

Abstract
Key points
Low‐volume high‐intensity exercise training promotes muscle mitochondrial adaptations that resemble those associated with high‐volume moderate‐intensity exercise training. These training‐induced mitochondrial adaptations stem from the cumulative effects of transient transcriptional responses to each acute exercise bout.
However, whether metabolic stress is a key mediator of the acute molecular responses to high‐intensity exercise is still incompletely understood.
Here we show that, by comparing different work‐matched low‐volume high‐intensity exercise protocols, more marked metabolic perturbations were associated with enhanced mitochondrial biogenesis‐related muscle mRNA responses.
Furthermore, when compared with high‐volume moderate‐intensity exercise, only the low‐volume high‐intensity exercise eliciting severe metabolic stress compensated for reduced exercise volume in the induction of mitochondrial biogenic mRNA responses.
The present results, besides improving our understanding of the mechanisms mediating exercise‐induced mitochondrial biogenesis, may have implications for applied and clinical research that adopts exercise as a means to increase muscle mitochondrial content and function in healthy or diseased individuals.
Abstract
The aim of the present study was to examine the impact of exercise‐induced metabolic stress on regulation of the molecular responses promoting skeletal muscle mitochondrial biogenesis. Twelve endurance‐trained men performed three cycling exercise protocols characterized by different metabolic profiles in a randomized, counter‐balanced order. Specifically, two work‐matched low‐volume supramaximal‐intensity intermittent regimes, consisting of repeated‐sprint (RS) and speed endurance (SE) exercise, were employed and compared with a high‐volume continuous moderate‐intensity exercise (CM) protocol. Vastus lateralis muscle samples were obtained before, immediately after, and 3 h after exercise. SE produced the most marked metabolic perturbations as evidenced by the greatest changes in muscle lactate and pH, concomitantly with higher post‐exercise plasma adrenaline levels in comparison with RS and CM. Exercise‐induced phosphorylation of CaMKII and p38 MAPK was greater in SE than in RS and CM. The exercise‐induced PGC‐1α mRNA response was higher in SE and CM than in RS, with no difference between SE and CM. Muscle NRF‐2, TFAM, MFN2, DRP1 and SOD2 mRNA content was elevated to the same extent by SE and CM, while RS had no effect on these mRNAs. The exercise‐induced HSP72 mRNA response was larger in SE than in RS and CM. Thus, the present results suggest that, for a given exercise volume, the initial events associated with mitochondrial biogenesis are modulated by metabolic stress. In addition, high‐intensity exercise seems to compensate for reduced exercise volume in the induction of mitochondrial biogenic molecular responses only when the intense exercise elicits marked metabolic perturbations.
Keywords: peroxisome proliferator‐activated receptor gamma coactivator 1‐alpha (PGC‐1α) mRNA, mitochondrial biogenesis and dynamics, intracellular signaling, sprint interval training
Key points
Low‐volume high‐intensity exercise training promotes muscle mitochondrial adaptations that resemble those associated with high‐volume moderate‐intensity exercise training. These training‐induced mitochondrial adaptations stem from the cumulative effects of transient transcriptional responses to each acute exercise bout.
However, whether metabolic stress is a key mediator of the acute molecular responses to high‐intensity exercise is still incompletely understood.
Here we show that, by comparing different work‐matched low‐volume high‐intensity exercise protocols, more marked metabolic perturbations were associated with enhanced mitochondrial biogenesis‐related muscle mRNA responses.
Furthermore, when compared with high‐volume moderate‐intensity exercise, only the low‐volume high‐intensity exercise eliciting severe metabolic stress compensated for reduced exercise volume in the induction of mitochondrial biogenic mRNA responses.
The present results, besides improving our understanding of the mechanisms mediating exercise‐induced mitochondrial biogenesis, may have implications for applied and clinical research that adopts exercise as a means to increase muscle mitochondrial content and function in healthy or diseased individuals.
Introduction
Endurance exercise training is a powerful stimulus for the expansion of the skeletal muscle mitochondrial network. This adaptive mechanism includes mitochondrial biogenesis, the cellular process promoting increases in mitochondrial volume and potential changes in mitochondrial composition. Consistent with the metabolic adaptations originating from the enlargement of the muscle mitochondrial pool (Holloszy & Coyle, 1984; Dudley et al. 1987), exercise training‐induced mitochondrial biogenesis is thought to be a crucial adaptive event for the prevention or treatment of a range of metabolic disorders (Hood et al. 2011; Russell et al. 2014; Hesselink et al. 2016) and for improving exercise capacity (Yan et al. 2012; Lundby & Jacobs, 2016).
High‐intensity interval training, defined as repeated intense work bouts separated by recovery periods, is broadly recognized as a time‐efficient alternative to traditional endurance exercise training for promoting skeletal muscle remodelling and enhancing exercise performance (Laursen & Jenkins, 2002; MacInnis & Gibala, 2017). In recent years, a growing body of research has focused on specific forms of intense interval training characterized by short‐duration maximal/supramaximal efforts (Bishop et al. 2011; Hostrup & Bangsbo, 2017). In this context, speed endurance exercise training, depicted as multiple prolonged “all‐out” bouts (<40 s) separated by comparatively long resting periods (Iaia & Bangsbo, 2010), has been shown to promote increments in the activity and/or content of muscle mitochondrial enzymes together with improvements in endurance exercise performance (Burgomaster et al. 2005; Little et al. 2010; Hostrup et al. 2018). Likewise, repeated‐sprint exercise training, characterized by shorter supramaximal efforts (<10 s) interspersed with relatively short (<60 s) recovery periods (Spencer et al. 2005), has recently been reported to upregulate the content of a series of mitochondrial proteins in human skeletal muscle (Serpiello et al. 2012). Therefore, it appears that both speed endurance and repeated‐sprint exercise training promote muscle mitochondrial adaptations that resemble those induced by high‐volume aerobic exercise training, implying that the high‐metabolic demand elicited by intense intermittent exercise plays a putative role in the mitochondrial adaptive processes that occur within the exercising skeletal muscle.
The long‐term mitochondrial adaptations to exercise training have been suggested to stem from the cumulative effects of the transient transcriptional responses to each acute exercise bout (Williams & Neufer, 1996). The peroxisome proliferator‐activated receptor γ coactivator 1‐α (PGC‐1α) is one factor proposed to contribute to initiating these early adaptive molecular responses by coactivating a broad range of transcription factors (Kelly & Scarpulla, 2004). Muscle‐specific overexpression of PGC‐1α in transgenic rodent models increased the mRNA and protein content of multiple mitochondrial proteins as well as elevated muscle mitochondrial enzyme activity and mitochondrial DNA (mtDNA) content (Lin et al. 2002; Calvo et al. 2008), supporting the purported role of PGC‐1α as a key regulator of mitochondrial biogenesis. The observation that a single bout of exercise upregulates PGC‐1α transcription and mRNA content in human skeletal muscle (Pilegaard et al. 2003), and that these transcriptional responses precede increases in muscle mitochondrial proteins (Perry et al. 2010), substantiates the possibility that PGC‐1α also mediates mitochondrial biogenesis in humans.
The molecular mechanisms through which PGC‐1α is thought to coordinate exercise‐induced mitochondrial biogenesis include regulation of mtDNA transcription and mitochondrial remodelling dynamics (Hood et al. 2016). Specifically, mtDNA transcription and replication are driven by the nuclear‐encoded mitochondrial transcription factor A (TFAM), the expression of which is controlled by nuclear transcription factors, counting nuclear respiratory factor 1 and 2 (NRF‐1 and ‐2) (Virbasius & Scarpulla, 1994). Consistent with a PGC‐1α‐mediated coactivation of these transcription factors, increased gene expression of muscle NRF‐1/2 and TFAM has been reported in concert with a higher PGC‐1α mRNA content following acute exercise (Baar et al. 2002; Pilegaard et al. 2003; Perry et al. 2010; Saleem & Hood, 2013). However, other human studies failed to observe exercise‐induced changes in the mRNA of these transcription factors despite an increase in PGC‐1α mRNA (Stepto et al. 2012; Scribbans et al. 2017), suggesting that more research is needed to clarify the exercise‐induced regulation of these mRNAs in human skeletal muscle. In addition, mtDNA integrity is preserved by the ability of mitochondria to constantly undergo structural changes via fusion and fission events, which alter mitochondrial morphology and contribute to mitochondrial quality control (Youle & van der Bliek, 2012; Hood et al. 2015). Despite the emerging importance of mitochondrial remodelling for the maintenance of a functional mitochondrial network, only a few human studies have investigated the impact of acute exercise on the mRNA response of proteins involved in mitochondrial fusion and fission dynamics (Cartoni et al. 2005; Ding et al. 2010; Granata et al. 2017).
PGC‐1α gene expression has been reported to be regulated by multiple intracellular signalling kinases, including AMP‐activated protein kinase (AMPK), Ca2+/calmodulin‐dependent protein kinase II (CaMKII) and p38 mitogen‐activated protein kinase (p38 MAPK) (Puigserver et al. 2001; Jager et al. 2007; Zhang et al. 2014). Given that the contractile activity‐induced activation of these signalling transduction pathways is governed by alterations in the AMP:ATP ratio, cytosolic Ca2+ and reactive oxygen species (ROS), metabolic stress is expected to be a key modulator of the signalling cascades converging on PGC‐1α. In this direction, more marked induction of AMPK and CaMKII phosphorylation with corresponding greater PGC‐1α mRNA abundance has been reported following high‐intensity compared with isocaloric low‐intensity continuous exercise (Egan et al. 2010). These differences were associated with greater plasma lactate accumulation and muscle glycogen depletion in the high‐intensity compared with the low‐intensity exercise protocol. In accordance, the exercise intensity‐dependent increase in PGC‐1α mRNA observed by Nordsborg et al. (2010) may be related to various degrees of metabolic challenges. Further evidence supporting the sensitivity of muscle PGC‐1α transcription to the metabolic environment has been provided by studies using either blood flow‐restricted exercise (Norrbom et al. 2004; Christiansen et al. 2018), nutritional means (Edge et al. 2015) or dietary interventions (Pilegaard et al. 2005) to manipulate the muscle metabolic state during or after exercise.
The intermittent pattern of interval exercise per se may also play a key role in evoking the signalling cascade towards mitochondrial biogenesis (Combes et al. 2015). On the other hand, the metabolic fluctuations characterizing interval exercise appeared not to have a putative role on the PGC‐1α mRNA response following “all‐out” exercise performed in either an intermittent or a continuous manner (Cochran et al. 2014; Taylor et al. 2016), implying that the pulsatile nature of exercise is not of importance when the exercise intensity is supramaximal. Consistent with these results, multiple studies highlighted the potency of supramaximal‐intensity intermittent exercise to promote mitochondrial biogenesis (Gibala et al. 2009; Little et al. 2011; Serpiello et al. 2012) and generate similar (Psilander et al. 2010) or greater (Skovgaard et al. 2016) muscle PGC‐1α mRNA responses compared with high‐volume submaximal exercise, likely due to the effectiveness of intense exercise in exacerbating metabolic stress and thereby inducing enhanced molecular responses. Nevertheless, the exercise‐induced elevation in PGC‐1α mRNA has been observed to be similar after work‐matched high‐intensity interval and continuous exercise characterized by different post‐exercise blood lactate levels (Bartlett et al. 2012), as well as following four diverse high‐volume exercise protocols associated with distinct alterations in muscle metabolism and plasma adrenaline (Brandt et al. 2016). This suggests that the role of metabolic stress in the initial mitochondrial biogenic responses to acute exercise in human skeletal muscle needs to be further elucidated. Furthermore, there is limited information regarding the impact of intense exercise‐associated metabolic stress on the regulation of genes modulating mtDNA transcription and mitochondrial remodelling dynamics.
