

Abstract
A novel bioconjugation strategy leading to ultrastable gold nanoparticles (AuNPs), utilizing DNA linkers and diluents in place of traditional self-assembled monolayers, is reported. The protective capacity of DNA confers straightforward biomolecular attachment and multistep derivatization capabilities to these nanoparticles and, more significantly, substantially enhances their stability in demanding and complex sensing environments. The DNA/AuNPs were assembled through pH-assisted thiol-gold bonding of single stranded DNA and salt aging, with preconjugated biotin moieties facing outward from the gold surface. These nanoparticles remain a stable colloidal suspension under a wide range of buffers and ionic strengths and can endure multiple rounds of lyophilization while retaining high biological activity. Furthermore, the high stability of the DNA/AuNPs allows for multiple reactions and conjugations to be performed within the colloidal suspensions (i.e., Protein A and antibody binding) for tailored and specific recognition to take place. We have demonstrated the applications of the DNA/AuNPs for colorimetric assays and ELISA feasibility; additionally, SPR imaging analysis of a supported membrane microarray shows excellent results with DNA/AuNPs as the enhancing agent. Together, the properties imparted by this interface render the material suitable for clinical and point-of-care applications where stability, throughput, and extended shelf lives are needed.
Graphic abstract
Gold nanoparticles have been established as ideal signaling agents and enhancement tools for a wide variety of sensing schemes and detection modalities,1–4 yet their practical use can be limited by low colloidal stabilities, especially within high salt media and complex biological samples and through extreme storage conditions.5 While the gold cores are resistant to oxidation, even after being aerosolized or heated,6 shielding of surface charges or application of mechanical forces may lead to irreversible aggregation into larger clusters, followed by their eventual precipitation from solution. Thiolated monolayers have been broadly used for functionalization of gold nanoparticles (AuNPs) and to impart some measure of stability, as they leave the cores coated with, and relatively protected by, a covalently bound alkane or PEG chain.7 However, a systematic investigation conducted by Gao et al. of commonly used thiolated monolayers, including a nonionic PEG-thiol, glutathione, mercaptopropionic acid, cysteine, cysteamine, and dihydrolipoic acid, has revealed that all the monolayers studied, with the exception of the PEG-thiol, resulted in AuNP conjugates that exhibited very little tolerance to pH or ionic strength,8 both of which may vary substantially in analytical samples. While PEG-thiols are clearly preferable for their increased tolerance to these conditions, they are not infallible; nonionic PEG-thiols have been shown to leave AuNPs subject to aggregation following freezing and lyophilization, both of which have been established as common, and sometimes necessary, storage techniques.9
Various biologically inspired and synthetic coatings have been utilized for the stabilization of gold nanoparticles against exposure to high ionic strengths and freeze-drying. Direct adsorption of proteins to the nanoparticle surface represents one of the most straightforward methods to functionalize and stabilize AuNPs and takes place via electrostatic interactions between the protein and the gold surface.10,11 Human serum albumin, for example, has been shown to stabilize AuNPs in up to 0.3 M NaCl,11 though control of protein orientation on the surface can be problematic, as are further ligand derivatizations. Simpler pentapeptide capping ligands, terminated with a functional carboxyl group, have been shown to have sequence-dependent effects on stability, producing colloids that are stable in up to 1 M NaCl and capable of being redispersed after lyophilization.12 Lipid bilayers, while not tested for their ability to shield against high ionic strengths, were also shown to protect AuNPs from the freezing process.13 Some of the most successful protective ligands are synthetic compounds, with large polymers and zwitterionic terminals conferring exceptionally high stability to gold colloids (up to 5 M NaCl and capable of being lyophilized).9,14–16 However, each of these has required in-house synthesis and purification of the ligands prior to attachment, thereby limiting accessibility.
The attachment of thiolated DNA to gold nanoparticles has been extensively explored for drug delivery and sensing, and the fabrication process has undergone multiple iterations to be made widely accessible and controllable.17–20 Similar to peptide capping ligands,12 the stability they confer is also sequence dependent,21 with thymine polynucleotides providing AuNPs that are stable in up to 6.1 M NaCl.22 Moreover, thiolated oligonucleotides, terminated with a wide variety of functional groups, are commercially available from a number of sources. With the above in mind, we set out to utilize DNA linkers and diluents as an alternative to alkane and PEG linkers, for the conjugation of proteins and small molecules to gold nanoparticle surfaces. Herein, we have fabricated DNA/AuNPs conjugated to biorecognition molecules that are exceptionally stable in high ionic strength solutions and complex media (Figure 1). Additionally, they can be lyophilized and resuspended multiple times with retention of biological activity. The DNA/AuNPs are shown to function well for surface plasmon resonance signal enhancement, are capable of being utilized within colorimetric microplate assays, and are robust enough to remain a stable colloid after multiple derivatizations at the nanoparticle surface toward orientation-controlled antibody loading. Taken together, the stability and versatility that this DNA interface offers for effective nanoparticle bioconjugation pathways are unparalleled by most conventional ligands.
Figure 1.
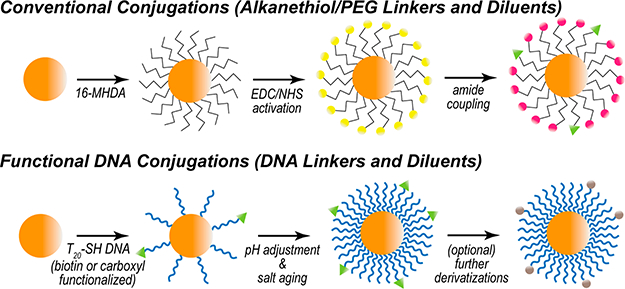
Gold nanoparticle conjugation schemes. (Top) Alkanethiol (16-MHDA) attachment and amide coupling through carbodiimide cross-linking. (Bottom) DNA (T20) attachment, facilitated through pH adjustment and salt aging. Further derivatizations may follow DNA attachment.
