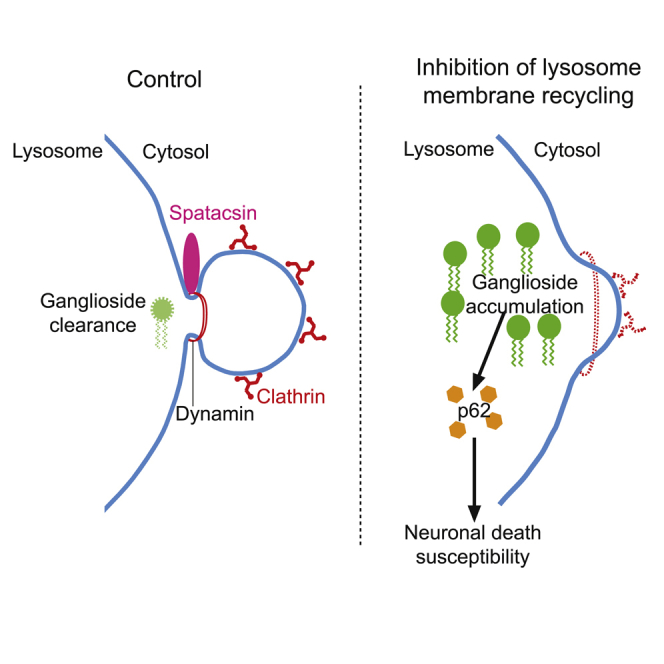
Highlights
-
•
Loss of spatacsin promotes accumulation of simple gangliosides in lysosomes
-
•
Inhibition of lysosome membrane recycling leads to accumulation of gangliosides
-
•
Gangliosides promote accumulation of autophagy markers in lysosomes
-
•
Gangliosides contribute to neurodegeneration when lysosome recycling is compromised
Boutry et al. show that inhibition of lysosome membrane recycling leads to lysosomal accumulation of some glycosphingolipids (simple gangliosides), which contributes to neuronal death.
Introduction
Lysosomes are essential organelles necessary for the degradation of cellular components by the action of hydrolytic enzymes. They are involved in endocytosis and autophagy pathways to promote the degradation of the content of late endosomes and autophagosomes. The degradation step is followed by recycling of structural components into newly forming lysosomes. Under normal conditions, recycling of lysosomal membranes has been proposed to result from the budding of small vesicle carriers from lysosomes (Sridhar et al., 2013). However, under starvation conditions, tubulation occurs in autolysosomes to promote the reformation of new functional lysosomes, a process called autophagic lysosome reformation (ALR) (Yu et al., 2010). The proteins involved in the recycling process include kinesin KIF5B (Du et al., 2016), clathrin, the lipid-modifying enzymes PIP5K1B and PIP5K1A (Rong et al., 2012), spatacsin and spastizin (Chang et al., 2014), members of the AP-2 and AP-4 complex (Rong et al., 2012), dynamin (Schulze et al., 2013), and SPNS1 (Rong et al., 2011). Mutations in the genes encoding the subunits of the AP-4 complex, as well as those encoding spatacsin and spastizin account for severe neurodegenerative disorders, hereditary spastic paraplegias (Tesson et al., 2015). In addition, loss of function of spinster, the Drosophila homolog of SPNS1, leads to neurodegeneration (Nakano et al., 2001). These data highlight the importance of the lysosome recovery pathway for neuron survival. The accumulation of autolysosomes in neurons of a knockout mouse lacking spatacsin was proposed as evidence that defective autophagic lysosome recovery can contribute to neurodegeneration (Varga et al., 2015). However, the mechanism of ALR has so far only been investigated in cultured non-neuronal cells.
Several of the proteins involved in the recycling of lysosome membranes are directly implicated in membrane curvature, such as clathrin and AP-2 and AP-4 complexes (Rong et al., 2012). Recently, we implicated the loss of spatacsin, a protein required for the initiation of ALR (Chang et al., 2014), in lysosomal accumulation of lipids in a knockout mouse model (Branchu et al., 2017), therefore linking lysosome membrane recycling to lipid metabolism. Spatacsin is encoded by the SPG11 gene (Stevanin et al., 2007). Mutations in this gene lead to a spastic gait disorder variably associated with cognitive impairment, peripheral neuropathy, cerebellar ataxia, parkinsonism, and retinal degeneration (Hehr et al., 2007, Stevanin et al., 2007). Knocking out Spg11 in mice is responsible for early cognitive and motor deficits, consistent with the symptoms observed in the majority of patients with mutations in the SPG11 gene (Branchu et al., 2017). The accumulation of lipids that was observed in the brain of Spg11 knockout mice was also detected in post-mortem brain samples of a SPG11 patient (Branchu et al., 2017). However, the mechanisms underlying lipid accumulation and neurodegeneration are unknown.
Here, we show that loss of spatacsin leads to the accumulation of simple gangliosides in lysosomes. The accumulation of gangliosides is a consequence of impaired recycling of lysosomes caused by the absence of functional spatacsin. Decreasing ganglioside levels prevented the accumulation of autophagy markers in lysosomes in cultured neurons, improved neuron survival in vitro, and prevented motor phenotype caused by inactivation of zspg11 in a zebrafish SPG11 model. Our work therefore highlights the importance of lysosome membrane recycling to prevent the detrimental role of gangliosides in neuronal death.
Results
Loss of Spatacsin Promotes the Progressive Accumulation of Gangliosides in Lysosomes in Neurons
Loss of spatacsin in mice leads to lysosomal accumulation of lipids in neurons (Branchu et al., 2017). We investigated the nature of the lipids that accumulate in the cerebral cortex of 8-month-old Spg11−/− mice by performing a lipidomic analysis. The levels of free fatty acids, ceramides, and lysophospholipids were significantly lower in the cortex of Spg11−/− mice than control mice. Only GM2 ganglioside levels were significantly higher in the cortex of Spg11−/− mice than in control mice (Table S1). Immunostaining with a specific antibody showed that GM2 colocalized with lysosomes in Spg11−/− mice, from the age of 6 weeks, whereas it was seen as rare small punctae in neurons of control mice (Figure 1A). The proportion of neurons with GM2 staining in lysosomes was higher in Spg11−/− than in Spg11+/+ mice at the age of 6 weeks, and by the age of 8 months, most neurons in motor cortex of Spg11−/− mice showed GM2 accumulation (Figure 1B). Quantification of the fluorescence intensity showed that GM2 levels were higher in Spg11−/− than Spg11+/+ cortical neurons at all ages (Figure 1C). GM2 accumulated in large lysosomes in knockout mice as indicated by the colocalization of GM2 and Lamp1 staining (Figure 1A). These data showed that the loss of spatacsin function leads to early and progressive accumulation of the GM2 ganglioside in the lysosomes of neurons in the cerebral cortex. Activity of hexosaminidase A, an enzyme involved in lysosomal catabolism of GM2, was not decreased in cortex extracts obtained from Spg11−/− mice compared to controls, suggesting that accumulation of GM2 was not due to an impaired activity of this enzyme (Figure S1A).
Figure 1.

Spatacsin Loss Promotes Lysosomal Accumulation of Gangliosides in Neurons of the Cortex
(A and B) GM2 (green) and Lamp1 (magenta) immunostaining of Spg11+/+ and Spg11−/− neurons of the layer V of the motor cortex from 6-week-old and 8-month-old mice observed by confocal microscopy with a 60× (A) or 20× (B) objective. Images showing the accumulation of GM2-positive staining in lysosomes surrounded by Lamp1 staining in 6-week-old and 8-month-old Spg11−/− animals. Insets show the view along the z axis (A). Scale bars: 10 μm (A) and 50 μm (B).