Therefore, the main aim of the present study was to assess whether different exercise‐induced metabolic perturbations were associated with distinct intracellular signalling and mRNA responses of genes implicated in mitochondrial biogenesis in human skeletal muscle. This was investigated by applying low‐volume repeated‐sprint and speed endurance exercise, as two work‐matched supramaximal‐intensity intermittent regimes inducing pronounced but different metabolic stress levels, and high‐volume submaximal‐intensity continuous exercise generating a mild but constant bioenergetic stress. It was hypothesized that speed endurance exercise promotes more marked mRNA responses than repeated‐sprint exercise due to more profound muscle metabolic perturbations, while continuous moderate‐intensity exercise, by virtue of a prolonged metabolic flux, evokes acute molecular responses analogous to those elicited by low‐volume intense exercise.
Methods
Human subjects and ethics
Thirteen healthy trained men were initially included of which 12 completed the study. Prior to inclusion, subjects were informed of risks and discomforts associated with the experimental procedures. Each subject gave his oral and written informed consent. Inclusion criteria were males, aged 18–40 years, a weekly training volume above 3 h, a maximal oxygen consumption () above 50 mL min−1 kg−1, and a body mass index below 30 kg m−2. In addition, only subjects who regularly performed cycling‐based training for the past 6 years were included to limit the possibility that the cycling exercise‐induced early adaptive responses occurred solely as a consequence of a new mechanical stimulus within the exercising muscle. Exclusion criteria were smoking and chronic disease. Subjects were amateur cyclists or triathletes, and their characteristics are presented in Table 1. The study was approved by the regional research ethics committee of Copenhagen, Denmark (H‐16000378) and adheres to the principles of the Declaration of Helsinki, except for registration in a database.
Table 1.
Subject characteristics
| Age (years) | 31.9 ± 2.5 |
| Height (cm) | 177 ± 2 |
| Weight (kg) | 74.8 ± 2.7 |
| (mL min−1) | 4541 ± 130 |
| (mL kg−1 min−1) | 61.1 ± 1.9 |
, maximal oxygen consumption. Values are means ± SEM (n = 12).
Study design
A randomized, counter‐balanced crossover design was used to compare the acute effects of three different exercise protocols conducted on experimental days separated by at least 1 week.
Prior to the commencement of the experimental period, an incremental test to exhaustion was performed on a mechanically braked cycle ergometer (LC6; Monark Exercise AB, Vansbro, Sweden) for determination of . The test protocol consisted of two submaximal 4 min bouts at 150 and 225 W followed by an incremental ramp test with increments of 25 W min−1 until volitional exhaustion. Pulmonary oxygen consumption () was measured breath‐by‐breath using an on‐line gas analysis system (Oxycon Pro, Viasys Healthcare, Hoechberg, Germany). was determined as the highest value achieved during a 30 s period. Criteria used for achievement of were a plateau in despite an increase in workload and a respiratory exchange ratio above 1.10. Heart rate was monitored throughout the test (Polar Team2, Polar Electro Oy, Kempele, Finland) and maximal heart rate established as the highest value achieved during the test checked for spikes. A regression equation of vs. power output was derived from the two submaximal workloads and was used to determine the individual workloads to be sustained during the exercise protocols.
Experiments
Exercise protocols
The exercise protocols were performed on a mechanically braked cycle ergometer (894E; Monark Exercise AB, Vansbro, Sweden). Saddle and handlebar height were recorded during the first trial and reused in subsequent tests. The exercise protocols consisted of either repeated‐sprint (RS) or speed endurance (SE) exercise at supramaximal intensity or continuous exercise at moderate intensity (CM) (Fig. 1). In RS the subjects performed 18 × 5 s “all‐out” efforts interspersed with 30 s of passive recovery, while SE comprised 6 × 20 s “all‐out” efforts interspersed with 120 s of passive recovery, with an average mean power output of 902 ± 33 and 669 ± 26 W (mean ± SEM) during RS and SE, respectively. CM consisted of 50 min of continuous exercise at a relative intensity corresponding to 70% (218 ± 7 W).
Figure 1. Schematic presentation of the experimental trials.
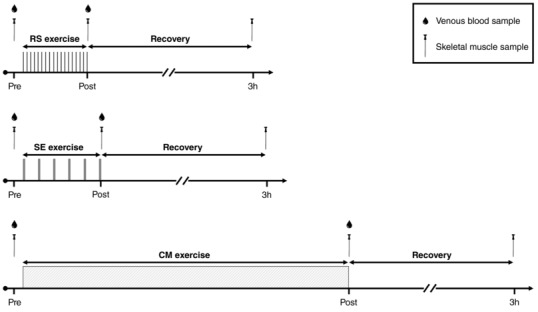
Schematic presentation of the three experimental trials consisting of cycling‐based repeated‐sprint exercise (RS, 18 × 5 s “all‐out” sprints against a breaking force of 0.90 N kg−1 interspersed with 30 s of recovery), speed endurance exercise (SE, 6 × 20 s “all‐out” sprints against a breaking force of 0.75 N kg−1 interspersed with 120 s of recovery) and continuous moderate‐intensity exercise (CM, 50 min at 70% ). Muscle metabolites, pH and protein content were determined in the biopsies obtained at rest (Pre) and immediately after exercise (Post). Muscle mRNA content was determined in the biopsies obtained at Pre and 3 h after exercise (3h). Plasma catecholamines were determined in blood samples taken at Pre and Post.
The rationale behind the choice of these protocols was to induce different metabolic perturbations in the myocellular milieu by using three common training routines. CM was employed as traditional high‐volume (640 ± 21 kJ) endurance exercise leading to mild, but prolonged metabolic disturbances, whereas RS and SE were selected as low‐volume intense intermittent exercise regimes matched for work volume (RS: 79 ± 3 kJ; SE: 78 ± 3 kJ) and work‐to‐rest ratio (1:6), but provoking distinct degrees of metabolic stress. Specifically, muscle lactate and hydrogen ion (H+) accumulation are expected to occur with different patterns and magnitudes during RS and SE. Indeed, while multiple short sprints (≤6 s) result in a gradual increase in muscle and blood lactate and a progressive drop in blood pH (Balsom et al. 1992; Gaitanos et al. 1993), a single long sprint (≥20 s) markedly increases muscle lactate levels, which remain elevated after 2–4 min of recovery (Bogdanis et al. 1996, 1998). Thus, for the same work volume, a higher degree of metabolic acidosis is more likely to be induced by SE than by RS.
All protocols were preceded by a standardized warm‐up consisting of 7 min of continuous cycling at a workload corresponding to 65% (199 ± 7 W) followed by 5 min at rest.
Prior to sprinting in RS and SE, subjects were instructed to begin pedalling as fast as possible and against no resistance for ∼2 s. The cycle ergometer was interfaced with a dedicated software (Monark Anaerobic Test Software 3.3, Monark Exercise AB, Vansbro, Sweden) set to apply the workload and start the timer once a cadence of 100 r.p.m. was reached. Braking forces corresponding to 0.90 and 0.75 N kg−1 body mass were used for RS and SE, respectively. Subjects were encouraged to maintain maximum pedalling speed throughout each sprint. During CM, the braking force was 0.33 ± 0.01 N kg−1 body mass and the subjects kept a cadence of 85 r.p.m. throughout the exercise.
Control procedures
To minimize potential variation in the exercise‐induced response due to external factors, subjects were instructed to refrain from caffeine, alcohol and exercise for 24 h before each trial. In addition, before commencing the experimental period, subjects reported their food habits in a questionnaire. Then, an individual diet plan was developed to standardize food intake during the 48 h preceding each experimental trial. Daily carbohydrate, protein and fat intake in the 48 h prior to the experimental trials were 4.7 ± 0.2, 1.7 ± 0.1 and 0.8 ± 0.0 g kg−1 body mass, respectively.
During the experimental period, subjects continued their usual training routine, and their weekly training load was kept constant. Time commitment and distance covered during endurance‐based training in the 6 days prior to each experimental trial were 455 ± 170 min and 222 ± 64 km (RS), 415 ± 173 min and 204 ± 76 km (SE), and 409 ± 182 min and 204 ± 79 km (CM), respectively.
Experimental procedures
On the experimental days, subjects reported to the laboratory 120 min after ingesting their last meal. After 15 min of rest in the supine position, two 3 mm incisions were made over the lateral portion of the left thigh under local anaesthesia (1 mL lidocaine without epinephrine, 20 mg mL−1 Xylocain, AstraZeneca). A biopsy was obtained from the vastus lateralis muscle at rest before warm‐up (Pre) and immediately after exercise (Post) using a percutaneous Bergstrom needle with suction through separate incisions. An additional muscle biopsy was obtained from the right thigh 3 h into recovery (3h), during which subjects remained in the laboratory and were allowed to consume only water ad libitum. The muscle biopsy samples were immediately frozen in liquid nitrogen and stored at −80°C until further analyses. A blood sample was taken from a catheter inserted in the right antecubital vein prior to and immediately after exercise.
Blood analyses
Blood samples were collected in 2 mL syringes and transferred to an Eppendorf tube containing 30 μL EDTA (0.2 M), after which they were spun at 20,000 g for 3 min to collect plasma, which was stored at −20°C until analysis. Plasma catecholamines were determined by using an enzyme immunoassay kit (2‐CAT Plasma ELISA High Sensitive BA E‐4500; LDN, Nordhorn, Germany).
Muscle analyses
Approximately 20 mg of the muscle samples were frozen separately for mRNA analyses. The remaining part of the muscle sample was freeze‐dried for 48 h and dissected free of blood, fat and connective tissue. Dissection was performed under a stereo microscope with an ambient temperature of ∼18°C and a relative humidity <30%. After dissection, muscle tissue was weighed and separated into different tubes for analysis. Muscle metabolites, pH, protein content and phosphorylation were determined in the biopsies sampled before (Pre) and immediately after (Post) exercise, while mRNA content was determined in the muscle samples obtained before (Pre) and 3 h after (3h) exercise.
Muscle metabolites and pH
Muscle ATP, phosphocreatine (PCr) and lactate were determined on freeze‐dried muscle tissue (∼2.0 mg dry weight (DW)). Determination was made by extraction in 3 M perchloric acid, neutralization to pH 7.0 with 2.2 M KHCO3, followed by fluorometric analyses as previously described (Lowry & Passonneau, 1972). Muscle glycogen was measured in DW muscle tissue (∼1.5 mg) by acid hydrolysis at 100°C for 3 h followed by determination of glycosyl units as previously described (Lowry & Passonneau, 1972). Muscle pH was measured by a small glass electrode (Radiometer GK2801, Copenhagen, Denmark) after homogenizing ∼2 mg DW in a non‐buffering solution containing 145 mM KCl, 10 mM NaCl and 5 mM iodoacetic acid.