EXPERIMENTAL SECTION
Materials and Reagents.
Gold(III) chloride trihydrate, trisodium citrate dihydrate, N-hydroxysuccinimide (NHS), 1- (3-(dimethylamino)propyl)-3-ethylcarbodiimide hydrochloride (EDC), 2-(2’-aminoethoxy) ethanol (AEE), Tween 20, 16- mercaptohexadecanoic acid (16-MHDA), 3,3’,5,5’-tetramethylbenzidine (TMB), horseradish peroxidase (HRP, Type VI), cholera toxin from Vibrio cholerae (CT), anticholera toxin antibody from rabbit (whole antiserum), and Protein A from Staphylococcus aureus were from Sigma-Aldrich (St. Louis, MO). Streptavidin and (+)-biotinyl-3,6,9-trioxaundecanediamine (BA) were from Thermo Scientific (Rockford, IL). 1-Oleoyl-2-palmitoyl-sn-glycero-3-phosphocholine (POPC) and 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(biotinyl) sodium salt (biotin-PE) were from Avanti Polar Lipids (Alabaster, AL). Monosialoganglioside receptor GM1 was from Matreya (Pleasant Gap, PA). Functionalized oligonucleotides were obtained from Integrated DNA Technologies (Coralville, IA), and their sequences are provided in Table 1. Detailed compositions of all buffers used (1× PBS, PBT, PCB, and MES) may be found in the Supporting Information. Nanopure water (≥18 MΩ·cm), purified through a Barnstead E-Pure filtration system (Thermo Scientific, Rockford, IL), was used for all reagent preparations.
Table 1.
Functionalized DNA Oligonucleotide Sequences
| name | 5’ modification | sequence |
| T20 | none | 5’-T20-(CH2)3-SH-3’ |
| bT20 | biotin | 5’-biotin-NH2-(CH2)2O(CH2)2-PO4-T20-(CH2)3-SH-3’ |
| cT20 | carboxyl | 5’-COOH-T-T20-(CH2)3-SH-3’ |
Instrumentation.
Absorbance spectra were obtained using a Cary 50 UV-vis spectrophotometer (Agilent Technologies, Santa Clara, CA), and microplate data was acquired through a PowerWave X-340 (Biotek Instruments, Winooski, VT). Transmission electron microscopy (TEM) was conducted on a Phillips FEI Tecnai 12 TEM (Andover, MA) in UCR CFAMM. Zeta potential analysis was performed using a Delsa Nano C particle analyzer (Beckman Coulter, Brea, CA). Surface plasmon resonance (SPR) spectroscopy and imaging were conducted at room temperature (ca. 23 °C) on a NanoSPR6– 321 (NanoSPR, Addison, IL) and a home-built SPR imaging setup,23 respectively, both using 1× PBS as the running buffer set to a flow rate of 5 mL/h (ca. 83 μL min−1). Nanoglassified gold sensor chips were fabricated in a Class 1000 cleanroom facility (UCR Center for Nanoscale Science & Engineering) according to established methods.24,25
Gold Nanoparticle Functionalization.
Citrate stabilized gold nanoparticles were fabricated by standard citrate reduction and stored in amber bottles at room temperature. Regarding sample handling for all functionalization and cleanup procedures, smaller nanoparticles were separated via centrifugation at 14000 rpm and larger nanoparticles at 8000 rpm. Biotin functionalization via 16-MHDA monolayer coupling to produce biotin/MHDA/AuNPs was performed according the procedure outlined by Aslan et al.26 and is detailed in the Supporting Information. Functionalization of AuNPs with thiolated oligonucleotides followed the methods of Hurst et al.18 and Zhang et al.19 In brief, 2.5 nmol of thiolated DNA (100 μΜ, H2O) was added to 1 mL of as-prepared 13 nm AuNPs (ca. 11 nM). For larger AuNPs (ca. 0.5 nM), 10 nmol of DNA was added.20 Immediately, 20 μL of citrate-HCl (500 mM, pH 3.0) was mixed into the solution, which was thoroughly stirred, sonicated for 20 s, and allowed to rest for 20 min. Thereafter, the ionic strength was adjusted to 1 M NaCl in one step to ensure maximum surface coverage of DNA and, again, stirred and sonicated for 20 s. After incubating at 4 °C overnight, the functionalized AuNPs were cleaned up from excess salts and oligonucleotides through centrifugal filtration (Amicon, MWCO 50 kDa).
Amide coupling of proteins to the cT20/AuNP surfaces was facilitated through carbodiimide cross-linking. The cT20/ AuNPs were suspended in 1 mL of a 50 mM NHS/200 mM EDC mixture in MES for 20 min, after which, the nanoparticles were centrifuged, resuspended in 1.5 mL of PBT, and centrifuged again. The supernatant was discarded, and 50 μg of protein (i.e., HRP or Protein A) was added directly to the pellet and incubated for 2 h at room temperature. Thereafter, the AuNPs were cleaned up from excess protein through centrifugal filtration (Amicon, MWCO 50 or 100 kDa). For the attachment of anti-CT to the Protein A/cT20/AuNP surface, the Protein A-conjugated cT20/AuNPs were centrifuged down, and 50 μg of anti-CT was added directly to the pellet, which was incubated for 4 h at room temperature. Excess antibody was removed from the mixture through multiple cycles (n ≥ 3) of centrifugation/resuspension of the AuNPs in PBT or 1× PBS.