(C–F) Quantification of the mean of the GM2 (C), GM3 (D), GD2 (E), and GD3 (F) immunostaining intensity per neuron. Quantification was performed in neurons of the layer V of motor cortex that were detected by their large soma. The graphs show the mean ± SEM values. N = 10–15 neurons quantified per cortex slices in five cortex slices of five independent mice. Differences between Spg11+/+ and Spg11−/− were analyzed by a Kruskal-Wallis test at each time point; ∗ p ≤ 0.05, ∗∗∗p ≤ 0.0001.
See also Table S1 and Figures S1 and S2.
Our lipidomic analysis was performed on the whole cortex. It is thus possible that other lipids may accumulate in lysosomes despite the absence of a global change in their levels. Therefore, we purified fractions enriched in lysosomes from the brains of Spg11+/+ and Spg11−/− mice (Figures S1B and S1C), and extracted lipids using the Folch procedure (Folch et al., 1957). Lipidomic analysis was performed on the Folch upper phase, which contains gangliosides. The levels of gangliosides GM2, GM3, GD2, and GD3 were markedly higher in lysosomal fractions obtained from Spg11−/− mouse brains than those of control brains (Table 1). We confirmed the enrichment of these gangliosides in lysosomes by immunostaining with specific antibodies (Figures S2A–S2C). Quantification of fluorescence intensity showed that the levels of the four lipid species increased with age and were higher in Spg11−/− mouse brains than control brains (Figures 1C–1F). The gangliosides GM3, GD2, and GD3 were colocalized with lysosomes, as GM2 was, consistent with their accumulation in lysosomes. The levels of complex gangliosides (GM1, GD1, and GT1) were slightly, but not significantly higher in the lysosome-enriched fractions (Table 1). Accordingly, there was no difference in the localization of GM1 between Spg11+/+ and Spg11−/− mouse brain as assessed by immunostaining (Figures S2D and S2E).
Table 1.
Relative Amounts of Various Classes of Gangliosides in Lysosome-Enriched Fractions Obtained from the Brains of 8-Month-Old Spg11+/+ and Spg11−/− Mice
| Spg11+/+ | Spg11−/− | Fold Change | |
|---|---|---|---|
| GM3 | N = 8 | N = 7 | – |
| GM3 (d18:1/18:1) | 14.2 ± 3.1 | 42.3 ± 5.3a | 2.98 |
| GM3 (d18:1/18:0) | 518.2 ± 89.5 | 1,316.6 ± 194.1a | 2.54 |
| GM3 (d18:1/20:0) | 51.1 ± 8.7 | 170.3 ± 24.5a | 3.34 |
| GM2 | |||
| GM2 (d18:1/18:0) | 286.5 ± 46.3 | 957.6 ± 119.5a | 3.34 |
| GM2 (d18:1/20:0) | 68.9 ± 12.3 | 296.9 ± 40.9a | 4.31 |
| GD3 | |||
| GD3 (d18:1/18:0) | 155.8 ± 23.9 | 321.0 ± 45.2a | 2.06 |
| GD3 (d18:1/18:0) | 139.8 ± 26.7 | 307.7 ± 41.0a | 2.20 |
| GD3 (d18:1/20:0) | 39.9 ± 8.5 | 87.1 ± 12.3a | 2.18 |
| GD2 | |||
| GD2 (d18:1/18:0) | 53.1 ± 8.4 | 145.3 ± 22.7a | 2.73 |
| GD2 (d18:1/20:0) | 54.7 ± 10.9 | 159.9 ± 24.2a | 2.93 |
| GM1 | |||
| GM1 (d18:1/18:1) | 52.8 ± 11.7 | 84.9 ± 10.7 | 1.61 |
| GM1 (d18:1/18:0) | 478.8 ± 87.2 | 778.7 ± 91.5 | 1.63 |
| GM1 (d18:1/20:0) | 198.6 ± 40.5 | 322.3 ± 42.7 | 1.62 |
| GD1 | |||
| GD1 (d18:1/18:1) | 280.2 ± 65.6 | 338.4 ± 40.1 | 1.21 |
| GD1 (d18:1/18:0) | 4,025.6 ± 1221.1 | 4,648.3 ± 615.8 | 1.15 |
| OAc-GD1 (d18:1/18:0) | 174.3 ± 39.3 | 222.2 ± 19.0 | 1.27 |
| GD1 (d18:1/20:0) | 1,653.3 ± 404.0 | 1,781.7 ± 225.9 | 1.08 |
| GT1 | |||
| GT1 (d18:1/18:0) | 493.4 ± 174.0 | 535.5 ± 74.3 | 1.09 |
| OAc-GT1 (d18:1/18:0) | 192.9 ± 60.9 | 243.4 ± 34.1 | 1.26 |
| GT1 (d18:1/20:0) | 220.6 ± 74.7 | 230.1 ± 32.5 | 1.04 |
| OAc-GT1 (d18:1/20:0) | 140.8 ± 37.8 | 163.7 ± 23.7 | 1.16 |
Arbitrary units, normalized to lysosomal protein concentration.
p < 0.05, t test with Benjamini-Hochberg procedure to correct for multiple testing.
Loss of Spatacsin Promotes the Early Accumulation of Gangliosides in Lysosomes in Human Neurons
We tested whether the accumulation of gangliosides is also relevant for human pathology using induced pluripotent stem cells (iPSCs) derived from fibroblasts of two independent SPG11 patients and two sex- and age-matched controls. iPSCs of SPG11 patients and healthy subjects that were validated with markers of pluripotency (Figure S3A) were differentiated into brain organoids with predominant cortical identity using a free-floating tridimensional culture method (Paşca et al., 2015). After 90 days of differentiation, the organoids were organized in layers of radial glial cells labeled by Pax6 and Nestin, and peripheral layers of neurons that expressed βIII-tubulin and NeuN (Figure 2A; data not shown). We examined by immunostaining whether gangliosides accumulated in lysosomes in the peripheral layer of neurons (Figures 2B–2F). GM2, GM3, and GD3 showed a punctate immunostaining pattern in the neurons of the peripheral layer of control and SPG11 brain organoids, and the staining largely colocalized with lysosomes in the neurons of the SPG11 brain organoids (Figures 2B and S3B–S3D). Quantification of fluorescence intensity showed GM2 and GM3 levels to be higher in SPG11 cortical organoids than control organoids (Figures 2C–2F). Furthermore, the variance of the fluorescence intensity of GM2, GM3, and GD3 staining was also higher in organoids derived from SPG11 patients than those derived from healthy controls, consistent with their accumulation in lysosomes. There was no difference in GD2 staining between organoids derived from SPG11 patients and healthy controls, in agreement with the lack of a difference in GD2 staining in the cortices of Spg11+/+ and Spg11−/− mice at early stages (Figure 1E). Overall, these data show that simple gangliosides accumulated in lysosomes of the human neurons, starting at early stages of development represented by the cortical organoid model (Paşca et al., 2015). This prompted us to investigate how spatacsin regulates the levels of gangliosides in lysosomes.
Figure 2.

Spatacsin Loss Promotes Lysosomal Accumulation of Gangliosides in Neurons Derived from SPG11 Patients
(A) Immunostaining of brain organoids differentiated for 90 days in vitro with antibodies against the progenitor marker Pax6 and the neuron-specific marker βIII-tubulin. Note that neuronal cells are concentrated at the periphery of the organoids. Scale bar: 50 μm.
