

Abstract
The inheritance of mitochondrial DNA (mtDNA) from mother to child is complicated by differences in the stability of the mitochondrial genome. Although the germ line mtDNA is protected through the minimization of replication between generations, sequence variation can occur either through mutation or due to changes in the ratio between distinct genomes that are present in the mother (known as heteroplasmy). Thus, the unpredictability in transgenerational inheritance of mtDNA may cause the emergence of pathogenic mitochondrial and cellular phenotypes in offspring. Studies of the role of mitochondrial metabolism in cancer have a long and rich history, but recent evidence strongly suggests that changes in mitochondrial genotype and phenotype play a significant role in the initiation, progression and treatment of cancer. At the intersection of these two fields lies the potential for emerging mtDNA mutations to drive carcinogenesis in the offspring. In this review, we suggest that this facet of transgenerational carcinogenesis remains underexplored and is a potentially important contributor to cancer.
Keywords: transplacental, mitochondrion, mutagenesis, heteroplasmy, cancer
Introduction to mitochondria and its genome
Mitochondria are thought to have arisen from an endosymbiotic event to form the first organelle in the cell [Margulis 1970]. Over millennia, the double membraned organelle has become the site of many key biosynthetic and metabolic processes that require compartmentalization. In this partnership with the cell, most of the mitochondrial-localized gene products have become encoded in the nucleus, where they are transcribed, cytoplasmically expressed, and then imported into the mitochondria. The most well known of the mitochondrial functions is oxidative phosphorylation, a process of efficient conversion of carbohydrates and fats with oxygen into energy in the form of ATP and carbon dioxide.
Oxidative phosphorylation (OXPHOS) complexes are unique in that subunits are encoded by either the nuclear or mitochondrial genome, making this essential function bigenomic in origin [Scarpulla 2008]. The mitochondrial DNA (mtDNA) is a small 16.5kb, multi-copy genome that encodes 37 genes and is located in the mitochondrial matrix (Figure 1). The mitochondrial genes include 13 protein-coding genes, plus two ribosomal RNAs and 22 tRNA required for protein translation. All of the protein machinery required for genome replication, transcription, RNA processing and translation are nuclear encoded. The proteins encoded by the mtDNA are core, hydrophobic subunits of four of the five OXPHOS complexes, and the sequences have been potentially retained due to complications of encoding these proteins in the nucleus. The bigenomic nature also creates the potential for interplay between the nuclear and mitochondrial compartments to affect mitochondrial respiratory function [Lane 2011].
Figure 1.
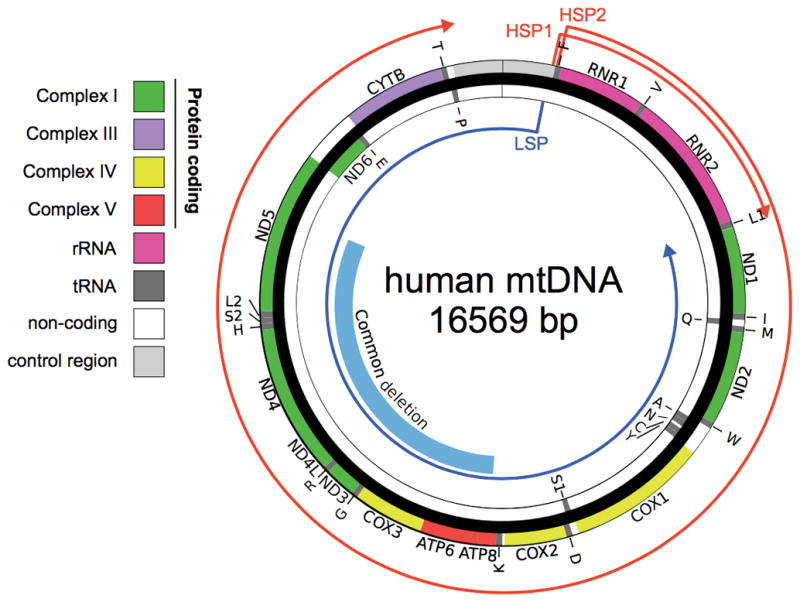
Map of the human mtDNA with genes and transcripts marked. The mitochondrial genome is a 16,569 b.p. circular DNA. The region eliminated in the “common deletion” is shown in blue. Genes are encoded on either strand, shown separated into inner and out strand for clarity. The nascent transcript originating from heavy strand promoters (HSP) 1 and 2 are shown in red, from the light strand promoter (LSP) in blue. Regions of each strand are color coded by oxidative phosphorylation complex (protein coding), ribosomal RNA (rRNA), transfer RNA (tRNA), non-coding, and control region.
