

Abstract
Mitochondria are pivotal organelles in calcium (Ca2+) handling and signalling, constituting intracellular checkpoints for numerous processes that are vital for cell life. Alterations in mitochondrial Ca2+ homeostasis have been linked to a variety of pathological conditions and are critical in the aetiology of several human diseases. Efforts have been taken to harness mitochondrial Ca2+ transport mechanisms for therapeutic intervention, but pharmacological compounds that direct and selectively modulate mitochondrial Ca2+ homeostasis are currently lacking. New avenues have, however, emerged with the breakthrough discoveries on the genetic identification of the main players involved in mitochondrial Ca2+ influx and efflux pathways and with recent hints towards a deep understanding of the function of these molecular systems. Here, we review the current advances in the understanding of the mechanisms and regulation of mitochondrial Ca2+ homeostasis and its contribution to physiology and human disease. We also introduce and comment on the recent progress towards a systems‐level pharmacological targeting of mitochondrial Ca2+ homeostasis.
Keywords: mitochondria, calcium, drug screening, calcium signaling, mitochondrial calcium uniporter, drug discovery
Introduction
In virtually all eukaryotic cells, mitochondria are primed organelles for the regulation of intracellular Ca2+ signalling. They can take up and release Ca2+ ions through the concerted action of influx and efflux pathways to shape the spatio‐temporal dynamics of cytosolic Ca2+ transients (Nicholls, 1978; Nicholls & Scott, 1980; Zoccarato & Nicholls, 1982). In addition, mitochondrial Ca2+ entry is coupled to an increase in energy production (Denton & McCormack, 1980) to meet the rising energy demand of signalling cells (Szabadkai & Duchen, 2008). Mitochondrial Ca2+ homeostasis has been extensively studied for decades and functionally linked to the regulation of diverse physiological processes such as energy metabolism, tissue growth and development, neurotransmitter release, muscle contraction, autophagy, and cell death. More than half a century ago, it was shown that Ca2+ uptake by isolated mammalian mitochondria was a rapid, electrogenic and relatively selective process that was driven by the negative potential (∼−180 mV) across the inner mitochondrial membrane (IMM) and inhibited by lanthanide cations and ruthenium red (RuR) (Deluca & Engstrom, 1961; Vasington & Murphy, 1962; Chance, 1965; Brand et al. 1976). At the same time, it was demonstrated that the presence of Na+ in the medium and changes in pH were able to stimulate the efflux of Ca2+ accumulated in isolated mitochondria (Carafoli et al. 1974). It was thus concluded that both mitochondrial Ca2+ influx and mitochondrial Ca2+ efflux occurred through specific carriers located in the IMM, a putative and ubiquitous mitochondrial Ca2+ uniporter and either a Na+/Ca2+ or a H+/Ca2+ exchanger, in excitable and non‐excitable tissues, respectively. However, although much progress has been made to elucidate the biophysical properties of mitochondrial Ca2+ entry and release, the molecular identities of these transporters remained elusive until 2010. Then, the mitochondrial Ca2+ uniporter 1 (MICU1; Perocchi et al. 2010) was discovered as the founding member of the mitochondrial Ca2+ uniporter complex and the Li‐permeable Na+/Ca2+ exchanger (NCLX; Palty et al. 2010) as a new member of the Na+/Ca2+ exchanger superfamily, opening a new era in the mitochondria and Ca2+ signalling fields. These findings allowed targeted genetic loss‐ and gain‐of‐function analyses in vitro and in vivo towards a comprehensive assessment of the physiological and pathophysiological role of mitochondrial Ca2+ homeostasis in cells, tissues and whole organisms (De Stefani et al. 2016). Moreover, the identification of MICU1 loss‐of‐function mutations in human patients with neuromuscular disorders (Logan et al. 2014; Lewis‐Smith et al. 2016) has confirmed that modulating mitochondrial Ca2+ homeostasis might constitute a promising therapeutic approach.
This review attempts to provide an overview of the latest knowledge on mitochondrial Ca2+ homeostasis and its modulation for therapeutic intervention in human diseases. First, we summarize the findings that led to the identification and characterization of the molecular components involved in mitochondrial Ca2+ uptake and efflux. We then discuss the relevance of mitochondrial Ca2+ homeostasis in physiology and disease and the so far available pharmacological approaches to modulate mitochondrial Ca2+ uptake and release pathways. Finally, we address recent progresses on pharmacological targeting of mitochondrial Ca2+ homeostasis and present a newly established high‐throughput chemical screening approach for the systematic identification of modulators of the mitochondrial Ca2+ uniporter (Arduino et al. 2017).
The mitochondrial Ca2+ uniporter
Many attempts have been made to identify the components of the mitochondrial Ca2+ uniporter, for example by purifying Ca2+‐binding proteins from isolated mitochondria and then reconstituting Ca2+ uptake in vitro. The group of Lehninger reported on the purification of a soluble heat‐labile protein of over 150 kDa from rat liver mitochondria subjected to osmotic shock (Lehninger, 1971). This factor was able of binding Ca2+ with high affinity and it was sensitive to lanthanides. Prompted by the serendipitous finding that mitochondria were stained by the polysaccharide stain RuR and that this completely inhibited mitochondrial Ca2+ uptake, Sottocasa and colleagues focused on the extraction of glycoproteins as possible active centres of Ca2+ binding and transport (Sottocasa et al. 1972). From ox and rat liver mitochondria they isolated a major band with a molecular mass of approximately 30 kDa by PAGE, which was stained by murexidine, a Ca2+ dye, and whose high‐affinity binding (K d of approximately 100 nm) was affected by both lanthanides and RuR. This proteinaceous factor, named ‘calvectin’, was found primarily in the intermembrane space in a free soluble form, but could become membrane bound in a Ca2+‐dependent manner. Calvectin was able to increase Ca2+ conductance in artificial lipid bilayers and its antisera specifically inhibited Ca2+ transport by rat liver mitoplasts (mitochondria stripped of their outer membrane) without affecting respiration. This protein was therefore suggested to represent a major component of the mitochondrial Ca2+ uniporter. Other laboratories also suggested that the components responsible for Ca2+ transport in mitochondria were not of protein but rather of lipid nature. A lipid moisture isolated from the Ca2+‐transported glycoprotein was shown to form channels in lipid bilayers with an electroconductivity between 100 and 200 times higher in the presence of Ca2+ and the ability to transport Ca2+ was lost in delipidated mitochondria (Medvedev et al. 1982). Similarly, Sokolove and Brenza (1983) demonstrated that cardiolipin, an abundant lipid of mitochondrial membranes, binds Ca2+ with an apparent affinity of 700 nm in the absence of other competitive divalent cations such as Mn2+, Zn2+ and Mg2+. Calcium binding to cardiolipin was also inhibited by lanthanides and RuR. After these pioneering reports, various Ca2+ binding glycoproteins and peptides were isolated, all of which were able to bind Ca2+ and were inhibited by lanthanides and RuR (Mironova & Utesheva Zh, 1989; Zazueta et al. 1998). Several of these factors have been dismissed as components of the Ca2+ uniporter, as they were then found to be either cytosolic or microsomal contaminants and the majority of those observations faded away with lack of follow‐up studies.
Much later, electrophysiological measurements of Ca2+ currents in single mitoplasts demonstrated that the mitochondrial Ca2+ uniporter is an inward rectifying channel, highly selective for Ca2+, and with a remarkable high capacity for Ca2+ transport (half‐saturation at ∼20 mm) (Kirichok et al. 2004), and several proteins, such as the mitochondrial type 1 ryanodine receptor (mRyR1) (Beutner et al. 2005) and the isoforms 2 and 3 of the ion‐transporting uncoupling protein UCP (UCP2 and UCP3) (Trenker et al. 2007), were initially proposed to form this channel. However, the direct involvement of these proteins in mediating RuR‐sensitive Ca2+ uptake has been questioned. Thus, even after so much effort spanning several decades of research and when the majority of the new genomics and proteomics approaches were already available, the molecular nature of the uniporter had remained a mystery to molecular biologists, biochemists and physiologists alike. In 2010, Perocchi et al. identified MICU1 as the first genuine member of the uniporter channel based on the conceptual integration of physiological and biophysical clues from 50 years of literature on mitochondrial Ca2+ homeostasis in different organisms with comparative genomics and mitochondrial proteomics analyses (Perocchi et al. 2010). Analyses performed during the 1960s and ’70s had shown that uniporter‐dependent Ca2+ uptake is not a prerogative of all eukaryotic organisms, as it can be measured in mitochondria of mammals and kinetoplastids but not in fungi. Therefore, human genes encoding core components of the uniporter should be conserved in the mitochondrial proteome of organisms that are competent for Ca2+ uptake but not in other species devoid of such ability. Of the thousand plus human proteins localized to mitochondria, the authors narrowed down the search to just 18 that fulfilled the evolutionary signature of the uniporter, were associated with the IMM and were widely expressed in mitochondria from several mouse tissues. RNA interference (RNAi) against these 18 genes highlighted one, CBARA1, which was thereafter renamed MICU1 (Perocchi et al. 2010). Silencing MICU1 did not disrupt mitochondrial respiration or membrane potential but impaired mitochondrial Ca2+ entry in intact and permeabilized cells, as well as in mitochondria isolated from mouse liver. It also attenuated the metabolic coupling between cytosolic Ca2+ transients and stimulation of energy metabolism. MICU1 had two predicted EF hands that were found to be essential for regulating the activity of the uniporter, suggesting a role in Ca2+ sensing. Building on this discovery, the same authors and the group of Rizzuto (Baughman et al. 2011; De Stefani et al. 2011) identified a 40 kDa coiled‐coiled protein, known as CCDC109A, that coevolved with MICU1 and had two transmembrane domains, which they proved to be the bona fide, pore‐forming subunit of the mitochondrial calcium uniporter and threrefore renamed it mitochondrial Ca2+ uniporter (MCU). First, RNAi‐mediated silencing of MCU was found to strongly inhibit mitochondrial Ca2+ uptake in cultured cells and in purified mouse liver mitochondria, whereas MCU overexpression enhanced RuR‐sensitive mitochondrial Ca2+ uptake in intact and permeabilized cells. Single‐point mutations of conserved acidic residues (E257A, D261A and E263A) within the short loop linking the two transmembrane domains of MCU on the intermembrane space side, termed the DIME motif, abrogated the ability of mitochondria to take up Ca2+, indicating these residues were required for Ca2+ transport (Baughman et al. 2011; De Stefani et al. 2011; Chaudhuri et al. 2013). Expression of S259A (Baughman et al. 2011; Chaudhuri et al. 2013) and D261A MCU (Arduino et al. 2017) mutants conferred reduced RuR sensitivity, highlighting a possible role in RuR‐dependent MCU inhibition. Second, expression of pure MCU in planar lipid bilayers was shown to be sufficient to reconstitute ion channel activity, generating a single channel Ca2+ current consistent with that of the uniporter (De Stefani et al. 2011). Finally, heterologous expression of the MCU orthologous protein from Dictyostelium dyscodium was sufficient to reconstitute mitochondrial Ca2+ uptake capacity in the yeast Saccharomyces cerevisiae, which lacks all uniporter components (Kovacs‐Bogdan et al. 2014). Altogether, these observations confirmed that MCU is the Ca2+‐conducting pore of the long‐sought mitochondrial uniporter. Moreover, recent studies on the structural biology (Oxenoid et al. 2016), coordination chemistry and molecular dynamics (Cao et al. 2017) of MCU from Caenorhabditis elegans suggested that a functional channel results from the pentameric assembly of MCU multimers stabilized by a coiled‐coil motif protruding into the mitochondrial matrix. In this structural model, the DIME motif forms the selectivity filter of the channel, whereby ion permeation and inhibitor binding occur. These insights have been recently exploited for in silico modelling of MCU and binding of mitoxantrone, a novel inhibitor of the uniporter channel (Arduino et al. 2017).
