

Abstract
The differential diagnosis of young-onset progressive dementia is an issue that requires effort. Recording the family history and careful clinical evaluation are useful tools in the diagnosis. In case of genetic bases, definitive diagnosis requires molecular analysis. We report consanguineous two patients presenting with young-onset progressive dementia characterized by behavioral changes and with bone cysts. Concomitant bone pathology and inheritance pattern directed us to investigate TREM2 gene, for differential diagnosis, which resulted with the identification of a causative mutation that confirmed the diagnosis of Nasu Hakola disease. The mutation (c.113A>G) is the same for a Turkish family with Nasu Hakola disease reported before. But the presence of bone cysts and absence of epilepsy in our patients are the different findings. Molecular analysis should be considered in patients with young age onset behavioral and cognitive deficits, with white matter lesions in brain magnetic resonance imaging, if especially associated with cystic bone lesions.
Keywords: Young onset progressive dementia, bone cysts, Nasu-Hakola disease
INTRODUCTION
Nasu-Hakola disease (MIM#221770), also known as PLOSL (polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy), is characterized by unique combination of young onset progressive dementia, associated with sclerosing leukoencephalopathy, and systemic bone cysts with onset in the second to fourth decade (1, 2).
In recent years, studies have demonstrated that Nasu-Hakola disease is caused by a mutation in the TROBP gene (tyro protein tyrosine kinase binding protein; in other name; DAP12 gene, DNAX-activating protein 12) or the TREM2 gene (triggering receptor expressed on myeloid cells 2) (3). Formally, in the natural progression of this disorder the following four different stages may be identified: i) latent disease (asymptomatic); ii) bone involvement (pathological fractures); iii) early neurological symptoms (patient’s personality changes and first dementia symptoms begin to arise); and iv) late neurological stage in which patients show symptoms of profound dementia and begin to lose their motility (4).
The early neurological stage begins in the second to fourth decade of life and manifests as frontal lobe syndrome. Personality change such as loss of judgment and social inhibition, euphoria, disturbance of concentration, lack of insight and libido are typical (5).
CASE PRESENTATION
We report two dementia patients who are third degree relatives (uncle-nephew) with symptoms, signs, and imaging findings highly correlated with probable behavioral variant type criteria of frontotemporal dementia (FTD) (6).
We have taken written informed consent from the patients and their families for the clinical evaluation, genetic testing and publication. The pedigree of the family shows the other effected persons in the family, suggestive of autosomal recessive inheritance (Figure 1). Upon consideration of all properties of the patients including molecular analysis, we diagnosed the cases as Nasu-Hakola disease.
Figure 1.

Pedigree of the family showing case 1 (III: 3), case 2 (II: 10) and other affected persons.
Case 1
A previously well 35-year-old, primary school educated housewife presented with cognitive deficits progressing for two years, especially over the past three months. She was complaining about nothing, feeling very happy, and euphoric. But her family noticed her forgetfulness, disorganization, and problems in her logics. She was not able to do any work at home. Her personality was changed, she behaved and spoke inappropriately, became unresponsive and lost her warmth to others, and easily became nervous. She had binge eating which is mostly sweet, and had loss of libido (Table 1).
Table 1.
Clinical properties of the cases
| Clinical properties | Case 1 | Case 2 |
|---|---|---|
| Behavioral Problems | + | + |
| Euphoria | + | + |
| Disorganization | + | + |
| Memory problems | + | + |
| Bone pain | + | - |
| Binge eating | + | - |
| Loss of libido | + | + |
The patient had no significant medical history other than foot and leg pain with repetitive fractures during the preceding 10 years. The family reported that her uncle and aunt died at the age of 45 (II: 8) and 42 (II: 9) because of young onset progressive dementia (Figure 1). Additionally, another uncle of her (case 2; II: 10) started to behave inappropriately.
On admission to outpatient neurology clinic of Erciyes University, neurological examination findings were unremarkable except mental examination. The patient’s neuropsychological tests revealed that digit span, verbal fluency, Stroop, motor programming tasks (including alternating programs using square and pointed pictures, Luria’s loop, and serial hand sequences), face recognition, and trail making tests were severely impaired showing problems in attention, organization, planning, and executive functions. Agraphia, acalculia and alexia were not detected. Mini mental state examination score was 28/30 (memory and copying parts), clock drawing test score was 3/7, and Montreal cognitive assessment score was 17/30 (losses in all parts except orientation) (Table 2). Her psychiatric symptoms and memory deficits minimally progressed following a six months’ duration of treatment with memantine, donepezil, citalopram, and vitamin B12.