Mitochondrial genome stability
The mitochondrial genome is in close proximity to the OXPHOS complexes, which are major sites of reactive oxygen species (ROS) generation in the form of superoxide. Superoxide is converted by dismutases to hydrogen peroxide, which is membrane diffusible. However, through Fenton chemistry, dependent upon the iron which is present at high levels in mitochondria [Seo et al. 2008], hydrogen peroxide can be converted to hydroxide ions [Imlay et al. 1988], which cause single or double strand breaks in DNA [Cassano et al. 2002]. This proximity to the source of reactive oxygen species is thought to contribute to the increased rates of mtDNA damage [Yakes and Van Houten 1997] and potentially mutation [Shokolenko et al. 2009] relative to the nucleus. Furthermore, mtDNA lacks many of the nuclear DNA repair pathways, and is slower to recover from the insult [Yakes and Van Houten 1997; Liu and Demple 2010]. The accumulation of damage and mutations negatively impacts mtDNA sequence integrity, primarily with age [Li et al. 2017; Neuhaus et al. 2017; Sondheimer et al. 2011], but the inheritance of existing mutations or the acquisition of mutations during oocyte maturation and embryogenesis is the primary cause of mtDNA-borne diseases [Alston et al. 2017].
Unlike nuclear DNA, which is contributed to equally by both mother and father, mtDNA is exclusively transferred from the mother [Giles et al. 1980]. The prevention of paternal transmission occurs through several mechanisms, including low incorporation of sperm mitochondria into the zygote, ubiquitination and destruction of the sperm mitochondria, and paternal mtDNA destruction by nucleases [Sato and Sato 2013]. Direct evidence as to the advantage for this clonal transmission has recently been suggested in model organisms. Paternal mitochondrial elimination is thought to aid in maintaining a single mitochondrial population of normal sequence, as interfering with paternal mtDNA destruction interferes with embryo survival in C. elegans [Wang et al. 2016]. Although the cause and consequence of this process is an area of on-going research, it is clear that the matrilineal transmission drives clonal mtDNA transmission.
The selective transmission of mtDNA provides stability to the genome sequence, which is extremely useful for tracking ancestry and migration. The evolutionary clock of mtDNA is fast enough to detect changes during recent human evolution, along the time scale of the emergence from Africa [Rito et al. 2013]. The co-inherited polymorphisms that help define modern day populations and their origins are called “haplogroups”. These variations have been thought to be stable, neutral changes in the genome, but increasingly, evidence suggests that these sequences interact with the sequences of the nuclear genome to modify overall mitochondrial function [Suissa et al. 2009; Cohen et al. 2016].
Heteroplasmy and its consequences
Maternal transmission from an ostensibly healthy parent does not eliminate the risk of disease because of heteroplasmy. Heteroplasmy is common feature of organellar genomes. It refers to the state that occurs when more than one allelic variant of the mitochondrial sequence is present. Because of their variable copy number, the nuclear concept of heterozygosity is inapplicable. Heteroplasmy is very commonly observed in human mtDNA samples, with progressively greater levels present with increasing age [Sondheimer et al. 2011].
When mitochondrial mutations have functional consequences, the degree of heteroplasmy (often referred to as the “load” of the variant or pathogenic allele) is one of the factors that determine disease phenotype. This has been extensively demonstrated in studies of mitochondrial mutations causing classical mitochondrial pathologies [Chinnery et al. 1997]. For example, the m.3243A>G mutation, most often associated with mitochondrial encephalopathy, lactic acidosis and stroke-like episodes (MELAS), is associated with earlier ages of onset at higher loads of heteroplasmy. Furthermore, the phenotype expressed can be different at different loads of mutation, possibly resulting from differences in nuclear gene expression responses with increasing mutation load [Picard et al. 2014]. Lower loads might be associated with maternally inherited diabetes, whereas higher loads cause the more lethal MELAS phenotype.
Heteroplasmy is also challenging to evaluate at an organismal level because it is unstable. If the tissues of a heteroplasmic individual are separately evaluated, it is apparent that the load of mutation can be widely variable [He et al. 2010]. This is particularly evident in pre-cancerous and cancerous tissues [Sui et al. 2006]. Heteroplasmy is also variable as a function of time. Multiple studies have demonstrated increasing heteroplasmy in individuals as they age [Barker and Murthy 2009; Sondheimer et al. 2011; Murdock et al. 2000].
Perhaps the most perplexing aspect of heteroplasmy (and indeed mitochondrial genetics) is the shifts in heteroplasmy between generations. The birth of children with mtDNA-dependent mitochondrial disorders from mothers with no evidence of disease implies that changes in heteroplasmy or novel mutations can and do occur. In a series of three children born with fatal and infantile-onset Leigh syndrome due to m.10158T>C mutation, the mutation was only detectable in only one of the three mothers [McFarland et al. 2004]. In the mother where it was observed, the level of mutation had progressed from 7% in her fibroblasts to >98% in tissues tested in her offspring.
Thus, although mutations in mtDNA can arise de novo during oocyte generation, observed shifts in heteroplasmy between mothers and their children are a variance due to sampling of maternal mtDNA through the “bottleneck” of oocyte formation, leading to altered distribution of heteroplasmy in the offspring (Figure 2). This bottleneck occurs because of a reduction in the number of mitochondrial genomes that are transmitted between generations through the oocyte line. Despite the fact that there can be ~106 copies of mtDNA in some cell types, the effective number of genomes that makes it through this bottleneck is orders of magnitude smaller [Marchington et al. 1998], although strikingly difficult to calculate and even distinct for specific mutations [Wilson et al. 2016].
Figure 2.