MCU was initially found to reside in the IMM as a large complex of roughly 450 kDa (Baughman et al. 2011). Over the past few years, the macromolecular nature of this channel was confirmed and several additional subunits have been characterized. MCU can form hetero‐oligomers with its paralogue, MCUb, a 33 kDa protein with very similar amino acid sequence and topological features (Raffaello et al. 2013). Co‐expression studies of MCU and MCUb in planar lipid bilayers and intact cells demonstrated that the presence of MCUb decreases MCU‐dependent Ca2+ uptake activity, suggesting that MCUb is a dominant‐negative pore‐forming component of the uniporter. The regulation of MCU activity by extramitochondrial Ca2+ is dictated by a duet of EF‐hand containing proteins, MICU1 and its paralogue EFHA1 (termed MICU2), interacting with MCU from the mitochondrial intermembrane space (Plovanich et al. 2013; Patron et al. 2014; Liu et al. 2016; Kamer et al. 2017). A related protein termed MICU3 was also proposed to be a member of the uniporter based on its sequence similarity to MICU2, but neither its mitochondrial localization nor its involvement in Ca2+ uptake have been confirmed so far. Following a similar strategy to Perocchi et al. (2010), Mallilarankaraman and colleagues identified the mitochondrial Ca2+ uniporter regulator 1 (MCUR1), formerly known as CCDC90A, as an essential membrane component of the uniporter (Mallilankaraman et al. 2012; Vais et al. 2015). Interestingly, MCUR1 was shown to have an opposite membrane topology to MCU, whereby both N‐ and C‐termini project into the intermembrane space, while the majority of the protein, including its coil‐coil domain, faces the mitochondrial matrix, suggesting that unlike MCU and MCUb, MCUR1 does not act as a pore‐forming subunit. In this regard, MCUR1 was recently suggested to function as a scaffold factor for the assembly of the uniporter channel (Tomar et al. 2016), although other independent roles have also been ascribed to it, such as the assembly of complex IV (Paupe et al. 2015) and the regulation of Ca2+‐induced mitochondrial permeability transition (Chaudhuri et al. 2016). Finally, a 10 kDa integral IMM protein termed essential MCU regulator (EMRE) was found to be indispensable for uniporter‐mediated Ca2+ uptake and together with MCU was sufficient to reconstitute Ca2+ uptake in yeast mitochondria (Sancak et al. 2013; Kovacs‐Bogdan et al. 2014). EMRE is thought to be a multifunctional element of the uniporter that is required for inwardly rectifying Ca2+ currents, bridging of MCU and its regulators MICU1 and MICU2 (Sancak et al. 2013; Kovacs‐Bogdan et al. 2014; Petrungaro et al. 2015; Tsai et al. 2016) and sensing of mitochondrial matrix Ca2+ (Vais et al. 2016). In contrast to its essential role in higher eukaryotes, EMRE expression is absent in some organisms where MCU is conserved and functional, such as D. discoideum (Kovacs‐Bogdan et al. 2014). To date, the precise role of this protein in both function and regulation of the uniporter remains to be clearly elucidated. In addition to this plethora of structural and regulatory subunits, both uniporter activity and composition can also vary across tissues through the expression of tissue‐specific components. For example, MICU2 and MICU3 were found to be mostly expressed in visceral organs and central nervous system, respectively (Plovanich et al. 2013; Murgia & Rizzuto, 2015), whereas a recently found MICU1 alternative splice variant, MICU1.1, is skeletal muscle‐specific (Vecellio Reane et al. 2016). The discovery of the major molecular players of mitochondrial Ca2+ homeostasis constitutes a great accomplishment for the mitochondrial biology and Ca2+ fields, and additional elements might be on the research horizon.
Mitochondrial Ca2+ efflux
Outward Ca2+ flux from mitochondria is mainly mediated by two transport systems, mitochondrial Na+/Ca2+ (mNCLX) and H+/Ca2+ (mHCX) exchangers. By a genome‐wide siRNA screen in Drosophila cells, the leucine zipper and EF‐hand containing transmembrane protein 1 (LETM1) was found to regulate RuR‐sensitive mitochondrial Ca2+ uptake at cytosolic concentrations below, but not above, 1 um and to couple the movement of Ca2+ in exchange for H+ both in intact cells and in proteoliposomes (Jiang et al. 2009). Thus, LETM1 was proposed to function as the mitochondrial electroneutral H+/Ca2+ antiporter. However, previous studies had characterized the same protein as a member of the mitochondrial K+/H+ exchanger and shown that it was essential to control mitochondrial volume (Nowikovsky et al. 2004; McQuibban et al. 2010). The hypothesis that LETM1 could mediate both extrusion and uptake of Ca2+ independently of K+ was also put forward (Jiang et al. 2013; Tsai et al. 2014) and later challenged based on evidence for electroneutral exchange of H+ with K+ in vivo, thus disclaiming the contribution of LETM1 to mitochondrial Ca2+ efflux (De Marchi et al. 2014; Nowikovsky & Bernardi, 2014). Despite the renowned relevance of mitochondrial outward shuttling mechanisms for cell physiology, the molecular identity of mHCX still remains unclear.
In a seminal study by Palty et al. (2010), it was found that the gene FLJ22233 (also known as SLC8B1) encodes an IMM‐located protein that mediates both Li+‐ and Na+‐dependent Ca2+ clearance from the mitochondrial matrix. The authors named this protein mNCLX and demonstrated that it constitutes the mitochondrial Ca2+ antiporter. Similarly to MCU‐dependent mitochondrial Ca2+ uptake, the action of mNCLX is electrogenic, with 3 Na+/1 Ca2+ ions per transport cycle. Consequently, mNCLX constitutes the main pathway for Na+ influx into mitochondria, and its activity is finely tuned to sense changes in intracellular Na+ concentrations within the physiological range. The discovery of the molecular identity of mNCLX (Palty et al. 2010) was instrumental for understanding the molecular mechanisms of mitochondrial Ca2+ homeostasis. Accordingly, a functional crosstalk between mNCLX and other Na+ and Ca2+ transporters located on both mitochondrial and plasma membranes has been described (Nita et al. 2015; Ben‐Kasus Nissim et al. 2017), underscoring the significant physiological role of mNCLX for the regulation of both Ca2+ and Na+ signalling pathways.
Physiopathological role of mitochondrial Ca2+ homeostasis
Genetic and biochemical strategies modulating mitochondrial Ca2+ homeostasis have already demonstrated that privileged functional relationships exist between mitochondrial Ca2+ homeostasis, cell bioenergetics and cell fate‐determination pathways. Calcium accumulation in functional mitochondria, mediated by the MCU complex (MCUC) and modulated by mNCLX, regulates mitochondrial ATP generation as well as cytosolic NAD+/NADH metabolism, thus sustaining the energy requirements of the cell. Ca2+ loading in the mitochondrial matrix stimulates mitochondrial generation of NADH, which can be shuttled to the cytosol where it initiates retrograde signalling, resulting in inhibition of sirtuins activity and alterations in protein acetylation (Marcu et al. 2014). Moreover, Ca2+ constitutively released from the ER to mitochondria is essential for efficient oxygen consumption, maintenance of cell bioenergetics and inhibition of autophagy (Cardenas et al. 2010). Conversely, mitochondrial Ca2+ overload, or perturbations in mitochondrial Ca2+ homeostasis, can sensitize cells to distinct modes of cell death induced by different stimuli, and in a cell type‐specific way (Curry et al. 2013; Qiu et al. 2013; Liao et al. 2015; Cardenas et al. 2016).