Table 2.
Neuropsychiatric test results of the cases
| Neuropsychiatric Tests | Case 1 | Case 2 |
|---|---|---|
| Montreal Cognitive Assessment | Losses in all parts except orientation | Losses in all parts except naming |
| Mini Mental State Examination | Losses in memory and copying parts | Losses in orientation and memory parts |
| Frontal lobe test battery and clock drawing test | Problems in attention, organization, planning and executive functions | Problems in attention, organization, planning and executive functions |
| Examinations of writing, calculation and reading | No agraphia, acalculia, alexia | No agraphia, acalculia, alexia |
The results of blood tests including biochemistry, complete blood count: CBC, thyroid hormone, vitamin B12, vitamin D, folic acid, serologic tests related to autoimmune encephalitis, VDRL, and HIV tests were normal except low vitamin B12. Peripheral smear, cerebrospinal fluid (CSF), electroencephalography, and electromyography examinations revealed normal results.
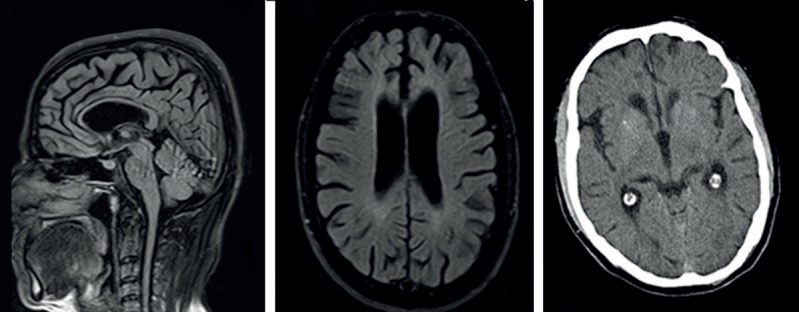
Brain Magnetic Resonance Imaging (MRI) (1.5 Tesla, Achieva; Philips Medical Systems, Best, The Netherlands) revealed periventricular signal alterations in the white matter, diffuse cerebral atrophy with prominent frontal and temporal lobe, diffusely thinned corpus callosum, and reduced anterior cingulate gyrus volume (Figure 2). [(18)F] fluorodeoxyglucose (FDG) positron emission tomography (PET) analysis (Philips Medical Systems, Cleveland, Ohio, USA) showed bilateral hypometabolic pattern, involving basal nuclei, parietotemporal cortex and anterior cingulate gyrus (Figure 3). X-ray imaging of both talus bones revealed osteoporotic and cystic lesions.
Figure 2.

Brain MRI and computed tomography imaging revealed diffuse signal alterations in the subcortical frontal and parietal white matters with global brain atrophy, thin corpus callosum and mild basal ganglia calcification in Case 1.
Figure 3.

[(18)F] fluorodeoxyglucose (FDG) positron emission tomography (PET) analysis showed bilateral hypometabolic pattern, involving basal nuclei, temporal cortex and anterior cingulate gyrus in Case 1.
Case 2
The patient, who was a high school educated 41-year-old male worker, applied to the outpatient neurology clinic of Erciyes University with personality and behavioral changes lasting for one year, described by family members. He had inappropriate behaviors, poor judgment, and was talking too much without inhibition, making inappropriate jokes. He couldn’t comply with his job because of his irresponsibility and forgetfulness (Table 1). The patient had no significant medical history. He didn’t mention about any fractures but complained impotence. His family history indicated affected persons (III: 3 presented as Case 1, II: 8, II: 9) (Figure 1).
Neurological examination findings were unremarkable except cognitive dysfunction. The Mini-Mental State Examination score was 26/30 (losses in orientation and memory parts). Frontal lobe test battery revealed problems in attention, organization, planning, and executive functions. Clock drawing test score was 3/7, and Montreal cognitive assessment score was 18/30 (losses in all parts except naming). Agraphia, acalculia, and alexia were not detected (Table 2). His symptoms remained stable with the treatment with donepezil, memantine, and citalopram in six months’ period.