Protein content in muscle lysate
Protein content was determined by SDS‐PAGE and western blotting analyses. Briefly, freeze‐dried muscle samples (∼3 mg DW) were homogenized for 1 min at 29 Hz (TissueLyser II, Qiagen, Valencia, CA, USA) in a fresh batch of ice‐cold buffer (10% glycerol, 20 mM sodium pyrophosphate, 150 mM NaCl, 50 mM HEPES (pH 7.5), 1% NP‐40, 20 mM β‐glycerophosphate, 2 mM Na3VO4, 10 mM NaF, 2 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM EDTA (pH 8), 1 mM EGTA (pH 8), 10 μg mL−1 aprotinin, 10 μg mL−1 leupeptin and 3 mM benzamidine). Afterwards, samples were rotated end over end for 1 h at 4°C followed by centrifugation at 17,500 g for 20 min at 4°C. The supernatant (lysate) was collected and total protein concentration in each sample was determined by a bovine serum albumin (BSA) standard kit (Thermo Scientific, Waltham, MA, USA), assayed in triplicate. Then, each lysate sample was mixed with 6 × Laemmli buffer (7 mL 0.5 M Tris‐base, 3 mL glycerol, 0.93 g DTT, 1 g SDS and 1.2 mg bromophenol blue) and ddH2O to reach equal protein concentration. Equal amounts of total protein (range: 12–24 μg) were loaded for each sample in precast gels (Bio‐Rad Laboratories, Hercules, CA, USA). All samples from each subject were loaded on the same gel, with the sample from before exercise (Pre) being placed adjacent to the sample after exercise (Post) for each exercise protocol. Proteins were separated by SDS‐PAGE and semi‐dry transferred to polyvinylidene difluoride (PVDF) membranes (Bio‐Rad Laboratories). The membranes were blocked in either 2% skimmed milk or 3% BSA in Tris‐buffered saline with Tween 20 buffer (TBST) before being incubated overnight at 4°C in primary antibody diluted either in 2% skimmed milk: AMP‐activated protein kinase α (AMPKα) (no. 2532, Cell Signaling Technology, Leiden, The Netherlands), Ca2+/calmodulin‐dependent protein kinase II (CaMKII) (611293, BD Transduction, San Jose, CA, USA), p38 mitogen‐activated protein kinase (p38 MAPK) (no. 9212, Cell Signaling Technology), acetyl‐CoA carboxylase (ACC)Ser79 phosphorylation (07‐303, Merck Millipore, Darmstadt, Germany), CaMKIIThr286 phosphorylation (no. 3361, Cell Signaling Technology); or in 3% BSA: ACC (p0397, Agilent Dako, Santa Clara, CA, USA), activating transcription factor‐2 (ATF‐2) (no. 9226, Cell Signaling Technology), class II histone deacetylase (HDAC) 4 (no. 2072, Cell Signaling Technology), HDAC5 (no. 20458, Cell Signaling Technology), AMPKαThr172 phosphorylation (no. 2531, Cell Signaling Technology), ATF‐2Thr71 phosphorylation (no. 9221, Cell Signaling Technology), HDAC4/5/7Ser632/Ser661/Ser486 phosphorylation (no. 3424, Cell Signaling Technology), p38 MAPKThr180/Tyr182 phosphorylation (no. 9211, Cell Signaling Technology).
After washing in TBST, membranes were incubated with a secondary horseradish peroxidase‐conjugated antibody for ∼1 h at room temperature. The membrane staining was visualized by incubation with a chemiluminescent horseradish peroxidase substrate (Millipore, Denmark) before image digitalization on a Chemi Doc MP (Bio‐Rad Laboratories). Western blot band intensity was determined by densitometry quantification (total band intensity adjusted for background intensity) using Image Lab v.4.0 (Bio‐Rad Laboratories). Phosphorylation levels and protein content were determined on separate membranes in separate analyses and none of the membrane was stripped before protein quantification.
Phosphorylated and total protein content were determined from duplicates, i.e. the biopsies were divided and kept in two parts after dissection, giving two results for the same muscle biopsy sample. The two samples were run on the same gel, and the mean signal intensity of the two samples was used as the result. Exercise‐induced changes in phosphorylated protein content are presented in graphical form as the geometric mean of the fold changes at Post normalized to their respective Pre values.
RNA isolation, reverse transcription and real‐time PCR
Total RNA was isolated from 15–20 mg of muscle tissue (wet weight) by a modified guanidinium thiocyanate–phenol–chloroform extraction method as described previously (Pilegaard et al. 2000) except for the use of a TissueLyser (TissueLyser II, Qiagen) for homogenization.
Superscript II RNase H−system and Oligo dT (Invitrogen, Carlsbad, CA, USA) were used to reverse transcribe the mRNA to cDNA. Quantification of cDNA as a measure of mRNA content of a given gene was performed by real‐time PCR using an ABI 7900 sequence‐detection system (Applied Biosystems, Foster City, CA, USA). Primers and TaqMan probes were designed from human specific databases from ensemble (http://www.ensembl.org/Homo_sapiens/Info/Index) and Primer Express (Applied Biosystems) and are presented in Table 2. Self‐designed TaqMan probes were labelled with 5′‐6‐carboxyfluorescein (FAM) and 3′‐6‐carboxy‐N,N,N′,N′‐tetramethylrhodamine (TAMRA) and obtained from TAG Copenhagen (Copenhagen, Denmark). Cyclophilin A, β‐actin and glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) mRNA were amplified using a 5′‐VIC‐ and 3′‐TAMRA‐labelled predeveloped assay reagent (Applied Biosystems). Real‐time PCR was performed in triplicates in a total reaction volume of 10 μL using Universal Mastermix with uracil‐N glycosylase (UNG; Applied Biosystems). The obtained cycle threshold values reflecting the initial content of the specific transcript in the samples were converted to a relative amount by using standard curves constructed from serial dilution of a pooled sample made from all samples. Cyclophilin A, β‐actin and GAPDH were analysed as reference genes. β‐Actin and GAPDH changed with exercise, whereas cyclophilin A was unaffected by exercise and did not change between trials (Table 3). Thus, cyclophilin A was used as an endogenous control, i.e. for each cDNA sample, the mRNA content of the given target mRNA was normalized to cyclophilin A mRNA. Exercise‐induced changes in mRNA content are presented in graphical form as the geometric mean of the fold changes at 3 h of recovery normalized to their respective Pre values.
Table 2.
Primers and TaqMan probe sequences used for real‐time PCR
| Gene | Forward primer | Reverse primer | TaqMan probe |
|---|---|---|---|
| PGC‐1α | 5' CAAGCCAAACCAACAACTTTATCTCT 3' | 5' CACACTTAAGGTGCGTTCAATAGTC 3' | 5' AGTCACCAAATGACCCCAAGGGTTCC 3' |
| NRF‐2 | 5' GAAATTGAGATTGATGGAACAGAGAA 3' | 5' TATGGCCTGGCTTACACATTCA 3' | 5' AAGCATTGTAGAACAAACCTACGCGCCAG 3' |
| TFAM | 5' AGATTCCAAGAAGCTAAGGGTGATT 3' | 5' TTTCAGAGTCAGACAGATTTTTCCA 3' | 5' CCGCAGGAAAAGCTGAAGACTGTAAAGGA 3' |
| MFN2 | 5' CCTCTCTGTACTGGTGGACGATT 3' | 5' TGGCGGTGCAGCTCATT 3' | 5' CCAGATGGACTTCCACCCTTCTCCAGTAG 3' |
| DRP1 | 5' CGGCAAATCAAACGTCTAGAAGA 3' | 5' AAGTTTAGGAAATCGTAACAATTCCT 3' | 5' CCCAGCCTCCGCTGTGTGGAAC 3' |
| SOD2 | 5' CGCGGCCTACGTGAACAA 3' | 5' CTATCTGGGCTGTAACATCTCCCTT 3' | 5' CACCGAGGAGAAGTACCAGGAGGCGTT 3' |
| HO‐1 | 5' GCCAGCAACAAAGTGCAAGAT 3' | 5' AGTGTAAGGACCCATCGGAGAA 3' | 5' AGAGGGAAGCCCCCACTCAACACC 3' |
| HSP72 | 5' GCGTGATGACTGCCCTGAT 3' | 5' CGCCCTCGTACACCTGGAT 3' | 5' TCCCCACCAAGCAGACGCAGATCT 3' |
| Cyc A | 5' GGTCTCTTTGGGCGGAAGAC 3' | 5' CTCTCCCCAGATGATGCCTTT 3' | 5' CCCTGGATACTCTTACACAGCCGCCAA 3' |
PGC‐1α, peroxisome proliferator‐activated receptor‐γ coactivator‐1α; NRF‐2, nuclear respiratory factor 2; TFAM, mitochondrial transcription factor A; MFN2, mitofusin‐2; DRP1, dynamin‐related protein 1; SOD2, superoxide dismutase 2; HO‐1, heme oxygenase‐1; HSP72, heat shock protein 72; Cyc A, cyclophilin A.
Table 3.
mRNA content of reference genes before (Pre) and 3 h after (3h) repeated‐sprint (RS), speed endurance (SE) and continuous moderate‐intensity (CM) exercise
| Pre | 3h | |||||
|---|---|---|---|---|---|---|
| RS | SE | CM | RS | SE | CM | |
| Cyc A | 0.47 ± 0.03 | 0.49 ± 0.04 | 0.44 ± 0.04 | 0.54 ± 0.06 | 0.53 ± 0.03 | 0.51 ± 0.05 |
| β‐Actin | 0.34 ± 0.05 | 0.31 ± 0.04 | 0.23 ± 0.04 | 0.48 ± 0.05† | 0.62 ± 0.07* | 0.65 ± 0.12* |
| GAPDH | 0.53 ± 0.08† | 0.41 ± 0.07 | 0.29 ± 0.07 | 0.53 ± 0.09 | 0.60 ± 0.09* | 0.56 ± 0.11* |
Values are means ± SEM (n = 12), *Significantly different from Pre (P < 0.05), †Significantly different from CM (P < 0.05).
Statistics
A priori sample size determination was performed for the primary outcome measure “PGC‐1α mRNA” based on data from a previous study with a similar experimental design (Brandt et al. 2016). Power was set to 0.8 and significance level was set to 0.05.