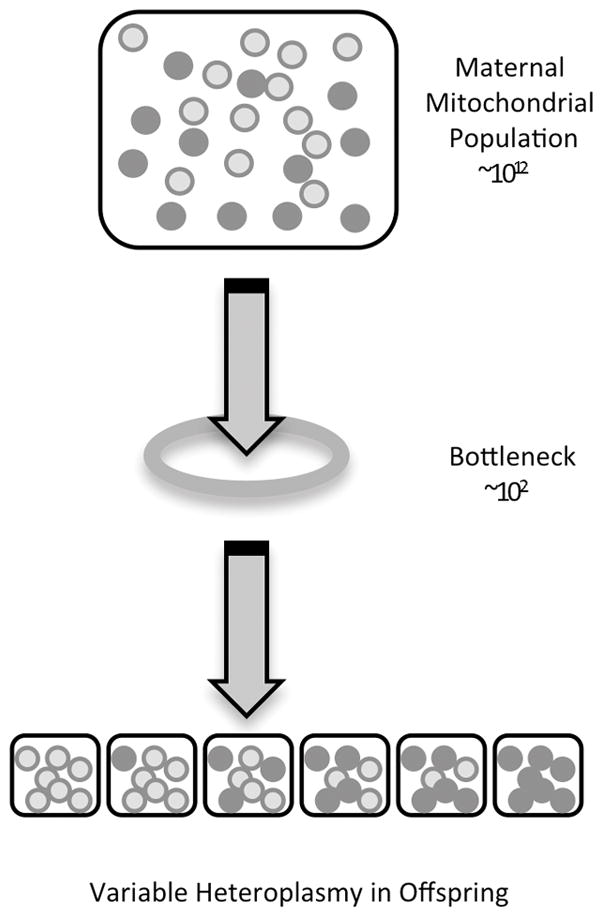
The mitochondrial bottleneck between generations allows for a distribution of heteroplasmy in offspring. The small number of molecules at the bottleneck limits the number maternal mtDNA inherited to produce varied heteroplasmy outcomes.
The existence of the bottleneck was most elegantly proven in studies of oocytes obtained at oophorectomy from a known carrier of the m.3243A>G mutation [Brown et al. 2001] and another of a woman carrying a mitochondrial deletion [Marchington et al. 1998]. These studies demonstrated that even for pathogenic mutations was no bias present in the selection process and that the derived heteroplasmies in the oocytes were effectively a normal distribution of the mother’s own heteroplasmy. Work in animal models has suggested that only very severe and high heteroplasmy mutations can be counter-selected by purifying selection over generations [Fan et al. 2008]. These studies and others also suggest that the number of effective mitochondrial genomes transmitted across the bottleneck is extremely small compared to the number of mitochondrial genomes present in the oocyte at fertilization, allowing considerable shifts in heteroplasmic ratios from mother to child.
Furthermore, once inherited, certain pathogenic mtDNA mutation levels can exhibit vast differences in the degree of observed heteroplasmy by adulthood. For example, pairs of monozygotic twins were found to carry very similar m.3243A>G mutation levels across multiple tissues, where some tissues like blood contained <10% heteroplasmy whereas the heart and brain contained a mutational load >80% [Maeda et al. 2016]. Together with the bottleneck influencing maternal transmission of mtDNA mutations, tissue-specific factors during development can also influence the dynamics of heteroplasmy across the lifespan [Picard and Hirano 2016].
Mitochondrial DNA operates within a nuclear context
Although mtDNA is derived from an organism once capable of independent life, the mtDNA now works cooperatively with the nuclear genome. All of the mtDNA-encoded polypeptides function within the electron transport chain or as a component of the F0F1-ATPase (Complex V). In all cases, the encoded mitochondrial proteins physically interact with nuclear-encoded partners. Furthermore, the regulation of mtDNA transcription and replication are entirely under the control of nuclear proteins [Falkenberg et al. 2007].
The interaction between mitochondrial and nuclear encoded gene products, and the use of nuclear-encoded proteins in genome regulation, creates the potential for loss of fitness when the mitochondrial and nuclear genomes are not well co-adapted. Many of the polymorphic variants that define the human haplogroups have an impact on functions like mitochondrial transcription [Suissa et al. 2009; Cohen et al. 2016]. If these variants have negative interactions with poorly co-adapted nuclear variation, overall mitochondrial activity could be impacted.
This loss of fitness due to differing ancestry of nuclear and mtDNA has been shown in studies of conplastic animals. A conplastic animal is created by transferring the mitochondrial genome of one strain into the nuclear context of another by repeatedly backcrossing to males. Conplastic mice have been found to have a range of problems including defects in behavior, susceptibility to autoimmune disorders, susceptibility to metabolic syndrome and changes in longevity [Yu et al. 2009; Latorre-Pellicer et al. 2016; Betancourt et al. 2014]. Many of these defects may be initiated by changes in the production of free radicals by mitochondrial with poorly aligned genetic input [Fetterman et al. 2013].
It is uncertain whether differences between human haplogroup and human nuclear variation will have a similar effect. One of us has recently developed a system for quantifying the discrete ancestry of mitochondrial and nuclear information, based upon genotyping performed on genome wide single-nucleotide polymorphism arrays. These studies showed that spontaneous preterm birth was more likely in infants where discrete mitochondrial ancestry was present [Crawford et al. 2017]. Future studies will be required to identify the range of phenotypes that may be associated with larger evolutionary distance between inherited nuclear and mitochondrial DNA.