Blood tests (biochemistry, CBC, thyroid hormone, vitamin B12, vitamin D, folic acid, serologic tests related to autoimmune encephalitis, VDRL, and HIV tests), examination of peripheral smear, CSF, electroencephalography, and electromyography all revealed normal results.
Computed tomography (CT) examination showed cerebral atrophy and mild basal ganglia calcification. Brain MRI revealed diffuse signal alterations in the subcortical frontal and parietal white matter with global brain atrophy with diffusely thinned corpus callosum, and reduced anterior cingulate gyrus volume. PET CT exhibited bilateral hypometabolism on anterior cingulate gyrus, basal nuclei, and parietotemporal cortex. X-ray and CT imagings of bilateral ankles revealed reduction in trabecular structure, and multiple lytic lesions with sclerotic rims in talus (Figure 4).
Figure 4.
CT imagings of bilateral ankles revealed reduction in trabecular structure and multiple lytic lesions with sclerotic rims in talus of Case 2.
Molecular Analyses
The molecular analysis was performed at The Medical Genetic Laboratory of İstanbul University. Genomic DNA was isolated from Case 2 and Case 1 (Figure 1, II: 10 and III: 3), from the collection of 2 ml venous blood in K3EDTA tubes by kit (MagNA Pure Compact Nucleic Acid Isolation Kit-Large Volume; Roche). Exon 2 of TREM2 (NM_018965.3) was sequenced by Sanger sequencing (ABI 3500). Analysis of the sequencing electropherograms revealed the same homozygous mutation in both, c.113A>G (p.Tyr38Cys).
DISCUSSION
We presented two consanguineous young patients with symptoms resembling, even fulfilling the criteria of probable behavioral variant of frontotemporal dementia (bvFTD). Nevertheless, there were some additional and controversial findings in their evaluation. Considering these findings and molecular analyses of the patients, we diagnosed a rare genetic disease named as Nasu Hakola. The warning findings in these patients were presence of history and/or of bone involvement, and MR and CT (7). Additionally, the pedigree was in conformity with autosomal recessive inheritance that was incompatible with autosomal dominant inherited FTD. Upon our evaluations, we realized that the genetic mutation detected in our patients was similar to an earlier reported Turkish case, but the clinical presentations differ with respect to the presence of bone pathology. This is an interesting finding, showing presence of phenotype variation in a defined genetic mutation of the disease.
Nasu Hakola disease is a rare genetic young onset progressive dementia caused by loss-of function mutations or deletions in TREM2 or TYROBP genes, resulting in loss of function of the TREM2-TYROBP immunoreceptor signaling complex (2, 3). This complex mediates myeloid cells, including microglia and osteoclasts, development, activation, and function. In the bone, it regulates the differentiation and function of osteoclasts, which are bone-resorbing cells; while it regulates microglia phagocytosis of neuronal debris and amyloid deposits in the brain (8). In the absence of this complex, microglia becomes excessively activated and dysfunctional causing accumulation of amyloid, loss of myelin, induction of inflammation, and possibly dysfunction in synaptic regulation of the brain (9). In recent years, TREM2 is also reported to be a related factor for the most common dementias, Alzheimer’s disease, and FTD (8).
Underlying factor responsible for this brain disease is mostly accepted to be related to white matter changes. However, gray matter pathologies as neuronal loss and gliosis in the thalamus and basal ganglia (10), and possibly dysfunction in synaptic regulation are other explanatory mechanisms (9). In our cases, as observed from the brain images, there are both white and gray matter pathologies.
TYROB mutations are the cause of Nasu Hakola disease predominate in Finland and Japan, while TREM2 mutations are more widely distributed in the world (11). All the identified TREM2 mutations associated with Nasu Hakola is presented in exon 2 of the gene encoding V-set domain by codons 19–129 (Q9NZC2). Up till now, there have been reported 14 different TREM2 mutations of missense, nonsense, and deletion type (12). The homozygous missense mutation detected in our patients was first identified in a Turkish case with FTD-like disease without bone involvement (13). In addition to the absence of bone involvement, the patient had generalized tonic clonic seizures beginning from the early period of the disease, and akinetic-rigid syndrome without any calcification of basal ganglia on computed tomography scan, in contradiction to our cases. In the literature, there are reported cases without bone involvement, epilepsy, akinetic rigid syndrome, or calcification of basal ganglia, presenting with various mutations (12–18). However, it is interesting to see different clinical findings in the patients with the same mutation, which can be attributed to phenotype variation, or some more unidentified genetic factors.