RESULTS AND DISCUSSION
Traditionally, alkane- or PEG-thiols are used in the preparation of gold nanoparticle bioconjugates. These ligands carry the advantages of commercial availability from a large number of sources, and their end terminals can be specifically tailored by the supplier for further desired chemical attachment schemes. Their attachment to gold or silver nanoparticles is met with relative ease, as these compounds exhibit a high packing density upon self-assembly onto gold and silver surfaces. Thiolated DNA, on the other hand, natively exhibits a much lower packing density on these surfaces, owing to high electrostatic repulsion of the phosphate backbone between adjacent strands.17 However, recent innovations in fabrication have rendered the quantitative attachment of DNA to nanoparticles fast and convenient.18,19 Here, we utilize a pH-assisted approach coupled with high salt incubation and sonication to ensure maximal loading of thiolated DNA onto the AuNP surfaces (Figure 1). Given that multiple strands can be attached at user designated ratios in this manner,19 we have decided to exploit this feature toward their use as both a chemical linker and diluent (dependent on their end terminal groups), analogous to how alkane- and PEG-thiols have been used.
Our initial studies targeted the incorporation of a biotinterminus on the DNA linkers (bT20) with a 1:1 ratio of T20 diluent strands, allowing for a universal nanoparticle probe in multiplexed analysis.27 Previous work has demonstrated that biotin may be attached to silver nanoparticles through a short DNA linker;28 here, we utilize a 20-nucleotide polythymine linker for maximum colloidal stability21,22 and attach biotin in conjunction with a diluent strand for improved binding sterics, as previously exemplified.26 The capacity of this nanoparticle conjugate to specifically bind its intended target, streptavidin, is demonstrated through the surface plasmon resonance (SPR) results in Figure 2. A supported lipid bilayer (SLB) platform was chosen for this analysis due to its ability to incorporate specific recognition elements while minimizing nonspecific binding.29 Phosphocholine (POPC) SUVs incorporating 5% (n/n) biotin-PE were flowed over nanoglassified gold sensor chips to form a biotinylated SLB in the analytical channel, while 100% POPC bilayers were formed in the reference channel (I in Figure 2). Thereafter, streptavidin (250 μg mL−1) was added to both channels (II in Figure 2), followed by the bT20/AuNPs (5 nM, III in Figure 2). Each injection and incubation was followed by a 10 min rinse with 1× PBS to remove any unbound molecules. The streptavidin and bT20/AuNPs bind specifically to the biotinylated membrane, whereas the signal changes from either of these complexes binding to the 100% POPC membrane are negligible, thus confirming that the bT20 attachment chemistry was successful and rendered the AuNP recognition specific toward streptavidin. This specificity is applicable to streptavidin not only over the POPC lipids but also over other proteins, as demonstrated in a toxin assay in which the bT20/AuNPs would not bind to the cholera toxin protein until a streptavidin linker was introduced (Figure S1). The results match those obtained from biotin/MHDA/AuNPs fabricated through traditional alkanethiol SAM attachment,26 for which the binding patterns to streptavidin were comparable (Figure S2).
Figure 2.
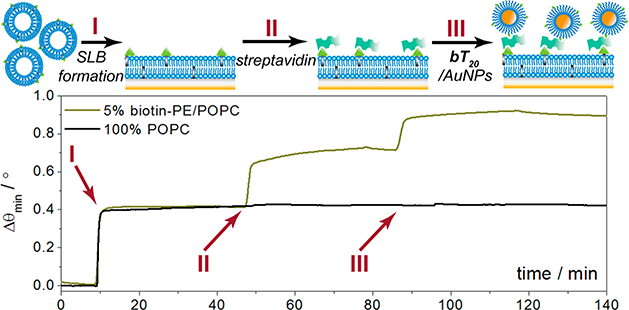
Surface plasmon resonance sensorgram depicting formation of a biotinylated supported lipid bilayer, followed by streptavidin and AuNP recognition. A 100% POPC bilayer is formed in the reference channel and exposed to streptavidin and AuNPs for control purposes.
Stability Against High Salt Concentrations.
The significant advantage of utilizing DNA linkers and diluents in place of conventional monolayers is the enhanced colloidal stability they append to gold nanoparticles. We used a modified protocol of previously established methods to compare the colorimetric shift, indicative of aggregation, of bare nanoparticles (with adsorbed citrate), biotin/MHDA/AuNPs, and bT20/AuNPs when exposed to increasing concentrations of NaCl.9,12,21,22 For each solution, the AuNP concentration was set to 1.5 nM, and designated concentrations of NaCl were added immediately before monitoring the absorbance at 620 nm over the course of 5 min (for details, see the Supporting Information and Figure S3). The absorbance spectra for each of these nanoparticles in nanopure water are shown in Figure 3A, while their aggregation-induced changes in absorbance at 620 nm (ΔΑ620) are shown in Figure 3B. There is a minor increase in the full width at half-maximum (fwhm) for the absorbance peak of the biotin/MHDA/AuNPs in nanopure water compared to the other nanoparticles (Figure 3A), which is likely due to inhomogeneity from the MHDA coupling procedure. As expected, bare nanoparticles exhibited the lowest tolerance to NaCl, with a critical ionic strength parameter (μc), defined as the midpoint between the minimum and maximum ΔΑ620,21 of ca. 35 mM NaCl. The biotin/MHDA/AuNPs fared significantly better than the bare AuNPs (μc ~ 1.5 M NaCl), though the bT20/AuNPs performed best, exhibiting no observable aggregation even when suspended in 4 M NaCl (Figure 3B).