To estimate between‐trial differences in the exercise‐induced changes across sampling time, as well as within‐ and between‐trial differences, a linear mixed model was used with trial and sampling time included as fixed factors and subjects as random factor for a full factorial design. Baseline (i.e. Pre) values were included as covariates in the mixed model. Model checking was based on residual and Q–Q plots. In the case of heteroscedasticity (i.e. unequal variance), log‐transformation was applied prior to analysis. Model‐based t tests were used in pairwise comparisons to identify between‐trial differences in the exercise‐induced changes across sampling time. In addition, within‐ and between‐trial differences were compared pairwise using model‐based t tests. Multiple linear regression analysis was used to determine the individual relationships between change of metabolic and molecular variables with low‐volume supramaximal‐intensity intermittent exercise. Fold changes in muscle ATP, PCr, lactate, [H+] and glycogen, and plasma adrenaline, were initially included in the model as predictors of the fold change in mRNA and phosphorylated protein content. Then, for each mRNA and protein, the stepwise backward method was used to determine the metabolic variables that best predicted the exercise‐induced response (i.e. best‐fitting model). Multicollinearity was assessed using the variance inflation factor (VIF), which indicates whether a predictor has a strong linear relationship with another predictor. The coefficient of determination of the multiple linear regression is presented as the R 2 and adjusted R 2. The level of significance for all analyses was defined as P < 0.05. The statistical analysis was carried out with R version 3.4.1 and the extension packages lme4 and multcomp. Exercise‐induced changes in protein and mRNA content are presented as geometric means ± 95% confidence interval (CI) of the geometric mean, while all other results are reported as means ± SEM.
Results
Muscle metabolites and pH
Muscle metabolite concentrations and pH levels before and immediately after exercise are reported in Table 4. Exercise reduced muscle ATP levels in RS and SE (P < 0.001). Muscle PCr decreased with exercise in all trials (P < 0.001), with greater PCr depletion in RS and SE than in CM (P = 0.003 and P = 0.004).
Table 4.
Muscle metabolites (mmol kg−1 DW) and pH before (Pre) and immediately after (Post) repeated‐sprint (RS), speed endurance (SE) and continuous moderate‐intensity (CM) exercise
| Pre | Post | Δ | |||||||
|---|---|---|---|---|---|---|---|---|---|
| RS | SE | CM | RS | SE | CM | RS | SE | CM | |
| ATP | 25.8 ± 1.8 | 25.1 ± 1.5 | 24.4 ± 2.3 | 19.2 ± 1.8*† | 16.7 ± 1.4*† | 21.9 ± 2.0 | −6.6 ± 1.9† | −8.4 ± 1.5† | −2.6 ± 0.5 |
| PCr | 79.8 ± 5.8 | 79.8 ± 3.0 | 79.0 ± 4.7 | 38.4 ± 5.3*† | 39.3 ± 6.9*† | 62.4 ± 4.5* | −41.4 ± 7.7† | −40.5 ± 6.9† | −16.7 ± 3.5 |
| Lactate | 5.8 ± 1.1 | 5.5 ± 0.7 | 5.0 ± 0.5 | 41.4 ± 7.2*† | 79.9 ± 8.4*§† | 11.4 ± 1.9* | 35.6 ± 6.4† | 74.4 ± 8.5§† | 6.4 ± 1.7 |
| Glycogen | 783 ± 39 | 722 ± 45 | 708 ± 48 | 652 ± 35*† | 576 ± 38*† | 513 ± 49* | −130 ± 29 | −146 ± 19 | −194 ± 26 |
| pH | 7.26 ± 0.02 | 7.25 ± 0.02 | 7.27 ± 0.02 | 7.02 ± 0.05*† | 6.74 ± 0.03*§† | 7.25 ± 0.04 | (47 ± 10) × 10−7 † | (132 ± 16) × 10−7 § † | (5 ± 5) × 10−7 |
Values are means ± SEM (n = 12). Δ, difference between Post and Pre. Differences in pH are presented as changes in [H+], *Significantly different from Pre (P < 0.05, †Significantly different from CM (P < 0.05), §Significantly different from RS (P < 0.05).
Muscle lactate increased with exercise in all trials (P < 0.001), with lactate accumulation being larger in SE than in RS and CM (P < 0.001), and larger in RS than in CM (P < 0.001). Muscle pH decreased in RS and SE (P < 0.001), and was unchanged in CM (P = 0.478). Moreover, muscle pH decreased more in SE than in RS and CM (P < 0.001), and more in RS than in CM (P < 0.001).
Muscle glycogen was lowered in all trials (P < 0.001), with post‐exercise glycogen levels being lower in CM than in RS (P = 0.003) and SE (P = 0.040).
Plasma catecholamines
Plasma adrenaline was increased by exercise in RS and SE (P < 0.001) (Fig. 2 A). Post‐exercise adrenaline was ∼2.5‐ and 3.5‐fold higher in RS and SE, respectively, than in CM (P = 0.005 and P < 0.001), and was higher in SE than in RS (P = 0.011). Furthermore, the exercise‐induced change in plasma adrenaline was greater in RS and SE than in CM (P = 0.042 and P = 0.001).
Figure 2. Plasma catecholamines.
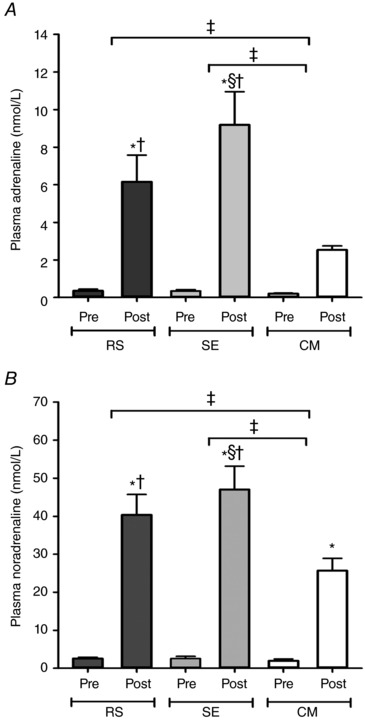
Venous plasma adrenaline (A) and noradrenaline (B) concentration before (Pre) and immediately after (Post) repeated‐sprint (RS), speed endurance (SE) and continuous moderate‐intensity (CM) exercise. Values are mean ± SEM (n = 12). *Significantly different from Pre (P < 0.05). †Significantly different from CM (P < 0.05). §Significantly different from RS (P < 0.05). ‡Significant difference in the exercise‐induced change (P < 0.05).
Plasma noradrenaline increased with exercise in all trials (P < 0.001) (Fig. 2 B). In RS and SE, post‐exercise noradrenaline was higher than in CM (P = 0.001 and P < 0.001), with concomitant differences in the magnitude of change between RS and CM (P = 0.027), and between SE and CM (P = 0.001).
Muscle protein phosphorylation
Representative blots for each protein analysed are presented in Fig. 3.
Figure 3. Representative western blots.
Representative western blots corresponding to phosphorylated and total protein content measured before (Pre) and immediately after (Post) repeated‐sprint (RS), speed endurance (SE) and continuous moderate‐intensity (CM) exercise. The molecular mass of band migration of the given protein has been included.
Phosphorylated AMPKα increased immediately after exercise in all trials (RS, P = 0.006; SE, P = 0.001; CM, P < 0.001), with no difference between trials (Fig. 4 A). Similarly, phosphorylated ACC increased to a similar extent in all trials (P < 0.001) (Fig. 4 B).
Figure 4. Exercise‐induced skeletal muscle intracellular signalling.
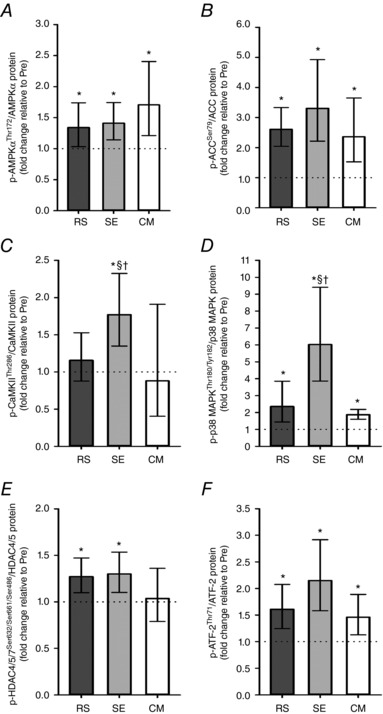
Repeated‐sprint (RS), speed endurance (SE) and continuous moderate‐intensity (CM) exercise‐induced alterations in phosphorylated AMP‐activated protein kinase alpha (AMPKα) protein, acetyl‐CoA carboxylase (ACC) protein, Ca2+/calmodulin‐dependent protein kinase II (CaMKII) protein, p38 mitogen‐activated protein kinase (p38 MAPK) protein, class II histone deacetylase 4/5/7 (HDAC4/5/7) protein and activating transcription factor‐2 (ATF‐2) protein normalized to protein content of the given protein. Values are geometric means ± 95% CI (n = 12). *Significantly different from Pre (P < 0.05). †Significantly different from CM (P < 0.05). §Significantly different from RS (P < 0.05).
Phosphorylated CaMKII content was elevated by SE (P < 0.001), but remained unchanged in RS (P = 0.310) and CM (P = 0.373), resulting in a higher level of phosphorylation in SE than in RS (P < 0.036) and CM (P = 0.006) (Fig. 4 C).
Phosphorylated p38 MAPK increased in all trials (RS, P < 0.001; SE, P < 0.001; CM, P = 0.004), but the change within SE was larger than observed within RS (P = 0.002) and CM (P < 0.001) (Fig. 4 D).
Phosphorylated HDAC4/5/7 increased in RS (P = 0.005) and SE (P = 0.002), but was unaltered in CM (P = 0.676), with no difference in the change between SE and CM (P = 0.059) (Fig. 4 E).
Phosphorylated ATF‐2 increased with exercise in all trials (RS, P = 0.001; SE, P < 0.001; CM, P = 0.018), with no difference in the change between SE and CM (P = 0.066) (Fig. 4 F).
Muscle mRNA
Muscle PGC‐1α mRNA content was elevated 3 h after exercise relative to Pre in all trials (P < 0.001), with SE and CM inducing a greater response than RS (P = 0.006 and P < 0.001) (Fig. 5 A). Muscle NRF‐2 mRNA levels were elevated in SE (P = 0.004) and CM (P < 0.001), but were unchanged in RS (P = 0.640), with the response being larger in CM than in RS (P < 0.001) (Fig. 5 B). TFAM mRNA content was upregulated by SE (P = 0.043) and CM (P = 0.010), while it did not change in RS (P = 0.667), and CM induced a more marked response than RS (P = 0.033) (Fig. 5 C).
Figure 5. Exercise‐induced skeletal muscle mRNA responses.