Until now there does not exist any specific therapy, and if present, prevention of seizure recurrence is necessary (5). It is crucial to inform family members properly about the disease in order to prevent possible socio-economic and legal problems. Molecular analysis should be considered in patients with young age onset behavioral and cognitive deficits resembling the phenotype of bvFTD, especially if associated with white matter abnormalities, and thin corpus callosum seen on MRI, even if cystic bone lesions don’t exist. Nasu Hakola disease is an interesting one with complex genotype-phenotype relations. The pathologic molecular mechanisms underlying the disease can reveal important clues in the pathogenesis and treatment of dementia.
Footnotes
Informed Consent: Written consent was obtained from the patients.
Peer-review: Externally peer-reviewed.
Author Contributions: Concept - EK, FT, MFY, OU, AE, ÜA, HH; Design - EK, FT, MFY, OU, AE, ÜA, HH; Supervision - EK, FT, MFY, OU, AE, ÜA, HH; Resource - FT, OU, AE, ÜA; Materials - EK, FT, MFY, OU; Data Collection and/ or Processing - EK, FT, MFY, OU, AE, ÜA, HH; Analysis and/or Interpretation - EK, HH; Literature Search - EK, MFY; Writing - EK; Critical Reviews - EK, FT, MFY, OU, AE, ÜA, HH.
Conflict of Interest: No conflict of interest was declared by the authors.
Financial Disclosure: The authors declared that this study has received no financial support.
REFERENCES
- 1.Hakola HP. Neuropsychiatric and genetic aspects of a new hereditary disease characterized by progressive dementia and lipomembranous polycystic osteodysplasia. Acta Psychiatr Scand Suppl. 1971;232:1–173. [PubMed] [Google Scholar]
- 2.Nasu T, Tsukahara Y, Terayama K. A lipid metabolic disease-“membranous lipodystrophy”-an autopsy case demonstrating numerous peculiar membrane-structures composed of compound lipid in bone and bone marrow and various adipose tissues. Acta Pathol Jpn. 1973;23:539–558. doi: 10.1111/j.1440-1827.1973.tb01223.x. [DOI] [PubMed] [Google Scholar]
- 3.Kaneko M, Sano K, Nakayama J, Amano N. Nasu-Hakola disease:The first case reported by Nasu and review:The 50th Anniversary of Japanese Society of Neuropathology. Neuropathology. 2010;30:463–470. doi: 10.1111/j.1440-1789.2010.01127.x. [DOI] [PubMed] [Google Scholar]
- 4.Giuliano S, Agresta AM, De Palma A, Viglio S, Mauri P, Fumagalli M, Iadarola P, Montalbetti L, Salvini R, Bardoni A. Proteomic analysis of lymphoblastoid cells from Nasu-Hakola patients:a step forward in our understanding of this neurodegenerative disorder. PloS One. 2014;9:e110073. doi: 10.1371/journal.pone.0110073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bock V, Botturi A, Gaviani P, Lamperti E, Maccagnano C, Piccio L, Silvani A, Salmaggi A. Polycystic Lipomembranous Osteodysplasia with Sclerosing Leukoencephalopathy (PLOSL):a new report of an Italian woman and review of the literature. J Neurol Sci. 2013;326:115–119. doi: 10.1016/j.jns.2013.01.021. [DOI] [PubMed] [Google Scholar]
- 6.Rascovsky K, Hodges JR, Kipps CM, Johnson JK, Seeley WW, Mendez MF, Knopman D, Kertesz A, Mesulam M, Salmon DP, Galasko D, Chow TW, Decarli C, Hillis A, Josephs K, Kramer JH, Weintraub S, Grossman M, Gorno-Tempini ML, Miller BM. Diagnostic criteria for the behavioral variant of frontotemporal dementia (bvFTD):current limitations and future directions. Alzheimer Dis Assoc Disord. 2007;21:S14–18. doi: 10.1097/WAD.0b013e31815c3445. [DOI] [PubMed] [Google Scholar]