Figure 3.
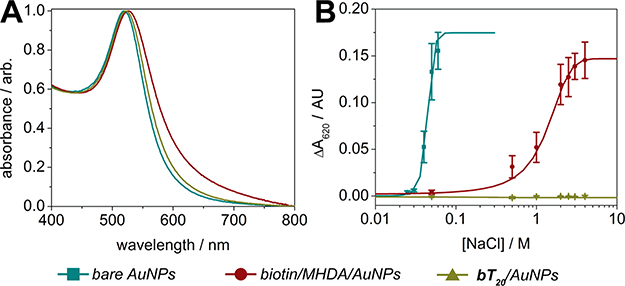
Ionic strength stability assays. (A) Absorbance spectra of AuNPs with various surface functionalizations in nanopure water. (B) Aggregation induced changes in nanoparticle absorbance at 620 nm resulting from varying ionic strength (±SEM, n = 5).
The highly enhanced stability of the bT20/AuNPs is likely due to the high density of negative charge along the DNA phosphate backbone, in line with the high electronegativity of thymine compared to other nucleobases,30 and increased steric repulsion when comparing T20 and hexadecanethiol (lengths measure approximately 6.8 and 2 nm, respectively).31 The correlation between surface charge and colloidal stability is further evidenced in the measured 𝜁-potentials of these nanoparticles, with the T20/AuNPs exhibiting the most negative value (−53 mV) of the conjugated nanoparticles investigated (Table S1). To further understand how the bT20/AuNPs would tolerate a range of electrolytes over longer periods of time, these nanoparticles were separately incubated for 1 h with representative cations from the Hofmeister series (each 4 M, with Cl- as counterion), ranked for their ability to salt out proteins from aqueous solutions.32 The monovalent cations studied appeared to follow standard Hofmeister trends, with the kosmotropes (i.e., Na+ and NH4+) causing no change in bT20/AuNP absorbance over 1 h and the representative chaotrope, guanidinium, resulting in a slight change in ΔΑ620 of ~0.025 AU (Figure S4). The divalent cation, Mg2+, exhibited a larger ΔΑ620 (~0.05 AU), indicating moderate aggregation of the bT20/AuNPs. This is in fact expected as Mg2+ is an effective chelator of the phosphate backbone, and the aggregation effect has been reported in other studies of DNA/AuNPs within extreme levels of Mg2+.21 Pooled human serum and wastewater samples sourced from the Los Angeles River, representing clinical and environmental matrices, were also investigated for their impact on stability and aggregation of the bT20/AuNPs, though neither caused any significant change in the AuNP absorbance over 1 h (Figure S4). Altogether, these data indicate that a remarkably stable and biofunctionalized nanoparticle conjugate can be formed after quantitative attachment of selective DNA strands to the AuNP surface, which in turn can be utilized within a wide variety of matrices and environments.
Lyophilization Endurance.
Freeze-drying of biological materials has been established as an ideal method to preserve structure and activity during long-term storage and transport. While lyophilization of native proteins may be met with some batch variability,33 the technique has overall proved robust enough for clinical and point-of-care applications where stable storage conditions are not guaranteed. Unfortunately, it is significantly challenging to preserve nanaoparticles through lyophilization as they exhibit a strong tendency to irreversibly aggregate and precipitate during freezing.34 While the use of cryoprotective matrices (e.g., sucrose, trehalose) has been presented as one method to preserve the colloidal stability of nanoparticles throughout the process,13,35 direct protection by the nanoparticle’s capping ligand is more desirable as it lends to simplified assay constituents and has been explored with varied degrees of success.9,12,14,35 As the cryoprotectant capacity of conjugated DNA has not been investigated for metal nanoparticles, the bT20/AuNPs and HRP/cT20/AuNPs fabricated here were chosen to be studied for their ability to be resuspended and utilized postlyophilization.
As expected, bare gold nanoparticles (5 nM) could not be effectively reconstituted in a monodisperse manner, exhibiting a massive decrease in absorbance and a colorimetric shift from red to pale blue after lyophilization overnight and resuspension in nanopure water (Figure 4A). The bT20/AuNPs (5 nM), on the other hand, were capable of being lyophilized and resuspended multiple times with minimal or no change in their absorbance (Figure 4B). Moreover, their streptavidin binding capability was left intact postlyophilization, resulting in specific SPR angular shifts that match those of the non-lyophilized bT20/AuNPs (Figure S5). This not only was possible for the smaller nanoparticles (da ~ 13 nm) but also could be accomplished for larger nanoparticles (da ~ 30 nm), which are inherently less stable in solution (Figure S6).
Figure 4.