(B) GM2 (green) and Lamp1 (magenta) immunostaining in the neuronal layer of organoids derived from healthy subjects or SPG11 patients. βIII-Tubulin (yellow) shows the neuronal identity of the cells that were analyzed. Confocal microscopy images showing the accumulation of GM2-positive staining in lysosomes labeled by Lamp1 staining of organoids derived from SPG11 patients (arrowheads). Scale bar: 10 μm.
(C–F) Quantification of the mean of the GM2 (C), GM3 (D), GD2 (E), and GD3 (F) immunostaining intensity per neuron. The graph shows mean ± SEM values. N = 10–15 neurons quantified per slices in five slices obtained from three independent organoids. One-way ANOVA; ∗∗∗p ≤ 0.001.
See also Figure S3.
Spatacsin Is Recruited to Lysosomes Containing Gangliosides in Primary Cultures of Cortical Neurons
We analyzed whether gangliosides accumulated in lysosomes of primary cultures of cortical neurons derived from Spg11−/− mouse embryos. By immunostaining, GM2, GM3, GD2, and GD3 significantly accumulated in lysosomes in cultured neurons derived from Spg11−/− embryos (Figures 3A–3E and S4A–S4C). Primary cultures of cortical neurons are thus a good model to investigate the mechanisms leading to ganglioside accumulation in lysosomes. GM2 was the ganglioside that accumulated the most in lysosomes, and lipidomic analysis showed higher levels of GM2 in Spg11−/− than in Spg11+/+ neurons (Figure S4D). We therefore used it as a marker for ganglioside accumulation in subsequent experiments.
Figure 3.

Spatacsin Loss Induces Lysosomal Accumulation of Gangliosides in Primary Cultures of Mouse Cortical Neurons
(A) GM2 (green) and Lamp1 (magenta) immunostaining of Spg11+/+ and Spg11−/− neurons cultured for 6 days in vitro. Confocal microscopy images showing the accumulation of GM2-positive staining of lysosomes labeled by Lamp1 staining. Scale bar: 10 μm.
(B–E) Quantification of the proportion of GM2 (B), GM3 (C), GD2 (D), and GD3 (E) staining that is localized in lysosomes. The graphs show the mean ± SEM values. N = 58–71 neurons quantified in three independent neuron preparations. t test; ∗∗p ≤ 0.01 and ∗∗∗p ≤ 0.001.
(F) Immunostaining of GM2 ganglioside (green), Lamp1 (cyan), and Spatacsin-V5 (magenta) in wild-type neurons co-transfected with vectors allowing expression of Spatacsin-V5 and either control miRNA or miRNA downregulating Neu1. Spatacsin is enriched around GM2-positive vesicles (arrowheads). Scale bar: 10 μm.
(G) Quantification of the number of lysosomes detected with Lamp1 immunostaining and with a diameter larger than 1 μm in Spg11+/+ and Spg11−/− neurons cultured for 6 days in vitro and treated or not with miglustat (100 μM). The graphs show the mean ± SEM values. N = 62–64 neurons quantified in four independent neuron preparations (control); N = 24–34 neurons quantified in three independent neuron preparations (miglustat). One-way ANOVA, followed by Holm-Sidak post hoc test; ∗∗∗p ≤ 0.001.
See also Figure S4.
We investigated the role of spatacsin in ganglioside metabolism by transfecting neurons with a V5-tagged spatacsin. In control conditions, the protein had a diffuse localization throughout the cytoplasm. However, it was partially colocalized with the few GM2-positive lysosomes in these cells (Figure 3F). To investigate the link between gangliosides and spatacsin, we enforced accumulation of gangliosides in lysosomes by downregulating the expression of neuraminidase 1 (Neu1), an enzyme involved in the lysosomal degradation of gangliosides (Bonten et al., 1996). We used two independent microRNA (miRNA) sequences that efficiently downregulated Neu1 expression (Figure S4E). Downregulation of Neu1 promoted significant accumulation of GM2 in lysosomes in transfected Spg11+/+ and Spg11−/− neurons (Figures S4F and S4G), suggesting that neuraminidase activity and lysosome membrane recycling are independent pathways to clear gangliosides. In wild-type neurons that were transfected with both a miRNA targeting Neu1 and expressing V5-tagged spatacsin, the latter was strongly enriched in GM2-positive lysosomes (Figure 3F), suggesting that spatacsin is recruited to lysosomes containing gangliosides.
Lysosome Membrane Recycling Promotes Ganglioside Clearance from Lysosomes
Loss of spatacsin in fibroblasts is responsible for the presence of large lysosomes (Renvoisé et al., 2014). This phenotype is the consequence of impaired lysosome recycling analyzed in cell lines by monitoring the formation of tubules emanating from Lamp1-positive vesicles by live imaging (Chang et al., 2014). In cultured neurons, we did not detect tubules emanating from lysosomes by live imaging (Figure S5A), even with treatment such as dynasore, a dynamin inhibitor that enhances the number of lysosomal tubules in fibroblasts (Chang et al., 2014). However, we observed that the average number of lysosomes immunostained with Lamp1 and with an apparent diameter larger than 1 μm was higher in Spg11−/− neurons than in control neurons (Figure 3G). We evaluated the role of gangliosides in the formation of large lysosomes using miglustat, which inhibits glucosylceramide synthase, an early step in glycosphingolipid synthesis, and is known to decrease GM2 levels in a model of Sandhoff disease (Jeyakumar et al., 1999). In primary cultures of cortical neurons, miglustat significantly decreased the levels of GM2 in a dose-dependent manner (Figures S5B and S5C), but it did not change the number of large lysosomes (Figure 3G). To confirm these data, we used two independent miRNAs that target GM3 synthase, the enzyme producing the first ganglioside in the biosynthetic pathway, and from which all other sialylated gangliosides are generated (Xu et al., 2010). Expression of these miRNAs significantly decreased the expression of GM3 synthase mRNA and GM2 levels (Figures S5D–S5F). The number of large lysosomes was unchanged when GM3 synthase was downregulated in Spg11+/+ and Spg11−/− neurons (Figure S5G). This indicates that accumulation of large lysosomes does not depend on de novo synthesis of gangliosides. We thus hypothesized that the accumulation of GM2 in lysosomes occurs as a consequence of inhibition of lysosome membrane recycling in Spg11−/− cells.
To test this hypothesis, we analyzed the role of clathrin, one of the proteins required for recycling of lysosome membrane (Rong et al., 2012). Downregulation of clathrin levels using two independent miRNAs (Figures S5H and S5I) led to higher number of large lysosomes per neurons (Figure 4A). Inhibition of lysosome membrane recycling following clathrin downregulation was associated with a higher amount of GM2 ganglioside in lysosomes (Figure 4B), suggesting that inhibition of lysosome membrane recycling prevented or slowed down clearance of gangliosides. In Spg11−/− neurons, clathrin colocalized with lysosomes containing high amounts of the GM2 ganglioside (Figure 4C). The proportion of cellular clathrin colocalized with the GM2 was higher in Spg11−/− neurons than control neurons (Figure 4D). Accordingly, clathrin staining was high for large autofluorescent lysosomes that contain high amounts of GM2 (Figure 1A) in the brains of Spg11−/− mice (Figure 4E). This suggests that clearance of gangliosides was blocked downstream of clathrin recruitment in the absence of spatacsin.
Figure 4.

Spatacsin Promotes Clearance of Gangliosides from Lysosomes
(A) Quantification of the number of lysosomes with a diameter larger than 1 μm in Spg11+/+ and Spg11−/− neurons expressing control miRNA or miRNA downregulating clathrin heavy chain (CHC). The graphs show the mean ± SEM values. N = 35–50 neurons quantified in two independent neuron preparations. One-way ANOVA, followed by Holm-Sidak post hoc test; ∗p = 0.03 and ∗∗∗p ≤ 0.001.
