

Abstract
Key points
Cigarette smoke components directly alter muscle fatigue resistance and intracellular muscle fibre Ca2+ handling independent of a change in lung structure.
Changes in muscle vascular structure are associated with a depletion of satellite cells.
Sarcoplasmic reticulum Ca2+ uptake is substantially impaired in myofibres during fatiguing contractions in mice treated with cigarette smoke extract.
Abstract
Cigarette smokers exhibit exercise intolerance before a decline in respiratory function. In the present study, the direct effects of cigarette smoke on limb muscle function were tested by comparing cigarette smoke delivered to mice by weekly injections of cigarette smoke extract (CSE), or nose‐only exposure (CS) 5 days each week, for 8 weeks. Cigarette smoke delivered by either route did not alter pulmonary airspace size. Muscle fatigue measured in situ was 50% lower in the CSE and CS groups than in control. This was accompanied by 34% and 22% decreases in soleus capillary‐to‐fibre ratio of the CSE and CS groups, respectively, and a trend for fewer skeletal muscle actin‐positive arterioles (P = 0.07). In addition, fewer quiescent satellite cells (Nes+Pax7+) were associated with soleus fibres in mice with skeletal myofibre VEGF gene deletion (decreased 47%) and CS exposed (decreased 73%) than with control fibres. Contractile properties of isolated extensor digitorum longus and soleus muscles were impaired. In flexor digitorum brevis myofibres isolated from CSE mice, fatigue resistance was diminished by 43% compared to control and CS myofibres, and this was accompanied by a pronounced slowing in relaxation, an increase in intracellular Ca2+ accumulation, and a slowing in sarcoplasmic reticulum Ca2+ uptake. These data suggest that cigarette smoke components may impair hindlimb muscle vascular structure, fatigue resistance and myofibre calcium handling, and these changes ultimately affect contractile efficiency of locomotor muscles independent of a change in lung function.
Keywords: cigarette smoke, vascular, skeletal muscle
Key points
Cigarette smoke components directly alter muscle fatigue resistance and intracellular muscle fibre Ca2+ handling independent of a change in lung structure.
Changes in muscle vascular structure are associated with a depletion of satellite cells.
Sarcoplasmic reticulum Ca2+ uptake is substantially impaired in myofibres during fatiguing contractions in mice treated with cigarette smoke extract.
Introduction
Patients with chronic obstructive pulmonary disease (COPD) usually present skeletal muscle wasting and an impairment in skeletal muscle function. These adverse changes in peripheral muscles are thought to contribute to exercise intolerance and a decreased quality of life (Degens et al. 2015). It has been extensively demonstrated that the main risk factor for COPD is the use of cigarettes. Cigarette smoke contains about 2256 chemical compounds known to be hazardous to skeletal muscles (Talhout et al. 2011) and with various types of properties, such as carcinogenic and pro‐oxidant (Pryor & Stone, 1993). Most of these compounds can diffuse through the blood‐gas barrier in the lungs, be carried in the bloodstream and reach peripheral tissues (Colombo et al. 2014). However, the effects of these compounds on peripheral muscle function, before COPD has reached advanced stages, are poorly understood.
Symptoms of muscle weakness have been reported in smokers who have not yet shown a decline in respiratory function (Maltais et al. 2014), and this observation also brings up the possibility that cigarette smoke components may directly alter limb muscle function. This is supported by a carefully controlled study reported by Wust et al. (2008) in which quadriceps function was tested using an electrical stimulation protocol in smokers without COPD and non‐smokers that were matched for age and physical activity levels. Interestingly, non‐COPD smokers, both men and women, revealed a decrease in fatigue resistance but no statistical decrement in maximal strength (Wust et al. 2008). Factors that could alter fatigue resistance in smokers include a change in oxygen delivery due to impaired vasodilatation (Mayhan & Patel, 1997), increased carboxyhaemoglobin (COHb) (Morse et al. 2008), decreased oxygen extraction due to fewer capillaries supplying myofibres (Tang et al. 2010), inefficient mitochondrial respiration (Naimi et al. 2011), or a change in myofibre function during fatiguing contractions (Stubbings et al. 2008).
Reports demonstrating capillary regression in humans with COPD have been mixed, but capillary loss due to smoking may preferentially occur in oxidative type I or IIa fibres (Larsson & Orlander, 1984). Analysis of mice exposed to cigarette smoke for up to 4 months by our laboratory showed that the highly oxidative soleus, containing type I and IIa fibres, but not the more glycolytic extensor digitorum longus (EDL; type IIb fibres), has a decrease in the ratio of capillaries to skeletal myofibres, decreased vascular endothelial growth factor (VEGF) expression, fibre‐type transition, and reduced fatigue resistance (Tang et al. 2010).
Several studies have also demonstrated that chronic smoking has negative impacts on skeletal muscle structure and function due to activation of muscle‐wasting signals (Tang et al. 2010), mitochondrial dysfunction (Aravamudan et al. 2014), oxidative modifications of metabolic proteins which affect intracellular energy metabolism (Barreiro et al. 2012) and sarcoplasmic reticulum (SR) Ca2+ release (Robison et al. 2017). These changes have the potential to produce atrophy and reduce force development and fatigue resistance and, thus, ultimately also contribute to exercise intolerance. Systemic inflammatory cytokines that are expressed in cigarette smoke‐exposed lungs, in particular tumour necrosis factor α (TNF‐α), have also been shown to be responsible for at least a part of the contractile dysfunction of peripheral locomotor muscles (Zuo et al. 2011; Tang et al. 2013).
Although it is well established that cigarette smoking produces muscle contractile dysfunction, the data showing that this may be due to altered intracellular Ca2+ handling have not been fully investigated (Robison et al. 2017). Changes in myofilament calcium sensitivity and SR Ca2+‐ATPase (SERCA) enzyme activity have been shown to be impaired in biopsies from COPD patients (Green et al. 2008), and intracellular Ca2+ handling is a key determinant of contractility and fatigue resistance (Allen et al. 2008).
Thus, to test the direct effect of cigarette smoke components on skeletal muscle vascular and myofibre function, C57BL/6J mice were administered cigarette smoke extract (i.p.) or exposed to cigarette smoke over the course of 8 weeks. Changes in lung structure were estimated from histological sections using the mean linear intercept index. Limb muscle fatigue was evaluated by electrically stimulating the gastrocnemius–plantaris–soleus complex in situ. Oxygen availability to peripheral skeletal muscle was evaluated by measuring the number of capillaries and satellite cells associated with soleus myofibres. Myofibre contractile function was assessed ex vivo in soleus, in EDL, and in isolated flexor digitorum brevis (FDB) fibres. The role of cigarette smoke in altering intracellular Ca2+ handling was evaluated in real time under non‐fatiguing and, importantly, during periods of fatiguing contractions in intact FDB fibres.
Methods
Ethical approval
This study was approved by the University of California, San Diego (Protocol S01144), and the Federal University of Rio de Janeiro (Protocol no. 039/17) Animal Care and Use Committees and conducted in accordance with the guidelines outlined by Grundy (2015). Male C57BL/6J mice (10 weeks of age) were housed three to four per cage in a pathogen‐free vivarium, maintained on a 12 h:12 h day–night cycle and provided standard chow (Harlan Tekland 8604, Madison, WI, USA) and tap water ad libitum. Nestin‐green fluorescent protein (GFP) mice were generated and kindly provided by Dr M. Yamaguchi (Rich et al. 2017). VEGFLoxP mice were from Genentech (Oceanside, CA, USA) and HSA‐Cre‐ERT2 mice were generated and kindly provided by Dr Daniel Metzger (Institut de Génétique et de Biologie Moléculaire et Cellulaire, Illkirch‐Graffenstaden, France) as previously described (Knapp et al. 2016). After all functional assessments were completed mice were killed by surgical removal of the heart while under anaesthesia (ketamine:xylazine, 10:1 mg kg−1, i.p.).
Cigarette smoke delivery
Non‐filtered 3R4F research cigarettes, purchased from Kentucky Tobacco Research and Development Center at the University of Kentucky (Lexington, KY, USA), were used for both forms of cigarette smoke delivery. For nose‐only exposure to cigarette smoke (CS group), the InExpose system (SCIREQ, Montreal, QC, Canada) was used. Mice were exposed twice each day (5 cigarettes/period, 5 days/week) with a minimum of a 30 min fresh air between sessions for eight consecutive weeks. Mice were restrained in SoftRestraints from SCIREQ that are made of nylon‐coated stainless steel wire. During the 30 min fresh air period the mice were removed from the exposure tower and maintained on a 37°C warm pad. Cigarette smoke extract (CSE) was prepared by passing cigarette smoke through sterile PBS with the use of a rodent ventilator to draw on a lit cigarette. The solution was “bubbled” with smoke until an optical density of 0.86 at 320 nm was reached. CSE was filtered (0.22 μm, EMD Millipore, Burlington, MA, USA) before being administered once each week (100 μl, i.p.) for eight consecutive weeks (Zhang et al. 2013). Control mice were the same strain and age and remained in room air under normal housing conditions. At the end of the 8‐week CS or CSE delivery period, plasma cotinine levels were assessed by ELISA (BQKITS, Inc., San Diego, CA, USA) following the manufacturer's protocols.