The role of the mitochondrion in the cancer cell
A hallmark of cancer cell is a shift away from oxidative phosphorylation as major source of energy, in favor of glycolysis [Hanahan and Weinberg 2011]. This phenomenon was first recognized by Otto Warburg [Warburg 1956] and thereafter named the Warburg effect. Although the initial assumption from this observation was that mitochondria became “unimportant” or dispensable in cancer cells, this view has changed substantially in recent years due to evidence that mitochondria – even if not the powerhouse of the cancer cell – contribute significantly to growth [Ward and Thompson 2012]. Indeed, the loss of respiratory chain function causes proliferative failure of cancer cells [Birsoy et al. 2015; Dong et al. 2017]. Together, these data demonstrate that even if mitochondria are no longer providing most of the cellular ATP in cancer cells, mitochondria are generally essential for oncogenesis.
One reason for this phenomenon is that in the transformed cell, mitochondria are actively involved in anabolic pathways required for rapid cell growth [Ward and Thompson 2012]. Glucose and glutamine oxidation generate citrate and acetyl-CoA for the synthesis of lipids [Vander Heiden et al. 2009], a process termed reductive carboxylation that favors cell growth [Mullen et al. 2011]. Mitochondria also release soluble chemical signals, including reactive oxygen species, that contribute to the activation of hypoxia-inducible genes (i.e., pseudohypoxic state) [Al-Mehdi et al. 2012] and act as mitogenic signals [Weinberg et al. 2010]. The antineoplastic effects of certain drugs, such as metformin, may also act in part by preventing reductive carboxylation, citrate production, and de novo lipid biosynthesis [Griss et al. 2015]. The production of oncometabolites by cells with mutations in genes encoding the Krebs cycle enzymes succinate dehydrogenase subunits B, C, and D (SDHB, SDHC, SDHD) [Ishii et al. 2005; van Gisbergen et al. 2015], and isocitrate dehydrogenase (IDH1/2) may also favor oncogenic transformation and be responsible for a small group of central nervous system tumors including astrocytomas, oligodendrogliomas, and secondary glioblastomas [M Gagné et al. 2017; Wallace 2012]. Thus, overall, clinical and pre-clinical models suggest that mitochondria actively contribute to tumorigenesis via intermediate metabolism and metabolic reprogramming.
The mitochondrion, genetics and cancer
Three major lines of evidence implicate the mitochondrial genome in cancer. The least understood is the integration of nuclear mitochondrial DNA segments (NUMTs) into the nuclear genome [Hazkani-Covo et al. 2010]. The first report to bring attention to this issue identified large mtDNA sequences – some almost the entire mtDNA sequence (16,556 bp) integrated into the tumor nuclear genome [Ju et al. 2015]. The presence of mtDNA-derived DNA sequences was extensively validated by PCR across the nuclear-mitochondrial genome junction, and sequencing of the NUMT to confirm concordance of the mtDNA sequence polymorphisms with those of blood cells from the same donor. In another study, compared to blood DNA, colorectal adenocarcinoma tumor DNA of the same individuals showed a 4-fold elevation in NUMTs [Srinivasainagendra et al. 2017]. It will be informative in future studies to quantify and compare the NUMT status of tumors with surrounding tissues rather than blood. The cause of increased NUMT insertions is unclear, as is their impact on oncogenesis, although NUMTs may increase genomic instability. Little is known about the regulation of these clandestine retrotransposed genetic elements and their functional significance in human cancer.
The second line of evidence suggests that mtDNA sequence variants can confer susceptibility to certain cancers [Seyfried 2015]. Controversial evidence suggests that mtDNA haplogroups can predispose to cancer. In epidemiological studies, it has been reported that mtDNA haplogroup U is associated with a ~2–2.5-fold higher risk for renal and prostate cancer [Canter et al. 2006] (see [van Gisbergen et al. 2015] for a review). However, some limitations must be noted: some studies have reported negative findings, we cannot rule out publication bias (studies with null results are not published, but positive findings are consistently published), and cross-sectional observational study designs can be confounded in many ways precluding causal inference. More work is thus required to overcome these caveats. Nevertheless, converging evidence from experimental studies indicate that mild mtDNA mutations may also promote tumor cell growth [Cruz-Bermúdez et al. 2015]. This implies that functional mtDNA variants (i.e., mutations) could drive and/or favor oncogenic transformation, although this point remains contentious and in need of further validation in humans.
Finally, a body of evidence has demonstrated an increased prevalence of mtDNA mutations in tumors. mtDNA sequence analysis of 1,675 tumors revealed a mutational pattern with heavy strand bias, predominantly C>T and A>G mutations [Ju et al. 2014]. Whereas synonymous mutations were often observed in homoplasmy, indicative of clonal expansion without selection, deleterious mutations causing protein truncation showed evidence of negative selection in this and another study [Strakova et al. 2016], suggesting that even in cancer cells mutations that affect the electron transport chain have a deleterious effects on cell growth, consistent with the notion that functional mitochondria are required for cancerous cell growth.