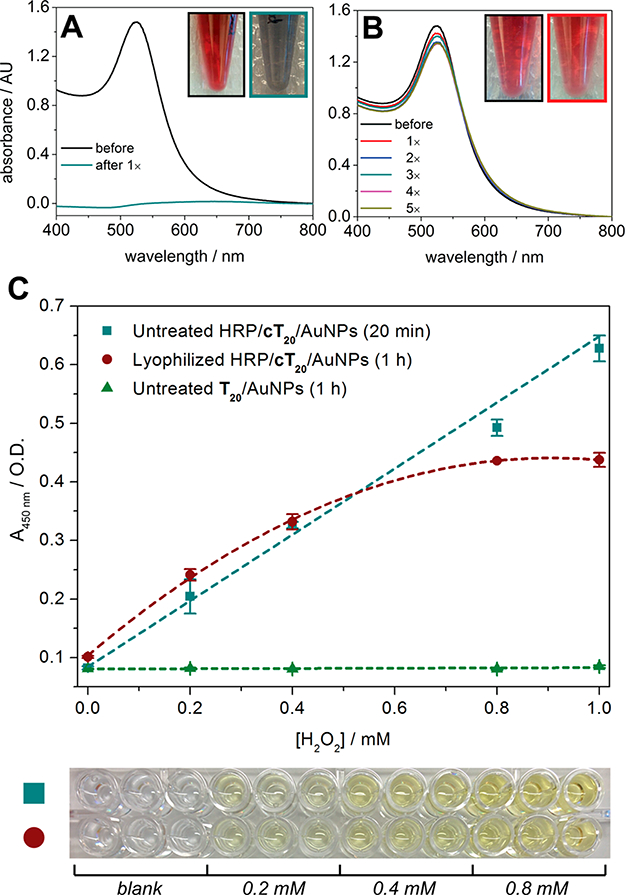
Lyophilization of DNA/AuNPs. (A) Absorbance spectra of bare AuNPs (citrate-capped) before and after lyophilization. (B) Absorbance spectra of bT20/AuNPs before and after five lyophilization cycles. (C) Hydrogen peroxide assay utilizing TMB and HRP/cT20/AuNPs (±SEM, n =3).
Gupta et al. recently demonstrated that the avidin protein could be covalently attached to a custom-synthesized zwitterionic PEG linker on the AuNP surface, which in turn could be lyophilized and functionally resuspended.9 However, retention of enzymatic activity on a nanoparticle surface after lyophilization has yet to be explored. We chose to investigate the conjugation of horseradish peroxidase (HRP), an enzyme that has previously been attached to AuNPs and utilized for ELISA, increasing target sensitivities by multiple orders of magnitude compared to free HRP.36–39 Toward this end, HRP/cT20/AuNPs were fabricated and applied to an enzyme-linked colorimetric microplate assay (see the Supporting Information for the protocol). The HRP/cT20/AuNPs (~333 pM, da ~ 13 nm) exhibited a hydrogen peroxide concentration-dependent response in their ability to generate the oxidized form of TMB (Figure 4C), consistent with results obtained for free HRP (Figure S7). While the nanoparticles remained stable in solution postlyophilization (Figure S8), the enzymatic activity of conjugated HRP was slightly compromised, with the turnover of TMB taking a 3-fold greater length of time and the maximum signal plateauing at a lower substrate concentration (Figure 4C). Interestingly, when free HRP was lyophilized, its activity was affected to a far greater extent than the AuNP-conjugated HRP, generating almost no colorimetric signal over the greater length of time, even for the highest concentrations (0.8–1 mM) of hydrogen peroxide tested (Figure S7). To confirm whether nanozyme activity (i.e., the inherent property of metal nanoparticles to exhibit peroxidase-mimicking characteristics) could explain the postlyophilization activity of the HRP/cT20/AuNPs,40,41 untreated T20/AuNPs were tested. No signal was generated for any substrate concentration (Figure 4C), thus excluding any nanozyme contribution from the observation. This is likely due to inaccessibility of TMB or hydrogen peroxide to the metal surface, as the HRP/cT20/AuNPs and T20/AuNPs were fully coated with DNA from the attachment procedure used.
The results suggest that, while AuNP-conjugated proteins are still subject to some degree of secondary structural changes that may lead to altered activity after freeze-drying,42,43 some level of protection resulting from the surface conjugation is imparted. The presence of covalently attached HRP or biotin does not appear to negatively affect the stability of the nanoparticles themselves, as evidenced from their ability to be lyophilized and resuspended with no colorimetric shift, providing a unique system to study the biological stability effects of ligand attachment. Beyond this proof-of-concept study indicating that bT20/AuNPs and HRP/cT20/AuNPs may be lyophilized and used thereafter, we believe the DNA conjugated particles reported here could potentially find broad applications in enzymatic assays where long-term storage of nanoparticle/enzyme conjugates is critical.
Enhanced SPR Imaging Analysis with Multistep Conjugation Products.
Control of protein orientation during surface attachment has attracted great attention for nanoparticle bioconjugates. Most attachment schemes may result in undesired or highly variable orientations, as covalent couplings are dependent upon accessible functional groups on the protein surface (e.g., free primary amines, carboxyls, thiols) and electrostatic adsorption depends on the localized areas of charge on the surface. In a worst-case scenario for antibodies, these complexes may be bound through the Fab region, blocking the antigen recognition site and rendering the nanoparticle nonselective toward the intended target. Various solutions for controlling the orientation have included altering the surface density of protein,44 reversing the surface charge of the nanoparticle,45 and attachment of antibodies through Protein G,46 which binds to the Fc region of antibodies. In the study in which Protein G was used, the protein had to be engineered to express a cysteine residue at the n-terminus, which was attached to an aminated DNA linker offline and bound to the gold surface through complementary DNA base pairing. Due to the high stability of the cT20/AuNPs, we were able to directly attach unmodified Protein A (PA), which also specifically targets antibody Fc regions, to the DNA/AuNP interface and subsequently bind an antibody of choice. Both 13 and 30 nm cT20/AuNPs remained stable through multiple rounds of derivatization and cleanup, as exhibited through the absorbance spectra in Figure 5. Indicative of surface attachment and consistent with the LSPR mechanism, the absorbance peaks also shifted toward higher wavelengths as Protein A and anticholera toxin (anti-CT) were immobilized to the cT20/ AuNPs.
Figure 5.