Mean linear intercept index
Air space enlargement was estimated by measuring the mean of the linear intercept lengths from lungs fixed with paraformaldehyde at an airway pressure of 25 cmH2O and embedded in paraffin (Tang et al. 2013).
Skeletal muscle in situ fatigue
The protocol to stimulate the gastrocnemius–plantaris–soleus (GPS) complex via the sciatic nerve to evaluate the fatigue resistance of the vascular myofibre system has been previously described (Knapp et al. 2016). Mice were anaesthetized with ketamine and xylazine (10:1 mg kg−1 , i.p.). Oxygen saturation levels were monitored before and immediately after the stimulation period by pulse oximeter, which cannot distinguish between oxyhaemoglobin (O2Hb) and COHb (Mouse PulseOx Plus, Starr Life Science, Oakmont, PA, USA), and remained above 95% throughout the fatigue test. The length allowing generated maximal twitch force (L 0) was set by evoking single twitch stimulations (monophasic pulses, 8 V, 0.5 ms pulse duration) using a S88X stimulator (Grass Technologies, Quincy, MA, USA). The GPS complex was then stimulated to evoke full tetanic contractions (8 V, 0.5 ms pulses, 80 Hz, 200 ms train duration), repeatedly contracted (0.25 trains s−1) until the force developed fell to 50% of the initial force output and the time was recorded.
Skeletal muscle morphology
In the non‐stimulated leg, the GPS complex was surgically removed and capillaries detected in 10 μm cryosections using the capillary lead‐ATPase method (Rosenblatt et al. 1987). For detection of smooth muscle‐positive arterioles (SMA+) and M2‐macrophage (CD206+) cells, 10 μm cryosections were fixed with 4% paraformaldehyde for 6 min at room temperature, rinsed with PBS, and incubated overnight with primary monoclonal antibody for α‐smooth muscle actin (1:200, Sigma‐Aldrich, St. Louis, MO, USA, cat. no. A2547). Signals were detected with anti‐mouse Alexa Fluor 488 (1:1000, Invitrogen, Molecular Probes, Carlsbad, CA, USA) and mounted with Fluoro‐gel II antifade reagent with 4′,6‐diamidino‐2‐phenylindole (DAPI) (Electron Microscopy Sciences, Hatfield, PA, USA, cat. no. 17985‐50). Images were scanned on an Hamamatsu Nanozoomer Slide Scanning System (Hamamatsu Photonics, Hamamatsu City, Japan). Capillary density and fibre number were counted with the aid of FIJI software and used to calculate the capillary‐to‐fibre ratio (C:F). SMA+ vessels per area of the entire GPS cross‐section was calculated.
Skeletal muscle ex vivo contractility
In another group of mice, the EDL and soleus from one hindlimb were dissected under anaesthesia (ketamine:xylazine, 10:1 mg kg−1, i.p.), and mice were then immediately killed by surgical removal of the heart. Each muscle was mounted in an experimental chamber (Model 1500A with a 402A force transducer, Aurora Scientific Inc., Aurora, ON, Canada) and electrically stimulated (S88X stimulator, Grass Technologies) using square‐wave pulses (16 V; EDL: 250 ms train duration, 0.5 ms pulses, 22°C; soleus: 500 ms train duration, 0.5 ms pulses, 28°C). Muscles were constantly perfused with Tyrode solution (121 mM NaCl, 5 mM KCl, 1.8 mM CaCl2, 0.5 mM MgCl2, 0.4 mM NaH2PO4, 24 mM NaHCO3, 5.5 mM glucose, 0.1 mM EGTA, containing 25 μM d‐tubocurarine) continuously bubbled with 95% O2–5% CO2 (final pH 7.4). L 0 was determined with single twitches and muscles allowed to rest for 15 min. Contractile function was evaluated by stimulating the muscle at different frequencies (1–150 Hz) with 100 s intervals. After the contractile protocol, muscles were blotted dry and weighed to determine the muscle cross‐sectional area (CSA). Force development was normalized with respect to the muscle CSA (kPa) (Zuo et al. 2011).
In vivo quiescent satellite cell number
Male HSA‐CRE‐ERT2 × VEGFLoxP × Nes‐GFP mice (Rich et al. 2017) (5 weeks of age) were injected with tamoxifen (1 mg/mouse, i.p. for 5 days) to delete the skeletal myofibre VEGF gene. Additional mice (4 weeks of age) were exposed to whole body cigarette smoke (5 days a week, two 30 min exposure periods, 10 cigarettes total) for 4 weeks. VEGF protein levels in the gastrocnemius were measured by ELISA (R&D Systems). Fibres were isolated from the soleus by incubation with collagenase (2.5 mg ml−1 Type I collagenase, Worthington Biochemical Corporation, Lakewood, NJ, USA) in Dulbecco's modified Eagle's medium for 1 h at 37°C, followed by manual dissociation with fire‐polished pipettes according to modifications of the method of Day et al. (2007). Fibres were allowed to attach to Matrigel‐coated slides for 1 h in a 37°C, 5% CO2 tissue culture incubator, before fixing with 4% paraformaldehyde and 2% sucrose in PBS at 4°C for 10 min. Slides were washed with Tris‐buffered saline (TBS), incubated with 20% sucrose for 30 min at 4°C, washed again with TBS, and permeabilized with 0.3% Triton X‐100 for 5 min at room temperature. The slides were washed and blocked with 3% bovine serum albumin–3% normal goat serum for 1 h and incubated with primary antibodies in blocking solution overnight at 4°C. Primary antibody dilutions were 1:10,000 for anti‐rabbit GFP (Invitrogen, Molecular Probes, Carlsbad, CA, USA) and 1:1000 for anti‐Pax7 (Cat# AB_528428, Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA, USA). Slides were TBS‐washed, incubated with secondary Alexa Fluor 488 and 546 antibodies, mounted with ProLong™ Gold Antifade Mountant with DAPI (Cat# P36931; Thermo Scientific, Carlsbad, CA, USA) and viewed by confocal microscopy.
Intact single fibre isolation from FDB muscle and FURA‐2 loading
Mice were killed by surgical removal of the heart while under anaesthesia (ketamine: xylazine, 10:1 mg kg−1, i.p.) followed by immediate dissection of the FDB muscles from both feet. Individual, intact single muscle fibres were dissected under dark field illumination in the presence of dissection solution (in mM: 136.5 NaCl, 5 KCl, 1.8 CaCl2, 0.5 MgCl2, 0.4 NaH2PO4, 11.9 NaHCO3, 5 glucose, 0.1 K2EGTA). Single fibres (one at a time) were placed on the stage of an inverted microscope for epifluorescence (Nikon Eclipse Ti‐S with a 40× long distance Fluor objective and integrated with a Photon Technology International (Birmingham, NJ, USA) illumination and detection system (DeltaScan model)) and fibre autofluorescence was measured (see details in “Intracellular Ca2+ ([Ca2+]c) measurements” section below). The fibre was then pressure injected with FURA‐2 (Life Technologies, Carlsbad, CA, USA) and mounted in a chamber (model 1500A with a 400A force transducer, Aurora Scientific Inc., Aurora, ON, Canada). The fibre was superfused with Tyrode solution (in mM: 121 NaCl, 5 KCl, 1.8 CaCl2, 0.5 MgCl2, 0.4 NaH2PO4, 24 NaHCO3, 5.5 glucose, 0.1 K2EGTA) that was constantly bubbled with 2% O2, 5% CO2 (for solution pH 7.4), balance N2 for the entire experimental procedure. The solution was measured with a fibre optic oxygen sensor (Oxymicro, World Precision Instruments, Sarasota, FL, USA) and maintained ∼15 Torr, which is not limiting to mitochondrial respiration during maximal work in mouse fibres (Gandra et al. 2018), as also determined in preliminary results from our laboratory in intact single mouse fibres. Only one fibre was used from each mouse, therefore the number of fibres used reflects the number of mice. All experimental procedures were performed at 22°C.
Measurements of contractility, fatigue resistance and intracellular Ca2+ transients
Each fibre was adjusted to L 0 (350 ms trains, 0.5 ms square‐wave pulses, 8 V, 100 Hz) using parallel platinum plates and a S88X stimulator (Grass Technologies, Quincy, MA, USA). A BIOPAC Systems MP100WSW (Santa Barbara, CA, USA) A–D converter was used, and the data were analysed with AcqKnowledgeIII 3.2.6 software (BIOPAC Systems). After a 30 min rest period, force–frequency measurements were collected; FF; 1–150 Hz; 350 ms trains, 0.5 ms pulses, 8 V; 100 s rest between trains. Immediately after the FF protocol, the perfusion solution was switched to a Tyrode solution supplemented with 10 mM caffeine, followed by a tetanic stimulation at 120 Hz, then re‐perfused back with Tyrode solution (no caffeine added). This procedure was performed to develop force at maximal Ca2+ release from the SR enhanced by the presence of caffeine (Westerblad & Allen, 1993; Nogueira & Hogan, 2010). After caffeine was washed out, each fibre rested for additional 10 min, followed by a fatigue‐inducing contraction protocol, which consisted of repetitive contractions evoked by 100 Hz pulse‐stimulations (350 ms trains, 0.5 ms pulses) with an initial train rate of 0.25 trains per second for 2 min followed by increases in train rate of 20% each 2 min (0.3, 0.36, 0.43, 0.52, 0.62, 0.75, 0.9, 1.1) until peak force had decreased 50% of the initial contraction (fatigue point). Force development (in mN) was normalized to the cross‐sectional area (in mm2) determined from the diameter of the fibre (31 ± 1 μm diameter, 794 ± 74 μm2; n = 12 fibres, mean ± SEM), and data are reported as kilopascals.