Cancer susceptibility (or lack thereof) in primary disorders of mtDNA
If mtDNA mutations that cause primary mitochondrial disorders could also trigger oncogenic transformation or support the progression, one would expect a higher prevalence of cancer among patients with inherited mtDNA mutations. However, there is no clinical or epidemiological evidence to support this possibility. To our knowledge, there has not been a formal study of the incidence of cancer in patients with mitochondrial disease. One exception is the identification of a rare form of lipoma in patients with the m.8344A>G mutation [Holme et al. 1993]. The general dissociation of these disease states could reflect a biological incompatibility between cancer and highly pathological forms of mitochondrial dysfunction.
Alternatively, this apparent clinical dissociation may arise from the fragmented medical architecture that has traditionally treated these diseases quite distinctly. Except for rare severe pediatric cases, mitochondrial disease is likely under diagnosed [Gorman et al. 2015]. Moreover, there is substantially greater medical and general public awareness around cancer that primary mitochondrial disorders. Thus it is possible that adults with both mitochondrial diseases are not uncommon, but fail to be recognized. On the other hand, some adult patients with confirmed and pre-existing mtDNA disease have been followed for many years, with few reports of malignancy. However, this represents a fairly small number of individuals and sampling bias could be at the origin of this apparent dichotomous state between cancer and mitochondrial disease. More research is needed in this area.
Hypothesis: Transgenerational Alterations in Mitochondrial Genetic Potential May Predispose to Cancer
We suggest that transgenerational alterations in mtDNA are a potentially under-recognized contributor to pro-oncogenic states. It is now well recognized that mitochondrial metabolism plays a critical role in supporting many types of cancer, and that adaptations to typical mitochondrial function play roles in both transformation and metastasis. A growing body of evidence suggests that these changes in mitochondrial metabolism are not bystanders to the cancer phenotype, but are rather essential aspects of the transformed state [Mullen et al. 2011; Wang et al. 2013; Spinelli et al. 2017]. Nuclear mutations in genes encoding mitochondrial function are known to be a potential oncogenic “hit,” and it is reasonable that mtDNA mutations could act in the same fashion.
Changes in mtDNA between generations, either as shifts in heteroplasmy or through the generation of new mutations, are not rare. Because of the dense coding of the mitochondrial genome, random mutations have a high potential to impact mitochondrial function. Additionally, the highly reactive environment of mitochondrial matrix creates the potential for positive feedback loops. Mutations that lead to higher free radical burden may create a more mutagenic milieu, a process with the potential to accelerate as the mutational burden grows. Even non-mutated transmission of mtDNA has the potential to result in altered mitochondrial capacity in offspring if the nuclear-encoded repertoire of the developing embryo is sufficiently distinct from the mother. We have seen in animal models [Betancourt et al. 2014; Latorre-Pellicer et al. 2016] and now in some human studies [Crawford et al. 2017] that the nuclear context of the mtDNA, and the interactions between mitochondrial and nuclear proteins, ultimately determine how effectively the mitochondrial functions.
What type of mutations would we expect to act as drivers for mutation? With few anecdotal exceptions, it is not likely that highly pathogenic, disease-causing mutations will end up with strong associations with cancer. Were this the case, the links between mitochondrial disorders and the risk for cancer would be well established, as these patients are highly medically involved. Instead we find only scattered reports of patients with obvious mitochondrial syndromes and cancer, potentially below the expected level due to random association, even given the shortened life span associated with these disorders that could reduce the window for observing cancer initiation and progression. This suggests that highly disruptive mutations may actually act as suppressors of cancer by compromising the reprogramming of mitochondrial function. Likewise, how would we consider the risk imposed by mtDNA operating in a distinct nuclear context within offspring as compared to their mothers?
If we hypothesize that transplacental mutagenesis or change in nuclear context can create the opportunity for elevated cancer risk, how would this be observable? An ideal data set would include high-resolution genome-level sequencing with accurate heteroplasmy detection and quantification, encompassing mothers and their children, obtained on DNA samples prior to the detection of cancer. It is clear that the use of mitochondrial sequencing from cancers themselves cannot be used because it will be impossible to determine whether individual mutations are drivers of cancer initiation and progression or merely passenger mutations that developed within the mutagenic environment of the cancer and have been clonally expanded. The population under study would need to be unselected and of sufficient size so that the study would be powered to detect differences between affected individuals with specific cancer types and controls who were free from cancer at similar ages. Clearly, a data set of this magnitude does not currently exist, although it might potentially be collected through mass population screening approaches, such as the newborn screen or other widespread population based sequencing approaches [Sondheimer 2013; Sankar and Parker 2017]. Alternative approaches may be possible with some large, well-genotyped cancer pedigrees, since mitochondrial sequence data is often inadvertently collected and available for analysis.
The determination that transplacental changes in mtDNA sequence were associated with the potential for cancer development would place a new emphasis on understanding mtDNA sequence changes in newborns. As we approach an era where sequencing is universally applied, this information could be used to guide screening approaches and improve health by managing cancer susceptibility risk.
Acknowledgments
The authors acknowledge the support of the March of Dimes Transdisciplinary Center for Preterm Birth Research at The University of Pennsylvania (B.A.K. and N.S.) and National Institutes of Health Grant GM119793-01 (M.P.).