A large body of evidence from experimental models and human subjects corroborates the notion that mitochondrial Ca2+ deregulation is a relevant feature of several human disorders. As a remarkable example, its role in cardiovascular pathologies has been supported by several reports demonstrating that in heart failure, elevated cytosolic levels of Na+, which stimulates mitochondrial Ca2+ efflux via the mNCLX, reduce mitochondrial bioenergetic responses and promote mitochondrial oxidative stress in cardiomyocytes (Maack et al. 2006). In contrast, mitochondrial Ca2+ overload caused by sarcoplasmic reticulum (SR) Ca2+ leakage elicits mitochondrial dysfunction and contributes to impaired cardiac function in postischaemic heart failure (Santulli et al. 2015). Also in this pathological context, loss‐of‐function mutations and genetic ablation of the major players in mitochondrial Ca2+ influx and efflux pathways in in vivo models have demonstrated that mitochondrial Ca2+ homeostasis is a determining factor in cardiac physiology. Cardiac‐specific deletion of mNCLX was recently found to elicit severe heart failure, predisposing mice to sudden death (Luongo et al. 2017). Moreover, adult inducible cardiac‐specific deletion of MCU and transgenic expression of a dominant‐negative MCU (DN‐MCU) isoform impaired the ability of the heart to adapt to certain stresses that require increases in mitochondrial metabolism, for example the heart's ‘fight‐or‐flight’ stress response associated to an increased cardiac contractile performance (Kwong et al. 2015; Luongo et al. 2015; Wu et al. 2015). In addition, inducible MCU ablation conferred significant protection against ischaemia–reperfusion injury in adult heart (Kwong et al. 2015), as expected from the anticipated role of mitochondrial Ca2+ overload on induction of cell death. In contrast to these findings, mice constitutively lacking MCU or expressing the DN‐MCU did not exhibit marked cardiac deficits, but a pathological response to ischaemia–reperfusion injury and only a slightly impaired skeletal muscle metabolism and peak performance (Pan et al. 2013; Holmstrom et al. 2015; Rasmussen et al. 2015). The discrepancies observed between the phenotypes of different MCU knockout mouse models as well as between in vivo and in vitro cellular models may result from both timing and duration of the genetic perturbation (e.g. acute versus chronic MCU deletion). Moreover, with regard to the mouse models, while MCU deletion at the whole‐organism level is embryonically lethal in a pure C57BL/6 mouse strain, an MCU knockout mouse model could, however, be established in a mixed C57BL/6xCD1 line (Pan et al. 2013). This paradox can be explained by an increase in the genetic variability of the outbred C57BL/6xCD1 strain that may result in compensatory mechanisms for the lack of MCU, and thus in milder phenotypes. However, the nature of those compensatory mechanisms is still unknown. Moreover, whole‐body deletion of MICU1 in mice was perinatally lethal, though no major anatomical deficiencies were observed. Instead, conditional MICU1 knock down in liver resulted in extensive inflammation, tissue damage and failure of the liver's regeneration ability (Antony et al. 2016). In an independent mouse model established in a different strain with a slightly different background, homozygous MICU1 deletion caused a significant but partial postnatal mortality (Liu et al. 2016). Similar to human patients carrying loss‐of‐function MICU1 mutations, viable MICU1 knockout mice revealed a strong phenotype, including atypical mitochondrial morphology, reduced muscle ATP levels and features of neuromuscular disorders such as ataxia (Logan et al. 2014). Reminiscent of these features, silencing of MCU and MICU1 in Drosophila melanogaster during development in a brain region essential for memory resulted in impaired memory formation in adulthood without affecting the learning ability (Drago & Davis, 2016). In contrast to the lethality phenotype observed for the MICU1‐null mice, constitutive loss of MICU2 in pure C57BL/6 background mice was recently found to be compatible with life and development, although leading to alterations in cardiovascular homeostasis associated with diastolic heart failure (Bick et al. 2017). All the above‐mentioned findings together with further evidence from other studies (Table 1) suggest that mitochondrial Ca2+‐handling proteins are relevant for drug targeting.
Table 1.
Phenotypic analysis of the mitochondrial Ca2+ uniporter complex components after genetic manipulation in the context of a whole organism or a specific tissue
| Gene | Specie | Organ/tissue | Genetic perturbation | Phenotype | Refs/source |
|---|---|---|---|---|---|
| Mitochondrial Ca2+ influx | |||||
| MCU | M. musculus | Whole organism (C57BL/6) | KO | Embryonic mortality | IMPC |
| Whole organism (C57BL/6xCD1) | KO |
|
Pan et al. (2013) | ||
| Whole organism | KO, single allele |
|
IMPC | ||
| Skeletal muscle | OE | Skeletal muscle hypertrophy | Mammucari et al. (2015) | ||
| KD | Skeletal muscle atrophy | ||||
| Heart | DN – MCU expression | Impaired cathecolamine‐induced heart rate increase | Wu et al. (2015) | ||
| KO |
|
||||
| D. melanogaster | Mushroom body neurons | KD (during pupation) | Impaired adult memory formation | Drago & Davis (2016) | |
| D. rerio | Whole organism | KD | Alterations during gastrulation | Prudent et al. (2013) | |
| C. elegans | Whole organism | KO | Impaired wound healing | Xu & Chisholm (2014) | |
| T. brucei | Whole organism | KD/KO | Defective growth | Huang et al. (2013) | |
| OE | Decreased infectivity to mice | ||||
| MCUb | M. musculus | Whole organism | KO |
|
IMPC |
| MCUR1 | M. musculus | Heart | KO | Partial perinatal mortality | Tomar et al. (2016) |
| Vasculature | KO |
|
|||
| MICU1 | H. sapiens | Whole organism | Loss‐of‐function mutations |
|
Logan et al. (2014) |
| Whole organism | Deletion (exon 1) |
|
Lewis‐Smith et al. (2016) | ||
| M. musculus | Whole organism (C57BL/6) | KO | Perinatal mortality | Antony et al. (2016) | |
| Whole organism (C57BL/6N or C57BL/6NxJF1) | KO |
|
Liu et al. (2016) | ||
| Liver | KD |
|
Antony et al. (2016) | ||
| MICU2 | M. musculus | Whole organism | KO |
Abnormal active cardiac relaxation Lethal abdominal aortic aneurisms (in hypertensive mice) |
Bick et al. (2017) |
| EMRE | M. musculus | Whole organism | KO | Embryonic mortality | IMPC |
| M. musculus (MICU1 −/−) | Whole organism | KO, single allele | Survival (rescue of high perinatal mortality) | Liu et al. (2016) | |
| Mitochondrial Ca2+ efflux | |||||
| NCLX | M. musculus | Heart | KO |
|
Luongo et al. (2017) |
C. elegans, Caenorhabditis elegans; D. melanogaster, Drosophila melanogaster; DN‐MCU, dominant negative MCU; D. rerio, Danio rerio; EMRE, essential MCU regulator; H. sapiens, Homo sapiens; IMPC, International Mouse Phenotyping Consortium; KD, knockdown; KO, knockout; MCU, mitochondrial calcium uniporter; MCUb, mitochondrial calcium uniporter b; MCUR1, mitochondrial calcium uniporter regulator 1; MICU1, mitochondrial calcium uptake 1; MICU2, mitochondrial calcium uptake 2; M. musculus, Mus musculus; NCLX, Li‐permeable Na+/Ca2+ exchanger; OE, overexpression; T. brucei, Trypanosoma brucei.
Pharmacological modulation of mitochondrial Ca2+ homeostasis
For more than 30 years, the only approaches and tools available to address the physiopathological importance of mitochondrial Ca2+ homeostasis relied on the use of pharmacological compounds that impinge on mitochondrial Ca2+ uptake and release processes.
Since the late 1960s, pioneer reports have demonstrated that lanthanides (particularly La3+) and the transition metal derivative RuR are powerful inhibitors of Ca2+‐associated responses in mitochondria (Mela, 1968, 1969). Studies by Vasington et al. (1972) and Reed & Bygrave (1974) established RuR and lanthanides as tightly binding inhibitors of Ca2+ transport in mitochondria, competing for different binding sites, which differ from the Ca2+‐transport site. Years later, an oxygen‐bridged dimeric Ru amine complex, purified from RuR, which absorbs light at 360 nm (named Ru360), was reported to exert a more specific inhibition of mitochondrial Ca2+ uptake in vitro (Ying et al. 1991; Matlib et al. 1998; Zazueta et al. 1998) and in situ in intact cardiomyocytes (Matlib et al. 1998). Other compounds that directly target additional mitochondrial functions, by dissipating the ΔΨm (e.g. uncouplers such as carbonyl cyanide‐p‐trifluoromethoxyphenylhydrazone (FCCP)/carbonyl cyanide m‐chlorophenyl hydrazone (CCCP) or dinitrophenol), inhibiting the ability to synthetize or transport ATP (oligomycin), and blocking the respiratory chain complexes (e.g. rotenone, antimycin A, potassium cyanide), have also an inhibitory effect on mitochondrial Ca2+ uptake. Several compounds are also known to inhibit mitochondrial Ca2+ extrusion through the mNCLX. Among those are benzothiazepines and benzodiazepines, including diltiazem, clonazepam and amiloride, as well as related compounds such as semotiadil, verapamil, trifluoroperazine, tetraphenilphosphonium and cyclosporine A (Jurkowitz et al. 1983; Matlib et al. 1983; Wolkowicz et al. 1983; Wingrove & Gunter, 1986; Wei et al. 2011). Of this class, the benzothiazepine CGP‐37157 was shown to be the most potent and selective inhibitor of mNCLX (Matlib et al. 1983; Chiesi et al. 1988), with a 10‐fold higher affinity than any other inhibitor.
