

Abstract
Key points
Chronic obstructive pulmonary disease (COPD) is largely caused by smoking, and patient limb muscle exhibits a fast fibre shift and atrophy.
We show that this fast fibre shift is associated with type grouping, suggesting recurring cycles of denervation–reinnervation underlie the type shift.
Compared to patients with normal fat‐free mass index (FFMI), patients with low FFMI exhibited an exacerbated fibre type shift, marked accumulation of very small persistently denervated muscle fibres, and a blunted denervation‐responsive transcript profile, suggesting failed denervation precipitates muscle atrophy in patients with low FFMI.
Sixteen weeks of passive tobacco smoke exposure in mice caused neuromuscular junction degeneration, consistent with a key role for smoke exposure in initiating denervation in COPD.
Abstract
A neurological basis for the fast fibre shift and atrophy seen in limb muscle of patients with chronic obstructive pulmonary disease (COPD) has not been considered previously. The objective of our study was: (1) to determine if denervation contributes to fast fibre shift and muscle atrophy in COPD; and (2) to assess using a preclinical smoking mouse model whether chronic tobacco smoke (TS) exposure could initiate denervation by causing neuromuscular junction (NMJ) degeneration. Vastus lateralis muscle biopsies were obtained from severe COPD patients [n = 10 with low fat‐free mass index (FFMI), 65 years; n = 15 normal FFMI, 65 years) and healthy age‐ and activity‐matched non‐smoker control subjects (CON; n = 11, 67 years), to evaluate morphological and transcriptional markers of denervation. To evaluate the potential for chronic TS exposure to initiate these changes, we examined NMJ morphology in male adult mice following 16 weeks of passive TS exposure. We observed a high proportion of grouped fast fibres and a denervation transcript profile in COPD patients, suggesting that motor unit remodelling drives the fast fibre type shift in COPD patient limb muscle. A further exacerbation of fast fibre grouping in patients with low FFMI, coupled with blunted reinnervation signals, accumulation of very small non‐specific esterase hyperactive fibres and neural cell adhesion molecule‐positive type I and type II fibres, suggests denervation‐induced exhaustion of reinnervation contributes to muscle atrophy in COPD. Evidence from a smoking mouse model showed significant NMJ degeneration, suggesting that recurring denervation in COPD is probably caused by decades of chronic TS exposure.
Keywords: denervation, cachexia, sarcopenia, muscle atrophy, smoking mouse
Key points
Chronic obstructive pulmonary disease (COPD) is largely caused by smoking, and patient limb muscle exhibits a fast fibre shift and atrophy.
We show that this fast fibre shift is associated with type grouping, suggesting recurring cycles of denervation–reinnervation underlie the type shift.
Compared to patients with normal fat‐free mass index (FFMI), patients with low FFMI exhibited an exacerbated fibre type shift, marked accumulation of very small persistently denervated muscle fibres, and a blunted denervation‐responsive transcript profile, suggesting failed denervation precipitates muscle atrophy in patients with low FFMI.
Sixteen weeks of passive tobacco smoke exposure in mice caused neuromuscular junction degeneration, consistent with a key role for smoke exposure in initiating denervation in COPD.
Introduction
Chronic tobacco smoke (TS) exposure is the leading cause of preventable disease in the USA, including cancer, cardiovascular disease and chronic obstructive pulmonary disease (COPD) (Warren et al. 2014). Not only are smokers at risk, but this also affects non‐smokers exposed to second‐hand smoke (Warren et al. 2014). Indeed, worldwide the economic burden associated with smoking is nearly 2% of the global gross domestic product (Goodchild et al. 2017), demonstrating the continuing widespread impact of the so‐called tobacco epidemic. The aforementioned TS‐related diseases are each associated with skeletal muscle deterioration that worsens clinical outcomes, including increasing the risk of death (Marquis et al. 2002; Swallow et al. 2007; Jones et al. 2015). Furthermore, there is a large body of evidence showing that TS directly affects skeletal muscle in a dose‐dependent manner (Degens et al. 2015). Finally, many of the muscle changes seen in different TS‐related diseases are similar (e.g. fast fibre type shift, erosion of oxidative capacity, muscle atrophy) (Kitzman et al. 2014; Maltais et al. 2014; Toth et al. 2016) and are also seen in smokers who are apparently free of disease (Orlander et al. 1979; van den Borst et al. 2011).