Intracellular Ca2+ ([Ca2+]c) measurements
During contractions [Ca2+]c was measured by illuminating the fibre with excitation wavelengths of 340 and 380 nm that were rapidly alternated (200 Hz). Fluorescence signals were collected at an emission of 510 nm and the ratio of signals excited at 340 nm to 380 nm were used to calculate the Ca2+‐dependent signal (Nogueira et al. 2013). The fluorescence:excitation ratio (340 nm/380 nm; R) was converted to [Ca2+]c according to eqn (1).
| (1) |
From eqn (1), K D, the dissociation constant for Ca2+‐FURA‐2, was set to 224 nM (Westerblad & Allen, 1991). β is the fluorescence ratio between high and no [Ca2+]c at 380 nm, and R min and R max are the fluorescence ratios when Ca2+–FURA‐2 binding is minimal (at low cytosolic Ca2+) and maximal (at high intracellular Ca2+), respectively. R min and R max were determined using an internal in vivo calibration described in Nogueira et al. (2013), but modified for mouse fibres. Briefly, to determine R min, a group of FURA‐2‐injected fibres (n = 3) were incubated for 30 min with a no‐calcium‐Tyrode–EGTA solution (121 mM NaCl, 5 mM KCl, 0.5 mM MgCl2, 0.4 mM NaH2PO4, 24 mM NaHCO3, 5.5 mM glucose, 10 mM K2EGTA) supplied with 30 mM caffeine. Then, caffeine was washed out and switched to 100 μM BAPTA‐AM (acetoxymethyl ester form of BAPTA), incubated for 30 min, then BAPTA‐AM was washed out. R min was determined as the smallest R value observed (0.24 ± 0.02; n = 3 fibres, mean ± SEM) and set as 0.24 for all fibres used in the present study. R max was determined for each of the contracting fibres and was set to 132% of the peak ratio obtained when fibres were contracted at 120 Hz in the presence of 10 mM caffeine (R max was 3.54 ± 0.24; n = 12 fibres, mean ± SEM) (Westerblad & Allen, 1991). β was also determined for each of the contracting fibres (5.5 ± 0.6; n = 12 fibres, mean ± SEM) as described previously (Gandra et al. 2018). Calculations of peak and basal [Ca2+]c were previously described in Nogueira et al. (2013).
In order to determine the myofilament Ca2+ sensitivity for each fibre, force development at each peak [Ca2+]c during the FF protocol was fitted using the sigmoidal eqn (2) (Nogueira & Hogan, 2010):
| (2) |
P is the force developed at different [Ca2+]c values, P 0 is the maximal force obtained by stimulating the fibre at 120 Hz in the presence of 10 mM caffeine. Ca50 is the mid‐point of the force–[Ca2+]c curve, and n H is the Hill coefficient.
In order to determine whether the treatments (CS or CSE) would alter relaxation, time to decay peak force by 80% (Relaxation time) was determined. Relaxation was also determined when Ca2+ measurements were converted to force (i.e. Ca2+‐derived force) by using the data obtained in the force–Ca2+ relationship (i.e. sensitivity of the myofilaments to Ca2+ and the slope of Ca2+–force relationship) for each fibre as described in Westerblad & Allen (1993).
Sarcoplasmic reticulum [Ca2+]c pumping rate
In order to measure the function of SR Ca2+ pumping during relaxation (Nogueira et al. 2013), the elevated long tail of [Ca2+]c decline (from 100 ms to 3.0 s after the end of the stimulation period during a single tetanic contraction) was used to determine the rate of [Ca2+]c decline (−d[Ca2+]c/dt) and the rate of SR Ca2+ pumping (A, eqn (3)).
| (3) |
From eqn (3), A, N and L are three adjustable parameters representing, respectively, the rate of SR Ca2+ pumping (in μMN −1 s−1), the power function, and the SR Ca2+ leak. In order to compare directly the SR Ca2+ pumping (in μM−3 s−1) between groups (control vs. CS vs. CSE) at the same time points of the fatigue protocols (i.e. first contraction, 116 s and 200 s of contractions), the curves were fitted setting N = 4 and L = 24 (N = 3.9 ± 0.2 and L = 24 ± 3, obtained from individual experiments, n = 8 fibres, mean ± SEM), with a maximum increase in least‐squares error of 15%.
Statistics
For comparisons between multiple groups, one‐way ANOVA followed by the Tukey test or two‐way ANOVA followed by the Bonferroni test was used, as indicated. Analyses were carried out using PRISM 4 software (GraphPad Software, La Jolla, CA, USA) and P < 0.05 was considered to represent a significant difference.
Results
Evaluation of cigarette smoke delivery
Plasma cotinine levels, measured at the end of the 8‐week CS or CSE delivery period, were increased above control values to the same extent in both mice exposed to cigarette smoke with a “nose‐only” exposure system and mice injected with cigarette smoke extract (i.p.) at the 8 week time point (control, 0.04 ± 0.02 ng ml−1; CS, 0.81 ± 0.41 ng ml−1, P < 0.05; CSE, 1.19 ± 0.14 ng ml−1, P < 0.05).
Air space size is not altered by 8 weeks of CS exposure or CSE delivery
Pulmonary air space size was estimated from paraffin‐embedded lung sections using the mean linear intercept index (Fig. 1 A and B). After 8 weeks of exposure to CS or weekly injections of CSE, the average chord lengths were not different between the experimental groups (control: 28.42 ± 0.97 μm, CS: 28.91 ± 0.59 μm, CSE: 29.68 ± 0.26 μm, n.s.).
Figure 1. Lung airspace size after 8 weeks of exposure to daily periods of cigarette smoke or weekly delivery of cigarette smoke extract.
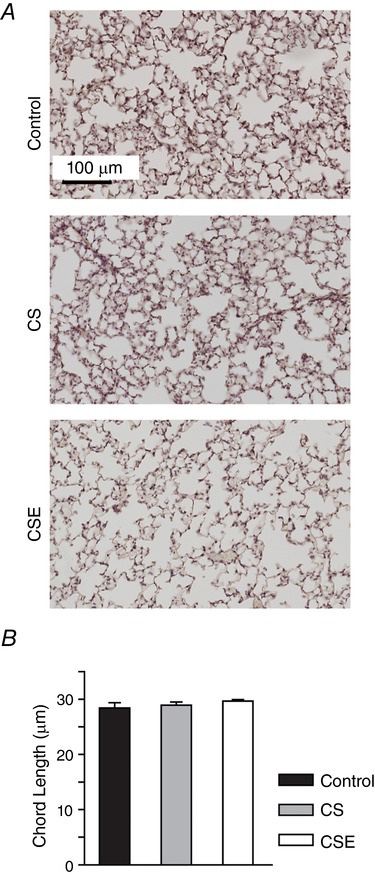
Mice remained in room air (control) or were exposed to daily periods of cigarette smoke (CS) or administered cigarette smoke extract (CSE) each week (i.p.). Mean linear intercept values (B) were estimated from paraffin‐embedded, 7 μm lung sections (A). Values are presented as the mean ± SD, n = 5 control, n = 6 CS, n = 4 CSE. [Color figure can be viewed at http://wileyonlinelibrary.com]
CS exposure, but not CSE, attenuates weight gain
Mice in the control group and CSE group both gained weight at a rate of 0.40 g week−1. In contrast, mice in the CS exposure group increased their body weight at an average rate of 0.07 g week−1. Figure 2 shows the percentage change in body weight from week 1 to week 8. The percentage body weight change compared to week 1 was lower in the CS group than in the CSE group at week 7 (P < 0.001) and lower than in both the control and CSE groups at week 8 (P < 0.01). The muscle mass to body weight ratio was not different between the groups for the soleus, EDL, plantaris and gastrocnemius (data not shown).
Figure 2. Mice weight gain.
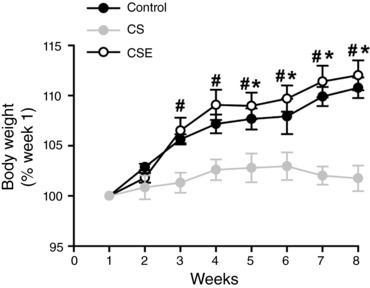
Mice in the control, CS and CSE groups were weighed weekly. Body weights are represented by the percentage of the value at week 1; data are presented as mean ± SEM for the control (n = 13), CS (n = 9) and CSE (n = 19) groups. At each time point # indicates that the CS group differs from the CSE group (P < 0.05) and * indicates that the CS group differs from control (P < 0.05).
CS exposure and CSE administration impaired hindlimb muscle fatigue resistance
In situ measurements (with intact vascular system) of fatigue resistance from the GPS complex stimulated by the sciatic nerve are shown in Fig. 3. There was a more rapid time to fatigue (i.e. less fatigue resistance of the GPS complex) in both CS and CSE groups than the control group at the 8 week time point (P < 0.05).
Figure 3. In situ fatigue resistance in response to both cigarette smoke exposures.