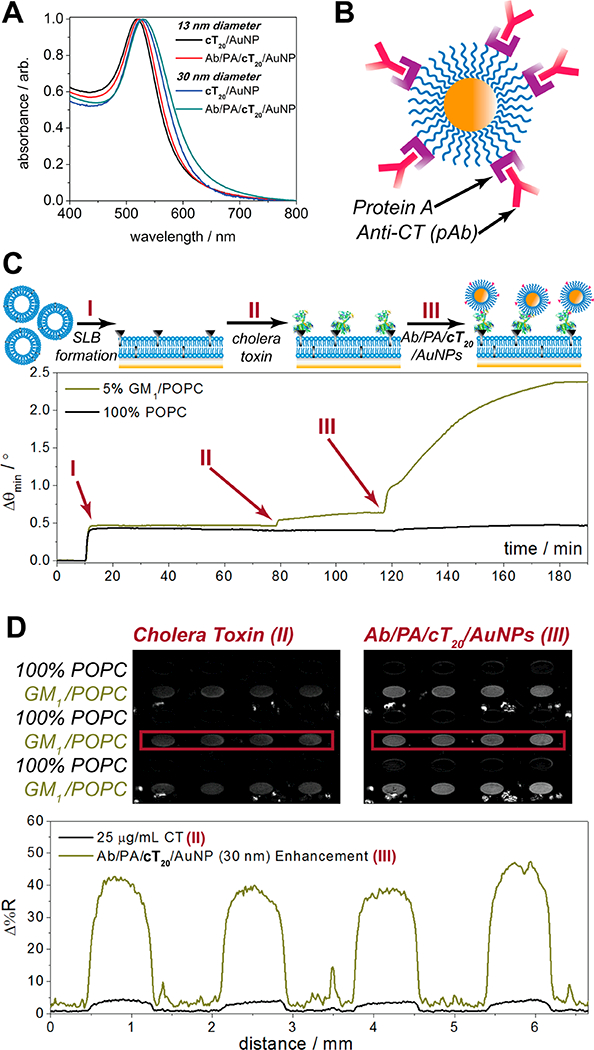
Multistep conjugation of cT20/AuNPs with Protein A and anticholera toxin. (A) Absorbance spectra of 13 and 30 nm cT20/AuNPs before and after Protein A and antibody conjugation. (B) Cartoon illustration of the final anti-CT/PA/cT20/AuNP bioconjugate. (C) Surface plasmon resonance sensorgram depicting formation of a ganglioside-impregnated supported lipid bilayer, followed by cholera toxin and AuNP recognition, with a 100% POPC bilayer in the reference channel. (D) SPR imaging results of cholera toxin and AuNP recognition across a lipid membrane array.
We tested the use of anti-CT/PA/cT20/AuNP toward the nanoparticle-enhanced detection of cholera toxin by SPR (Figure 5C) and SPR imaging (Figure 5D) in a standard sandwich assay format. Similar to the bT20/AuNP SPR binding assays, a supported lipid bilayer was used to host a membrane-bound ganglioside receptor, GM1, which is specific to cholera toxin. Following the formation of a POPC bilayer containing 5% (n/n) GM1 in the analytical channel and 100% POPC in the reference channel (I in Figure 5C), 20 μg mL−1 cholera toxin was incubated for 30 min on both membrane surfaces (II in Figure 5C). After a 10 min rinse with 1× PBS, 2 nM anti-CT/PA/cT20/AuNPs (da ~ 30 nm) were introduced to both channels (III in Figure 5C), which were incubated for 1 h before a final rinse with 1× PBS. The enhancement seen here is strong and specific for the GM1 impregnated membranes, with no binding of cholera toxin or the anti-CT/PA/cT20/AuNPs exhibited in the reference channel. This high level of signal amplification for cholera toxin is in line with those reported previously from our group,47,48 though here could be achieved in far fewer assay steps due to the majority of the constituents (i.e., antibodies and linkers) being preconjugated to the AuNP surface. Furthermore, the use of these nanoparticles was scalable toward their specific enhancement in a microarray format. For SPR imaging analysis, the difference images of a 4 × 6 SLB array are presented in Figure 5D. The individual array elements were formed through trehalose-assisted vesicle deposition, with alternating rows of 5% (n/n) GM1/POPC SUVs and 100% POPC SUVs protected from intermixing and the effects of desiccation through a sacrificial, anhydrobiotic trehalose matrix.25,49 Upon rehydration, the array was rinsed under a 1× PBS flow for 30 min to remove the trehalose and form spatially defined SLBs, prior to conducting the same SPR sandwich assay for cholera toxin as described above. The difference images, obtained by digitally subtracting a prebinding image from a postbinding image, show similar patterns to the SPR sensorgrams in Figure 5C, with a significant increase in contrast between the 5% (n/n) GM1/POPC and 100% POPC array elements after the anti-CT/PA/cT20/AuNPs were introduced (corresponding line profiles shown in the lower panel of Figure 5D). The nanoparticle enhanced signals for the GM1/POPC array elements exhibited >10-fold amplification, consistent with previous CT signal amplification work from our group,48 with values of 3.8 ± 0.5 Δ%R prior to nanoparticle enhancement and 41.8 ± 6.4 Δ%R after nanoparticle enhancement. By combining this level of amplification with trehalose-mediated vesicle preservation, the large-scale analysis and signal enhancement across an entire array of varied constituents can take place in ~2 h. Moreover, the specificity exhibited in this nanoparticle-enhanced microarray platform may be generally applied beyond the cholera toxin/GM1 system, as Protein A conjugated to the cT20/AuNPs can bind most antibodies of choice for tailored, high-throughput, and ultrasensitive assay development.