The inhibitory properties of these compounds on mitochondrial Ca2+ dynamics are especially relevant in pathological situations where mitochondrial Ca2+ overload has been shown to be detrimental. In fact, protective effects of approaches that moderate mitochondrial Ca2+ accumulation have been demonstrated for different disease contexts. Chemical inhibition by RuR and Ru360 improved the functional recovery of hearts after ischaemia and suppressed arrhythmias and haemodynamic dysfunction elicited by reperfusion in vivo (Garcia‐Rivas Gde et al. 2006). Similarly, modest depolarization of ΔΨm, by transient pharmacological uncoupling of mitochondrial oxidative phosphorylation, was also found to be a potential therapeutic strategy for several human disorders that involve metabolic and mitochondrial oxidative stress, including Parkinson's and Alzheimer's diseases (Wu et al. 2011; Geisler et al. 2017), cerebral ischaemia (Korde et al. 2005), heart failure (Brennan et al. 2006), and metabolic diseases such as diabetes and obesity (Parascandola, 1974; Perry et al. 2013; Tao et al. 2014). Pharmacological inhibition of mNCLX with CGP‐37157 conferred protection in hippocampal slices against veratridine‐induced Ca2+ and Na+ overload by regulating oxidative stress and p38 mitogen‐activated protein kinase‐linked activation of cell death (Nicolau et al. 2010). In addition, by enhancing mitochondrial oxidative metabolism, CGP‐37157 promoted ATP generation and increased glucose‐stimulated insulin secretion in rat islets and INS‐1 cells (Lee et al. 2003). These initial results illustrated the potential utility of drugs that inhibit mitochondrial Ca2+ release as neuroprotective tools and insulin secretagogues, underscoring mNCLX as a novel drug target in neurological and metabolic disorders.
However, all of the pharmacological approaches and tools to modulate mitochondrial Ca2+ homeostasis listed above have major drawbacks when applied in vivo. Classical MCU and mNCLX inhibitors (1) lack specificity, as they also inhibit other channels and transporters in the cell; (2) show several side effects, interfering with other organelle functions and/or several extra‐mitochondrial targets; and (3) reveal suboptimal cellular targeting, as most of them are poorly or not cell‐permeant. The most commonly used mitochondrial Ca2+ uptake inhibitors, RuR, Ru360 and its derivatives (Nathan et al. 2017), are potent and effective on isolated mitochondria, but their use in intact cells or in vivo is inadequate due to their limited permeance across the plasma membrane (Hajnoczky et al. 2006). In addition, RuR, due to its affinity for ryanodine receptors, was also shown to inhibit Ca2+ release from the SR (Chamberlain et al. 1984). Moreover, positive and negative inotropic responses to RuR were observed in isolated rat hearts in a dose‐dependent manner. These effects were attributed to its concomitant capacity to inhibit SR Ca2+ release or sarcolemmal Na+/Ca2+ exchange (Gupta et al. 1988).
Mitochondrial Ca2+ uptake inhibition is often indirectly achieved by depolarizing the ΔΨm. Nevertheless, in most cells types, compounds that inhibit the electron transport chain (ETC) also increase mitochondrial reactive oxygen species production (rotenone, antimycin, myxothiazol) and inhibit the plasma membrane Na+/K+‐ATPase (oligomycin) (Plesner & Plesner, 1991). In addition, the protonophore activity of uncouplers (FCCP/CCCP or dinitrophenol) is not limited to mitochondria, as they are also able to dissipate proton gradients across the plasma membrane and other membranous organelles at concentrations comparable to the ones used for mitochondria. Therefore, these chemical strategies lead to numerous undesired side effects, including plasma membrane depolarization, inhibition of ATP production, alterations in intracellular pH, and ultimately cytotoxicity (Juthberg & Brismar, 1997; Buckler & Vaughan‐Jones, 1998; Park et al. 2002).
Major concerns were also raised regarding the use of the known inhibitors of mNCLX owing to their additional interaction with many Ca2+ channels and transporters in the cell. The benzodiazepine and benzothiazepine analogues diltiazem and clonazepam were found to inhibit the L‐type Ca2+ channel in the plasma membrane (Koidl et al. 1997) and other receptor proteins in mitochondria (Kinnally et al. 1993). Even the classical blocker CGP‐37157, at low micromolar concentration, was more recently shown to affect Ca2+ fluxes through the ryanodine receptor and block the sarco/endoplasmic reticulum Ca2+‐ATPase pump in striated muscle (Neumann et al. 2011). Many other studies also demonstrated the pleiotropic nature of this compound in different cell types. Resembling the effects of the other described inhibitors, CGP37157 also inhibits the L‐type Ca2+ channel in β‐cells at concentrations considered specific for mNCLX (Luciani et al. 2007), and modulates intracellular Ca2+ entry through voltage gated Ca2+ channels in cortical neurons, attenuating NMDA‐induced cytosolic and mitochondrial Ca2+ overload (Ruiz et al. 2014). As these channels constitute an integral part of the cellular machinery that regulates Ca2+ homeostasis, this constitutes a confounding factor to inhibition of mitochondrial Ca2+ efflux mechanisms. Furthermore, this evidence further compromised the role of mNCLX in the regulation of insulin secretion and prevention of neuronal cell death.
Taken together, these comprehensive studies demonstrated that caution should be taken regarding the use of these chemical inhibitors of mitochondrial influx and efflux pathways, especially in an in vivo context. Therefore, over the past years there has been an urge to find drugs and therapeutic strategies that directly and specifically modulate mitochondrial Ca2+ homeostasis.
Novel drug‐screening strategies to identify direct modulators of mitochondrial Ca2+ homeostasis
The molecular characterization of mitochondrial Ca2+ pathways has opened the possibility of systematic and large‐scale drug screenings to identify novel means to target MCUC and mNCLX. The right approach to drug discovery depends on the availability of a robust, affordable and highly selective assay compatible with high‐throughput screening (HTS) (Walters & Namchuk, 2003). Common methods used to monitor dynamic changes in mitochondrial Ca2+ levels, such as cell‐based Ca2+ imaging assays and patch‐clamp recordings in mitoplasts, have not been optimized so far for HTS. Furthermore, the close functional interconnection between intracellular Ca2+ signalling, energy production, ΔΨm and Ca2+ uptake and release constitute a major challenge in the design of robust and effective high‐throughput assays for the discovery of specific modulators of MCUC and mNCLX. As mentioned above, the entry and exit of Ca2+ in mitochondria is dependent on the same ΔΨm used to produce ATP and, therefore, any compound with inhibitory effects on mitochondrial bioenergetics and ion gradients will indirectly affect mitochondrial Ca2+ homeostasis. Moreover, MCUC and mNCLX are intracellular targets and their activities depend on increases of cytoplasmic ionic concentrations by signalling events upstream of mitochondria. Accordingly, there is the potential in cell‐based assays for numerous false‐positive hits.
Very recently, a novel systematic chemical biology approach was developed by our group to identify small molecule drugs that directly modulate MCU (Arduino et al. 2017). We used mitochondria isolated from the yeast S. cerevisiae, wherein mitochondrial Ca2+ uptake activity was reconstituted by heterologous expression of the human MCU and its essential regulator EMRE, and MCU‐mediated Ca2+ uptake was quantitatively measured by the Ca2+‐sensitive photoprotein aequorin, stably expressed in the mitochondrial matrix. This approach takes advantage of several key properties of yeast mitochondria. First, they do not possess any intrinsic mitochondrial Ca2+ permeability (Carafoli & Lehninger, 1971). Second, they have a simplified OXPHOS system lacking complex I but including a d‐lactate:cytochrome c oxidoreductase (DLD), which is sensitive to d‐lactate and enables the direct transfer of electrons to cytochrome c oxidase (complex IV). Thus, when mitochondria are energized with d‐lactate there is a bioenergetic shunt pathway that bypasses the majority of the ETC complexes and NADH generating pathways while it is still sufficient to build up ΔΨm (Gregolin & D'Alberton, 1964; Pajot & Claisse, 1974). In our study, Ca2+ uptake in mitochondria fuelled with succinate remained sensitive to blockers of all the ETC complexes, whereas the use of d‐lactate as a respiratory substrate rescued the inhibitory effects of ETC blockers upstream of complex IV (Arduino et al. 2017). Likewise, yeast mitochondria developed and maintained a CCCP‐insensitive ΔΨm when assayed in an isosmotic mannitol–sucrose medium. In contrast, in a nearly isotonic KCl‐based medium, CCCP treatment resulted in a dramatic reduction of mitochondrial Ca2+ uptake (Arduino et al. 2017). Finally, yeast mitochondria reconstituted with coelenterazine can be frozen without losing the ability to generate a ΔΨm that is sufficiently high to allow Ca2+ fluxes as well as other mitochondrial processes such as, for example, protein import (Hartl & Neupert, 1990; Koll et al. 1992; Izawa & Unger, 2017). Altogether, these properties make the use of reconstituted yeast mitochondria a powerful high‐throughput drug screening strategy to identify specific and direct modulators of MCU activity, by minimizing false discovery rate due to confounding effects of drug‐mediated inhibition of ΔΨm and bioenergetics, ETC and signalling events upstream of mitochondria.
In a screen of ∼700 Food and Drug Administration (FDA)‐approved drugs, our group identified mitoxantrone as a specific inhibitor of MCU (Arduino et al. 2017). We confirmed that mitoxantrone inhibits mitochondrial Ca2+ uptake in mammalian cells without affecting mitochondrial bioenergetics. In addition, patch‐clamp electrophysiology in human mitoplasts treated with mitoxantrone showed a direct and reversible inhibition of MCU‐dependent Ca2+ currents when the drug was applied on the cytosolic but not on the matrix side. Although Ca2+ channel blockers are often reported to have off‐target effects on other ion channels, mitoxantrone exhibited an exquisite selectivity for MCU when tested on ER and plasma membrane channels. Based on our structure ‐ activity relationship (SAR) analysis, we found that the antineoplastic and anti‐MCU properties of mitoxantrone are not interrelated. Accordingly, the quinizarin moiety, which is common to other related chemotherapeutic drugs and mediates DNA intercalation and topoisomerase II binding, is dispensable for the inhibition of MCU. Instead, the positively charged side chains of mitoxantrone at positions 5 and 8 played a key role as they mediate the binding to highly conserved aspartate residues in the selectivity filter of the uniporter channel (Arduino et al. 2017).