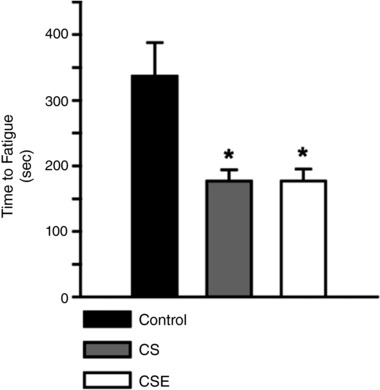
The time to reach 50% of initial force (fatigue) was measured in the gastrocnemius–plantaris–soleus (GPS) complex electrically stimulated to repeatedly contract. The average fatigue time is presented ± SEM for control (n = 10), CS (n = 6) and CSE (n = 6) mice. * indicates a difference from the control time, P < 0.05.
Capillary regression and a trend for fewer arterioles were detected in locomotor muscles of CS and CSE mice
The calculated capillary‐to‐fibre ratio (C:F) was 22% (P < 0.05) lower in the CS soleus than in the control soleus (Fig. 4 A). The C:F was 34% lower in the CSE soleus than control values (P < 0.05). There was no difference in C:F between the CS and CSE groups. Smooth muscle actin‐positive arterioles, measured in cross‐sections from the entire GPS complex, revealed a trend for fewer arterioles in the CS and CSE groups (Fig. 4 B).
Figure 4. Vascular regression correlates with a trend for fewer arterioles and M2‐like macrophages, and loss of satellite cells.
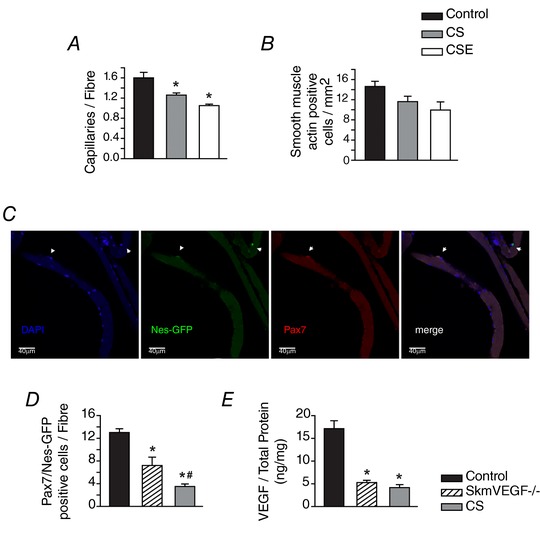
The capillary‐to‐fibre ratio was detected in soleus (A), and α‐smooth muscle actin‐positive arterioles detected in the GPS complex (B) of control, CS and CSE mice. Values are presented as the mean ± SEM. * indicates a difference from control, P < 0.05. n = 6–9 control, n = 5–6 CS and n = 3–5 CSE. C, confocal immunofluorescence image of soleus fibres. D, Pax7/Nestin‐positive satellite cells associated with isolated soleus fibres were detected in fibres from control, skmVEGF−/− mice and mice exposed to 8 weeks of whole body CS. Values are the mean number of satellite cells per fibre. * indicates a difference from control fibres. # indicates a difference from both control and skmVEGF−/− fibres. Values are the mean ± SEM, n = 5–14 fibres per group. E, amount of VEGF normalized by the amount of total protein in gastrocnemius homogenates.
CS reduces the number of quiescent satellite cells associated with soleus myofibres
Soleus fibres were isolated to HSA‐Cre‐ERT2 × Nes‐GFP × VEGFLoxP mice exposed to whole body cigarette smoke or treated with tamoxifen to induce deletion of the VEGF gene in skeletal myofibres (skmVEGF−/−). Figure 4 C shows confocal images of a representative soleus fibre immunostained for muscle stem cell markers Pax7+ and Nestin‐GFP. Nestin is a marker for quiescent muscle stem cells (Rich et al. 2017). Figure 4 D shows a lower number of Pax7+/Nes+ cells associated with soleus fibres from skmVEGF−/− mice than control (P < 0.05) that was even lower in soleus fibres exposed to CS (P < 0.01). To confirm efficient VEGF gene deletion, VEGF levels were measured in the gastrocnemius, and were 70% and 75% lower in the skmVEGF−/− and CS muscles, respectively, than in control muscles (P < 0.01; Fig. 4 E).
CSE administration decreased soleus, but not EDL, muscle mass and cross‐sectional area
Locomotor muscles from hindlimbs (EDL and soleus) were dissected from control and CSE groups of mice, and wet weights and cross‐sectional areas were measured (Table 1). There was no change in muscle weight and cross‐sectional area for EDL muscles between groups. However, soleus from CSE revealed a slightly, but statistically significant, smaller mass and cross‐sectional area (∼10% decrease) than the control muscles (P < 0.05).
Table 1.
Morphometric data
| Control | CSE | P value (Student's t test) | |
|---|---|---|---|
| Body weight (g) | 29.5 ± 1.0 | 28.4 ± 0.5 | 0.303 |
| EDL muscle | |||
| L 0 (mm) | 14.0 ± 0.2 | 14.1 ± 0.2 | 0.922 |
| Mass (mg) | 13.6 ± 0.6 | 12.6 ± 0.5 | 0.231 |
| CSA (mm2) | 0.91 ± 0.03 | 0.84 ± 0.03 | 0.153 |
| Soleus muscle | |||
| L 0 (mm) | 13.2 ± 0.2 | 13.2 ± 0.2 | 0.959 |
| Mass (mg) | 11.0 ± 0.5 | 9.8 ± 0.2 | 0.039* |
| CSA (mm2) | 0.78 ± 0.03 | 0.70 ± 0.02 | 0.024* |
L 0, optimal muscle length; CSA, cross‐sectional area of the muscle, *statistically different (P < 0.05) from Control.
In isolated EDL and soleuys, there was a decrease in force development over a range of frequencies in the CSE group compared to control (Fig. 5 A and B), but when submaximal forces were normalized by the maximal frequency of stimulation (150 Hz), there were no statistical differences between control and CSE groups (data not shown).
Figure 5. Force development measured at different frequencies of pulse‐stimulation in isolated EDL and soleus muscles after cigarette smoke extract treatments.
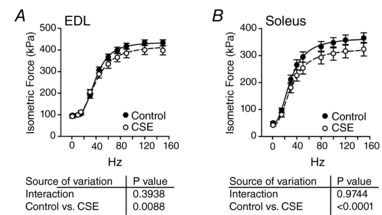
Force–frequency protocol normalized by the cross‐sectional area (kPa) in EDL (A) or in soleus (B) muscles. Mean ± SEM; n = 6 for control group and n = 7 for CSE group.
Intact fibres from CSE‐treated, but not CS‐exposed, mice show impaired fatigue resistance and slower Ca2+ uptake into the SR
During the FF protocol in isolated fibres, the CSE group showed higher force at submaximal frequencies than CS or control fibres (Fig. 6 A). The mid‐point of the FF relationship was significantly decreased only in the CSE group (27 ± 4 Hz vs. 22 ± 2 Hz vs. 10 ± 2 Hz, for control vs. CS vs. CSE, respectively; P < 0.05). Maximal force was not affected (control, 480 ± 107 kPa; CS, 572 ± 93 kPa; CSE, 457 ± 43 kPa; P > 0.05). The peak [Ca2+]c at submaximal frequencies was overall increased in the CSE fibres (P = 0.0003, two‐way ANOVA) compared to control and CS fibres, but the Bonferroni post‐test did not show statistical difference at individual frequencies (P > 0.05; Fig. 6 B). Maximal Ca2+ transients, evoked by 120 Hz contractions in the presence of 10 mM caffeine, were very similar between the groups (Fig. 6 B). Nonetheless, there was no difference in myofibril Ca2+ sensitivity (i.e. the contractile response to intracellular Ca2+ transients) between groups. The Ca50 was 539 ± 160 nM for control, 498 ± 83 nM for CS exposure, and 461 ± 59 nM for CSE (P > 0.05, Fig. 6 C). When relaxation after tetanic forces was evaluated during the FF protocol, there was a clear delay in force decay in the CSE group compared to control and CS groups (Fig. 6 D) after a tetanic contraction (i.e. evoked by a train of 100 Hz pulse‐stimulations). It was confirmed by the mean data of rate of relaxation at the linear phase (∼43% decrease; Fig. 6 E, P < 0.05). Cytosolic Ca2+ changes during contractions (Fig. 6 F) were converted to force (Ca2+‐derived force) and relaxation times (80% decay) were determined in real force recordings and in Ca2+‐derived force. As shown in Fig. 6 F, there was a clear delay in Ca2+‐derived force decay in the CSE group compared to CS and control groups, which was less evident in real force traces. This was confirmed by an increase in relaxation time in the CSE compared to CS and control groups (Fig. 6 G, P < 0.05). In order to detect whether the slowing in relaxation was due to the slowing in Ca2+ pumping into the SR, the tail of elevated [Ca2+]c after the stimulation period (300 ms to 1 s) when intracellular Ca2+ decay is primarily due to SR Ca2+ uptake (Klein et al. 1991) was used to generate the rate of [Ca2+]c decay (−d[Ca2+]c/dt). The plots shown in Fig. 6 H revealed a right‐shift in the CSE group. The rate of Ca2+ pumping (the parameter A in eqn (3)) was significantly smaller (P < 0.05) in the CSE group compared to control and CS groups (in μM−3 s−1: 2562 ± 696 vs. 2130 ± 680 vs. 822 ± 252, for control vs. CS vs. CSE, respectively, Fig. 6 I).