Footnotes
Statement of Author Contributions
Drs. Kaufman, Picard and Sondheimer wrote the manuscript and agreed to the final version.
References
- Al-Mehdi AB, Pastukh VM, Swiger BM, et al. Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Science signaling. 2012;5(231):ra47. doi: 10.1126/scisignal.2002712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alston CL, Rocha MC, Lax NZ, Turnbull DM, Taylor RW. The genetics and pathology of mitochondrial disease. The Journal of pathology. 2017;241(2):236–250. doi: 10.1002/path.4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker PE, Murthy M. Biomarker Validation for Aging: Lessons from mtDNA Heteroplasmy Analyses in Early Cancer Detection. Biomarker insights. 2009;4:165–179. doi: 10.4137/bmi.s2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betancourt AM, King AL, Fetterman JL, et al. Mitochondrial-nuclear genome interactions in non-alcoholic fatty liver disease in mice. The Biochemical journal. 2014;461(2):223–32. doi: 10.1042/BJ20131433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M, Sabatini DM. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell. 2015;162(3):540–51. doi: 10.1016/j.cell.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DT, Samuels DC, Michael EM, Turnbull DM, Chinnery PF. Random genetic drift determines the level of mutant mtDNA in human primary oocytes. The American Journal of Human Genetics. 2001;68(2):533–536. doi: 10.1086/318190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canter JA, Kallianpur AR, Fowke JH. Re: North American white mitochondrial haplogroups in prostate and renal cancer. The Journal of urology. 2006;176(5):2308–9. doi: 10.1016/j.juro.2006.07.067. author reply 2309. [DOI] [PubMed] [Google Scholar]
- Cassano AG, Anderson VE, Harris ME. Evidence for direct attack by hydroxide in phosphodiester hydrolysis. Journal of the American Chemical Society. 2002;124(37):10964–5. doi: 10.1021/ja020823j. [DOI] [PubMed] [Google Scholar]
- Chinnery PF, Howell N, Lightowlers RN, Turnbull DM. Molecular pathology of MELAS and MERRF. The relationship between mutation load and clinical phenotypes. Brain: a journal of neurology. 1997;120(Pt 1):1713–1721. doi: 10.1093/brain/120.10.1713. [DOI] [PubMed] [Google Scholar]
- Cohen T, Levin L, Mishmar D. Ancient Out-of-Africa Mitochondrial DNA Variants Associate with Distinct Mitochondrial Gene Expression Patterns. PLOS Genetics. 2016;12(11):e1006407. doi: 10.1371/journal.pgen.1006407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford N, Prendergast D, Oehlert JW, et al. Divergent patterns of mitochondrial and nuclear ancestry are associated with the risk for preterm birth. Journal of Pediatrics. 2017 doi: 10.1016/j.jpeds.2017.10.052. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Bermúdez A, Vallejo CG, Vicente-Blanco RJ, et al. Enhanced tumorigenicity by mitochondrial DNA mild mutations. Oncotarget. 2015;6(15):13628–13643. doi: 10.18632/oncotarget.3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong L-F, Kovarova J, Bajzikova M, et al. Horizontal transfer of whole mitochondria restores tumorigenic potential in mitochondrial DNA-deficient cancer cells. eLife. 2017;6 doi: 10.7554/eLife.22187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkenberg M, Larsson NG, Gustafsson CM. DNA replication and transcription in mammalian mitochondria. Annual Review of Biochemistry. 2007;76:679–699. doi: 10.1146/annurev.biochem.76.060305.152028. [DOI] [PubMed] [Google Scholar]
- Fan W, Waymire KG, Narula N, et al. A mouse model of mitochondrial disease reveals germline selection against severe mtDNA mutations. Science (New York, NY ) 2008;319(5865):958–962. doi: 10.1126/science.1147786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fetterman JL, Zelickson BR, Johnson LW, et al. Mitochondrial genetic background modulates bioenergetics and susceptibility to acute cardiac volume overload. Biochem J. 2013;455:157–167. doi: 10.1042/BJ20130029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles RE, Blanc H, Cann HM, Wallace DC. Maternal inheritance of human mitochondrial DNA. Proceedings of the National Academy of Sciences of the United States of America. 1980;77(11):6715–9. doi: 10.1073/pnas.77.11.6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gisbergen MW, Voets AM, Starmans MHW, et al. How do changes in the mtDNA and mitochondrial dysfunction influence cancer and cancer therapy? Challenges, opportunities and models. Mutation research Reviews in mutation research. 2015;764:16–30. doi: 10.1016/j.mrrev.2015.01.001. [DOI] [PubMed] [Google Scholar]