- 7.Kilic SA, Oner AY, Yuce C, Ozlu IC. Imaging findings of Nasu-Hakola disease:a case report. Clin Imaging. 2012;36:877–880. doi: 10.1016/j.clinimag.2012.01.036. [DOI] [PubMed] [Google Scholar]
- 8.Lue LF, Schmitz C, Walker DG. What happens to microglial TREM2 in Alzheimer's disease:Immunoregulatory turned into immunopathogenic? Neuroscience. 2015;302:138–150. doi: 10.1016/j.neuroscience.2014.09.050. [DOI] [PubMed] [Google Scholar]
- 9.Bianchin MM, Martin KC, de Souza AC, de Oliveira MA, Rieder CR. Nasu-Hakola disease and primary microglial dysfunction. Nat Rev Neurol. 2010;6:2–523. doi: 10.1038/nrneurol.2010.17-c1. [DOI] [PubMed] [Google Scholar]
- 10.Aoki N, Tsuchiya K, Togo T, Kobayashi Z, Uchikado H, Katsuse O, Suzuki K, Fujishiro H, Arai T, Iseki E, Anno M, Kosaka K, Akiyama H, Hirayasu Y. Gray matter lesions in Nasu-Hakola disease:a report on three autopsy cases. Neuropathology. 2011;31:135–143. doi: 10.1111/j.1440-1789.2010.01152.x. [DOI] [PubMed] [Google Scholar]
- 11.Sasaki A, Kakita A, Yoshida K, Konno T, Ikeuchi T, Hayashi S, Matsuo H, Shioda K. Variable expression of microglial DAP12 and TREM2 genes in Nasu-Hakola disease. Neurogenetics. 2015;16:265–276. doi: 10.1007/s10048-015-0451-3. [DOI] [PubMed] [Google Scholar]
- 12.MalaCards Human Disease Database. Nasu-Hakola Disease. Available from: http://www.malacards.org/card/nasu_hakola_disease?search=nasu+hakola+disease .
- 13.Guerreiro R, Bilgic B, Guven G, Brás J, Rohrer J, Lohmann E, Hanagasi H, Gurvit H, Emre M. A novel compound heterozygous mutation in TREM2 found in a Turkish frontotemporal dementia-like family. Neurobiol Aging. 2013;34:2890.e1–2890.e5. doi: 10.1016/j.neurobiolaging.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chouery E, Delague V, Bergougnoux A, Koussa S, Serre JL, Megarbane A. Mutations in TREM2 lead to pure early-onset dementia without bone cysts. Hum Mutat. 2008;29:E194–204. doi: 10.1002/humu.20836. [DOI] [PubMed] [Google Scholar]
- 15.Giraldo M, Lopera F, Siniard AL, Corneveaux JJ, Schrauwen I, Carvajal J, Muñoz C, Ramirez-Restrepo M, Gaiteri C, Myers AJ, Caselli RJ, Kosik KS, Reiman EM, Huentelman MJ. Variants in triggering receptor expressed on myeloid cells 2 are associated with both behavioral variant frontotemporal lobar degeneration and Alzheimer's disease. Neurobiol Aging. 2013;34:2077.e11–e18. doi: 10.1016/j.neurobiolaging.2013.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klünemann H, Ridha B, Magy L, Wherrett J, Hemelsoet D, Keen R, De Bleecker J, Rossor M, Marienhagen J, Klein H, Peltonen L, Paloneva J. The genetic causes of basal ganglia calcification, dementia, and bone cysts DAP12 and TREM2. Neurology. 2005;64:1502–1507. doi: 10.1212/01.WNL.0000160304.00003.CA. [DOI] [PubMed] [Google Scholar]
- 17.Montalbetti L, Ratti M, Greco B, Aprile C, Moglia A, Soragna D. Neuropsychological tests and functional nuclear neuroimaging provide evidence of subclinical impairment in Nasu-Hakola disease heterozygotes. Funct Neurol. 2005;20:71–75. [PubMed] [Google Scholar]
- 18.Paloneva J, Autti T, Raininko R, Partanen J, Salonen O, Puranen M, Hakola P, Haltia M. CNS manifestations of Nasu-Hakola disease:a frontal dementia with bone cysts. Neurology. 2001;56:1552–1558. doi: 10.1212/wnl.56.11.1552. [DOI] [PubMed] [Google Scholar]