Figure 6. Contractility and calcium measurements in isolated single FDB fibres during the force–frequency protocol.
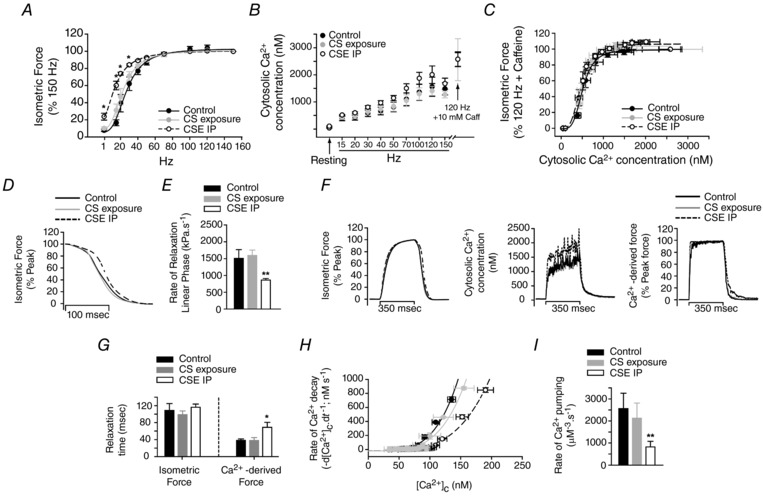
A, force–frequency relationship obtained from the three groups of mice (* P < 0.05 vs. control). B, cytosolic Ca2+ concentration ([Ca2+]c) during the force–frequency protocol. The peak [Ca2+]c detected during a 120 Hz pulse‐stimulation in the presence of 10 mM caffeine and the basal [Ca2+]c (during resting) are indicated by arrows. C, isometric force development (data in panel A) was plotted against the respective basal and peak [Ca2+]c (data in panel B) during the force–frequency protocol. D, trace recordings of isometric force during relaxation (average of 12 independent contractions evoked by 100 Hz pulse‐stimulations for each experimental group). E, rate of force decay during the linear phase of the relaxation (** P < 0.01 vs. control and CS exposure groups). F, trace recordings of isometric force (left), cytosolic Ca2+ concentration (middle) and Ca2+‐derived force (right) (average of 4 independent contractions for each group evoked by 100 Hz pulse‐stimulations). G, time to decay 80% of the peak force after the stimulation period (relaxation time) obtained from real force measurements (isometric force) and from converted force from real Ca2+ measurements (Ca2+‐derived force) (* P < 0.05 vs. control and CS exposure groups). H, relationship between the rate of [Ca2+]c decay (−d[Ca2+]c/dt) and the [Ca2+]c at the tail (see Methods for details). I, rate of Ca2+ pumping determined by eqn (3) described in Methods, but fixing the parameters N and L to 4 and 24, respectively. These parameters were determined by the individual fitting for each fibre (** P < 0.01 vs. control and CS exposure groups). Values are presented as the mean ± SEM, n = 4 fibres for each group.
After the FF protocol, each fibre was repetitively contracted until the fatigue time point. Time to fatigue was significantly reduced in CSE fibres (243 ± 15 s) compared to control (306 ± 23 s) or CS (364 ± 13 s) groups (P < 0.01, Fig. 7 A). Since the Ca2+ pumping rate is slowed in fibres from the CSE group under non‐fatiguing conditions (i.e. during the FF protocol), and fatigue resistance was impaired in the CSE fibres, relaxation and Ca2+ pumping function were investigated under fatiguing contractions. As demonstrated in trace recordings of force in Fig. 7 B, the slowing in relaxation at the fatigue time point was clearly pronounced in CSE fibres in relation to the control and CS groups. The relaxation times during the time course of the fatigue protocol were significantly higher in the CSE group compared to CS and control groups after 120 s of contractions (Fig. 7 C, P < 0.01).
Figure 7. Contractility and calcium measurements in isolated single FDB fibres during a fatigue protocol.
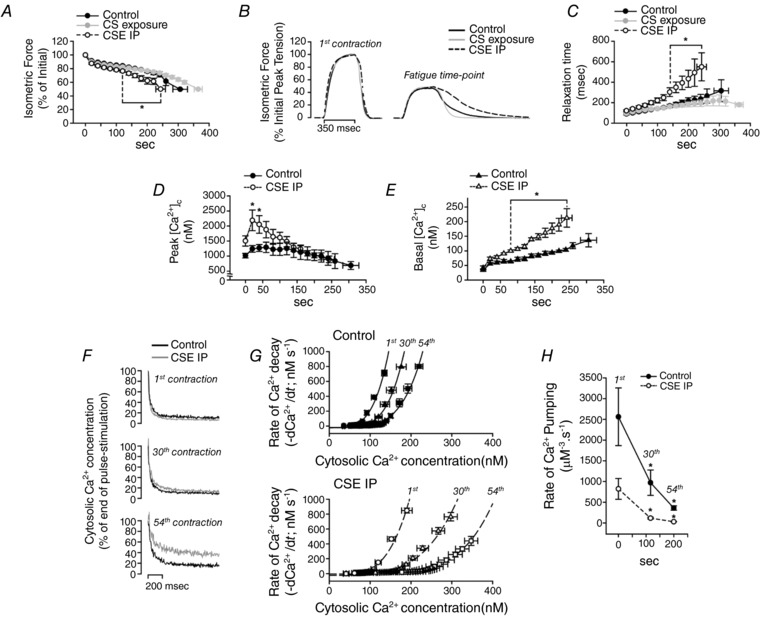
A, time course of force development (* P < 0.01 vs. control and CS exposure). B, trace recordings of force developed at the first contraction and at the fatigue time point for each group of fibres. The data are an average of 4 different fibres from each group. C, time to force decay by 80% after the stimulation period (relaxation time) (* P < 0.01 vs. control and CS exposure). D and E, peak [Ca2+]c (D) and basal [Ca2+]c (E) (* P < 0.05 vs. control and CS exposure). F, representative trace recordings of cytosolic Ca2+ concentration decay during relaxation at selected time points of the fatigue protocol (1st contraction; contraction at 116 s (30th contraction); contraction at 200 s (54th contraction)). G, relationship between the velocity of [Ca2+]c decay (−d[Ca2+]c/dt) and the [Ca2+]c at the tail (see Methods for details) from the same time points shown in F. Top graph, control group; lower graph, CSE group. H, rate of Ca2+ pumping determined by eqn (3) described in Methods, but fixing the parameters N and L to 4 and 24, respectively. Time points are the same as shown in F (* P < 0.05 vs. control and CS exposure at the same time points). Values are presented as the mean ± SEM, n = 4 fibres for each group.
Interestingly, there was a higher increase in peak [Ca2+]c in the beginning of the fatigue protocol (Fig. 7 D, P < 0.01). Also, the intracellular Ca2+ accumulation between contractions (basal [Ca2+]c) was significantly augmented in the CSE group compared to control group (Fig. 7 E, P < 0.001). This was due to a clear slowing in [Ca2+]c decay between contractions in the CSE group compared to control (i.e. trace [Ca2+]c recordings shown in Fig. 7 F), which may be due to a slowing in SR Ca2+ pumping. Measuring the elevated [Ca2+]c after the stimulation period at different time points of the fatigue protocol, CSE fibres showed a pronounced right‐shift in the rate of Ca2+ decay per [Ca2+]c compared to control fibres (Fig. 7 G). The rate of Ca2+ pumping (Fig. 7 H) was significantly decreased (P < 0.05) in both groups of fibres. However, the changes from the initial contraction occurred in greater increments (P < 0.05) in the CSE group (81 ± 6% decreased at 30th contraction and 95 ± 2% at 54th contraction) compared to control fibres (62 ± 1% decreased at 30th contraction and 82 ± 4% decreased at 54th contraction). Therefore, these data suggest that SR Ca2+ pumping rate impairment was pronounced in fibres from animals treated with CSE.
Discussion
In the present study, CSE was used in order to bypass the primary effects of cigarette smoke on the lungs and more directly study its effects on peripheral skeletal muscle (Zhang et al. 2013). No evidence of emphysema (increased airspace size) was detected in either the direct injection or exposure groups, and plasma cotinine levels were also not different between the cigarette smoke extract and exposure groups. Thus, mice are estimated to receive similar amounts of cigarette smoke components with the caveat that the exposures were 5 days a week and the injected extract was given only once each week. Administration of CSE or exposure to cigarette smoke also resulted in a similar decrement in fatigue resistance (∼48% decrease in time to fatigue) when measured in situ with the vascular system intact. Evidence of a change in O2 availability to the mitochondria is supported by a reduction in the numbers of capillaries surrounding soleus fibres and a trend for fewer arterioles (SMA+ cells) in the GPS complex. However, muscle fatigue resistance is multifactorial (Allen et al. 2008) and it is also known that chronic smoking could affect the muscle contractile apparatus (Rinaldi et al. 2012). In the present study, mice treated chronically with CSE for 8 weeks were shown to also have reduced fatigue resistance at the cellular level (FDB intact single fibres; Fig. 7). This rapid fatigue response was accompanied by a pronounced slowing in relaxation and SR Ca2+ pumping in the CSE group compared to control. These results suggest that CS components may directly affect the efficiency of muscle relaxation during exercise. Thus, CS‐induced decrements in both vascular and myofibre function can contribute to exercise intolerance in smokers.