(B) Quantification of the proportion of GM2 staining that is localized in lysosomes. The graphs show the mean ± SEM values. N = 36–49 neurons quantified in two independent neuron preparations. One-way ANOVA, followed by Holm-Sidak post hoc test; ∗∗p = 0.0017.
(C) Immunostaining of Spg11−/− neurons with Lamp1 (cyan), GM2 (yellow), and clathrin (magenta) antibodies. Arrowheads point to lysosomes positive for the GM2 marker that also show recruitment of clathrin. Scale bar: 5 μm.
(D) Quantification of the proportion of clathrin colocalized with GM2-positive vesicles. The graphs show the mean ± SEM values. N = 88 (Spg11+/+) and 102 (Spg11−/−) neurons quantified in three independent neuron preparations. t test; ∗∗p = 0.0023.
(E) Confocal microscopy images of autofluorescence (green), Lamp1 (magenta), and clathrin (cyan) immunostaining of Spg11+/+ and Spg11−/− cortical motor neurons from 8-month-old animals showing the accumulation of clathrin surrounding autofluorescent lysosomes. Scale bars: 10 μm.
(F) Western blot showing the interaction of GFP-spatacsin (domain 1943–2443) with dynamin in HeLa cells expressing GFP or GFP-spatacsin 1943–2443. Western blot signals were detected by chemiluminescence.
(G) Left: western blots showing the relative amount of Lamp1, p62, clathrin heavy chain (CHC), and dynamin in whole-brain lysates and lysosome-enriched fractions obtained from Spg11+/+ and Spg11−/− mouse brains. Western blot signals were detected using an infrared imaging system. Right: relative quantification of the band intensities. The graphs show the mean ± SEM. Preparations from N = 3 independent animals. One-tailed Mann-Whitney U test; ∗p = 0.05.
(H) Quantification of the proportion of GM2 staining that is localized in lysosomes in neurons treated with 40 μM dynasore. The graphs show the mean ± SEM values. N = 73–124 neurons quantified in three independent neuron preparations. One-way ANOVA, followed by Holm-Sidak post hoc test; ∗p = 0.02 and ∗∗p = 0.01.
(I) Quantification of the number of lysosomes with a diameter larger than 1 μm in neurons treated with 40 μM dynasore. The graphs show the mean ± SEM values. N = 49–54 neurons quantified in two independent neuron preparations. One-way ANOVA, followed by Holm-Sidak post hoc test; ∗∗p = 0.001 and ∗∗∗p ≤ 0.001.
(J) Immunostaining of GM2 ganglioside (yellow) and Spatacsin-V5 (magenta) in neurons transfected with vector allowing expression of Spatacsin-V5 and treated with either vehicle or dynasore (40 μM) for 2 hr. Spatacsin is enriched around GM2-positive vesicles (arrowheads). Scale bar: 10 μm.
See also Figure S5.
In a two-hybrid screen, we found that dynamin-1, a homolog of dynamin-2 that is important for termination of the lysosome recycling (Schulze et al., 2013), is a putative interactor of the C-terminal domain of spatacsin. We validated this interaction by co-immunoprecipitation in HeLa cells (Figure 4F). We analyzed whether the absence of spatacsin had an impact on dynamin recruitment to lysosomes. Western blot performed on the lysosome-enriched fractions that were used for lipidomic analysis (Table 1) showed that dynamin was markedly less abundant in lysosome fractions obtained from Spg11−/− than Spg11+/+ mouse brains (Figure 4G), while no difference was observed in whole-brain extracts. Immunostaining on cultured neurons showed a decreased amount of dynamin colocalized with Lamp1-positive lysosomes in Spg11−/− than Spg11+/+ neurons (Figure S5J). These results, together with the enrichment of spatacsin on lysosomes containing GM2 (Figure 3F), led us to hypothesize that spatacsin participates in recruitment of dynamin to lysosomes to clear gangliosides. To test this hypothesis, we inhibited dynamins using dynasore treatment, which promoted higher levels of GM2 in lysosomes (Figure 4H). Furthermore, dynasore treatment in control neurons increased the number of large lysosomes (Figure 4I), consistent with the role of dynamin in late step of lysosome membrane recycling. Dynasore also led to the enrichment of V5-spatacsin in lysosomes containing GM2 (Figure 4J), suggesting that spatacsin is implicated upstream of dynamin. Altogether, these data suggest that the machinery implicated in lysosome membrane recycling in fibroblasts is used by neurons to clear gangliosides from lysosomes.
High Levels of Gangliosides in Lysosomes Contribute to the Accumulation of Autophagy Markers in Lysosomes
We then investigated the consequences of ganglioside accumulation on lysosome function. Catalytic activity monitored by the Magic red cathepsin B substrate was not altered in Spg11−/− neurons (Figure S6A), suggesting that it was not altered by accumulation of gangliosides. Loss of spatacsin was shown to result in accumulation of autolysosomes (Chang et al., 2014, Varga et al., 2015). Accordingly, even if the global levels of the autophagy marker p62 were similar in total brain extracts of 8-month-old Spg11−/− and Spg11+/+ mice, p62 was more abundant in lysosome-enriched fractions obtained from Spg11−/− than Spg11+/+ mouse brains (Figure 4G). Similarly, in primary cultures of neurons, the proportion of lysosomes that were positive for the autophagic marker p62, was higher in Spg11−/− neurons than in control neurons, and this proportion increased over time in cultured Spg11−/− neurons (Figure 5A). We then investigated whether GM2 contributed to increase the proportion of lysosomes containing p62 marker. Close examination of GM2 staining in neurons showed that it mainly accumulated in a subset of lysosomes that were also stained with p62 (Figure 5B). The proportion of lysosomes stained with p62 and GM2 antibodies was higher in Spg11−/− neurons than in control neurons and increased over time (Figure 5C), suggesting that GM2 accumulation might contribute to the increase in lysosomes displaying autophagy markers.
Figure 5.

GM2 Accumulation in Lysosomes Promotes the Accumulation of Autophagy Marker p62 in Lysosomes
(A) Proportion of lysosomes stained with Lamp1 that were also positive for p62 in Spg11+/+ and Spg11−/− neurons after 3 and 6 days in vitro. The graph shows the mean ± SEM values. Two-way ANOVA, followed by Holm-Sidak post hoc test; ∗p = 0.04 and ∗∗∗p < 0.001.
(B) Immunostaining of Spg11−/− neurons with GM2 antibody (cyan), lysosomal marker Lamp1 (magenta), and the autophagy marker p62 (yellow). Arrowheads indicate lysosomes, which were positive for p62 and GM2 staining. Scale bar: 10 μm.
(C) Proportion of lysosomes stained with Lamp1 that were also positive for both p62 and GM2 in Spg11+/+ and Spg11−/− neurons after 3 and 6 days in vitro. The graph shows the mean ± SEM values. Two-way ANOVA, followed by Holm-Sidak post hoc test; ∗∗∗p < 0.001.
(D) Effect of miglustat treatment (100 μM) on the proportion of lysosomes stained with Lamp1 that were also positive for p62 in Spg11+/+ and Spg11−/− neurons after 6 days in vitro. The graph shows the mean ± SEM values. Two-way ANOVA, followed by Holm-Sidak post hoc test; ∗∗∗p < 0.001.
(E) Effect of the downregulation of GM3 synthase with two independent miRNAs (GM3S-1 and GM3S-2) on the proportion of lysosomes stained with Lamp1 that were also positive for p62 in control neurons after 6 days in vitro. The graph shows the mean ± SEM values. Two-way ANOVA, followed by Holm-Sidak post hoc test; ∗∗∗p < 0.001.