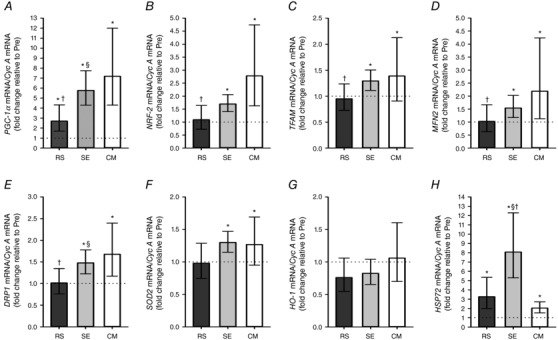
Repeated‐sprint (RS), speed endurance (SE) and continuous moderate‐intensity (CM) exercise‐induced mRNA responses of peroxisome proliferator‐activated receptor‐γ coactivator‐1α (PGC‐1α), nuclear respiratory factor 2 (NRF‐2), mitochondrial transcription factor A (TFAM), mitofusin‐2 (MFN2), dynamin‐related protein 1 (DRP1), superoxide dismutase 2 (SOD2), heme oxygenase‐1 (HO‐1) and heat shock protein 72 (HSP72) mRNA content normalized to cyclophilin A (Cyc A) mRNA content. Values are geometric means ± 95% CI (n = 12). *Significantly different from Pre (P < 0.05). †Significantly different from CM (P < 0.05). §Significantly different from RS (P < 0.05).
Mitofusin‐2 (MFN2) mRNA abundance increased in SE (P = 0.041) and CM (P < 0.001), with no changes observed in RS (P = 0.905), resulting in a greater mRNA response in CM than in RS (P = 0.012) (Fig. 5 D). Dynamin related protein 1 (DRP1) mRNA content increased with exercise in SE (P = 0.002) and CM (P < 0.001), while it was unaltered in RS (P = 0.935). Moreover, the DRP1 mRNA response was more marked in SE and CM than in RS (P = 0.037 and P = 0.005, respectively) (Fig. 5 E).
Muscle mRNA content of superoxide dismutase 2 (SOD2), the gene encoding the mitochondrial anti‐oxidant enzyme MnSOD, increased 3 h into recovery in SE (P = 0.017) and CM (P = 0.031), with no change observed in RS (P = 0.856) (Fig. 5 F). Muscle mRNA abundance of the ROS‐sensitive gene heme oxygenase‐1 (HO‐1) was not altered by exercise in either of the trials (Fig. 5 G). Muscle mRNA content of heat shock protein 72 (HSP72), a protein exerting a cyto‐protective function against oxidative stress, was upregulated by exercise in all trials (P < 0.001), and the change within SE was larger than the change within RS (P = 0.001) and CM (P < 0.001) (Fig. 5 H).
Relationships between change in metabolic and molecular variables
Multiple linear regression analysis showed that change in muscle PCr, [H+] and glycogen, and plasma adrenaline, predicted the PGC‐1α mRNA response to RS and SE (Table 5), with change in muscle glycogen (P = 0.032) and plasma adrenaline (P = 0.014) adding significantly to the model, whereas change in muscle PCr (P = 0.103) and [H+] (P = 0.055) did not contribute significantly to the model.
Table 5.
Individual relationships between change of metabolic and molecular variables with repeated‐sprint (RS) and speed endurance (SE) exercise
| Metabolic variables included in the model | Multiple R2 | Adjusted R 2 | P value | |
|---|---|---|---|---|
| mRNA | ||||
| PGC‐1α | PCr, [H+], glycogen, adrenaline | 0.41 | 0.28 | 0.034 |
| NRF‐2 | PCr, adrenaline | 0.32 | 0.25 | 0.018 |
| TFAM | PCr, [H+], glycogen, adrenaline | 0.54 | 0.45 | 0.004 |
| MFN2 | PCr, adrenaline | 0.45 | 0.39 | 0.002 |
| DRP1 | PCr, [H+], adrenaline | 0.46 | 0.38 | 0.005 |
| SOD2 | PCr, [H+], adrenaline | 0.27 | 0.15 | 0.071 |
| HO‐1 | ATP, PCr, adrenaline | 0.22 | 0.10 | 0.168 |
| HSP72 | Lactate | 0.34 | 0.31 | 0.003 |
| Phosphorylated protein | ||||
| AMPKα | PCr, [H+], glycogen | 0.12 | −0.02 | 0.474 |
| ACC | Adrenaline, glycogen | 0.19 | 0.11 | 0.114 |
| CaMKII | Lactate, [H+], glycogen | 0.39 | 0.30 | 0.017 |
| p38 MAPK | ATP, lactate | 0.58 | 0.54 | <0.001 |
| HDAC | [H+] | 0.01 | −0.03 | 0.639 |
| ATF‐2 | Lactate | 0.16 | 0.12 | 0.054 |
mRNA abbreviations are defined in Table 2. Phosphorylated protein: AMPKα, AMP‐activated protein kinase α; ACC, acetyl‐CoA carboxylase; CaMKII, Ca2+/calmodulin‐dependent protein kinase II; p38 MAPK, p38 mitogen‐activated protein kinase; HDAC, histone deacetylase; ATF‐2, activating transcription factor‐2.
Moreover, change in metabolic variables predicted change in NRF‐2, TFAM, MFN2 and DRP1 mRNA, as well as in phosphorylated CaMKII and phosphorylated p38 MAPK in response to RS and SE (Table 5).
Discussion
The major finding in the present study was that the PGC‐1α mRNA response to low‐volume intense intermittent exercise was greater when exercise induced the highest muscle lactate accumulation, the greatest drop in muscle pH, and the highest plasma adrenaline levels. On the other hand, although supramaximal‐intensity intermittent exercise resulted in more marked metabolic perturbations than continuous moderate‐intensity exercise (CM), PGC‐1α mRNA was elevated to a lower or similar extent in response to repeated‐sprint (RS) and speed endurance (SE) exercise, respectively, compared with CM. Moreover, the severe metabolic stress elicited by SE was associated with higher exercise‐induced phosphorylation of CaMKII and p38 MAPK, while no differences in the exercise‐induced signalling were detected between RS and CM. Only SE and CM upregulated the mRNA abundance of NRF‐2, TFAM, MFN2, DRP1 and SOD2. In accordance with the different exercise‐induced metabolic challenges, SE elevated the mRNA content of HSP72 more than RS and CM.
The present finding that both RS and SE upregulated PGC‐1α mRNA content highlights the potential impact of low‐volume intense intermittent exercise on mitochondrial biogenesis in well‐trained skeletal muscle. While the high responsiveness of PGC‐1α mRNA to SE has been demonstrated in trained individuals (Psilander et al. 2010; Skovgaard et al. 2016), the present study provides novel evidence that RS also increases muscle PGC‐1α mRNA abundance in well‐trained subjects. Moreover, the observation that PGC‐1α mRNA was elevated by only 90–120 s of active exercise time indicates that intense exercise of even shorter duration than that employed by Psilander et al. (2010) and Skovgaard et al. (2016) (i.e. 180–240 s) is sufficient to increase PGC‐1α mRNA, and therefore potentially induce transcriptional regulation of mitochondrial proteins, in endurance‐trained skeletal muscle.
The observation that SE was associated with a more marked increase in PGC‐1α mRNA than work‐matched RS, suggests that the greater metabolic perturbations with high muscle lactate and low muscle pH characterizing SE contributed to eliciting the enhanced PGC‐1α mRNA response. Accordingly, treatment with lactate elevated PGC‐1α mRNA in myotubes (Hashimoto et al. 2007) and mouse skeletal muscle (Kitaoka et al. 2016), and a restricted exercise‐induced decline in muscle pH blunted the PGC‐1α mRNA upregulation during the early recovery from high‐intensity exercise in humans (Edge et al. 2015).
The higher plasma adrenaline levels detected after SE compared with RS may also have caused the different PGC‐1α mRNA responses, as injections of adrenaline (Chinsomboon et al. 2009) or the β‐adrenergic agonist clenbuterol (Miura et al. 2007) have been shown to induce an increase in PGC‐1α mRNA content in mouse skeletal muscle. Conversely, the unaltered plasma adrenaline concentration found after CM was not associated with a less robust PGC‐1α mRNA response compared with intense intermittent exercise. This suggests that the time frame with elevated adrenaline during intense intermittent exercise may not have been sufficiently long to influence the exercise‐induced regulation of PGC‐1α mRNA, as also proposed by a recent human study (Brandt et al. 2016). In addition, muscle glycogen levels have been reported to affect the exercise‐induced regulation of PGC‐1α mRNA, with studies showing that exercise with low glycogen promoted a greater increase in PGC‐1α mRNA than exercise with normal or high muscle glycogen content (Bartlett et al. 2013; Psilander et al. 2013). Likewise, low muscle glycogen concentration during recovery from exercise has been reported to prolong the exercise‐induced elevation in PGC‐1α mRNA (Pilegaard et al. 2005). Thus, although pre‐exercise muscle glycogen content did not differ between the trials in the current study, the observation that post‐exercise muscle glycogen content was lower after high‐volume submaximal exercise than after low‐volume supramaximal exercise may partly explain the higher PGC‐1α mRNA response detected 3 h into recovery from CM compared with RS.
In summary, while the similar PGC‐1α mRNA responses to CM and SE are in line with previous findings (Psilander et al. 2010) and underline the ability of high‐intensity exercise‐induced metabolic stress to counteract a reduced volume of exercise, the lower elevation in PGC‐1α mRNA detected in response to RS than both SE and CM is novel and indicates that the metabolic perturbations induced by RS did not provide sufficient stimuli to compensate for the low work volume.
The finding that the mRNA content of genes controlling mtDNA transcription increased in response to CM is in accordance with other studies (Pilegaard et al. 2003; Saleem & Hood, 2013), whereas the significant NRF‐2 and TFAM mRNA response observed with SE but not with RS has not been shown before. Moreover, this exercise protocol‐specific regulation of NRF‐2 and TFAM mRNA corresponds with the PGC‐1α responses, which may suggest a common regulatory mechanism or a PGC‐1α‐mediated induction of these mRNAs, as previously proposed (Pilegaard et al. 2003). Likewise, the observation that MFN2 and DRP1 mRNA content was elevated after SE and CM, but not after RS, indicates that regulation of mitochondrial morphology depends on the nature of the exercise stimulus. The lack of response to RS may be related to the dampened PGC‐1α mRNA response, as recent evidence indicates a PGC‐1α‐dependent regulation of both MFN2 (Zechner et al. 2010) and DRP1 expression (Vainshtein et al. 2015). It should be noted that a study involving trained cyclists reported elevated MFN1 and MFN2 mRNA levels 24 h, but not 2 h after exercise (Cartoni et al. 2005), suggesting that an upregulation of MFN2 mRNA may have occurred at a later stage into recovery from RS in the current study. In accordance with the present observation, elevated MFN2 and DRP1 mRNA levels have been observed immediately after both speed endurance and continuous exercise (Granata et al. 2017), supporting both an exercise intensity‐ and volume‐dependent transcriptional regulation of mitochondrial fusion and fission proteins. This resembles the scenario depicted for PGC‐1α, indicating a close interplay between the complex mechanisms involved in the maintenance of a high‐quality mitochondrial network.
Interestingly, the results of multiple linear regression analysis suggest that change in muscle metabolic variables, along with change in plasma adrenaline, significantly contributed to the change in PGC‐1α, NRF‐2, TFAM, MFN2 and DRP1 mRNA induced by RS and SE. Thus, the relationship between metabolic stress and the mitochondrial biogenic molecular response to intense intermittent exercise was significant not only at the group but also at the individual level.