In this respect, several lines of evidence indirectly support the idea that neuromuscular junction impact with chronic TS exposure probably underlies the muscle affect. Firstly, chronic TS exposure is an established risk factor for exacerbating muscle deterioration in conditions where neuromuscular junction degeneration is well known, including normal ageing and amyotrophic lateral sclerosis (ALS) (van den Borst et al. 2011; de Jong et al. 2012; Hepple & Rice, 2016). Secondly, the TS‐induced fast fibre shift is irreversible following smoking cessation (Larsson & Orlander, 1984), as it would be if the fibre type shift is due to recurring cycles of denervation–reinnervation (motor unit remodelling) driven by TS‐induced neuromuscular junction degeneration. Thirdly, patients with the TS‐related disease COPD exhibit an accumulation of small angular fibres and myosin heavy chain co‐expression in limb muscle (Gosker et al. 2002), features that are well known in conditions associated with neuromuscular junction degeneration such as ALS (Baloh et al. 2007) and normal ageing (Hepple & Rice, 2016). However, there are as yet no data addressing the impact of chronic TS exposure on the neuromuscular junction, and no study has assessed TS‐related disease patient muscle for evidence of recurring cycles of denervation–reinnervation and the potential contribution of persistent muscle fibre denervation to muscle atrophy. The current study aims to address these gaps by first presenting an in‐depth histological analysis of muscle motor unit remodelling and pairing this with targeted analysis of transcripts related to denervation and reinnervation in patients with one of the most devastating smoking‐related diseases, COPD; and second, by using a smoking mouse model to facilitate direct investigation of the impact of chronic TS exposure on neuromuscular junction morphology.
We hypothesized that COPD patients would exhibit motor unit remodelling consistent with recurring cycles of denervation/reinnervation, an accumulation of persistently denervated fibres and perturbation of denervation‐responsive transcripts. We further hypothesized that these changes would be exacerbated in patients with low muscle mass based upon fat‐free mass index (FFMI). Finally, as chronic cigarette TS exposure is the primary cause of COPD and causes a fast fibre shift independent of pulmonary disease (Orlander et al. 1979; Larsson & Orlander, 1984), we hypothesized that TS would induce neuromuscular junction degeneration. The significance of validating these hypotheses is that it would implicate denervation as a probable player involved in driving the fast fibre shift and muscle atrophy in COPD. It would also implicate TS‐induced neuromuscular junction degeneration as an important initiating event causing denervation in COPD muscle.
Methods
Ethical approval
For all human procedures, this study was approved by the Institutional Review Board for human studies of the Montreal Chest Institute (Montréal, QC, Canada) and all participants provided written informed consent, in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments, except for registration in a database. All procedures involving mice were approved by the McGill University Animal Care Committee (protocol no. 5933 to C. Baglole) and adhered to regulations of the Canadian Council on Animal Care.
Study participants
Twenty‐five ambulatory male patients with severe to very‐severe COPD were recruited from the outpatient clinic of the Montreal Chest Hospital of the McGill University Health Centre. COPD disease severity was based on the Global Initiative for Chronic Obstructive Lung Disease classification, i.e. GOLD 3 and 4. Study participants were excluded if they: (1) reported an exacerbation requiring the use of corticosteroids or antibiotics within the preceding month; (2) were on long‐term oxygen therapy; (3) used oral corticosteroids; (4) participated in a pulmonary rehabilitation programme within the preceding year; or (5) had a known comorbidity that could interfere with outcome measures, including severe cardiac, diabetes, neurological, neuromuscular, cancer and/or orthopaedic disease/condition. COPD patients were subsequently subdivided into those patients with normal muscle mass and those with low muscle mass, as detailed below. All patients who responded to the question during screening (three patients abstained), were smokers or former smokers. Age‐matched (n = 11) non‐smoking control subjects were recruited from the Montreal community. All participants underwent, on two separate days, a clinical and physiological evaluation followed by a muscle needle biopsy.
Study procedures
Clinical and physiological evaluation
Anthropometric measurements were obtained followed by pulmonary function testing to assess the degree of airflow limitations and obstruction (Medisoft) according to The American Thoracic Society (ATS) guidelines (Wanger et al. 2005). Dual‐energy X‐ray absorptiometry (DEXA) was performed to measure body composition and lean and fat mass using the GE Lunar iDXA scanner (General Electric Healthcare, Piscataway, NJ, USA). Low muscle mass was defined as an FFMI (= fat‐free mass in kg/height in m2) less than the 25th percentile of the general population as per Schutz et al. (2002). The partial pressures of oxygen and carbon dioxide ( and ) at rest were obtained from an arterialized blood sample from a pre‐warmed earlobe as per Mollard et al. (2010). Thereafter, a symptom‐limited incremental cardiopulmonary cycle exercise test following ATS/American College of Chest Physicians (ACCP) guidelines (http://American Thoracic Society; http://American College of Chest Physicians, 2003) was conducted to measure peak work capacity. After a 1‐h rest period, peak isokinetic torque of the right quadriceps muscle was measured using a quantitative dynamometer (Biodex System 4 Pro, Biodex Medical Systems, New York, NY, USA), with subjects performing five sequential volitional maximal contractions over a range of motion from 15° to 100° at a velocity of 60° s−1, with the highest value used in analysis.