(F) Effect of Neu1 downregulation with two independent miRNAs (Neu1-1 and Neu1-2) on the proportion of lysosomes stained with Lamp1 that were also positive for p62 in control neurons after 6 days in vitro. The graph shows the mean ± SEM values. One-way ANOVA, followed by Holm-Sidak post hoc test; ∗∗∗p < 0.001.
In (A–D), day in vitro 3 (DIV3), N = 47–50 neurons quantified in two independent neuron preparations; DIV6, N = 173–193 neurons quantified in four independent neuron preparations. In (E), N = 7–29 neurons quantified in two independent neuron preparations. In (F), N = 57–68 neurons quantified in three independent neuron preparations.
To test this hypothesis, we decreased the amount of gangliosides in lysosomes using miglustat (100 μM), or by downregulating GM3 synthase (Figures S5D–S5F). This strongly decreased the proportion of lysosomes positive for the p62 marker in Spg11−/− neurons (Figures 5D and 5E). Overall, our data suggest that the accumulation of GM2 in lysosomes prevents the degradation of their content. Conversely, we enforced accumulation of GM2 in lysosomes by downregulation of the expression of Neu1. This resulted in a significant increase in the proportion of lysosomes positive for the autophagic marker p62 (Figure 5F). Together, these data suggest that ganglioside accumulation leads to the accumulation of the autophagy marker p62 in lysosomes.
Gangliosides Contribute to Neurodegeneration in Cultured Neurons
We then investigated whether accumulation of gangliosides contribute to neurodegeneration using primary cultures of mouse cortical neurons. Basal neuronal death was similar in Spg11+/+ and Spg11−/− neurons (Figures 6A and 6B). We thus evaluated neuronal death triggered by glutamate, which occurs in many models of neurodegenerative diseases (Lewerenz and Maher, 2015) and was shown to increase ganglioside levels in cultured neurons (Park et al., 2016). Accordingly, we observed a moderate, but statistically significant, increase in overall GM2 immunoreactivity, both in control and Spg11−/− neurons (Figure S6B).
Figure 6.

Decreasing Ganglioside Synthesis Prevents Neuronal Death and Improves Motor Dysfunction in Zebrafish Deficient for Spatacsin and Modeling SPG11
(A) Quantification of neuronal death 30 hr after incubation of neurons with glutamate (200 μM) in primary cultures of Spg11+/+ or Spg11−/− cortical neurons treated with miglustat. The graph shows the mean ± SEM values. N = 5–14 independent experiments. One-way ANOVA, followed by Holm-Sidak post hoc test; ∗p = 0.002 and ∗∗∗p < 0.001.
(B) Quantification of neuronal death 30 hr after incubation of neurons with glutamate (200 μM) in primary cultures of Spg11+/+ and Spg11−/− cortical neurons transfected with vectors expressing control miRNA or two different miRNAs against GM3 synthase. The graph shows the mean ± SEM. N = 3–8 independent experiments. One-way ANOVA, followed by Holm-Sidak post hoc test; ∗p < 0.02 and ∗∗∗p < 0.001.
(C) Quantification of neuronal death 30 hr after incubation of neurons with glutamate (200 μM) in primary cultures of Spg11+/+ and Spg11−/− cortical neurons transfected with vectors that downregulate Neu1 with two independent miRNAs (Neu1-1 and Neu1-2). The graph shows the mean ± SEM values. N = 6 independent experiments. One-way ANOVA, followed by Holm-Sidak post hoc test; ∗∗∗p < 0.001.
(D and E) Immunostaining (D) and quantification (E) of GM2 immunostaining in the telencephalon of morphants injected with 1.2-pmol zspg11spl antisense or 1.2-pmol mismatch (mm) morpholino (MO). Injection of zspg11spl morpholino increased the mean and variance of GM2 immunostaining intensity. This phenotype was corrected when morphants were treated with miglustat. N = 6–12 morphants analyzed in each condition. One-way ANOVA; ∗∗∗p < 0.001.
(F) Phenotype of morphants that were non-injected or injected with 1.2-pmol zspg11spl or 1.2-pmol mismatch (mm) morpholino. 48 hr post-fertilization, morphants were classified as normal phenotype, slowly swimming, paralyzed, or curly morphants. Injection of zspg11spl morpholino leads a large proportion of paralyzed or slowly swimming phenotypes. Treatment with miglustat decreased the proportion of paralyzed morphants. N = 102–366 morphants analyzed in each groups. Chi square test; ∗∗∗p < 0.0001.
(G and H) Tracking (G) and quantification of distance traveled (H) by larvae following a touch-evoked escape response. Injection of zspg11spl morpholino impaired the swimming of morphants, which was corrected when treated by miglustat. The graph shows the mean ± SEM. N = 12 morphants analyzed in each condition. One-way ANOVA; ∗∗∗p < 0.001.
See also Figure S6.
Neuronal death triggered by glutamate was significantly higher in neurons obtained from Spg11−/− embryos than control neurons (Figures 6A and 6B). Miglustat treatment of Spg11−/− neurons decreased glutamate-induced neuronal death in a dose-dependent manner (Figure 6A). Similar data were obtained when GM2 levels were decreased after downregulation of GM3 synthase (Figure 6B). These data suggest that GM2 accumulation contributes to neuronal death. We confirmed this hypothesis by inducing the accumulation of gangliosides in lysosomes, after downregulating Neu1, and monitoring neuronal death triggered by glutamate. Increasing ganglioside levels sensitized the neurons to glutamate-triggered cell death (Figure 6C).
We monitored p62 levels in control and Spg11−/− neurons treated with glutamate to determine whether ganglioside-mediated accumulation of autophagy markers in lysosomes contributed to glutamate-induced neuronal death. We observed no difference in p62 levels in untreated neurons. In contrast, glutamate treatment significantly increased p62 levels in Spg11−/− neurons, which was inhibited when ganglioside levels were decreased by miglustat treatment (Figure S6C). These data suggest that gangliosides contribute to neuronal death by promoting the accumulation of the autophagy marker p62.
Inhibition of Ganglioside Synthesis in Zebrafish Rescues the Motor Dysfunction Induced by Loss of Spatacsin
Finally, we tested the pathological role of ganglioside accumulation in an in vivo model. Since gangliosides accumulate in lysosomes at embryonic stages, we inhibited spatacsin expression in zebrafish larvae using injection of antisense morpholinos, which leads to a motor phenotype (Martin et al., 2012). Zebrafish larvae injected with zspg11 antisense morpholinos had a stronger GM2 staining in the telencephalon than larvae injected with mismatch morpholinos, as assessed by immunostaining. The stronger staining with GM2 antibody was corrected in larvae treated by miglustat (Figures 6D and 6E). We therefore were able to monitor the role of gangliosides in the motor phenotype in this model. Larvae injected with zspg11 antisense morpholinos presented with a motor phenotype that was characterized by either a loss of motility or a paralysis (Figure 6F) that was rarely or never observed in larvae injected with a mismatch morpholino or in uninjected larvae, respectively. When larvae were treated with miglustat, the proportion of paralyzed larvae was significantly reduced in a dose-dependent manner compared to controls (Figure 6F). To confirm these data, we monitored the distance traveled by larvae following a touch-evoked escape response. Morphants injected with zspg11 antisense morpholinos showed significantly shorter touch-induced escapes than larvae injected with control morpholinos (Videos S1 and S2). This phenotype was significantly corrected when larvae were treated with miglustat (Figures 6G and 6H; Video S3), while miglustat had no effect on morphants injected with mismatch morpholino. Together, these data demonstrate the deleterious role of gangliosides in an in vivo model of the SPG11 pathology.