The finding that both phosphorylated AMPKα and phosphorylated ACC, a sensitive marker of AMPK activation (Chen et al. 2003), were increased to the same extent by RS, SE and CM, implies that the current differences in the exercise‐induced PGC‐1α mRNA response may not be explained by a differential activation of the AMPK signalling pathway. The present exercise‐induced regulation of the AMPK pathway is consistent with the observed increase in AMPKα phosphorylation immediately after both speed endurance (Gibala et al. 2009) and endurance exercise (Wojtaszewski et al. 2000), and with the increased ACC phosphorylation reported immediately after repeated‐sprint exercise (Serpiello et al. 2012).
The observation that SE increased CaMKII and p38 MAPK phosphorylation more than both RS and CM, with no differences between these two protocols, does not support that CaMKII and p38 MAPK determined the different elevation in PGC‐1α mRNA. The greater CaMKII phosphorylation detected in response to SE compared with CM is in line with studies reporting an exercise intensity‐dependent phosphorylation of CaMKII (Rose et al. 2006; Egan et al. 2010), which in turn is consistent with the more marked alterations in sarcoplasmic Ca2+ concentration likely to occur during exercise characterized by a heavy recruitment of type II muscle fibres (Baylor & Hollingworth, 2003). In spite of the high‐intensity nature of repeated‐sprint exercise, the lack of change in phosphorylated CaMKII immediately after the RS trial may be explained by the very short‐lasting exercise stimulus. Similarly, Serpiello et al. (2012) observed no change in CaMKII phosphorylation immediately after repeated short sprints, while an increase was detected 1 h into recovery. The current finding that p38 MAPK phosphorylation was less robust after RS and CM than after SE is in line with the lack of response of phosphorylated CaMKII to RS and CM. Furthermore, these observations are in agreement with a study indicating that increments in Ca2+ flux are involved in p38 MAPK activation, and that p38 MAPK may be a downstream target of CaMKII (Wright et al. 2007). Notably, multiple linear regression analysis showed that intense intermittent exercise‐induced phosphorylation of CaMKII and p38 MAPK could be partly explained by change in muscle metabolic variables, suggesting a metabolic stress‐dependent activation of these kinases. As class II histone deacetylases (HDACs) and activating transcription factor‐2 (ATF‐2) have been proposed as intensity‐dependent mediators of the exercise‐induced PGC‐1α mRNA response (Egan et al. 2010), the phosphorylation of these proteins was examined. The present observation that phosphorylated HDAC4/5/7 was elevated by supramaximal‐intensity, but not by submaximal‐intensity exercise is in agreement with the previously reported exercise intensity‐dependent inactivation of HDACs (Egan et al. 2010). On the other hand, the lack of difference in HDAC phosphorylation between RS and SE does not seem to depend on CaMKII as shown in cell cultures (Backs et al. 2008). Phosphorylated ATF‐2 was upregulated to the same extent by all trials, which is in contrast to the observed differential phosphorylation of CaMKII and p38 MAPK, as these signalling kinases have been suggested to regulate ATF‐2 phosphorylation (Wright et al. 2007). Hence, the current data indicate that signalling events other than the ones formerly proposed may be responsible for the exercise‐induced phosphorylation of HDACs and ATF‐2 in human skeletal muscle.
Given the proposed importance of ROS in mediating the signalling cascades towards mitochondrial biogenesis (Gomez‐Cabrera et al. 2005; Irrcher et al. 2009; Kang et al. 2009; Morales‐Alamo et al. 2013; Place et al. 2015), the exercise‐induced mRNA responses of proteins implicated in muscle anti‐oxidant capacity were assessed. The finding that SOD2 mRNA abundance increased in response to SE and CM, but not following RS, may indicate different adaptive responses in mitochondrial ROS‐scavenging capacity. Furthermore, the lack of change in SOD2 mRNA content after RS is in accordance with the attenuated PGC‐1α mRNA response, as previous evidence has suggested a PGC‐1α‐mediated regulation of SOD2 (Leick et al. 2008; Geng et al. 2010). The observation that HO‐1 mRNA was unaffected by exercise is in contrast with the elevated HO‐1 mRNA levels reported after endurance exercise (Pilegaard et al. 2000). However, the well‐trained status of the subjects, the sample time point and the exercise protocols employed may explain the unresponsiveness of HO‐1 mRNA to acute exercise observed in the current study. Moreover, in agreement with the present findings, no alterations in HO‐1 mRNA content were observed in a moderately trained population after 90 min of interval exercise (Ballmann et al. 2014), suggesting that further studies are needed to clarify the exercise‐induced regulation of HO‐1 mRNA in human skeletal muscle. While increased levels of HSP72 mRNA have been demonstrated following traditional endurance exercise (Morton et al. 2009), the present study shows for the first time that low‐volume intense intermittent exercise upregulated HSP72 mRNA to a similar or even higher extent than high‐volume submaximal exercise. This finding may be explained not only by the greater muscle metabolic stress, but perhaps also by a higher degree of oxidative stress elicited by SE than RS and CM. On the other hand, similar HSP72 mRNA responses were reported following work‐matched high‐intensity interval and submaximal continuous exercise in spite of different metabolic demands (Bartlett et al. 2012), implying that the exercise‐induced HSP72 mRNA response may be further enhanced only by very high levels of metabolic stress, as occurred during SE in the current study.
A limitation of the present study is the different durations of the exercise protocols employed. Indeed, the long‐lasting continuous submaximal exercise may have induced a prolonged activation of the signalling pathways mediating PGC‐1α transcription. In fact, AMPK, ACC and CaMKII phosphorylation have been shown to increase shortly after the onset of submaximal exercise (Rose & Hargreaves, 2003; Rose et al. 2005), implying that the activation of these signalling pathways lasted longer with the long‐term continuous exercise than with the short‐term intermittent exercise. This might explain not only the smaller PGC‐1α mRNA response observed following RS than CM despite similar degrees of intracellular signalling, but also the lack of differences in the PGC‐1α mRNA response between SE and CM, although CaMKII and p38 MAPK phosphorylation levels were different. Moreover, post‐exercise mRNA levels were determined in the muscle biopsy sampled 3 h into recovery, which corresponded to a sampling time of 3 h 10 min, 3 h 12 min and 3 h 50 min after the effective onset of RS, SE and CM, respectively. The exercise‐induced mRNA response of regulators of genes encoding mitochondrial proteins has been shown to peak within 2–8 h into recovery from exercise (Pilegaard et al. 2003; Booth & Neufer, 2005), which supports our choice of measuring mRNA content 3 h after the cessation of exercise. However, further research is warranted to elucidate whether a time frame of ∼40 min may underlie significantly different degrees of elevation in mRNA abundance.
Although the current results have important practical implications for effective exercise training prescription, we recognize that mRNA transcription represents only the first step towards protein synthesis, and thus the mRNA response to acute exercise may not accurately predict the skeletal muscle adaptations to chronic exercise training (Cochran et al. 2014). In this context, the assessment of post‐transcriptional regulating mechanisms (e.g. microRNA) may improve the comprehension of the molecular events underlying the training‐induced adaptations (Miller et al. 2016). On the other hand, our findings are supported by long‐term studies showing that repeated‐sprint, speed endurance and continuous submaximal exercise training promote mitochondrial biogenesis in skeletal muscle from moderately trained individuals (MacDougall et al. 1998; Gibala et al. 2006; Serpiello et al. 2012). Nevertheless, more evidence is needed to confirm whether such long‐term mitochondrial adaptations are also evident in a well‐trained population.
In conclusion, the present study demonstrates that, for a given low‐volume of exercise, a high degree of exercise‐induced metabolic stress is associated with a greater PGC‐1α mRNA response. The exercise‐induced mRNA responses of proteins implicated in mtDNA transcription and mitochondrial remodelling dynamics seem to follow the same pattern delineated for PGC‐1α mRNA. In addition, mRNA responses related to mitochondrial turnover are induced by both low‐volume supramaximal‐intensity intermittent and high‐volume continuous moderate‐intensity exercise in skeletal muscle from well‐trained individuals, but increased intensity appears to compensate for reduced volume only when the intense exercise promotes remarkably pronounced alterations in the intracellular metabolic milieu, with a substantial activation of the CaMKII and p38 MAPK signalling pathway. Together these findings suggest a metabolic stress‐mediated regulation of the initial events that promote the development and maintenance of a high‐quality mitochondrial pool in human skeletal muscle.
Additional information
Competing interests
None declared.
Author contributions
M.F. designed the study, conducted the human experiments, participated in data collection, analysis and interpretation, and drafted the manuscript. T.P.G. participated in designing the study, as well as in data collection and interpretation, and in revising the manuscript. M.H. participated in data collection, analysis and interpretation, as well as in revising the manuscript. F.M.I. and F.S. participated in designing the study, as well as in revising the manuscript. H.P. participated in data analysis and interpretation, as well as in revising the manuscript. J.B. participated in designing the study, and drafted the manuscript. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed. All experiments were carried out at the Department of Nutrition, Exercise and Sports at the University of Copenhagen, Denmark.
Funding
The study was supported by a grant from the Danish Ministry of Culture and Team Denmark.
Acknowledgements
We express our gratitude for the technical assistance of Jens Jung Nielsen, Thomas Svare Ehlers and Yusuke Shirai throughout the study in the collection and analysis of data.
Biography
Matteo Fiorenza is a PhD student in exercise physiology at the Department of Nutrition, Exercise and Sports (NEXS), University of Copenhagen, Denmark and at the Department of Neurosciences, Biomedicine and Movement Sciences, University of Verona, Italy. His current research focuses on molecular, metabolic and physiological mechanisms underlying adaptations to intense exercise. His research interests include skeletal muscle physiology and exercise performance, with particular emphasis on the effects of exercise training on mitochondrial physiology in healthy and clinical populations.