Loss of vascular structure in cigarette smoke‐exposed mice
Exercise therapy has shown promise in rehabilitation programmes for those with COPD, but caution must be considered due the high chronic systemic inflammation that often includes high circulating TNF‐α levels (Schols et al. 1996). This inflammatory burden could interfere with the polarization or sequence of activation events that regulate macrophage subpopulations in skeletal muscle (Takeda et al. 2011). These regenerative M2 cells provide trophic factors, including VEGF, that participate in regeneration and repair in part by protecting cells, particularly satellite cells, from apoptosis (Takeda et al. 2011; Saclier et al. 2013). Satellite cells reside adjacent to capillary endothelial cells and proportionally change with the skeletal muscle capillaries resulting from exercise training‐induced angiogenesis or capillary regression (Langen et al. 2006; Christov et al. 2007; Tang et al. 2010). In the present study satellite cells associated with soleus fibres isolated from skeletal myofibre VEGF‐deficient mice revealed a decreased number per fibre, and smoke exposure resulted in an even greater loss. Thus, the added insult from cigarette smoke, possibly due to elevated apoptosis or increased reactive species, resulted in a further loss of satellite cells and capillaries (Christov et al. 2007) and this can be predicted to occur in patients who chronically smoke (Menon et al. 2012).
Direct effects of cigarette smoke components on skeletal muscle function
In isolated muscles from mice administered CSE, there was an overall decrease in force production that was more prominent in the oxidative soleus. The soleus, but not the EDL, also showed a decreased muscle mass in the CSE group compared to control group. These results are in accordance with previous data from our group showing the greater effect of 16 weeks of CS treatment on soleus contractile function compared to EDL (Tang et al. 2010). However, Tang et al. (2010) have also shown that 8 weeks of CS treatment did not produce any change in ex vivo EDL or soleus muscle contractile function. The main results obtained from measurements in isolated, single FDB fibres, mostly composed of fast‐oxidative type IIa fibres, were that CSE slowed myofibre relaxation rate (Fig. 6 D). Real‐time monitoring of intracellular Ca2+ in isolated single fibres suggests that this slowing in relaxation in fibres from CSE‐treated mice is due to a decreased rate of SR Ca2+ pumping (Fig. 6 F and G) (Nogueira & Hogan, 2010; Nogueira et al. 2013). This is supported by the reduced fatigue resistance (but not maximal force generation) detected in FDB fibres from the CSE group. This contractile profile is similar to that observed in human smokers (Wust et al. 2008) and characteristic of a slowing of relaxation during fatiguing contractions (Westerblad & Allen, 1993; Nogueira et al. 2013). Although the study of Wust et al. (2008) did not elucidate the mechanism by which relaxation was impaired in male smokers, there is evidence that vastus lateralis muscle biopsies from patients with COPD have lower SR Ca2+‐ATPase (SERCA) activity and SERCA–Ca2+ affinity compared to samples collected from healthy patients (Green et al. 2008). These results suggest that cigarette smoke components may directly affect fatigue resistance, in addition to a capillary‐dependent limitation in O2 availability, and could potentially alter one or more steps in excitation–contraction coupling or the metabolic energy supply systems.
Cigarette smoke extract enhances submaximal force development during non‐fatiguing contractions
During electrical stimulation of skeletal muscles, Ca2+ diffuses from the SR to the cytosol, raises the cytosolic Ca2+ concentration ([Ca2+]c) and rapidly binds to the regulatory sites on troponin C located in the thin filament. This orchestrated change in subcellular calcium results in the activation of the myofilament cross‐bridge cycling and the development of force (Allen et al. 2008). Also, and not less important, the rise in [Ca2+]c immediately activates SERCA, which starts to pump Ca2+ back into the SR. The equilibrium between the amount of Ca2+ released from the SR and the velocity of SR Ca2+ uptake results in a transient rise in [Ca2+]c that is dependent on the time period and the frequency of the electrical stimulation (Westerblad & Allen, 1996). After electrical stimulation ceases, Ca2+ stops diffusing from the SR, SERCA continues to pump Ca2+ into the SR, and [Ca2+]c drops, resulting in an inactivation of the myofilaments and force decay (i.e. relaxation) (Westerblad & Allen, 1996).
Under non‐fatiguing conditions (during the FF protocol), fibres from CSE mice had an increase in [Ca2+]c during contractions and this resulted in a higher submaximal force development (Fig. 6) (Nogueira & Hogan, 2010). However, we were not able to determine whether this increase in [Ca2+]c was due to more Ca2+ released from the sarcoplasmic reticulum. These data are different from a recent report (Robison et al. 2017) which showed, instead, a decrease in single twitch peak [Ca2+]c in collagen‐dissociated FDB fibres from mice exposed to whole body CS for 6 months. Although these data are contradictory, the methods used to treat mice with the CS components (inhaled CS vs. CSE), fibre isolation and the period of treatment (6 vs. 2 months) were notably different between the two studies.
In the present study, maximal force development and force responses to [Ca2+]c during contractions (i.e. myofilament Ca2+ sensitivity, Fig. 6 C; Westerblad & Allen, 1993) were measured and found to be not different between the groups of fibres. These observations suggest that the myofilament Ca2+‐binding function was not affected by CS or CSE treatments when the fibres were not fatigued (Pinto et al. 2008). These data also differ from studies of skinned diaphragm muscle fibres from COPD patients in which force development and myofilament Ca2+ sensitivity were diminished (Ottenheijm et al. 2005; Stubbings et al. 2008). Nonetheless, the mice in the present study did not develop COPD, which was expected within a 2‐month CS treatment period (Craig et al. 2017), and therefore, impairment in the myofilament Ca2+ binding function of hindlimb muscles may not be detected early during the progress of COPD.
One explanation that could account for the increase in force during non‐fatiguing contractions in the CSE group is the slowing in relaxation (Fig. 6 D and E). A slower decay of force in‐between pulse‐stimulations during unfused contractions could increase force development in the subsequent twitches during the train (Westerblad & Allen, 1996). The velocity of relaxation is a combined result of the Ca2+ dissociation off‐kinetics from troponin C, the cross‐bridge detachment rate, the Ca2+ loading rate by parvalbumin, and the Ca2+ uptake rate by SERCA (Poggesi et al. 2005). Our data showed that the rate of force decay during the initial phase of relaxation was slowed in the CSE group of fibres (Fig. 6 E). This observation suggests that cross‐bridge detachment during relaxation may be impaired, but also it may be due to a reduced rate of Ca2+ removal from the cytoplasm (Westerblad & Lannergren, 1991). To distinguish the contributions to the delayed relaxation in the CSE group between Ca2+ and cross‐bridge cycling responses, cytosolic Ca2+ was converted to force (Ca2+‐derived force) (Westerblad & Allen, 1993). The slowed Ca2+‐derived force decay during relaxation in the CSE group suggested that Ca2+ uptake kinetics were also impaired and could possibly be due to a slowing in Ca2+ uptake by the SR. In order to investigate whether the slowed relaxation was due to changes in Ca2+ pumping into the SR, a main contributing factor, we measured the Ca2+ decay rate after a single train stimulation, in a time period (0.1–3 s post‐stimulation) named “tail of Ca2+ decay” after a contraction (Klein et al. 1991; see Methods for details). Interestingly, the data in Fig. 6 G show that SR Ca2+ pumping is impaired in fibres from mice treated with CSE, thereby increasing peak [Ca2+]c and slowing the relaxation. The present study did not investigate the reasons why the SR Ca2+ pumping rate was slowed in the CSE group of fibres, but the literature shows that SERCA activity and Ca2+ pumping function may be inhibited by reactive oxygen and nitrogen species (Viner et al. 1999). These species can modify SERCA residues by tyrosine nitration or cysteine oxidation or S‐nitrosylation (Viner et al. 1999), and CS components have substantial amounts of pro‐oxidant compounds that could produce intracellular oxidative stress in muscle cells (Pryor & Stone, 1993). Therefore, additional investigation is needed to understand the underlying mechanisms of this impaired SR Ca2+ uptake in muscle fibres by CS compounds.
Cigarette smoke compounds alter sarcoplasmic reticulum Ca2+ uptake in skeletal muscle fibres and this effect is more pronounced during fatiguing contractions
To mimic high‐intensity exercise in intact single fibres, a series of repetitive contractions at near‐maximal frequencies of stimulation were evoked (i.e. fatigue protocol). These repetitive contractions lead to changes in contractile function (e.g. progressive reduction in the force development) and metabolism (e.g. accumulation of intracellular metabolites and depletion of metabolic substrates; Allen et al. 2008). Intact fibres from CSE‐treated animals had a faster fall in force development (i.e. less fatigue resistance) compared to fibres from the control and CS groups (Fig. 7 A). In the GPS complex during in situ contractions, fatigue resistance was diminished in both CS and CSE groups (Fig. 3). These results suggest that capillary regression after the 2‐month CS treatment (Fig. 4) was a major factor to reduce time to fatigue in this muscle group. These data correlate with previous data published by our group showing that isolated soleus function does not change until 4 months of daily CS exposure (Tang et al. 2010). Although under non‐fatiguing contractions fibres from CSE group had a slightly slower relaxation (Fig. 6), there was an excessive slowing in relaxation (∼3× higher relaxation time) during fatiguing contractions in CSE fibres compared to the other two groups (Fig. 7 C). Reduced fatigue resistance and slower relaxation under fatiguing conditions in the CSE group is in agreement with the faster time to fatigue and smaller rates of relaxation observed in quadriceps muscles from male smokers (non‐COPD) (Wust et al. 2008) and from isolated muscle obtained from COPD patients (Debigare et al. 2003).