Discussion
Using models of hereditary spastic paraplegia caused by mutations in the SPG11 gene, as well as inhibition of proteins implicated in ALR, we show that impairment of lysosome membrane recycling in neurons leads to accumulation of gangliosides in lysosomes, thereby contributing to neurodegeneration.
The recycling of lysosomal membranes has been mainly investigated in cultured cell lines or fibroblasts, and few data are available in neuronal models. Loss of spatacsin leads to accumulation of autolysosomes and depletion of lysosomes in the brain of an Spg11 knockout mouse model (Varga et al., 2015), but the molecular mechanisms underlying this phenomenon have not been entirely elucidated. ALR described in cell lines occurs after the induction of autophagy and leads to the formation of tubules extruding from autolysosomes (Chang et al., 2014, Yu et al., 2010). In neurons, we were unable to promote autophagy following starvation, as previously observed (Mizushima et al., 2004), and did not observe tubules extruding from lysosomes. In nutrient-rich conditions, lysosome recycling mainly occurs via vesiculation, a mechanism implicating notably the phosphoinositide kinase PI4KB and clathrin (Sridhar et al., 2013). Spatacsin interacts with PI4KB and was proposed to participate in lysosome vesiculation in nutrient-rich conditions (Chang et al., 2014). The presence of large lysosomes observed in neurons when spatacsin is absent or when clathrin or dynamin are inhibited could thus be a consequence of impaired recycling of lysosomal membrane mediated by vesiculation.
The loss of spatacsin leads to a strong accumulation of gangliosides in lysosomes in the cerebral cortex of mice, which increased with age, but was not due to impairment of hexosaminidase A activity. Gangliosides are normally degraded into ceramide and sialic acid in lysosomes, and these metabolites are transported to the endoplasmic reticulum (Tettamanti et al., 2003). Our lipidomic data showed that the accumulation of gangliosides is paralleled by a decrease in ceramide, which could be a consequence of impaired ganglioside clearance from lysosomes. The implication of spatacsin in initiation of lysosome membrane recycling (Chang et al., 2014) led us to hypothesize that recycling of lysosome membrane could contribute to clear gangliosides from lysosomes. This is supported by the accumulation of gangliosides observed when lysosome membrane recycling is inhibited by downregulation of clathrin or inhibition of dynamin. The coimmunoprecipitation of spatacsin with dynamin, and the decreased amount of dynamin in lysosomes in the absence of spatacsin, suggests that spatacsin recruits dynamin to this subcellular compartment. Thus, spatacsin, besides its role in initiation of lysosome membrane recycling (Chang et al., 2014), could contribute to the late stage of recycling by recruiting dynamin to promote fission and release membrane from lysosomes. Impaired vesicle fission and accumulation of clathrin-coated buds have been observed in cortical granules of oocytes in the souffle zebrafish mutant that has no spastizin (Kanagaraj et al., 2014). Spastizin is a spatacsin interactor implicated in lysosome membrane recycling (Chang et al., 2014). Its expression is strongly decreased in the brain of Spg11−/− mice (Branchu et al., 2017), which could also contribute to impaired lysosomal membrane recycling. Our data therefore suggest that, in the absence of spatacsin, impaired lysosome membrane recycling leads to the progressive accumulation of gangliosides in lysosomes as the disease progresses.
Inhibition of ganglioside synthesis prevented neuronal death in vitro but did not compensate for the formation of large lysosomes, suggesting that inhibition of lysosome membrane recycling itself is not responsible for neurodegeneration. In contrast, modulation of ganglioside synthesis showed that these molecules contribute to the accumulation of the autophagy marker p62 in lysosomes. The catalytic activity of the lysosomal protease cathepsin B was not altered in Spg11−/− neurons, but the accumulation of gangliosides could slightly impair other lysosomal activities leading to progressive accumulation of autophagic markers in lysosomes. In the long term, defect in autophagy clearance could contribute to neurodegeneration (Menzies et al., 2017). Accordingly, the accumulation of gangliosides in lysosomes paralleled the accumulation of p62 in lysosomes observed by Varga et al. (2015) and the progression of neurodegeneration that we observed in the Spg11−/− mice (Branchu et al., 2017). In vitro, glutamate treatment was proposed to inhibit the autophagic flux in hippocampal neurons leading to accumulation of p62 (Kulbe et al., 2014). Accordingly, decreasing ganglioside synthesis has a protective action against neuronal death induced by glutamate in Spg11−/− neurons, probably by preventing accumulation of the autophagy marker p62. Altogether, our results suggest that the accumulation of gangliosides and the subsequent accumulation of lysosomes containing autophagic markers are contributing to neuronal death in the absence of spatacsin.
Decreasing gangliosides synthesis therefore appears as a rational therapeutic strategy to prevent the symptoms caused by the accumulation of these glycosphingolipids in lysosomes. Consistently, diminution of gangliosides improved motor phenotype in an in vivo zebrafish model of SPG11. Such treatment should be started at very early stages of the disease as we showed early accumulation of gangliosides in lysosomes, both in the Spg11 knockout mouse model and human SPG11 iPSC-derived neurons. However, it was not possible to test this hypothesis in the Spg11−/− mouse model due to the controversial action of miglustat on brain glycosphingolipids (Ashe et al., 2011, Boudewyn et al., 2017).
In conclusion, we show that gangliosides accumulate in lysosomes following impairment of lysosome membrane recycling, and not only upon metabolic impairment as observed in lysosomal storage disorders (Walkley and Vanier, 2009). The accumulation of gangliosides was responsible for accumulation of autophagy markers in lysosomes and neurodegeneration. Since gangliosides are highly present in the brain (Xu et al., 2010), our work reveals why proteins implicated in lysosome membrane recycling are often associated with neurodegeneration, and provides a rational therapeutic strategy for neurodegenerative diseases linked to such lysosomal dysfunction. Accumulation of undigested material in lysosomes with abnormal morphology was also observed in SPG4, SPG8, SPG15, SPG31, and SPG48 models of hereditary spastic paraplegias (Allison et al., 2017, Hirst et al., 2015, Renvoisé et al., 2014). It might be informative to investigate lysosome membrane recycling and ganglioside accumulation in these diseases. In addition, gangliosides accumulate in lysosomes in models of quite prevalent neurodegenerative diseases, such as Alzheimer’s disease (Yang et al., 2014), and it would be interesting to test whether the recycling of lysosome membrane is altered in these conditions as well. This would allow a better understanding of the physiopathological pathway leading to neuronal death and indicate whether these patients can benefit from the same therapeutic approaches.
Experimental Procedures
Ethical Approval
The care and treatment of animals followed European legislation (no. 2010/63/UE) and national (Ministère de l’Agriculture, France) guidelines for the detention, use, and ethical treatment of laboratory animals. All experiments on animals were approved by the local ethics committee (approval no. A751319 and ce5/2012/057) and conducted by authorized personnel. Patient-derived materials were obtained through procedures approved by the ethics committee with the written, informed consent of the family (approval RBM-1-029).
Spg11-Knockout Mouse Immunohistochemistry
Spg11-knockout mice were described previously (Branchu et al., 2017). Immunohistochemistry was performed as previously described (Branchu et al., 2017). Images were obtained with an Olympus FV-1000 confocal microscope with a 60× objective (numerical aperture [NA], 1.35). For quantification, regions of interest (ROIs) corresponding to the cell body of individual neurons were surrounded using ImageJ, and the mean of fluorescence intensity of ganglioside immunostaining in each ROI was quantified using the measure tool. In each brain slice, we analyzed all neurons that were identified by their morphology and large cell body in the layer V of the motor cortex.