- Gorman GS, Schaefer AM, Ng Y, et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Annals of neurology. 2015;77(5):753–9. doi: 10.1002/ana.24362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griss T, Vincent EE, Egnatchik R, et al. Metformin Antagonizes Cancer Cell Proliferation by Suppressing Mitochondrial-Dependent Biosynthesis. PLoS Biology. 2015;13:12. doi: 10.1371/journal.pbio.1002309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hazkani-Covo E, Zeller RM, Martin W. Molecular poltergeists: Mitochondrial DNA copies (numts) in sequenced nuclear genomes. PLoS Genetics. 2010;6 doi: 10.1371/journal.pgen.1000834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Wu J, Dressman DC, et al. Heteroplasmic mitochondrial DNA mutations in normal and tumour cells. Nature. 2010;464(7288):610–614. doi: 10.1038/nature08802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science. 2009;324(5930):1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holme E, Larsson NG, Oldfors A, Tulinius M, Sahlin P, Stenman G. Multiple symmetric lipomas with high levels of mtDNA with the tRNA(Lys) A-->G(8344) mutation as the only manifestation of disease in a carrier of myoclonus epilepsy and ragged-red fibers (MERRF) syndrome. American journal of human genetics. 1993;52(3):551–6. [PMC free article] [PubMed] [Google Scholar]
- Imlay JA, Chin SM, Linn S. Toxic DNA damage by hydrogen peroxide through the Fenton reaction in vivo and in vitro. Science (New York, NY ) 1988;240(4852):640–2. doi: 10.1126/science.2834821. [DOI] [PubMed] [Google Scholar]
- Ishii T, Yasuda K, Akatsuka A, Hino O, Hartman PS, Ishii N. A mutation in the SDHC gene of complex II increases oxidative stress, resulting in apoptosis and tumorigenesis. Cancer research. 2005;65(1):203–9. [PubMed] [Google Scholar]
- Ju YS, Alexandrov LB, Gerstung M, et al. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. eLife. 2014;3 doi: 10.7554/eLife.02935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju YS, Tubio JMC, Mifsud W, et al. Frequent somatic transfer of mitochondrial DNA into the nuclear genome of human cancer cells. Genome research. 2015;25(6):814–24. doi: 10.1101/gr.190470.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane N. Mitonuclear match: optimizing fitness and fertility over generations drives ageing within generations. BioEssays: news and reviews in molecular, cellular and developmental biology. 2011;33(11):860–9. doi: 10.1002/bies.201100051. [DOI] [PubMed] [Google Scholar]
- Latorre-Pellicer A, Moreno-Loshuertos R, Lechuga-Vieco AV, et al. Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature. 2016;535(7613):561–5. doi: 10.1038/nature18618. [DOI] [PubMed] [Google Scholar]
- Li H, Shen L, Hu P, et al. Aging-associated mitochondrial DNA mutations alter oxidative phosphorylation machinery and cause mitochondrial dysfunctions. Biochimica et biophysica acta. 2017;1863(9):2266–2273. doi: 10.1016/j.bbadis.2017.05.022. [DOI] [PubMed] [Google Scholar]
- Liu P, Demple B. DNA repair in mammalian mitochondria: Much more than we thought? Environmental and molecular mutagenesis. 2010 doi: 10.1002/em.20576. NA-NA. [DOI] [PubMed] [Google Scholar]
- Maeda K, Kawai H, Sanada M, et al. Clinical Phenotype and Segregation of Mitochondrial 3243A>G Mutation in 2 Pairs of Monozygotic Twins. JAMA neurology. 2016;73(8):990–3. doi: 10.1001/jamaneurol.2016.0886. [DOI] [PubMed] [Google Scholar]
- Gagné ML, Boulay K, Topisirovic I, Huot M-É, Mallette FA. Oncogenic Activities of IDH1/2 Mutations: From Epigenetics to Cellular Signaling. Trends in cell biology. 2017 doi: 10.1016/j.tcb.2017.06.002. [DOI] [PubMed] [Google Scholar]
- Marchington DR, Macaulay V, Hartshorne GM, Barlow D, Poulton J. Evidence from human oocytes for a genetic bottleneck in an mtDNA disease. The American Journal of Human Genetics. 1998;63(3):769–775. doi: 10.1086/302009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulis L. Origin of eukaryotic cells: Evidence and research implications for a theory of the origin and evolution of microbial, plant and animal cells on the precambrian Earth. Yale University Press; New Haven: 1970. [Google Scholar]
- McFarland R, Kirby DM, Fowler KJ, et al. De Novo Mutations in the Mitochondrial ND3 Gene as a Cause of Infantile Mitochondrial Encephalopathy and Complex I Deficiency. Annals of Neurology. 2004;55(1):58–64. doi: 10.1002/ana.10787. [DOI] [PubMed] [Google Scholar]
- Mullen AR, Wheaton WW, Jin ES, et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2011;481(7381):385–8. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murdock DG, Christacos NC, Wallace DC. The age-related accumulation of a mitochondrial DNA control region mutation in muscle, but not brain, detected by a sensitive PNA-directed PCR clamping based method. Nucleic acids research. 2000;28(21):4350–4355. doi: 10.1093/nar/28.21.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuhaus JFG, Baris OR, Kittelmann A, Becker K, Rothschild MA, Wiesner RJ. Catecholamine Metabolism Induces Mitochondrial DNA Deletions and Leads to Severe Adrenal Degeneration during Aging. Neuroendocrinology. 2017;104(1):72–84. doi: 10.1159/000444680. [DOI] [PubMed] [Google Scholar]