Calcium handling during fatiguing contraction showed significant changes in the CSE fibres. There was an evident increase in the peak [Ca2+]c during the first 100 s of contractions, a pronounced accumulation of intracellular Ca2+ between contractions (Fig. 7 D and E), and a remarkable slowing in intracellular Ca2+ decay following each contraction (Fig. 7 F). Comparing the rate of Ca2+ pumping (Fig. 7 H) in isolated fibres between control and CSE groups at the same time points of the fatigue protocol, the velocity of Ca2+ pumping was progressively decreased in control fibres, as expected (Nogueira et al. 2013), as well as in CSE fibres. However, the decrease was more pronounced in the CSE fibres compared to control fibres, which explains the greater accumulation of Ca2+. This is the first time in which SR Ca2+ pumping function was investigated in mice treated with CS by using an experimental system to test single myofibre function, a relevant model of the “in vivo” environment. This intact single fibre preparation allows several metabolites known to impair SR Ca2+ pumping function, such as Pi, ADP, IMP, and reactive oxygen and nitrogen species (for review see Tupling, 2004), to be available at physiological concentrations during contractions.
This pronounced accumulation in cytosolic Ca2+ during contractions may produce mitochondrial calcium overload that positively correlates with intracellular oxidative stress and the activation of muscle atrophy and muscle wasting signalling pathways (Powers et al. 2016). The activation of atrophy pathways (e.g. increase in the expression of MuRF1, Atrogi1, FOXO3) as well as the inhibition of hypertrophic pathways (e.g. decrease in phospho‐Akt) have been shown for skeletal muscles obtained from mice treated with cigarette smoke before the development of COPD (2 months of CS treatment; Tang et al. 2010). Therefore, a slowing in Ca2+ uptake in muscle fibres from mice treated with CS extract may be a trigger for later adaptations in the muscles during the development of COPD.
Differences in myofibre function in CS‐exposed and CSE‐administered mice
In the present study, it is unclear why the changes in fatigue resistance and calcium handling were observed in fibres that were isolated from mice administered CSE and not those exposed to CS. Cigarette smoke capillary number and in situ fatigue resistance were both decreased independent of the CS delivery route. One possibility is that even though the cotinine levels measured each week were similar between the two CS groups, the mice exposed to the “nose‐only” delivery system received repeated CS components filtered through the lung each day. The CSE was administered only once a week. Thus, it is reasonable to surmise that CSE mice received an acute higher initial dose that dissipated over the week that may have had a greater muscle effect at the cellular level.
Summary
Cigarette smoke components whether delivered by inhalation or more directly into the circulation negatively impact the fatigue resistance of hindlimb skeletal muscle. This loss of muscle function is at least in part due to fewer capillaries and small arterioles that supply skeletal myofibres, but also to changes in intracellular Ca2+ handling, which are exacerbated during fatiguing contractions. Interestingly, these changes in vascular structure correlate with a loss of quiescent satellite cells associated with skeletal myofibres. These data suggest that cigarette smoke alters limb muscle function in part by limiting the number of capillaries and associated regulatory cells that allow adequate O2 for mitochondrial respiration and a dysregulation of excitation–contraction coupling that is mediated by slowed calcium uptake into the sarcoplasmic reticulum. Therefore, the results from the present investigation suggest that components present in cigarette smoke have direct effects on peripheral tissues before overt damage to the lungs.
Additional information
Competing interests
None declared.
Author contributions
These studies were performed in the Division of Physiology, Department of Medicine at the University of California, San Diego and at the Instituto de Bioquímica Médica Leopoldo de Meis, Universidade Federal do Rio de Janeiro, RJ, Brazil. L.N., B.M.T. and F.L.L.‐R. contributed to the design, collection, analysis and interpretation of the nose‐only and i.p. delivery of cigarette smoke and muscle function and morphometric data, and also contributed in drafting/revising the manuscript. J.J. and H.L.‐P. contributed to the design, collection and analysis of data from soleus fibres. M.Y. generated and provided the Nestin‐GFP mice and contributed with the interpretation of the satellite cells associated to myofibers data. E.C.B. and L.N. contributed to the overall concept, experimental design, analysis and interpretation of data and drafting and revision of the manuscript. All authors approved the final version of the manuscript and have agreed to be accountable for all aspects of the work. All persons designated as authors qualify for authorship and all those who qualify for authorship are listed.
Funding
This study was funded by NIH 1 PO1 HL091830‐01A1. L. Nogueira was supported by Brazilian grants from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES; Project no. 2603/2013) and Fundação Carlos Chagas Filho de Apoio à Pesquisa do Estado do Rio de Janeiro (FAPERJ‐APQ1; Project no. E‐26/111.256/2014). F. L. Lima‐Rosa was supported by a graduate fellowship from CAPES. The School of Medicine Light Microscopy Core Facility is supported by NIH (P30 NS047101).
Acknowledgements
Images were collected at the University of California, San Diego (UCSD) School of Medicine Light Microscopy Facility with the assistance of Jennifer Santini (http://neurosciences.ucsd.edu/research/microscopy-core/resources/Pages/microscopy.aspx). We would also like to thank Janelle Fine for the technical support in completing this study.
Biography
Leonardo Nogueira is Associate Professor at IBqM‐UFRJ, Brazil and Assistant Research Scientist at UCSD. His research is focused on the role of nitric oxide signaling on skeletal muscle function and metabolism, and muscle development and repair.

Edited by: Scott Powers & Paul Greenhaff
This is an Editor's Choice article from the 15 July 2018 issue.
References
- Allen DG, Lamb GD & Westerblad H (2008). Skeletal muscle fatigue: cellular mechanisms. Physiol Rev 88, 287–332. [DOI] [PubMed] [Google Scholar]
- Aravamudan B, Kiel A, Freeman M, Delmotte P, Thompson M, Vassallo R, Sieck GC, Pabelick CM & Prakash YS (2014). Cigarette smoke‐induced mitochondrial fragmentation and dysfunction in human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 306, L840–L854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreiro E, del Puerto‐Nevado L, Puig‐Vilanova E, Perez‐Rial S, Sanchez F, Martinez‐Galan L, Rivera S, Gea J, Gonzalez‐Mangado N & Peces‐Barba G (2012). Cigarette smoke‐induced oxidative stress in skeletal muscles of mice. Respir Physiol Neurobiol 182, 9–17. [DOI] [PubMed] [Google Scholar]
- Christov C, Chretien F, Abou‐Khalil R, Bassez G, Vallet G, Authier FJ, Bassaglia Y, Shinin V, Tajbakhsh S, Chazaud B & Gherardi RK (2007). Muscle satellite cells and endothelial cells: close neighbors and privileged partners. Mol Biol Cell 18, 1397–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo G, Clerici M, Giustarini D, Portinaro NM, Aldini G, Rossi R, Milzani A & Dalle‐Donne I (2014). Pathophysiology of tobacco smoke exposure: recent insights from comparative and redox proteomics. Mass Spectrom Rev 33, 183–218. [DOI] [PubMed] [Google Scholar]
- Craig JM, Scott AL & Mitzner W (2017). Immune‐mediated inflammation in the pathogenesis of emphysema: insights from mouse models. Cell Tissue Res 367, 591–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day K, Shefer G, Richardson JB, Enikolopov G & Yablonka‐Reuveni Z (2007). Nestin‐GFP reporter expression defines the quiescent state of skeletal muscle satellite cells. Dev Biol 304, 246–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debigare R, Cote CH, Hould FS, LeBlanc P & Maltais F (2003). In vitro and in vivo contractile properties of the vastus lateralis muscle in males with COPD. Eur Respir J 21, 273–278. [DOI] [PubMed] [Google Scholar]
- Degens H, Gayan‐Ramirez G & van Hees HW (2015). Smoking‐induced skeletal muscle dysfunction: from evidence to mechanisms. Am J Respir Crit Care Med 191, 620–625. [DOI] [PubMed] [Google Scholar]