Altogether, these results validate the yeast mitochondria‐based screening strategy as a tool to discover drug molecules directly targeting MCUC. Indeed, the assay represents a flexible, cost‐effective HTS solution, which can be exploited to reconstitute and target other subunits of the uniporter, such as tissue‐specific regulators (Fig. 1). This would address the need to tailor the modulation of MCUC activity to the tissue's physiology as well as to pathological consequences of loss or gain of function mutations. Additionally, drug delivery should ensure specificity to an intended tissue or cell type while simultaneously minimizing cytotoxicity by reducing side effects due to undesired drug accumulation on peripheral healthy tissues. This is normally achieved by active targeting, i.e. targeting unique receptors in specific cell types or tissues of interest by ligands present on the surface of drug delivery nanocarriers (e.g. polymeric nanoparticles or liposomes) (Cheng et al. 2015). Nowadays, the application of these drug delivery platforms constitutes a promising approach of demonstrated efficacy even in disease contexts where pharmacological targeting of MCUC has been proven to be beneficial, such as myocardial infarction and ischaemia–reperfusion injury (Takahama et al. 2009; Magruder et al. 2017).
Figure 1. Workflow of the drug screen assay in reconstituted yeast mitochondria.
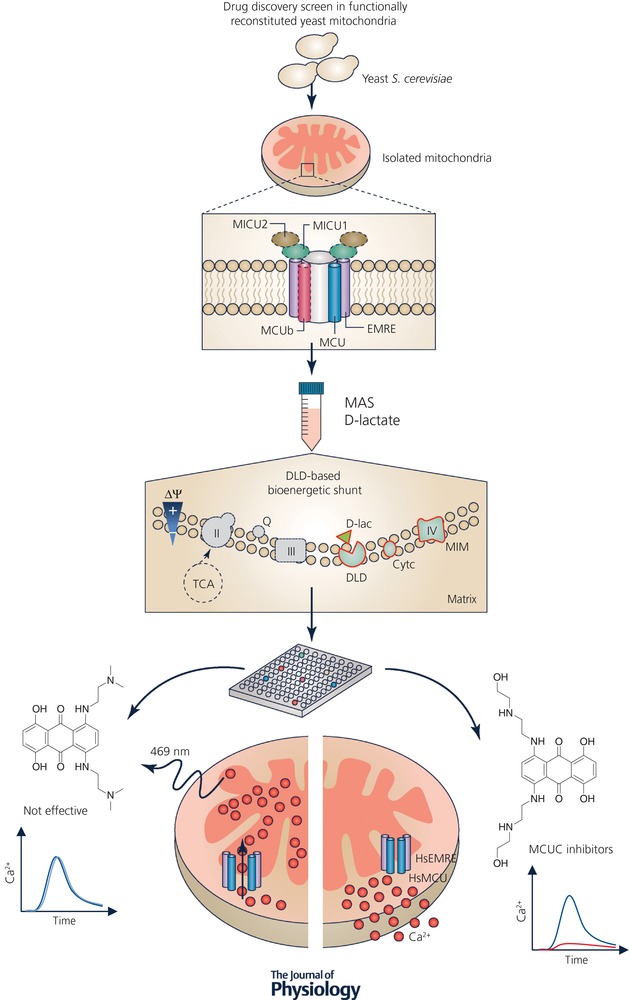
The yeast S. cerevisiae constitutes a versatile system that can be reconstituted with different components of the MCUC machinery (e.g. MCU and EMRE and additionally MCUb, MICU1 and/or MICU2, as represented with dashed lines). When D‐Lactate (D‐Lac) is supplied to mitochondria as the energy source, it provides a bioenergetic shunt pathway that minimizes the detection of false‐positive hits. This drug screen platform allows the quantification of mitochondrial Ca2+ uptake kinetics based on mitochondria‐targeted‐aequorin luminescence emitted at 469 nm. MCUC modulators are accurately identified based on their effects on mitochondrial Ca2+ uptake kinetics. MAS, mannitol‐sucrose buffer; DLD, D‐lactate:cytochrome c oxidoreductase; TCA, tricarboxylic acid cycle; Q, coenzyme Q; Cytc, cytochrome c; II, succinate dehydrogenase; III, coenzyme Q:cytochrome c‐oxidoreductase; IV, cytochrome c oxidase.
In the near future, in order to enhance the translational value of differently regulated proteins involved in mitochondrial Ca2+ homeostasis as biomarkers or disease‐targets, it is essential to integrate hints from different disciplines. Those can comprise results from drug screenings, detailed information from clinical studies (e.g. disease endophenotypes) and prediction of novel drug–target interactions. This strategy will not only allow the identification of novel drug molecules but also provide a broader knowledge on their application to treat or halt human diseases in which mitochondrial Ca2+ homeostasis deregulation and mitochondrial dysfunction are pathological hallmarks.
Additional information
Competing interests
None of the authors have any conflict of interests to declare.
Author contributions
Both authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by the German Research Foundation (DFG) under the Emmy Noether Programme (PE 2053/1‐1) and the Bavarian Ministry of Sciences, Research and the Arts in the framework of the Bavarian Molecular Biosystems Research Network (D2‐F5121.2‐10c/4822) to F.P. and D.M.A.
Acknowledgements
We thank all the members of the Perocchi laboratory for critical reading of the manuscript.
Biographies
Daniela Arduino obtained her PhD in ‘Biomedical Sciences’ from CNC‐UC, Portugal. Her studies in the laboratories of Prof. Cardoso and Prof. Cuervo uncovered a main role for mitochondrial metabolism in the regulation of autophagy–lysosomal pathways in Parkinson's disease. She is currently a postdoctoral fellow in Fabiana Perocchi's laboratory, where she has been developing novel high‐throughput chemical screening approaches to identify specific modulators of mitochondrial Ca2+ homeostasis as well as characterizing mitochondrial signalling networks and elucidating their potential roles in cell physiology and disease.

Fabiana Perocchi is group leader at LMU and HMGU in Munich. Throughout her graduate studies at EMBL and postdoctoral training at Harvard, she became an expert in the area of functional genomics applied to the study of mitochondrial biogenesis and calcium signalling. Her research interests have been on the development of integrative strategies that combine systematic approaches with genetic, biochemical and physiological studies of mitochondrial function.
Edited by: Ole Petersen & Maike Glitsch
This review was presented at the symposium ‘Intracellular Calcium Signals: Generation, Function and Therapeutic Intervention’, which took place at Gordon Research Conferences 2017, Lucca, Italy, 18–23 June 2017.
References
- Antony AN, Paillard M, Moffat C, Juskeviciute E, Correnti J, Bolon B, Rubin E, Csordas G, Seifert EL, Hoek JB & Hajnoczky G (2016). MICU1 regulation of mitochondrial Ca2+ uptake dictates survival and tissue regeneration. Nat Commun 7, 10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arduino DM, Wettmarshausen J, Vais H, Navas‐Navarro P, Cheng Y, Leimpek A, Ma Z, Delrio‐Lorenzo A, Giordano A, Garcia‐Perez C, Medard G, Kuster B, Garcia‐Sancho J, Mokranjac D, Foskett JK, Alonso MT & Perocchi F (2017). Systematic identification of MCU modulators by orthogonal interspecies chemical screening. Mol Cell 67, 711–723.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher‐Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V & Mootha VK (2011). Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben‐Kasus Nissim T, Zhang X, Elazar A, Roy S, Stolwijk JA, Zhou Y, Motiani RK, Gueguinou M, Hempel N, Hershfinkel M, Gill DL, Trebak M & Sekler I (2017). Mitochondria control store‐operated Ca2+ entry through Na+ and redox signals. EMBO J 36, 797–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutner G, Sharma VK, Lin L, Ryu SY, Dirksen RT & Sheu SS (2005). Type 1 ryanodine receptor in cardiac mitochondria: transducer of excitation‐metabolism coupling. Biochim Biophys Acta 1717, 1–10. [DOI] [PubMed] [Google Scholar]
- Bick AG, Wakimoto H, Kamer KJ, Sancak Y, Goldberger O, Axelsson A, DeLaughter DM, Gorham JM, Mootha VK, Seidman JG & Seidman CE (2017). Cardiovascular homeostasis dependence on MICU2, a regulatory subunit of the mitochondrial calcium uniporter. Proc Natl Acad Sci USA 114, E9096–E9104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand MD, Chen CH & Lehninger AL (1976). Stoichiometry of H+ ejection during respiration‐dependent accumulation of Ca2+ by rat liver mitochondria. J Biol Chem 251, 968–974. [PubMed] [Google Scholar]
- Brennan JP, Southworth R, Medina RA, Davidson SM, Duchen MR & Shattock MJ (2006). Mitochondrial uncoupling, with low concentration FCCP, induces ROS‐dependent cardioprotection independent of KATP channel activation. Cardiovasc Res 72, 313–321. [DOI] [PubMed] [Google Scholar]
- Buckler KJ & Vaughan‐Jones RD (1998). Effects of mitochondrial uncouplers on intracellular calcium, pH and membrane potential in rat carotid body type I cells. J Physiol 513, 819–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao C, Wang S, Cui T, Su XC & Chou JJ (2017). Ion and inhibitor binding of the double‐ring ion selectivity filter of the mitochondrial calcium uniporter. Proc Natl Acad Sci USA 114, E2846–E2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carafoli E & Lehninger AL (1971). A survey of the interaction of calcium ions with mitochondria from different tissues and species. Biochem J 122, 681–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carafoli E, Tiozzo R, Lugli G, Crovetti F & Kratzing C (1974). The release of calcium from heart mitochondria by sodium. J Mol Cell Cardiol 6, 361–371. [DOI] [PubMed] [Google Scholar]
- Cardenas C, Miller RA, Smith I, Bui T, Molgo J, Muller M, Vais H, Cheung KH, Yang J, Parker I, Thompson CB, Birnbaum MJ, Hallows KR & Foskett JK (2010). Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 142, 270–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenas C, Muller M, McNeal A, Lovy A, Jana F, Bustos G, Urra F, Smith N, Molgo J, Diehl JA, Ridky TW & Foskett JK (2016). Selective vulnerability of cancer cells by inhibition of Ca2+ transfer from endoplasmic reticulum to mitochondria. Cell Rep 15, 219–220. [DOI] [PubMed] [Google Scholar]
- Chamberlain BK, Volpe P & Fleischer S (1984). Inhibition of calcium‐induced calcium release from purified cardiac sarcoplasmic reticulum vesicles. J Biol Chem 259, 7547–7553. [PubMed] [Google Scholar]