CONCLUSIONS
In summary, we have shown that 20-nucleotide polythymine DNA sequences function effectively as a linker and diluent for preparing gold nanoparticle bioconjugates for analytical applications. The stability they append toward high ionic strengths and complex media is exceptional, exhibiting no traceable aggregation of nanoparticles in a number of testing media including 4 M NaCl, 4 M NH4Cl, Los Angeles River water, and human serum. Only a slight change in absorbance in 4 M guanidinium chloride and 4 M MgCl2 was noted, suggesting the DNA/AuNP suspensions are generally stable in these harsh conditions, and only mild aggregation had occurred. Lyophilization and resuspension of the DNA/AuNPs was possible, with retention of activity for horseradish peroxidase, offering a method of preservation that is not accessible for most nanoparticle conjugates. While there seems to be a protective effect provided by the nanoparticles to the conjugated enzyme during lyophilization, further studies need to be undertaken to understand this mechanism and its amenability to other proteins. Nevertheless, both this observation and the line of study are only made possible as a result of the high stability offered by the unique interface we have developed. Additionally, the cT20/AuNPs proved successful for microplate and microarray assays, two standard techniques for high-throughput detection and characterization, demonstrating the versatility and selectivity of this material. We envision wide adoption of this interface toward stable and specific nanoparticle bio-conjugates throughout multiplexed assay development for clinical, environmental, and point-of-care settings, given the unparalleled sensitivity offered by nanoparticle enhancement, and the convenience of these commercially available and accessible DNA ligands.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge financial support from the National Science Foundation (CHE-1413449). S.S.H. was supported by a University of California, Riverside (UCR) Dissertation Year Program (DYP) fellowship and an NIEHS T32 training grant (T32 ES018827). We give thanks to Prof. Cynthia K. Larive and her students for use of the lyophilizer, Prof. Yadong Yin for use of the particle analyzer, and Steven Pham for nanoparticle synthesis.
Footnotes
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.anal-chem.7b00341.
Experimental details including buffer compositions, sensor chip fabrication, vesicle preparation, gold nanoparticle syntheses, and modified assay protocols. Table providing conjugated AuNP zeta potentials. Figures including results for bT20/AuNP specificity (Figure S1), the biotin/MHDA/AuNP SPR experiment (Figure S2), ionic strength stability assays (Figure S3), stabilities within other salts and media (Figure S4), lyophilized 13 nm bT20/AuNP SPR experiment (Figure S5), lyophilized 30 nm bT20/AuNP experiments (Figure S6), free HRP assays (Figure S7), lyophilized HRP/cT20/AuNP absorbance (Figure S8), and representative TEM images of the synthesized nanoparticles (Figure S9) (PDF)
REFERENCES
- (1).Zhou W; Gao X; Liu DB; Chen XY Chem. Rev 2015, 115, 10575–10636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Kumar A; Kim S; Nam JM J. Am. Chem. Soc 2016, 138, 14509–14525. [DOI] [PubMed] [Google Scholar]
- (3).Li T; Wu X; Liu F; Li N Analyst 2017, 142, 248–256. [DOI] [PubMed] [Google Scholar]
- (4).Syedmoradi L; Daneshpour M; Alvandipour M; Gomez FA; Hajghassem H; Omidfar K Biosens. Bioelectron 2017, 87, 373–387. [DOI] [PubMed] [Google Scholar]
- (5).Zhou JF; Ralston J; Sedev R; Beattie DA J. Colloid Interface Sci 2009, 331, 251–262. [DOI] [PubMed] [Google Scholar]
- (6).Balasubramanian SK; Yang LM; Yung LYL; Ong CN; Ong WY; Yu LE Biomaterials 2010, 31, 9023–9030. [DOI] [PubMed] [Google Scholar]
- (7).Zeng SW; Yong KT; Roy I; Dinh XQ; Yu X; Luan F Plasmonics 2011, 6, 491–506. [Google Scholar]
- (8).Gao J; Huang XY; Liu H; Zan F; Ren JC Langmuir 2012, 28, 4464–4471. [DOI] [PubMed] [Google Scholar]
- (9).Gupta A; Moyano DF; Parnsubsakul A; Papadopoulos A; Wang LS; Landis RF; Das R; Rotello VM ACS Appl. Mater. Interfaces 2016, 8, 14096–14101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Shang L; Wang YZ; Jiang JG; Dong SJ Langmuir 2007, 23, 2714–2721. [DOI] [PubMed] [Google Scholar]
- (11).Canaveras F; Madueno R; Sevilla JM; Blazquez M; Pineda TJ Phys. Chem. C 2012, 116, 10430–10437. [Google Scholar]
- (12).Levy R; Thanh NTK; Doty RC; Hussain I; Nichols RJ; Schiffrin DJ; Brust M; Fernig DGJ Am. Chem. Soc 2004, 126, 10076–10084. [DOI] [PubMed] [Google Scholar]