- Gandra PG, Shiah AA, Nogueira L & Hogan MC (2018). A mitochondrial‐targeted antioxidant improves myofilament Ca2+ sensitivity during prolonged low frequency force depression at low J Physiol 596, 1079–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green HJ, Burnett M, Duhamel TA, D'Arsigny C, O'Donnell DE, Webb KA & Ouyang J (2008). Abnormal sarcoplasmic reticulum Ca2+‐sequestering properties in skeletal muscle in chronic obstructive pulmonary disease. Am J Physiol Cell Physiol 295, C350–C357. [DOI] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein MG, Kovacs L, Simon BJ & Schneider MF (1991). Decline of myoplasmic Ca2+, recovery of calcium release and sarcoplasmic Ca2+ pump properties in frog skeletal muscle. J Physiol 441, 639–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp AE, Goldberg D, Delavar H, Trisko BM, Tang K, Hogan MC, Wagner PD & Breen EC (2016). Skeletal myofiber VEGF regulates contraction‐induced perfusion and exercise capacity but not muscle capillarity in adult mice. Am J Physiol Regul Integr Comp Physiol 311, R192–R199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langen RC, Schols AM, Kelders MC, van der Velden JL, Wouters EF & Janssen‐Heininger YM (2006). Muscle wasting and impaired muscle regeneration in a murine model of chronic pulmonary inflammation. Am J Respir Cell Mol Biol 35, 689–696. [DOI] [PubMed] [Google Scholar]
- Larsson L & Orlander J (1984). Skeletal muscle morphology, metabolism and function in smokers and non‐smokers. A study on smoking‐discordant monozygous twins. Acta Physiol Scand 120, 343–352. [DOI] [PubMed] [Google Scholar]
- Maltais F, Decramer M, Casaburi R, Barreiro E, Burelle Y, Debigare R, Dekhuijzen PN, Franssen F, Gayan‐Ramirez G, Gea J, Gosker HR, Gosselink R, Hayot M, Hussain SN, Janssens W, Polkey MI, Roca J, Saey D, Schols AM, Spruit MA, Steiner M, Taivassalo T, Troosters T, Vogiatzis I & Wagner PD; ATS/ERS Ad Hoc Committee on Limb Muscle Dysfunction in COPD (2014). An official American Thoracic Society/European Respiratory Society statement: update on limb muscle dysfunction in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 189, e15–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayhan WG & Patel KP (1997). Effect of nicotine on endothelium‐dependent arteriolar dilatation in vivo. Am J Physiol 272, H2337–H2342. [DOI] [PubMed] [Google Scholar]
- Menon MK, Houchen L, Singh SJ, Morgan MD, Bradding P & Steiner MC (2012). Inflammatory and satellite cells in the quadriceps of patients with COPD and response to resistance training. Chest 142, 1134–1142. [DOI] [PubMed] [Google Scholar]
- Morse CI, Pritchard LJ, Wust RC, Jones DA & Degens H (2008). Carbon monoxide inhalation reduces skeletal muscle fatigue resistance. Acta Physiol (Oxf) 192, 397–401. [DOI] [PubMed] [Google Scholar]
- Naimi AI, Bourbeau J, Perrault H, Baril J, Wright‐Paradis C, Rossi A, Taivassalo T, Sheel AW, Rabol R, Dela F & Boushel R (2011). Altered mitochondrial regulation in quadriceps muscles of patients with COPD. Clin Physiol Funct Imaging 31, 124–131. [DOI] [PubMed] [Google Scholar]
- Nogueira L & Hogan MC (2010). Phenol increases intracellular [Ca2+] during twitch contractions in intact Xenopus skeletal myofibers. J Appl Physiol (1985) 109, 1384–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogueira L, Shiah AA, Gandra PG & Hogan MC (2013). Ca2+‐pumping impairment during repetitive fatiguing contractions in single myofibers: role of cross‐bridge cycling. Am J Physiol Regul Integr Comp Physiol 305, R118–R125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottenheijm CA, Heunks LM, Sieck GC, Zhan WZ, Jansen SM, Degens H, de Boo T & Dekhuijzen PN (2005). Diaphragm dysfunction in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 172, 200–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto JR, Veltri T & Sorenson MM (2008). Modulation of troponin C affinity for the thin filament by different cross‐bridge states in skinned skeletal muscle fibers. Pflugers Arch 456, 1177–1187. [DOI] [PubMed] [Google Scholar]
- Poggesi C, Tesi C & Stehle R (2005). Sarcomeric determinants of striated muscle relaxation kinetics. Pflugers Arch 449, 505–517. [DOI] [PubMed] [Google Scholar]
- Powers SK, Morton AB, Ahn B & Smuder AJ (2016). Redox control of skeletal muscle atrophy. Free Radic Biol Med 98, 208–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryor WA & Stone K (1993). Oxidants in cigarette smoke. Radicals, hydrogen peroxide, peroxynitrate, and peroxynitrite. Ann N Y Acad Sci 686, 12–27. [DOI] [PubMed] [Google Scholar]
- Rich B, Scadeng M, Yamaguchi M, Wagner PD & Breen EC (2017). Skeletal myofiber vascular endothelial growth factor is required for the exercise training‐induced increase in dentate gyrus neuronal precursor cells. J Physiol 595, 5931–5943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldi M, Maes K, De Vleeschauwer S, Thomas D, Verbeken EK, Decramer M, Janssens W & Gayan‐Ramirez GN (2012). Long‐term nose‐only cigarette smoke exposure induces emphysema and mild skeletal muscle dysfunction in mice. Dis Model Mech 5, 333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robison P, Sussan TE, Chen H, Biswal S, Schneider MF & Hernandez‐Ochoa EO (2017). Impaired calcium signaling in muscle fibers from intercostal and foot skeletal muscle in a cigarette smoke‐induced mouse model of COPD. Muscle Nerve 56, 282–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblatt JD, Kuzon WM Jr, Plyley MJ, Pynn BR & McKee NH (1987). A histochemical method for the simultaneous demonstration of capillaries and fiber type in skeletal muscle. Stain Technol 62, 85–92. [DOI] [PubMed] [Google Scholar]
- Saclier M, Cuvellier S, Magnan M, Mounier R & Chazaud B (2013). Monocyte/macrophage interactions with myogenic precursor cells during skeletal muscle regeneration. FEBS J 280, 4118–4130. [DOI] [PubMed] [Google Scholar]
- Schols AM, Buurman WA, Staal van den Brekel AJ, Dentener MA & Wouters EF (1996). Evidence for a relation between metabolic derangements and increased levels of inflammatory mediators in a subgroup of patients with chronic obstructive pulmonary disease. Thorax 51, 819–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbings AK, Moore AJ, Dusmet M, Goldstraw P, West TG, Polkey MI & Ferenczi MA (2008). Physiological properties of human diaphragm muscle fibres and the effect of chronic obstructive pulmonary disease. J Physiol 586, 2637–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda Y, Costa S, Delamarre E, Roncal C, Leite de Oliveira R, Squadrito ML, Finisguerra V, Deschoemaeker S, Bruyere F, Wenes M, Hamm A, Serneels J, Magat J, Bhattacharyya T, Anisimov A, Jordan BF, Alitalo K, Maxwell P, Gallez B, Zhuang ZW, Saito Y, Simons M, De Palma M & Mazzone M (2011). Macrophage skewing by Phd2 haplodeficiency prevents ischaemia by inducing arteriogenesis. Nature 479, 122–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talhout R, Schulz T, Florek E, van Benthem J, Wester P & Opperhuizen A (2011). Hazardous compounds in tobacco smoke. Int J Environ Res Public Health 8, 613–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang K, Murano G, Wagner H, Nogueira L, Wagner PD, Tang A, Dalton ND, Gu Y, Peterson KL & Breen EC (2013). Impaired exercise capacity and skeletal muscle function in a mouse model of pulmonary inflammation. J Appl Physiol (1985) 114, 1340–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang K, Wagner PD & Breen EC (2010). TNF‐α‐mediated reduction in PGC‐1α may impair skeletal muscle function after cigarette smoke exposure. J Cell Physiol 222, 320–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tupling AR (2004). The sarcoplasmic reticulum in muscle fatigue and disease: role of the sarco(endo)plasmic reticulum Ca2+‐ATPase. Can J Appl Physiol 29, 308–329. [DOI] [PubMed] [Google Scholar]
- Viner RI, Williams TD & Schoneich C (1999). Peroxynitrite modification of protein thiols: oxidation, nitrosylation, and S‐glutathiolation of functionally important cysteine residue(s) in the sarcoplasmic reticulum Ca‐ATPase. Biochemistry 38, 12408–12415. [DOI] [PubMed] [Google Scholar]
- Westerblad H & Allen DG (1991). Changes of myoplasmic calcium concentration during fatigue in single mouse muscle fibers. J Gen Physiol 98, 615–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerblad H & Allen DG (1993). The contribution of [Ca2+]i to the slowing of relaxation in fatigued single fibres from mouse skeletal muscle. J Physiol 468, 729–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerblad H & Allen DG (1996). Mechanisms underlying changes of tetanic [Ca2+]i and force in skeletal muscle. Acta Physiol Scand 156, 407–416. [DOI] [PubMed] [Google Scholar]
- Westerblad H & Lannergren J (1991). Slowing of relaxation during fatigue in single mouse muscle fibres. J Physiol 434, 323–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wust RC, Morse CI, de Haan A, Rittweger J, Jones DA & Degens H (2008). Skeletal muscle properties and fatigue resistance in relation to smoking history. Eur J Appl Physiol 104, 103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Cao J, Chen Y, Chen P, Peng H, Cai S, Luo H & Wu SJ (2013). Intraperitoneal injection of cigarette smoke extract induced emphysema, and injury of cardiac and skeletal muscles in BALB/C mice. Exp Lung Res 39, 18–31. [DOI] [PubMed] [Google Scholar]
- Zuo L, Nogueira L & Hogan MC (2011). Effect of pulmonary TNF‐α overexpression on mouse isolated skeletal muscle function. Am J Physiol Regul Integr Comp Physiol 301, R1025–R1031. [DOI] [PMC free article] [PubMed] [Google Scholar]