- Chance B (1965). The energy‐linked reaction of calcium with mitochondria. J Biol Chem 240, 2729–2748. [PubMed] [Google Scholar]
- Chaudhuri D, Artiga DJ, Abiria SA & Clapham DE (2016). Mitochondrial calcium uniporter regulator 1 (MCUR1) regulates the calcium threshold for the mitochondrial permeability transition. Proc Natl Acad Sci USA 113, E1872–E1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri D, Sancak Y, Mootha VK & Clapham DE (2013). MCU encodes the pore conducting mitochondrial calcium currents. Elife 2, e00704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CJ, Tietjen GT, Saucier‐Sawyer JK & Saltzman WM (2015). A holistic approach to targeting disease with polymeric nanoparticles. Nat Rev Drug Discov 14, 239–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiesi M, Schwaller R & Eichenberger K (1988). Structural dependency of the inhibitory action of benzodiazepines and related compounds on the mitochondrial Na+‐Ca2+ exchanger. Biochem Pharmacol 37, 4399–4403. [DOI] [PubMed] [Google Scholar]
- Curry MC, Peters AA, Kenny PA, Roberts‐Thomson SJ & Monteith GR (2013). Mitochondrial calcium uniporter silencing potentiates caspase‐independent cell death in MDA‐MB‐231 breast cancer cells. Biochem Biophys Res Commun 434, 695–700. [DOI] [PubMed] [Google Scholar]
- De Marchi U, Santo‐Domingo J, Castelbou C, Sekler I, Wiederkehr A & Demaurex N (2014). NCLX protein, but not LETM1, mediates mitochondrial Ca2+ extrusion, thereby limiting Ca2+‐induced NAD(P)H production and modulating matrix redox state. J Biol Chem 289, 20377–20385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stefani D, Raffaello A, Teardo E, Szabo I & Rizzuto R (2011). A forty‐kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stefani D, Rizzuto R & Pozzan T (2016). Enjoy the trip: calcium in mitochondria back and forth. Annu Rev Biochem 85, 161–192. [DOI] [PubMed] [Google Scholar]
- Deluca HF & Engstrom GW (1961). Calcium uptake by rat kidney mitochondria. Proc Natl Acad Sci USA 47, 1744–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton RM & McCormack JG (1980). On the role of the calcium transport cycle in heart and other mammalian mitochondria. FEBS Lett 119, 1–8. [DOI] [PubMed] [Google Scholar]
- Drago I & Davis RL (2016). Inhibiting the mitochondrial calcium uniporter during development impairs memory in adult Drosophila . Cell Rep 16, 2763–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Rivas Gde J, Carvajal K, Correa F & Zazueta C (2006). Ru360, a specific mitochondrial calcium uptake inhibitor, improves cardiac post‐ischaemic functional recovery in rats in vivo. Br J Pharmacol 149, 829–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler JG, Marosi K, Halpern J & Mattson MP (2017). DNP, mitochondrial uncoupling, and neuroprotection: A little dab'll do ya. Alzheimers Dement 13, 582–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregolin C & D'Alberton A (1964). On the properties of a D(−)lactic oxidase system in respiratory particles of yeast. Biochem Biophys Res Commun 14, 103–108. [DOI] [PubMed] [Google Scholar]
- Gupta MP, Innes IR & Dhalla NS (1988). Responses of contractile function to ruthenium red in rat heart. Am J Physiol Heart Circ Physiol 255, H1413–H1420. [DOI] [PubMed] [Google Scholar]
- Hajnoczky G, Csordas G, Das S, Garcia‐Perez C, Saotome M, Sinha Roy S & Yi M (2006). Mitochondrial calcium signalling and cell death: approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium 40, 553–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl FU & Neupert W (1990). Protein sorting to mitochondria: evolutionary conservations of folding and assembly. Science 247, 930–938. [DOI] [PubMed] [Google Scholar]
- Holmstrom KM, Pan X, Liu JC, Menazza S, Liu J, Nguyen TT, Pan H, Parks RJ, Anderson S, Noguchi A, Springer D, Murphy E & Finkel T (2015). Assessment of cardiac function in mice lacking the mitochondrial calcium uniporter. J Mol Cell Cardiol 85, 178–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G, Vercesi AE & Docampo R (2013). Essential regulation of cell bioenergetics in Trypanosoma brucei by the mitochondrial calcium uniporter. Nat Commun 4, 2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izawa T & Unger AK (2017). Isolation of mitochondria from Saccharomyces cerevisiae . Methods Mol Biol 1567, 33–42. [DOI] [PubMed] [Google Scholar]
- Jiang D, Zhao L & Clapham DE (2009). Genome‐wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science 326, 144–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D, Zhao L, Clish CB & Clapham DE (2013). Letm1, the mitochondrial Ca2+/H+ antiporter, is essential for normal glucose metabolism and alters brain function in Wolf‐Hirschhorn syndrome. Proc Natl Acad Sci USA 110, E2249–E2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurkowitz MS, Altschuld RA, Brierley GP & Cragoe EJ Jr (1983). Inhibition of Na+‐dependent Ca2+ efflux from heart mitochondria by amiloride analogues. FEBS Lett 162, 262–265. [DOI] [PubMed] [Google Scholar]
- Juthberg SK & Brismar T (1997). Effect of metabolic inhibitors on membrane potential and ion conductance of rat astrocytes. Cell Mol Neurobiol 17, 367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamer KJ, Grabarek Z & Mootha VK (2017). High‐affinity cooperative Ca2+ binding by MICU1‐MICU2 serves as an on‐off switch for the uniporter. EMBO Rep 18, 1397–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinnally KW, Zorov DB, Antonenko YN, Snyder SH, McEnery MW & Tedeschi H (1993). Mitochondrial benzodiazepine receptor linked to inner membrane ion channels by nanomolar actions of ligands. Proc Natl Acad Sci USA 90, 1374–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirichok Y, Krapivinsky G & Clapham DE (2004). The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427, 360–364. [DOI] [PubMed] [Google Scholar]
- Koidl B, Miyawaki N & Tritthart HA (1997). A novel benzothiazine Ca2+ channel antagonist, semotiadil, inhibits cardiac L‐type Ca2+ currents. Eur J Pharmacol 322, 243–247. [DOI] [PubMed] [Google Scholar]
- Koll H, Guiard B, Rassow J, Ostermann J, Horwich AL, Neupert W & Hartl FU (1992). Antifolding activity of hsp60 couples protein import into the mitochondrial matrix with export to the intermembrane space. Cell 68, 1163–1175. [DOI] [PubMed] [Google Scholar]
- Korde AS, Pettigrew LC, Craddock SD & Maragos WF (2005). The mitochondrial uncoupler 2,4‐dinitrophenol attenuates tissue damage and improves mitochondrial homeostasis following transient focal cerebral ischemia. J Neurochem 94, 1676–1684. [DOI] [PubMed] [Google Scholar]
- Kovacs‐Bogdan E, Sancak Y, Kamer KJ, Plovanich M, Jambhekar A, Huber RJ, Myre MA, Blower MD & Mootha VK (2014). Reconstitution of the mitochondrial calcium uniporter in yeast. Proc Natl Acad Sci USA 111, 8985–8990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, York AJ, Zhang J, Bers DM & Molkentin JD (2015). The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Rep 12, 15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B, Miles PD, Vargas L, Luan P, Glasco S, Kushnareva Y, Kornbrust ES, Grako KA, Wollheim CB, Maechler P, Olefsky JM & Anderson CM (2003). Inhibition of mitochondrial Na+‐Ca2+ exchanger increases mitochondrial metabolism and potentiates glucose‐stimulated insulin secretion in rat pancreatic islets. Diabetes 52, 965–973. [DOI] [PubMed] [Google Scholar]
- Lehninger AL ( 1971). A soluble, heat‐labile, high‐affinity Ca2+‐binding factor extracted from rat liver mitochondria. Biochem Biophys Res Commun 42, 312–318. [DOI] [PubMed] [Google Scholar]
- Lewis‐Smith D, Kamer KJ, Griffin H, Childs AM, Pysden K, Titov D, Duff J, Pyle A, Taylor RW, Yu‐Wai‐Man P, Ramesh V, Horvath R, Mootha VK & Chinnery PF (2016). Homozygous deletion in MICU1 presenting with fatigue and lethargy in childhood. Neurol Genet 2, e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Hao Y, Chen H, He Q, Yuan Z & Cheng J (2015). Mitochondrial calcium uniporter protein MCU is involved in oxidative stress‐induced cell death. Protein Cell 6, 434–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JC, Liu J, Holmstrom KM, Menazza S, Parks RJ, Fergusson MM, Yu ZX, Springer DA, Halsey C, Liu C, Murphy E & Finkel T (2016). MICU1 serves as a molecular gatekeeper to prevent in vivo mitochondrial calcium overload. Cell Rep 16, 1561–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan CV, Szabadkai G, Sharpe JA, Parry DA, Torelli S, Childs AM, Kriek M, Phadke R, Johnson CA, Roberts NY, Bonthron DT, Pysden KA, Whyte T, Munteanu I, Foley AR, Wheway G, Szymanska K, Natarajan S, Abdelhamed ZA, Morgan JE, Roper H, Santen GW, Niks EH, van der Pol WL, Lindhout D, Raffaello A, De Stefani D, den Dunnen JT, Sun Y, Ginjaar I, Sewry CA, Hurles M, Rizzuto R, Consortium UK, Duchen MR, Muntoni F & Sheridan E (2014). Loss‐of‐function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat Genet 46, 188–193. [DOI] [PubMed] [Google Scholar]