- (13).Zhang L; Li P; Li D; Guo S; Wang E Langmuir 2008, 24, 3407–3411. [DOI] [PubMed] [Google Scholar]
- (14).Mangeney C; Ferrage F; Aujard I; Marchi-Artzner V; Jullien L; Ouari O; Rekai ED; Laschewsky A; Vikholm I; Sadowski JW J. Am. Chem. Soc 2002, 124, 5811–5821. [DOI] [PubMed] [Google Scholar]
- (15).Rouhana LL; Jaber JA; Schlenoff JB Langmuir 2007, 23, 12799–12801. [DOI] [PubMed] [Google Scholar]
- (16).Yang W; Zhang L; Wang SL; White AD; Jiang SY Biomaterials 2009, 30, 5617–5621. [DOI] [PubMed] [Google Scholar]
- (17).Mirkin CA; Letsinger RL; Mucic RC; Storhoff JJ Nature 1996, 382, 607–609. [DOI] [PubMed] [Google Scholar]
- (18).Hurst SJ; Lytton-Jean AKR; Mirkin CA Anal. Chem 2006, 78, 8313–8318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Zhang X; Servos MR; Liu JW J. Am. Chem. Soc 2012, 134, 7266–7269. [DOI] [PubMed] [Google Scholar]
- (20).Zhang X; Gouriye T; Goeken K; Servos MR; Gill R; Liu JW J. Phys. Chem. C 2013, 117, 15677–15684. [Google Scholar]
- (21).Storhoff JJ; Elghanian R; Mirkin CA; Letsinger RL Langmuir 2002, 18, 6666–6670. [Google Scholar]
- (22).Heo JH; Cho HH; Lee JH Analyst 2014, 139, 5936–5944. [DOI] [PubMed] [Google Scholar]
- (23).Wilkop T; Wang ZZ; Cheng Q Langmuir 2004, 20, 11141–11148. [DOI] [PubMed] [Google Scholar]
- (24).Abbas A; Linman MJ; Cheng Q Anal. Chem 2011, 83, 3147–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Hinman SS; Ruiz CJ; Drakakaki G; Wilkop TE; Cheng Q ACS Appl. Mater. Interfaces 2015, 7, 17122–17130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Aslan K; Luhrs CC; Perez-Luna VH J. Phys. Chem. B 2004, 108, 15631–15639. [Google Scholar]
- (27).Scott AW; Garimella V; Calabrese CM; Mirkin CA Bioconjugate Chem 2017, 28, 203–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Cunningham JC; Kogan MR; Tsai YJ; Luo L; Richards I; Crooks R M. ACS Sensors 2016, 1, 40–47. [Google Scholar]
- (29).Phillips KS; Han JH; Martinez M; Wang ZZ; Carter D; Cheng Q Anal. Chem 2006, 78, 596–603. [DOI] [PubMed] [Google Scholar]
- (30).Zhang Q; Chen ECM Biochem. Biophys. Res. Commun 1995, 217, 755–760. [DOI] [PubMed] [Google Scholar]
- (31).Malinsky MD; Kelly KL; Schatz GC; Van Duyne RP J. Am. Chem. Soc 2001, 123, 1471–1482. [Google Scholar]
- (32).Xie WJ; Gao YQ J. Phys. Chem. Lett 2013, 4, 4247–4252. [DOI] [PubMed] [Google Scholar]
- (33).Wahl V; Khinast J; Paudel A TrAC, Trends Anal. Chem 2016, 82, 468–491. [Google Scholar]
- (34).Abdelwahed W; Degobert G; Stainmesse S; Fessi H Adv. Drug Delivery Rev 2006, 58, 1688–1713. [DOI] [PubMed] [Google Scholar]
- (35).Alkilany AM; Abulateefeh SR; Mills KK; Yaseen AIB; Hamaly MA; Alkhatib HS; Aiedeh KM; Stone JW Langmuir 2014, 30, 13799–13808. [DOI] [PubMed] [Google Scholar]
- (36).Ambrosi A; Airo F; Merkoci A Anal. Chem 2010, 82, 1151–1156. [DOI] [PubMed] [Google Scholar]
- (37).Zhou Y; Tian XL; Li YS; Pan FG; Zhang YY; Zhang JH; Yang L; Wang XR; Ren HL; Lu SY; Li ZH; Chen QJ; Liu ZS; Liu JQ Biosens. Bioelectron. 2011, 26, 3700–3704. [DOI] [PubMed] [Google Scholar]
- (38).Zhan L; Wu WB; Yang XX; Huang CZ New J. Chem 2014, 38, 2935–2940. [Google Scholar]
- (39).Guo Q; Han JJ; Shan S; Liu DF; Wu SS; Xiong YH; Lai WH Biosens. Bioelectron 2016, 86, 990–995. [DOI] [PubMed] [Google Scholar]
- (40).McKeating KS; Sloan-Dennison S; Graham D; Faulds K Analyst 2013, 138, 6347–6353. [DOI] [PubMed] [Google Scholar]
- (41).Wang XY; Hu YH; Wei H Inorg. Chem. Front 2016, 3, 41–60. [Google Scholar]
- (42).Griebenow K; Klibanov AM Proc. Natl. Acad. Sci. U. S. A 1995, 92, 10969–10976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Roy I; Gupta MN Biotechnol. Appl. Biochem 2004, 39, 165–177. [DOI] [PubMed] [Google Scholar]
- (44).Liu F; Wang L; Wang HW; Yuan L; Li JW; Brash JL; Chen H ACS Appl. Mater. Interfaces 2015, 7, 3717–3724. [DOI] [PubMed] [Google Scholar]
- (45).Lin W; Insley T; Tuttle MD; Zhu LY; Berthold DA; Kral P; Rienstra CM; Murphy CJ J. Phys. Chem. C 2015, 119, 21035–21043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Jung Y; Lee JM; Jung H; Chung BH Anal. Chem 2007, 79, 6534–6541. [DOI] [PubMed] [Google Scholar]
- (47).Liu Y; Dong Y; Jauw J; Linman MJ; Cheng Q Anal. Chem 2010, 82, 3679–3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Liu Y; Cheng Q Anal. Chem 2012, 84, 3179–3186. [DOI] [PubMed] [Google Scholar]
- (49).Wilkop TE; Sanborn J; Oliver AE; Hanson JM; Parikh AN J. Am. Chem. Soc 2014, 136, 60–63. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.