Edited by: Scott Powers & Bruno Grassi
References
- Baar K, Wende AR, Jones TE, Marison M, Nolte LA, Chen M, Kelly DP & Holloszy JO (2002). Adaptations of skeletal muscle to exercise: rapid increase in the transcriptional coactivator PGC‐1. FASEB J 16, 1879–1886. [DOI] [PubMed] [Google Scholar]
- Backs J, Backs T, Bezprozvannaya S, McKinsey TA & Olson EN (2008). Histone deacetylase 5 acquires calcium/calmodulin‐dependent kinase II responsiveness by oligomerization with histone deacetylase 4. Mol Cell Biol 28, 3437–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballmann C, McGinnis G, Peters B, Slivka D, Cuddy J, Hailes W, Dumke C, Ruby B & Quindry J (2014). Exercise‐induced oxidative stress and hypoxic exercise recovery. Eur J Appl Physiol 114, 725–733. [DOI] [PubMed] [Google Scholar]
- Balsom PD, Seger JY, Sjodin B & Ekblom B (1992). Physiological responses to maximal intensity intermittent exercise. Eur J Appl Physiol Occup Physiol 65, 144–149. [DOI] [PubMed] [Google Scholar]
- Bartlett JD, Hwa Joo C, Jeong TS, Louhelainen J, Cochran AJ, Gibala MJ, Gregson W, Close GL, Drust B & Morton JP (2012). Matched work high‐intensity interval and continuous running induce similar increases in PGC‐1xsα mRNA, AMPK, p38, and p53 phosphorylation in human skeletal muscle. J Appl Physiol (1985) 112, 1135–1143. [DOI] [PubMed] [Google Scholar]
- Bartlett JD, Louhelainen J, Iqbal Z, Cochran AJ, Gibala MJ, Gregson W, Close GL, Drust B & Morton JP (2013). Reduced carbohydrate availability enhances exercise‐induced p53 signaling in human skeletal muscle: implications for mitochondrial biogenesis. Am J Physiol Regul Integr Comp Physiol 304, R450–R458. [DOI] [PubMed] [Google Scholar]
- Baylor SM & Hollingworth S (2003). Sarcoplasmic reticulum calcium release compared in slow‐twitch and fast‐twitch fibres of mouse muscle. J Physiol 551, 125–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop D, Girard O & Mendez‐Villanueva A (2011). Repeated‐sprint ability – part II: recommendations for training. Sports Med 41, 741–756. [DOI] [PubMed] [Google Scholar]
- Bogdanis GC, Nevill ME, Boobis LH & Lakomy HK (1996). Contribution of phosphocreatine and aerobic metabolism to energy supply during repeated sprint exercise. J Appl Physiol (1985) 80, 876–884. [DOI] [PubMed] [Google Scholar]
- Bogdanis GC, Nevill ME, Lakomy HK & Boobis LH (1998). Power output and muscle metabolism during and following recovery from 10 and 20 s of maximal sprint exercise in humans. Acta Physiol Scand 163, 261–272. [DOI] [PubMed] [Google Scholar]
- Booth FW & Neufer PD (2005). Exercise controls gene expression. Am Sci 93, 28–35. [Google Scholar]
- Brandt N, Gunnarsson TP, Hostrup M, Tybirk J, Nybo L, Pilegaard H & Bangsbo J (2016). Impact of adrenaline and metabolic stress on exercise‐induced intracellular signaling and PGC‐1α mRNA response in human skeletal muscle. Physiol Rep 4, e12844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgomaster KA, Hughes SC, Heigenhauser GJF, Bradwell SN & Gibala MJ (2005). Six sessions of sprint interval training increases muscle oxidative potential and cycle endurance capacity in humans. J Appl Physiol (1985) 98, 1985–1990. [DOI] [PubMed] [Google Scholar]
- Calvo JA, Daniels TG, Wang X, Paul A, Lin J, Spiegelman BM, Stevenson SC & Rangwala SM (2008). Muscle‐specific expression of PPARγ coactivator‐1α improves exercise performance and increases peak oxygen uptake. J Appl Physiol (1985) 104, 1304–1312. [DOI] [PubMed] [Google Scholar]
- Cartoni R, Leger B, Hock MB, Praz M, Crettenand A, Pich S, Ziltener JL, Luthi F, Deriaz O, Zorzano A, Gobelet C, Kralli A & Russell AP (2005). Mitofusins 1/2 and ERRα expression are increased in human skeletal muscle after physical exercise. J Physiol 567, 349–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZP, Stephens TJ, Murthy S, Canny BJ, Hargreaves M, Witters LA, Kemp BE & McConell GK (2003). Effect of exercise intensity on skeletal muscle AMPK signaling in humans. Diabetes 52, 2205–2212. [DOI] [PubMed] [Google Scholar]
- Chinsomboon J, Ruas J, Gupta RK, Thom R, Shoag J, Rowe GC, Sawada N, Raghuram S & Arany Z (2009). The transcriptional coactivator PGC‐1α mediates exercise‐induced angiogenesis in skeletal muscle. Proc Natl Acad Sci U S A 106, 21401–21406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen D, Murphy RM, Bangsbo J, Stathis CG & Bishop DJ (2018). Increased FXYD1 and PGC‐1α mRNA after blood flow‐restricted running is related to fibre type‐specific AMPK signalling and oxidative stress in human muscle. Acta Physiol (Oxf) 223, e13045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran AJ, Percival ME, Tricarico S, Little JP, Cermak N, Gillen JB, Tarnopolsky MA & Gibala MJ (2014). Intermittent and continuous high‐intensity exercise training induce similar acute but different chronic muscle adaptations. Exp Physiol 99, 782–791. [DOI] [PubMed] [Google Scholar]
- Combes A, Dekerle J, Webborn N, Watt P, Bougault V & Daussin FN (2015). Exercise‐induced metabolic fluctuations influence AMPK, p38‐MAPK and CaMKII phosphorylation in human skeletal muscle. Physiol Rep 3, e12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H, Jiang N, Liu H, Liu X, Liu D, Zhao F, Wen L, Liu S, Ji LL & Zhang Y (2010). Response of mitochondrial fusion and fission protein gene expression to exercise in rat skeletal muscle. Biochim Biophys Acta 1800, 250–256. [DOI] [PubMed] [Google Scholar]
- Dudley GA, Tullson PC & Terjung RL (1987). Influence of mitochondrial content on the sensitivity of respiratory control. J Biol Chem 262, 9109–9114. [PubMed] [Google Scholar]
- Edge J, Mundel T, Pilegaard H, Hawke E, Leikis M, Lopez‐Villalobos N, Oliveira RS & Bishop DJ (2015). Ammonium chloride ingestion attenuates exercise‐induced mRNA levels in human muscle. PLoS One 10, e0141317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan B, Carson BP, Garcia‐Roves PM, Chibalin AV, Sarsfield FM, Barron N, McCaffrey N, Moyna NM, Zierath JR & O'Gorman DJ (2010). Exercise intensity‐dependent regulation of peroxisome proliferator‐activated receptor coactivator‐1 mRNA abundance is associated with differential activation of upstream signalling kinases in human skeletal muscle. J Physiol 588, 1779–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaitanos GC, Williams C, Boobis LH & Brooks S (1993). Human muscle metabolism during intermittent maximal exercise. J Appl Physiol (1985) 75, 712–719. [DOI] [PubMed] [Google Scholar]
- Geng T, Li P, Okutsu M, Yin X, Kwek J, Zhang M & Yan Z (2010). PGC‐1α plays a functional role in exercise‐induced mitochondrial biogenesis and angiogenesis but not fiber‐type transformation in mouse skeletal muscle. Am J Physiol Cell Physiol 298, C572–C579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibala MJ, Little JP, van Essen M, Wilkin GP, Burgomaster KA, Safdar A, Raha S & Tarnopolsky MA (2006). Short‐term sprint interval versus traditional endurance training: similar initial adaptations in human skeletal muscle and exercise performance. J Physiol 575, 901–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibala MJ, McGee SL, Garnham AP, Howlett KF, Snow RJ & Hargreaves M (2009). Brief intense interval exercise activates AMPK and p38 MAPK signaling and increases the expression of PGC‐1α in human skeletal muscle. J Appl Physiol (1985) 106, 929–934. [DOI] [PubMed] [Google Scholar]
- Gomez‐Cabrera MC, Borras C, Pallardo FV, Sastre J, Ji LL & Vina J (2005). Decreasing xanthine oxidase‐mediated oxidative stress prevents useful cellular adaptations to exercise in rats. J Physiol 567, 113–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granata C, Oliveira RS, Little JP, Renner K & Bishop DJ (2017). Sprint‐interval but not continuous exercise increases PGC‐1α protein content and p53 phosphorylation in nuclear fractions of human skeletal muscle. Sci Rep 7, 44227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T, Hussien R, Oommen S, Gohil K & Brooks GA (2007). Lactate sensitive transcription factor network in L6 cells: activation of MCT1 and mitochondrial biogenesis. FASEB J 21, 2602–2612. [DOI] [PubMed] [Google Scholar]
- Hesselink MK, Schrauwen‐Hinderling V & Schrauwen P (2016). Skeletal muscle mitochondria as a target to prevent or treat type 2 diabetes mellitus. Nat Rev Endocrinol 12, 633–645. [DOI] [PubMed] [Google Scholar]
- Holloszy JO & Coyle EF (1984). Adaptations of skeletal muscle to endurance exercise and their metabolic consequences. J Appl Physiol Respir Environ Exerc Physiol 56, 831–838. [DOI] [PubMed] [Google Scholar]
- Hood DA, Tryon LD, Carter HN, Kim Y & Chen CC (2016). Unravelling the mechanisms regulating muscle mitochondrial biogenesis. Biochem J 473, 2295–2314. [DOI] [PubMed] [Google Scholar]
- Hood DA, Tryon LD, Vainshtein A, Memme J, Chen C, Pauly M, Crilly MJ & Carter H (2015). Exercise and the regulation of mitochondrial turnover. Prog Mol Biol Transl Sci 135, 99–127. [DOI] [PubMed] [Google Scholar]
- Hood DA, Uguccioni G, Vainshtein A & D'Souza D ( 2011). Mechanisms of exercise‐induced mitochondrial biogenesis in skeletal muscle: implications for health and disease. Compr Physiol 1, 1119–1134. [DOI] [PubMed] [Google Scholar]
- Hostrup M & Bangsbo J (2017). Limitations in intense exercise performance of athletes – effect of speed endurance training on ion handling and fatigue development. J Physiol 595, 2897–2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hostrup M, Onslev J, Jacobson GA, Wilson R & Bangsbo J (2018). Chronic β2‐adrenoceptor agonist treatment alters muscle proteome and functional adaptations induced by high intensity training in young men. J Physiol 596, 231–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iaia FM & Bangsbo J (2010). Speed endurance training is a powerful stimulus for physiological adaptations and performance improvements of athletes. Scand J Med Sci Sports 20, Suppl. 2, 11–23. [DOI] [PubMed] [Google Scholar]
- Irrcher I, Ljubicic V & Hood DA (2009). Interactions between ROS and AMP kinase activity in the regulation of PGC‐1α transcription in skeletal muscle cells. Am J Physiol Cell Physiol 296, C116–C123. [DOI] [PubMed] [Google Scholar]
- Jager S, Handschin C, St‐Pierre J & Spiegelman BM (2007). AMP‐activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC‐1α . Proc Natl Acad Sci U S A 104, 12017–12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang C, O'Moore KM, Dickman JR & Ji LL (2009). Exercise activation of muscle peroxisome proliferator‐activated receptor‐γ coactivator‐1α signaling is redox sensitive. Free Radic Biol Med 47, 1394–1400. [DOI] [PubMed] [Google Scholar]
- Kelly DP & Scarpulla RC (2004). Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev 18, 357–368. [DOI] [PubMed] [Google Scholar]