- Picard M, Hirano M. Disentangling (Epi)Genetic and Environmental Contributions to the Mitochondrial 3243A>G Mutation Phenotype: Phenotypic Destiny in Mitochondrial Disease? JAMA neurology. 2016;73(8):923–5. doi: 10.1001/jamaneurol.2016.1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard M, Zhang J, Hancock S, et al. Progressive increase in mtDNA 3243A>G heteroplasmy causes abrupt transcriptional reprogramming. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(38):E4033–42. doi: 10.1073/pnas.1414028111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rito T, Richards MB, Fernandes V, et al. The first modern human dispersals across Africa. PloS one. 2013;8(11):e80031. doi: 10.1371/journal.pone.0080031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankar PL, Parker LS. The Precision Medicine Initiative’s All of Us Research Program: an agenda for research on its ethical, legal, and social issues. Genetics in medicine: official journal of the American College of Medical Genetics. 2017;19(7):743–750. doi: 10.1038/gim.2016.183. [DOI] [PubMed] [Google Scholar]
- Sato M, Sato K. Maternal inheritance of mitochondrial DNA by diverse mechanisms to eliminate paternal mitochondrial DNA. Biochimica et biophysica acta. 2013;1833(8):1979–84. doi: 10.1016/j.bbamcr.2013.03.010. [DOI] [PubMed] [Google Scholar]
- Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiological Reviews. 2008;88(2):611–638. doi: 10.1152/physrev.00025.2007. [DOI] [PubMed] [Google Scholar]
- Seo AY, Xu J, Servais S, et al. Mitochondrial iron accumulation with age and functional consequences. Aging Cell. 2008;7(5):706–716. doi: 10.1111/j.1474-9726.2008.00418.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyfried TN. Cancer as a mitochondrial metabolic disease. Frontiers in Cell and Developmental Biology. 2015;3 doi: 10.3389/fcell.2015.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shokolenko I, Venediktova N, Bochkareva A, Wilson GL, Alexeyev MF. Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Research. 2009;37(8):2539–2548. doi: 10.1093/nar/gkp100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondheimer N. Newborn screening by sequence and the road ahead. Clinical Chemistry. 2013;59(7):1011–1013. doi: 10.1373/clinchem.2013.205864. [DOI] [PubMed] [Google Scholar]
- Sondheimer N, Glatz CE, Tirone JE, Deardorff MA, Krieger AM, Hakonarson H. Neutral mitochondrial heteroplasmy and the influence of aging. Human molecular genetics. 2011;20(8):1653–1659. doi: 10.1093/hmg/ddr043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinelli JB, Yoon H, Ringel AE, Jeanfavre S, Clish CB, Haigis MC. Metabolic recycling of ammonia via glutamate dehydrogenase supports breast cancer biomass. Science. 2017;358(6365):941–946. doi: 10.1126/science.aam9305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasainagendra V, Sandel MW, Singh B, et al. Migration of mitochondrial DNA in the nuclear genome of colorectal adenocarcinoma. Genome Medicine. 2017;9(1):31. doi: 10.1186/s13073-017-0420-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strakova A, Leathlobhair MN, Wang GD, et al. Mitochondrial genetic diversity, selection and recombination in a canine transmissible cancer. eLife. 2016 May;5 doi: 10.7554/eLife.14552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui G, Zhou S, Wang J, et al. Mitochondrial DNA mutations in preneoplastic lesions of the gastrointestinal tract: a biomarker for the early detection of cancer. Molecular cancer. 2006;5:73. doi: 10.1186/1476-4598-5-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suissa S, Wang Z, Poole J, et al. Ancient mtDNA genetic variants modulate mtDNA transcription and replication. PLoS genetics. 2009;5(5):e1000474. doi: 10.1371/journal.pgen.1000474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC. Mitochondrial function and cancer. Nature Reviews: Cancer. 2012;12(10):1. doi: 10.1038/nrc3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Travins J, DeLaBarre B, et al. Targeted Inhibition of Mutant IDH2 in Leukemia Cells Induces Cellular Differentiation. Science (New York, NY) 2013 doi: 10.1126/science.1234769. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zhang Y, Chen L, et al. Kinetics and specificity of paternal mitochondrial elimination in Caenorhabditis elegans. Nature Communications. 2016 May;7:12569. doi: 10.1038/ncomms12569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. On respiratory impairment in cancer cells. Science (New York, NY ) 1956;124(3215):269–70. [PubMed] [Google Scholar]
- Ward PS, Thompson CB. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell. 2012;21(3):297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg F, Hamanaka R, Wheaton WW, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proceedings of the National Academy of Sciences. 2010;107(19):8788–8793. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson IJ, Carling PJ, Alston CL, et al. Mitochondrial DNA sequence characteristics modulate the size of the genetic bottleneck. Human Molecular Genetics. 2016;25(5):1031–1041. doi: 10.1093/hmg/ddv626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(2):514–9. doi: 10.1073/pnas.94.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Gimsa U, Wester-rosenlöf L, et al. Dissecting the effects of mtDNA variations on complex traits using mouse conplastic strains Dissecting the effects of mtDNA variations on complex traits using mouse conplastic strains. Genome Research. 2009:159–165. doi: 10.1101/gr.078865.108. [DOI] [PMC free article] [PubMed] [Google Scholar]