Human SPG11 Fibroblasts
Skin biopsies were collected from two healthy female subjects and two SPG11 female patients. Patient SPG11-1 (FSP-1123-004) carried two heterozygous truncating mutations in trans (c.2431 C>T, pGln811X; deletion of exon 29). This patient had normal intellectual development and experienced gait difficulties by age 14, gradually worsened, and became stick-dependent at age 20. Examination at age 23 showed that she could still walk with sticks. Spasticity and weakness was present in the lower limbs, while tone and strength were normal in the upper limbs. She had increased reflexes with ankle clonus and bilateral extensor plantar reflex as well as Hoffman sign in the upper limbs. Deep sensation was normal. She had postural tremor in the arms, normal eye gaze, and no cognitive impairment. There was no evident cerebellar sign. Patient SPG11-2 (FSP-1232-003) carried two heterozygous truncating mutations in trans (c.1951 C>T, pArg651X; c.5623 C>T, pGln1875X). This woman had onset of spastic gait at age 17. At age 27, she presented with moderate spastic gait, needing walking aids since age 26, and more recently a wheelchair. She had increased reflexes in lower limbs, including bilateral extensor plantar reflexes, and Hoffman signs were present in the upper limbs. She had moderate weakness in the legs and decreased deep sensation at the ankles. Bradykinesia was evident, and the finger-nose test was performed with mild tremor. Cognition was clinically normal, and she exhibited no abnormal eye movements. Cerebral imaging showed a thin corpus callosum.
Zebrafish Modeling
Modeling of SPG11 pathology in zebrafish was performed using morpholino oligonucleotides as described previously (Martin et al., 2012). After morpholino injection, embryos were maintained at 28°C in E3 medium, containing miglustat or DMSO for control groups. At 24 hr post-fertilization (hpf), they were manually dechorionated using fine forceps. The embryo morphology was observed at 48 hr post-fertilization.
Biochemical Analyses
Experimental procedures for lipidomic analysis, lysosome fraction purification, and enzymatic assays are detailed in Supplemental Experimental Procedures.
Cell Culture
Experimental procedures for cultures of primary neurons and iPSCs, as well as differentiation of organoids are detailed in Supplemental Experimental Procedures.
Statistics
Statistics were performed using GraphPad Prism software, using the Kruskal-Wallis, ANOVA, or t tests, depending on the experiments. Multiple comparisons after ANOVA were performed with the Holm-Sidak test using GraphPad Prism. For lipidomic analysis, the mcp package was used with R, version 3.0.2. Comparison of the means between phenotypes was performed using a t test with a Benjamini-Hochberg procedure to correct for multiple testing, with a false-discovery rate of 5%.
Acknowledgments
We thank Phenoparc, Histomics, IGenSeq, Celis, Celis-IPS, and ICM.quant facilities of the Institut du Cerveau et de la Moelle Épinière for their contributions. This work was supported by “Investissements d’Avenir” Program grants ANR-10-IAIHU-06 and ANR-11-INBS-0011 and received funding from the Verum Foundation (to A.B. and G.S.), the French Agency for Research (ANR) (13-ISV1-00002 to G.S. and MetaboHUB: ANR-11-INBS-0010), the GIS-Maladies Rares Foundation (06/GIS/PB/SJ/no.71 to G.S.), the Fondation Roger de Spoelberch (to A.B.), the European Union with the ANR (to A.B., Seventh Framework Programme–FP7, Omics Call [grant No. 305121]; to G.S., the E-Rare Programme Neurolipid [ANR-13-ERARE-0003-02]), and the European Research Council (European Research Council Starting Grant No. 311149 to F.D.). M.B. received a fellowship from the French Ministry of Research (Doctoral School ED3C). A.P. received an ARDoC fellowship (17012953) from the Région Ile de France (Doctoral School ED3C).
Author Contributions
M.B., J.B., A.B., F.M., K.H.E.H., G.S., and F.D. conceived and designed the experiments. M.B., J.B., C.L., C.P., J.P., R.M., A.S., M.P., E.C.-V., A.P., K.D., J.-P.P., C.C., A.D., B.C., K.H.E.H., and F.D. performed the experiments. M.B., J.B., C.L., C.P., J.P., R.M., A.S., M.P., B.C., F.M., K.H.E.H., and F.D. analyzed the data. M.B., J.B., G.S., and F.D. wrote the paper.
Declaration of Interests
M.B., J.B., F.M., G.S., and F.D. have a patent related to this work.
Published: June 26, 2018
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, six figures, one table, and three videos and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.05.098.
Contributor Information
Giovanni Stevanin, Email: giovanni.stevanin@upmc.fr.
Frédéric Darios, Email: frederic.darios@upmc.fr.
Supplemental Information
References
- Allison R., Edgar J.R., Pearson G., Rizo T., Newton T., Günther S., Berner F., Hague J., Connell J.W., Winkler J. Defects in ER-endosome contacts impact lysosome function in hereditary spastic paraplegia. J. Cell Biol. 2017;216:1337–1355. doi: 10.1083/jcb.201609033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashe K.M., Bangari D., Li L., Cabrera-Salazar M.A., Bercury S.D., Nietupski J.B., Cooper C.G., Aerts J.M., Lee E.R., Copeland D.P. Iminosugar-based inhibitors of glucosylceramide synthase increase brain glycosphingolipids and survival in a mouse model of Sandhoff disease. PLoS One. 2011;6:e21758. doi: 10.1371/journal.pone.0021758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonten E., van der Spoel A., Fornerod M., Grosveld G., d’Azzo A. Characterization of human lysosomal neuraminidase defines the molecular basis of the metabolic storage disorder sialidosis. Genes Dev. 1996;10:3156–3169. doi: 10.1101/gad.10.24.3156. [DOI] [PubMed] [Google Scholar]
- Boudewyn L.C., Sikora J., Kuchar L., Ledvinova J., Grishchuk Y., Wang S.L., Dobrenis K., Walkley S.U. N-Butyldeoxynojirimycin delays motor deficits, cerebellar microgliosis, and Purkinje cell loss in a mouse model of mucolipidosis type IV. Neurobiol. Dis. 2017;105:257–270. doi: 10.1016/j.nbd.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branchu J., Boutry M., Sourd L., Depp M., Leone C., Corriger A., Vallucci M., Esteves T., Matusiak R., Dumont M. Loss of spatacsin function alters lysosomal lipid clearance leading to upper and lower motor neuron degeneration. Neurobiol. Dis. 2017;102:21–37. doi: 10.1016/j.nbd.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J., Lee S., Blackstone C. Spastic paraplegia proteins spastizin and spatacsin mediate autophagic lysosome reformation. J. Clin. Invest. 2014;124:5249–5262. doi: 10.1172/JCI77598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du W., Su Q.P., Chen Y., Zhu Y., Jiang D., Rong Y., Zhang S., Zhang Y., Ren H., Zhang C. Kinesin 1 drives autolysosome tubulation. Dev. Cell. 2016;37:326–336. doi: 10.1016/j.devcel.2016.04.014. [DOI] [PubMed] [Google Scholar]