- Kitaoka Y, Takeda K, Tamura Y & Hatta H (2016). Lactate administration increases mRNA expression of PGC‐1α and UCP3 in mouse skeletal muscle. Appl Physiol Nutr Metab 41, 695–698. [DOI] [PubMed] [Google Scholar]
- Laursen PB & Jenkins DG (2002). The scientific basis for high‐intensity interval training: optimising training programmes and maximising performance in highly trained endurance athletes. Sports Med 32, 53–73. [DOI] [PubMed] [Google Scholar]
- Leick L, Wojtaszewski JF, Johansen ST, Kiilerich K, Comes G, Hellsten Y, Hidalgo J & Pilegaard H (2008). PGC‐1α is not mandatory for exercise‐ and training‐induced adaptive gene responses in mouse skeletal muscle. Am J Physiol Endocrinol Metab 294, E463–E474. [DOI] [PubMed] [Google Scholar]
- Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN, Lowell BB, Bassel‐Duby R & Spiegelman BM (2002). Transcriptional co‐activator PGC‐1α drives the formation of slow‐twitch muscle fibres. Nature 418, 797–801. [DOI] [PubMed] [Google Scholar]
- Little JP, Safdar A, Bishop D, Tarnopolsky MA & Gibala MJ (2011). An acute bout of high‐intensity interval training increases the nuclear abundance of PGC‐1α and activates mitochondrial biogenesis in human skeletal muscle. Am J Physiol Regul Integr Comp Physiol 300, R1303–R1310. [DOI] [PubMed] [Google Scholar]
- Little JP, Safdar A, Wilkin GP, Tarnopolsky MA & Gibala MJ (2010). A practical model of low‐volume high‐intensity interval training induces mitochondrial biogenesis in human skeletal muscle: potential mechanisms. J Physiol 588, 1011–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry OH & Passonneau JV (1972). A Flexible System of Enzymatic Analysis. Academic Press, New York. [Google Scholar]
- Lundby C & Jacobs RA (2016). Adaptations of skeletal muscle mitochondria to exercise training. Exp Physiol 101, 17–22. [DOI] [PubMed] [Google Scholar]
- MacDougall JD, Hicks AL, MacDonald JR, McKelvie RS, Green HJ & Smith KM (1998). Muscle performance and enzymatic adaptations to sprint interval training. J Appl Physiol (1985) 84, 2138–2142. [DOI] [PubMed] [Google Scholar]
- MacInnis MJ & Gibala MJ (2017). Physiological adaptations to interval training and the role of exercise intensity. J Physiol 595, 2915–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BF, Konopka AR & Hamilton KL (2016). The rigorous study of exercise adaptations: why mRNA might not be enough. J Appl Physiol (1985) 121, 594–596. [DOI] [PubMed] [Google Scholar]
- Miura S, Kawanaka K, Kai Y, Tamura M, Goto M, Shiuchi T, Minokoshi Y & Ezaki O (2007). An increase in murine skeletal muscle peroxisome proliferator‐activated receptor‐γ coactivator‐1α (PGC‐1α) mRNA in response to exercise is mediated by β‐adrenergic receptor activation. Endocrinology 148, 3441–3448. [DOI] [PubMed] [Google Scholar]
- Morales‐Alamo D, Ponce‐Gonzalez JG, Guadalupe‐Grau A, Rodriguez‐Garcia L, Santana A, Cusso R, Guerrero M, Dorado C, Guerra B & Calbet JA (2013). Critical role for free radicals on sprint exercise‐induced CaMKII and AMPKα phosphorylation in human skeletal muscle. J Appl Physiol (1985) 114, 566–577. [DOI] [PubMed] [Google Scholar]
- Morton JP, Kayani AC, McArdle A & Drust B (2009). The exercise‐induced stress response of skeletal muscle, with specific emphasis on humans. Sports Med 39, 643–662. [DOI] [PubMed] [Google Scholar]
- Nordsborg NB, Lundby C, Leick L & Pilegaard H (2010). Relative workload determines exercise‐induced increases in PGC‐1α mRNA. Med Sci Sports Exerc 42, 1477–1484. [DOI] [PubMed] [Google Scholar]
- Norrbom J, Sundberg CJ, Ameln H, Kraus WE, Jansson E & Gustafsson T (2004). PGC‐1α mRNA expression is influenced by metabolic perturbation in exercising human skeletal muscle. J Appl Physiol (1985) 96, 189–194. [DOI] [PubMed] [Google Scholar]
- Perry CG, Lally J, Holloway GP, Heigenhauser GJ, Bonen A & Spriet LL (2010). Repeated transient mRNA bursts precede increases in transcriptional and mitochondrial proteins during training in human skeletal muscle. J Physiol 588, 4795–4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilegaard H, Ordway GA, Saltin B & Neufer PD (2000). Transcriptional regulation of gene expression in human skeletal muscle during recovery from exercise. Am J Physiol Endocrinol Metab 279, E806–E814. [DOI] [PubMed] [Google Scholar]
- Pilegaard H, Osada T, Andersen LT, Helge JW, Saltin B & Neufer PD (2005). Substrate availability and transcriptional regulation of metabolic genes in human skeletal muscle during recovery from exercise. Metabolism 54, 1048–1055. [DOI] [PubMed] [Google Scholar]
- Pilegaard H, Saltin B & Neufer PD (2003). Exercise induces transient transcriptional activation of the PGC‐1α gene in human skeletal muscle. J Physiol 546, 851–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Place N, Ivarsson N, Venckunas T, Neyroud D, Brazaitis M, Cheng AJ, Ochala J, Kamandulis S, Girard S, Volungevicius G, Pauzas H, Mekideche A, Kayser B, Martinez‐Redondo V, Ruas JL, Bruton J, Truffert A, Lanner JT, Skurvydas A & Westerblad H (2015). Ryanodine receptor fragmentation and sarcoplasmic reticulum Ca2+ leak after one session of high‐intensity interval exercise. Proc Natl Acad Sci U S A 112, 15492–15497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psilander N, Frank P, Flockhart M & Sahlin K (2013). Exercise with low glycogen increases PGC‐1α gene expression in human skeletal muscle. Eur J Appl Physiol 113, 951–963. [DOI] [PubMed] [Google Scholar]
- Psilander N, Wang L, Westergren J, Tonkonogi M & Sahlin K (2010). Mitochondrial gene expression in elite cyclists: effects of high‐intensity interval exercise. Eur J Appl Physiol 110, 597–606. [DOI] [PubMed] [Google Scholar]
- Puigserver P, Rhee J, Lin J, Wu Z, Yoon JC, Zhang CY, Krauss S, Mootha VK, Lowell BB & Spiegelman BM (2001). Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARγ coactivator‐1. Mol Cell 8, 971–982. [DOI] [PubMed] [Google Scholar]
- Rose AJ, Broholm C, Kiillerich K, Finn SG, Proud CG, Rider MH, Richter EA & Kiens B (2005). Exercise rapidly increases eukaryotic elongation factor 2 phosphorylation in skeletal muscle of men. J Physiol 569, 223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose AJ & Hargreaves M (2003). Exercise increases Ca2+‐calmodulin‐dependent protein kinase II activity in human skeletal muscle. J Physiol 553, 303–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose AJ, Kiens B & Richter EA (2006). Ca2+‐calmodulin‐dependent protein kinase expression and signalling in skeletal muscle during exercise. J Physiol 574, 889–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell AP, Foletta VC, Snow RJ & Wadley GD (2014). Skeletal muscle mitochondria: a major player in exercise, health and disease. Biochim Biophys Acta 1840, 1276–1284. [DOI] [PubMed] [Google Scholar]
- Saleem A & Hood DA (2013). Acute exercise induces tumour suppressor protein p53 translocation to the mitochondria and promotes a p53‐Tfam‐mitochondrial DNA complex in skeletal muscle. J Physiol 591, 3625–3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scribbans TD, Edgett BA, Bonafiglia JT, Baechler BL, Quadrilatero J & Gurd BJ (2017). A systematic upregulation of nuclear and mitochondrial genes is not present in the initial postexercise recovery period in human skeletal muscle. Appl Physiol Nutr Metab 42, 571–578. [DOI] [PubMed] [Google Scholar]
- Serpiello FR, McKenna MJ, Bishop DJ, Aughey RJ, Caldow MK, Cameron‐Smith D & Stepto NK (2012). Repeated sprints alter signaling related to mitochondrial biogenesis in humans. Med Sci Sports Exerc 44, 827–834. [DOI] [PubMed] [Google Scholar]
- Skovgaard C, Brandt N, Pilegaard H & Bangsbo J (2016). Combined speed endurance and endurance exercise amplify the exercise‐induced PGC‐1α and PDK4 mRNA response in trained human muscle. Physiol Rep 4, e12864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer M, Bishop D, Dawson B & Goodman C (2005). Physiological and metabolic responses of repeated‐sprint activities: specific to field‐based team sports. Sports Med 35, 1025–1044. [DOI] [PubMed] [Google Scholar]
- Stepto NK, Benziane B, Wadley GD, Chibalin AV, Canny BJ, Eynon N & McConell GK (2012). Short‐term intensified cycle training alters acute and chronic responses of PGC1α and Cytochrome C oxidase IV to exercise in human skeletal muscle. PLoS One 7, e53080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor CW, Ingham SA, Hunt JE, Martin NR, Pringle JS & Ferguson RA (2016). Exercise duration‐matched interval and continuous sprint cycling induce similar increases in AMPK phosphorylation, PGC‐1α and VEGF mRNA expression in trained individuals. Eur J Appl Physiol 116, 1445–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainshtein A, Tryon LD, Pauly M & Hood DA (2015). Role of PGC‐1α during acute exercise‐induced autophagy and mitophagy in skeletal muscle. Am J Physiol Cell Physiol 308, C710–C719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virbasius JV & Scarpulla RC (1994). Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: a potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc Natl Acad Sci U S A 91, 1309–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RS & Neufer PD (1996). Regulation of gene expression in skeletal muscle by contractile activity In Handbook of Physiology, Exercise: Regulation and Integration of Multiple Systems, pp. 1124–1150. American Physiological Society, Bethesda, MD, USA. [Google Scholar]
- Wojtaszewski JF, Nielsen P, Hansen BF, Richter EA & Kiens B (2000). Isoform‐specific and exercise intensity‐dependent activation of 5'‐AMP‐activated protein kinase in human skeletal muscle. J Physiol 528, 221–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright DC, Geiger PC, Han DH, Jones TE & Holloszy JO (2007). Calcium induces increases in peroxisome proliferator‐activated receptor γ coactivator‐1α and mitochondrial biogenesis by a pathway leading to p38 mitogen‐activated protein kinase activation. J Biol Chem 282, 18793–18799. [DOI] [PubMed] [Google Scholar]
- Yan Z, Lira VA & Greene NP (2012). Exercise training‐induced regulation of mitochondrial quality. Exerc Sport Sci Rev 40, 159–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ & van der Bliek AM (2012). Mitochondrial fission, fusion, and stress. Science 337, 1062–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zechner C, Lai L, Zechner JF, Geng T, Yan Z, Rumsey JW, Collia D, Chen Z, Wozniak DF, Leone TC & Kelly DP (2010). Total skeletal muscle PGC‐1 deficiency uncouples mitochondrial derangements from fiber type determination and insulin sensitivity. Cell Metab 12, 633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Uguccioni G, Ljubicic V, Irrcher I, Iqbal S, Singh K, Ding S & Hood DA (2014). Multiple signaling pathways regulate contractile activity‐mediated PGC‐1α gene expression and activity in skeletal muscle cells. Physiol Rep 2, e12008. [DOI] [PMC free article] [PubMed] [Google Scholar]