- Luciani DS, Ao P, Hu X, Warnock GL & Johnson JD (2007). Voltage‐gated Ca2+ influx and insulin secretion in human and mouse beta‐cells are impaired by the mitochondrial Na+/Ca2+ exchange inhibitor CGP‐37157. Eur J Pharmacol 576, 18–25. [DOI] [PubMed] [Google Scholar]
- Luongo TS, Lambert JP, Gross P, Nwokedi M, Lombardi AA, Shanmughapriya S, Carpenter AC, Kolmetzky D, Gao E, van Berlo JH, Tsai EJ, Molkentin JD, Chen X, Madesh M, Houser SR & Elrod JW (2017). The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature 545, 93–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luongo TS, Lambert JP, Yuan A, Zhang X, Gross P, Song J, Shanmughapriya S, Gao E, Jain M, Houser SR, Koch WJ, Cheung JY, Madesh M & Elrod JW (2015). The mitochondrial calcium uniporter matches energetic supply with cardiac workload during stress and modulates permeability transition. Cell Rep 12, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maack C, Cortassa S, Aon MA, Ganesan AN, Liu T & O'Rourke B (2006). Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation‐contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ Res 99, 172–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magruder JT, Crawford TC, Lin YA, Zhang F, Grimm JC, Kannan RM, Kannan S & Sciortino CM (2017). Selective localization of a novel dendrimer nanoparticle in myocardial ischemia‐reperfusion injury. Ann Thorac Surg 104, 891–898. [DOI] [PubMed] [Google Scholar]
- Mallilankaraman K, Cardenas C, Doonan PJ, Chandramoorthy HC, Irrinki KM, Golenar T, Csordas G, Madireddi P, Yang J, Muller M, Miller R, Kolesar JE, Molgo J, Kaufman B, Hajnoczky G, Foskett JK & Madesh M (2012). MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat Cell Biol 14, 1336–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammucari C, Gherardi G, Zamparo I, Raffaello A, Boncompagni S, Chemello F, Cagnin S, Braga A, Zanin S, Pallafacchina G, Zentilin L, Sandri M, De Stefani D, Protasi F, Lanfranchi G & Rizzuto R (2015). The mitochondrial calcium uniporter controls skeletal muscle trophism in vivo. Cell Rep 10, 1269–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcu R, Wiczer BM, Neeley CK & Hawkins BJ (2014). Mitochondrial matrix Ca2+ accumulation regulates cytosolic NAD+/NADH metabolism, protein acetylation, and sirtuin expression. Mol Cell Biol 34, 2890–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matlib MA, Lee SW, Depover A & Schwartz A (1983). A specific inhibitory action of certain benzothiazepines and benzodiazepines on the sodium‐calcium exchange process of heart and brain mitochondria. Eur J Pharmacol 89, 327–328. [DOI] [PubMed] [Google Scholar]
- Matlib MA, Zhou Z, Knight S, Ahmed S, Choi KM, Krause‐Bauer J, Phillips R, Altschuld R, Katsube Y, Sperelakis N & Bers DM (1998). Oxygen‐bridged dinuclear ruthenium amine complex specifically inhibits Ca2+ uptake into mitochondria in vitro and in situ in single cardiac myocytes. J Biol Chem 273, 10223–10231. [DOI] [PubMed] [Google Scholar]
- McQuibban AG, Joza N, Megighian A, Scorzeto M, Zanini D, Reipert S, Richter C, Schweyen RJ & Nowikovsky K (2010). A Drosophila mutant of LETM1, a candidate gene for seizures in Wolf‐Hirschhorn syndrome. Hum Mol Genet 19, 987–1000. [DOI] [PubMed] [Google Scholar]
- Medvedev BI, Azarashvily TS, Evtodienko Ju V, Luk'yanenko AI & Yagushinskij LS (1982). Isolation of calcium‐transporting lipid from the mitochondrial glycolipoprotein. Mol Cell Biochem 48, 19–23. [DOI] [PubMed] [Google Scholar]
- Mela L (1968). Interactions of La3+ and local anesthetic drugs with mitochondrial Ca++ and Mn++ uptake. Arch Biochem Biophys 123, 286–293. [DOI] [PubMed] [Google Scholar]
- Mela L (1969). Inhibition and activation of calcium transport in mitochondria. Effect of lanthanides and local anesthetic drugs. Biochemistry 8, 2481–2486. [DOI] [PubMed] [Google Scholar]
- Mironova GD & Utesheva ZhA (1989). [Molecular mechanism of calcium ion transport in mitochondria. I. Glycoprotein‐peptide complex as a component of the electron transport system]. Ukr Biokhim Zh 61, 48–54. [PubMed] [Google Scholar]
- Murgia M & Rizzuto R (2015). Molecular diversity and pleiotropic role of the mitochondrial calcium uniporter. Cell Calcium 58, 11–17. [DOI] [PubMed] [Google Scholar]
- Nathan SR, Pino NW, Arduino DM, Perocchi F, MacMillan SN & Wilson JJ (2017). Synthetic methods for the preparation of a functional analogue of Ru360, a potent inhibitor of mitochondrial calcium uptake. Inorg Chem 56, 3123–3126. [DOI] [PubMed] [Google Scholar]
- Neumann JT, Diaz‐Sylvester PL, Fleischer S & Copello JA (2011). CGP‐37157 inhibits the sarcoplasmic reticulum Ca2+ ATPase and activates ryanodine receptor channels in striated muscle. Mol Pharmacol 79, 141–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG ( 1978). The regulation of extramitochondrial free calcium ion concentration by rat liver mitochondria. Biochem J 176, 463–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG & Scott ID (1980). The regulation of brain mitochondrial calcium‐ion transport. The role of ATP in the discrimination between kinetic and membrane‐potential‐dependent calcium‐ion efflux mechanisms. Biochem J 186, 833–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolau SM, Egea J, Lopez MG & Garcia AG (2010). Mitochondrial Na+/Ca2+ exchanger, a new target for neuroprotection in rat hippocampal slices. Biochem Biophys Res Commun 400, 140–144. [DOI] [PubMed] [Google Scholar]
- Nita II, Hershfinkel M, Lewis EC & Sekler I (2015). A crosstalk between Na+ channels, Na+/K+ pump and mitochondrial Na+ transporters controls glucose‐dependent cytosolic and mitochondrial Na+ signals. Cell Calcium 57, 69–75. [DOI] [PubMed] [Google Scholar]
- Nowikovsky K & Bernardi P (2014). LETM1 in mitochondrial cation transport. Front Physiol 5, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowikovsky K, Froschauer EM, Zsurka G, Samaj J, Reipert S, Kolisek M, Wiesenberger G & Schweyen RJ (2004). The LETM1/YOL027 gene family encodes a factor of the mitochondrial K+ homeostasis with a potential role in the Wolf‐Hirschhorn syndrome. J Biol Chem 279, 30307–30315. [DOI] [PubMed] [Google Scholar]
- Oxenoid K, Dong Y, Cao C, Cui T, Sancak Y, Markhard AL, Grabarek Z, Kong L, Liu Z, Ouyang B, Cong Y, Mootha VK & Chou JJ (2016). Architecture of the mitochondrial calcium uniporter. Nature 533, 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajot P & Claisse ML (1974). Utilization by yeast of D‐lactate and L‐lactate as sources of energy in the presence of antimycin A. Eur J Biochem 49, 275–285. [DOI] [PubMed] [Google Scholar]
- Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, Nolte C, Fishman D, Shoshan‐Barmatz V, Herrmann S, Khananshvili D & Sekler I (2010). NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Natl Acad Sci USA 107, 436–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, Aponte AM, Gucek M, Balaban RS, Murphy E & Finkel T (2013). The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol 15, 1464–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parascandola J ( 1974). Dinitrophenol and bioenergetics: an historical perspective. Mol Cell Biochem 5, 69–77. [DOI] [PubMed] [Google Scholar]
- Park KS, Jo I, Pak K, Bae SW, Rhim H, Suh SH, Park J, Zhu H, So I & Kim KW (2002). FCCP depolarizes plasma membrane potential by activating proton and Na+ currents in bovine aortic endothelial cells. Pflugers Archiv 443, 344–352. [DOI] [PubMed] [Google Scholar]
- Patron M, Checchetto V, Raffaello A, Teardo E, Vecellio Reane D, Mantoan M, Granatiero V, Szabo I, De Stefani D & Rizzuto R (2014). MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol Cell 53, 726–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paupe V, Prudent J, Dassa EP, Rendon OZ & Shoubridge EA (2015). CCDC90A (MCUR1) is a cytochrome c oxidase assembly factor and not a regulator of the mitochondrial calcium uniporter. Cell Metab 21, 109–116. [DOI] [PubMed] [Google Scholar]
- Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE & Mootha VK (2010). MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature 467, 291–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Kim T, Zhang XM, Lee HY, Pesta D, Popov VB, Zhang D, Rahimi Y, Jurczak MJ, Cline GW, Spiegel DA & Shulman GI (2013). Reversal of hypertriglyceridemia, fatty liver disease, and insulin resistance by a liver‐targeted mitochondrial uncoupler. Cell Metab 18, 740–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrungaro C, Zimmermann KM, Kuttner V, Fischer M, Dengjel J, Bogeski I & Riemer J (2015). The Ca2+‐dependent release of the Mia40‐induced MICU1‐MICU2 dimer from MCU regulates mitochondrial Ca2+ uptake. Cell Metab 22, 721–733. [DOI] [PubMed] [Google Scholar]