- Folch J., Lees M., Sloane Stanley G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- Hehr U., Bauer P., Winner B., Schule R., Olmez A., Koehler W., Uyanik G., Engel A., Lenz D., Seibel A. Long-term course and mutational spectrum of spatacsin-linked spastic paraplegia. Ann. Neurol. 2007;62:656–665. doi: 10.1002/ana.21310. [DOI] [PubMed] [Google Scholar]
- Hirst J., Edgar J.R., Esteves T., Darios F., Madeo M., Chang J., Roda R.H., Durr A., Anheim M., Gellera C. Loss of AP-5 results in accumulation of aberrant endolysosomes, defining a new type of lysosomal storage disease. Hum. Mol. Genet. 2015;24:4984–4996. doi: 10.1093/hmg/ddv220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyakumar M., Butters T.D., Cortina-Borja M., Hunnam V., Proia R.L., Perry V.H., Dwek R.A., Platt F.M. Delayed symptom onset and increased life expectancy in Sandhoff disease mice treated with N-butyldeoxynojirimycin. Proc. Natl. Acad. Sci. USA. 1999;96:6388–6393. doi: 10.1073/pnas.96.11.6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanagaraj P., Gautier-Stein A., Riedel D., Schomburg C., Cerdà J., Vollack N., Dosch R. Souffle/Spastizin controls secretory vesicle maturation during zebrafish oogenesis. PLoS Genet. 2014;10:e1004449. doi: 10.1371/journal.pgen.1004449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulbe J.R., Mulcahy Levy J.M., Coultrap S.J., Thorburn A., Bayer K.U. Excitotoxic glutamate insults block autophagic flux in hippocampal neurons. Brain Res. 2014;1542:12–19. doi: 10.1016/j.brainres.2013.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewerenz J., Maher P. Chronic glutamate toxicity in neurodegenerative diseases—what is the evidence? Front. Neurosci. 2015;9:469. doi: 10.3389/fnins.2015.00469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin E., Yanicostas C., Rastetter A., Alavi Naini S.M., Maouedj A., Kabashi E., Rivaud-Péchoux S., Brice A., Stevanin G., Soussi-Yanicostas N. Spatacsin and spastizin act in the same pathway required for proper spinal motor neuron axon outgrowth in zebrafish. Neurobiol. Dis. 2012;48:299–308. doi: 10.1016/j.nbd.2012.07.003. [DOI] [PubMed] [Google Scholar]
- Menzies F.M., Fleming A., Caricasole A., Bento C.F., Andrews S.P., Ashkenazi A., Füllgrabe J., Jackson A., Jimenez Sanchez M., Karabiyik C. Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron. 2017;93:1015–1034. doi: 10.1016/j.neuron.2017.01.022. [DOI] [PubMed] [Google Scholar]
- Mizushima N., Yamamoto A., Matsui M., Yoshimori T., Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano Y., Fujitani K., Kurihara J., Ragan J., Usui-Aoki K., Shimoda L., Lukacsovich T., Suzuki K., Sezaki M., Sano Y. Mutations in the novel membrane protein spinster interfere with programmed cell death and cause neural degeneration in Drosophila melanogaster. Mol. Cell. Biol. 2001;21:3775–3788. doi: 10.1128/MCB.21.11.3775-3788.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park D.H., Wang L., Pittock P., Lajoie G., Whitehead S.N. Increased expression of GM1 detected by electrospray mass spectrometry in rat primary embryonic cortical neurons exposed to glutamate toxicity. Anal. Chem. 2016;88:7844–7852. doi: 10.1021/acs.analchem.6b01940. [DOI] [PubMed] [Google Scholar]
- Paşca A.M., Sloan S.A., Clarke L.E., Tian Y., Makinson C.D., Huber N., Kim C.H., Park J.Y., O’Rourke N.A., Nguyen K.D. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods. 2015;12:671–678. doi: 10.1038/nmeth.3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renvoisé B., Chang J., Singh R., Yonekawa S., FitzGibbon E.J., Mankodi A., Vanderver A., Schindler A., Toro C., Gahl W.A. Lysosomal abnormalities in hereditary spastic paraplegia types SPG15 and SPG11. Ann. Clin. Transl. Neurol. 2014;1:379–389. doi: 10.1002/acn3.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong Y., McPhee C.K., Deng S., Huang L., Chen L., Liu M., Tracy K., Baehrecke E.H., Yu L., Lenardo M.J. Spinster is required for autophagic lysosome reformation and mTOR reactivation following starvation. Proc. Natl. Acad. Sci. USA. 2011;108:7826–7831. doi: 10.1073/pnas.1013800108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong Y., Liu M., Ma L., Du W., Zhang H., Tian Y., Cao Z., Li Y., Ren H., Zhang C. Clathrin and phosphatidylinositol-4,5-bisphosphate regulate autophagic lysosome reformation. Nat. Cell Biol. 2012;14:924–934. doi: 10.1038/ncb2557. [DOI] [PubMed] [Google Scholar]
- Schulze R.J., Weller S.G., Schroeder B., Krueger E.W., Chi S., Casey C.A., McNiven M.A. Lipid droplet breakdown requires dynamin 2 for vesiculation of autolysosomal tubules in hepatocytes. J. Cell Biol. 2013;203:315–326. doi: 10.1083/jcb.201306140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridhar S., Patel B., Aphkhazava D., Macian F., Santambrogio L., Shields D., Cuervo A.M. The lipid kinase PI4KIIIβ preserves lysosomal identity. EMBO J. 2013;32:324–339. doi: 10.1038/emboj.2012.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevanin G., Santorelli F.M., Azzedine H., Coutinho P., Chomilier J., Denora P.S., Martin E., Ouvrard-Hernandez A.M., Tessa A., Bouslam N. Mutations in SPG11, encoding spatacsin, are a major cause of spastic paraplegia with thin corpus callosum. Nat. Genet. 2007;39:366–372. doi: 10.1038/ng1980. [DOI] [PubMed] [Google Scholar]
- Tesson C., Koht J., Stevanin G. Delving into the complexity of hereditary spastic paraplegias: how unexpected phenotypes and inheritance modes are revolutionizing their nosology. Hum. Genet. 2015;134:511–538. doi: 10.1007/s00439-015-1536-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tettamanti G., Bassi R., Viani P., Riboni L. Salvage pathways in glycosphingolipid metabolism. Biochimie. 2003;85:423–437. doi: 10.1016/s0300-9084(03)00047-6. [DOI] [PubMed] [Google Scholar]
- Varga R.E., Khundadze M., Damme M., Nietzsche S., Hoffmann B., Stauber T., Koch N., Hennings J.C., Franzka P., Huebner A.K. In vivo evidence for lysosome depletion and impaired autophagic clearance in hereditary spastic paraplegia type SPG11. PLoS Genet. 2015;11:e1005454. doi: 10.1371/journal.pgen.1005454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkley S.U., Vanier M.T. Secondary lipid accumulation in lysosomal disease. Biochim. Biophys. Acta. 2009;1793:726–736. doi: 10.1016/j.bbamcr.2008.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y.H., Barnes S., Sun Y., Grabowski G.A. Multi-system disorders of glycosphingolipid and ganglioside metabolism. J. Lipid Res. 2010;51:1643–1675. doi: 10.1194/jlr.R003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D.S., Stavrides P., Saito M., Kumar A., Rodriguez-Navarro J.A., Pawlik M., Huo C., Walkley S.U., Saito M., Cuervo A.M., Nixon R.A. Defective macroautophagic turnover of brain lipids in the TgCRND8 Alzheimer mouse model: prevention by correcting lysosomal proteolytic deficits. Brain. 2014;137:3300–3318. doi: 10.1093/brain/awu278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L., McPhee C.K., Zheng L., Mardones G.A., Rong Y., Peng J., Mi N., Zhao Y., Liu Z., Wan F. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465:942–946. doi: 10.1038/nature09076. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.