- Plesner L & Plesner IW (1991). Kinetics of oligomycin inhibition and activation of Na+/K+‐ATPase. Biochim Biophys Acta 1076, 421–426. [DOI] [PubMed] [Google Scholar]
- Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, Girgis HS, Kuchimanchi S, De Groot J, Speciner L, Taneja N, Oshea J, Koteliansky V & Mootha VK (2013). MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS One 8, e55785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prudent J, Popgeorgiev N, Bonneau B, Thibaut J, Gadet R, Lopez J, Gonzalo P, Rimokh R, Manon S, Houart C, Herbomel P, Aouacheria A & Gillet G (2013). Bcl‐wav and the mitochondrial calcium uniporter drive gastrula morphogenesis in zebrafish. Nat Commun 4, 2330. [DOI] [PubMed] [Google Scholar]
- Qiu J, Tan YW, Hagenston AM, Martel MA, Kneisel N, Skehel PA, Wyllie DJ, Bading H & Hardingham GE (2013). Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat Commun 4, 2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffaello A, De Stefani D, Sabbadin D, Teardo E, Merli G, Picard A, Checchetto V, Moro S, Szabo I & Rizzuto R (2013). The mitochondrial calcium uniporter is a multimer that can include a dominant‐negative pore‐forming subunit. EMBO J 32, 2362–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen TP, Wu Y, Joiner ML, Koval OM, Wilson NR, Luczak ED, Wang Q, Chen B, Gao Z, Zhu Z, Wagner BA, Soto J, McCormick ML, Kutschke W, Weiss RM, Yu L, Boudreau RL, Abel ED, Zhan F, Spitz DR, Buettner GR, Song LS, Zingman LV & Anderson ME (2015). Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart. Proc Natl Acad Sci USA 112, 9129–9134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed KC & Bygrave FL (1974). The inhibition of mitochondrial calcium transport by lanthanides and ruthenium red. Biochem J 140, 143–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz A, Alberdi E & Matute C (2014). CGP37157, an inhibitor of the mitochondrial Na+/Ca2+ exchanger, protects neurons from excitotoxicity by blocking voltage‐gated Ca2+ channels. Cell Death Dis 5, e1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Markhard AL, Kitami T, Kovacs‐Bogdan E, Kamer KJ, Udeshi ND, Carr SA, Chaudhuri D, Clapham DE, Li AA, Calvo SE, Goldberger O & Mootha VK (2013). EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 342, 1379–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santulli G, Xie W, Reiken SR & Marks AR (2015). Mitochondrial calcium overload is a key determinant in heart failure. Proc Natl Acad Sci USA 112, 11389–11394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolove PM & Brenza JM (1983). Isolation of a fraction with Ca2+ ionophore properties from rat liver mitochondria. Arch Biochem Biophys 221, 404–416. [DOI] [PubMed] [Google Scholar]
- Sottocasa G, Sandri G, Panfili E, De Bernard B, Gazzotti P, Vasington FD & Carafoli E (1972). Isolation of a soluble Ca2+ binding glycoprotein from ox liver mitochondria. Biochem Biophys Res Commun 47, 808–813. [DOI] [PubMed] [Google Scholar]
- Szabadkai G & Duchen MR (2008). Mitochondria: the hub of cellular Ca2+ signaling. Physiology 23, 84–94. [DOI] [PubMed] [Google Scholar]
- Takahama H, Minamino T, Asanuma H, Fujita M, Asai T, Wakeno M, Sasaki H, Kikuchi H, Hashimoto K, Oku N, Asakura M, Kim J, Takashima S, Komamura K, Sugimachi M, Mochizuki N & Kitakaze M (2009). Prolonged targeting of ischemic/reperfused myocardium by liposomal adenosine augments cardioprotection in rats. J Am Coll Cardiol 53, 709–717. [DOI] [PubMed] [Google Scholar]
- Tao H, Zhang Y, Zeng X, Shulman GI & Jin S (2014). Niclosamide ethanolamine‐induced mild mitochondrial uncoupling improves diabetic symptoms in mice. Nat Med 20, 1263–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomar D, Dong Z, Shanmughapriya S, Koch DA, Thomas T, Hoffman NE, Timbalia SA, Goldman SJ, Breves SL, Corbally DP, Nemani N, Fairweather JP, Cutri AR, Zhang X, Song J, Jana F, Huang J, Barrero C, Rabinowitz JE, Luongo TS, Schumacher SM, Rockman ME, Dietrich A, Merali S, Caplan J, Stathopulos P, Ahima RS, Cheung JY, Houser SR, Koch WJ, Patel V, Gohil VM, Elrod JW, Rajan S & Madesh M (2016). MCUR1 is a scaffold factor for the MCU complex function and promotes mitochondrial bioenergetics. Cell Rep 15, 1673–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trenker M, Malli R, Fertschai I, Levak‐Frank S & Graier WF (2007). Uncoupling proteins 2 and 3 are fundamental for mitochondrial Ca2+ uniport. Nat Cell Biol 9, 445–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MF, Jiang D, Zhao L, Clapham D & Miller C (2014). Functional reconstitution of the mitochondrial Ca2+/H+ antiporter Letm1. J Gen Physiol 143, 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MF, Phillips CB, Ranaghan M, Tsai CW, Wu Y, Willliams C & Miller C (2016). Dual functions of a small regulatory subunit in the mitochondrial calcium uniporter complex. Elife 5, e15545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vais H, Mallilankaraman K, Mak DO, Hoff H, Payne R, Tanis JE & Foskett JK (2016). EMRE is a matrix Ca2+ sensor that governs gatekeeping of the mitochondrial Ca2+ uniporter. Cell Rep 14, 403–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vais H, Tanis JE, Muller M, Payne R, Mallilankaraman K & Foskett JK (2015). MCUR1, CCDC90A, is a regulator of the mitochondrial calcium uniporter. Cell Metab 22, 533–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasington FD, Gazzotti P, Tiozzo R & Carafoli E (1972). The effect of ruthenium red on Ca2+ transport and respiration in rat liver mitochondria. Biochim Biophys Acta 256, 43–54. [DOI] [PubMed] [Google Scholar]
- Vasington FD & Murphy JV (1962). Ca ion uptake by rat kidney mitochondria and its dependence on respiration and phosphorylation. J Biol Chem 237, 2670–2677. [PubMed] [Google Scholar]
- Vecellio Reane D, Vallese F, Checchetto V, Acquasaliente L, Butera G, De Filippis V, Szabo I, Zanotti G, Rizzuto R & Raffaello A (2016). A MICU1 splice variant confers high sensitivity to the mitochondrial Ca2+ uptake machinery of skeletal muscle. Mol Cell 64, 760–773. [DOI] [PubMed] [Google Scholar]
- Walters WP & Namchuk M (2003). Designing screens: how to make your hits a hit. Nat Rev Drug Discovery 2, 259–266. [DOI] [PubMed] [Google Scholar]
- Wei AC, Liu T, Cortassa S, Winslow RL & O'Rourke B (2011). Mitochondrial Ca2+ influx and efflux rates in guinea pig cardiac mitochondria: low and high affinity effects of cyclosporine A. Biochim Biophys Acta 1813, 1373–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingrove DE & Gunter TE (1986). Kinetics of mitochondrial calcium transport. II. A kinetic description of the sodium‐dependent calcium efflux mechanism of liver mitochondria and inhibition by ruthenium red and by tetraphenylphosphonium. J Biol Chem 261, 15166–15171. [PubMed] [Google Scholar]
- Wolkowicz PE, Michael LH, Lewis RM & McMillin‐Wood J (1983). Sodium‐calcium exchange in dog heart mitochondria: effects of ischemia and verapamil. Am J Physiol Heart Circ Physiol 244, H644–H651. [DOI] [PubMed] [Google Scholar]
- Wu Y, Rasmussen TP, Koval OM, Joiner ML, Hall DD, Chen B, Luczak ED, Wang Q, Rokita AG, Wehrens XH, Song LS & Anderson ME (2015). The mitochondrial uniporter controls fight or flight heart rate increases. Nat Commun 6, 6081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YN, Munhall AC & Johnson SW (2011). Mitochondrial uncoupling agents antagonize rotenone actions in rat substantia nigra dopamine neurons. Brain Res 1395, 86–93. [DOI] [PubMed] [Google Scholar]
- Xu S & Chisholm AD (2014). C. elegans epidermal wounding induces a mitochondrial ROS burst that promotes wound repair. Dev Cell 31, 48–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying WL, Emerson J, Clarke MJ & Sanadi DR (1991). Inhibition of mitochondrial calcium ion transport by an oxo‐bridged dinuclear ruthenium ammine complex. Biochemistry 30, 4949–4952. [DOI] [PubMed] [Google Scholar]
- Zazueta C, Zafra G, Vera G, Sanchez C & Chavez E (1998). Advances in the purification of the mitochondrial Ca2+ uniporter using the labeled inhibitor 103Ru360. J Bioenerg Biomembr 30, 489–498. [DOI] [PubMed] [Google Scholar]
- Zoccarato F & Nicholls D (1982). The role of phosphate in the regulation of the independent calcium‐efflux pathway of liver mitochondria. Eur J Biochem 127, 333–338. [DOI] [PubMed] [Google Scholar]