Muscle biopsy
The mid‐portion of the vastus lateralis muscle was sampled using the modified Bergstrom needle method with suction (Shanely et al. 2014). Approximately 40 mg portions were mounted in transverse orientation using triganth gum and frozen in liquid nitrogen‐cooled isopentane for in situ labelling, and 30 mg portions were fast frozen in liquid nitrogen for mRNA analysis, with both samples stored at −80°C until the time of analysis.
Quantification of gene transcript levels
Total RNA was extracted from human muscle samples according to manufacturer's instructions using the RNeasy Lipid Tissue Mini Kit (Qiagen Cat. No.74804, , Valencia, CA, USA). RNA concentration and purity (A 260/A 280 ratios >1.8) were assessed using a NanoDrop‐2000 spectrophotometer (Thermo Scientific, Waltham, MA, USA). RNA (1 μg) was reverse transcribed to cDNA using a qScript cDNA Synthesis Kit (Quanta biosciences Cat. No. 95047‐025, Beverly, CA, USA), according to the manufacturer's instructions. Real‐time PCR was performed using a StepOnePlus Real‐Time PCR system (Life Technologies, Carlsbad, CA, USA). To gain insights into denervation‐ and reinnervation‐related transcriptional responses, we examined mRNA levels of Agrin, Musk, Lrp4, rapsyn, FGFBP1 and the AChR subunits α, β, δ,γ and ε (Table 1). Primers were designed (Table 1) with a freely available software program (Primer 3 plus, Thermo Scientific), and Power SYBR Green PCR Master Mix (Life Technologies) was used to quantify the mRNA. TATA box binding protein (TBP) was used as an internal control. The cDNA was amplified at 95°C for 10 min followed by 40 cycles of 95°C for 15 s and 55°C for 60 s. All real time PCR experiments were performed in triplicate and melt curve analysis for each PCR experiment was performed to assess primer dimer formation or contamination. The comparative threshold cycle (CT) method was used to calculate fold changes in expression, where ΔCT = CT of gene of interest − CT of TBP. Relative differences in gene expression were determined as 2−ΔΔCT normalized to age‐matched control subjects. Due to limited tissue availability, only a subset of samples was used for mRNA analysis.
Table 1.
mRNA primers used in the study
| Gene name | Sequence | NCBI reference no. |
|---|---|---|
| MuSK |
|
PMID: 17192614 |
| Agrin |
|
PMID: 22379342 |
| Lrp4 |
|
NM_002334.3 |
| Rapsyn |
|
NM_005055.4 |
| FGFBP1 |
|
NM_005130.4 |
| AChRα |
|
NM_000079.3 |
| AChRβ |
|
NM_000747.2 |
| AChRδ |
|
NM_000751.2 |
| AChRγ |
|
NM_005199.4 |
| AChRε |
|
NM_000080.3 |
| TBP |
|
NM_001172085.1 |
PMID: primer sequence retrieved from corresponding publication.
In situ tissue labelling and image analysis
Muscle samples were cryosectioned at −18°C to 8 μm section thickness. Muscle cross‐sections used in fibre typing experiments were hydrated using PBS and blocked against non‐specific labelling using 10% normal goat serum (NGS). A cocktail of primary antibodies, myosin heavy chain (MHC) Type I (BA‐F8, DSHB, University of Iowa), IIa (Sc71, DSHB), IIx (6H1 DSHB) and laminin (L9393, Sigma, St Louis, MO, USA), was applied for 1 h at room temperature, followed by a series of PBS washes and incubation with fluorescence‐conjugated secondary antibodies for 1 h at room temperature. Sections were mounted with Prolong Gold and imaged the following day.
To identify denervated fibres we employed a clinical histological stain used in diagnosing neurogenic atrophies based upon non‐specific esterase activity, where denervated fibres appear as darkly staining cells (Goebel et al. 2013). Muscle cross‐sections were incubated in non‐specific esterase solution (4% sodium nitrite, 0.1 m sodium phosphate buffer, 1% alpha‐napthyl acetate dissolved in acetone) for 30 min at room temperature and then rinsed with tap water for 10 min. Sections were dehydrated in an ethanol series and cleared in xylene (Malicdan et al. 2009). We also assessed neural cell adhesion molecule (NCAM; aka CD56) labelling in tissue cross‐sections, noting that NCAM is a cytokine produced by denervated muscle fibres and motor neurons which comes in three distinct isoforms (180, 140 and 120 kDa) and which is essential for reinnervation and formation of stable motor neuron–muscle contacts (Hata et al. 2018). Briefly, sections were post‐fixed in 4% paraformaldehyde (ChemCruz, Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 20 min at room temperature, washed with 1× TBS, permeabilized with 0.1% Triton‐X for 15 min and washed with 1× TBS‐0.05% Tween 20 (TBST). Sections were blocked with 10% NGS and 1% bovine serum albumin (BSA) for 40 min at room temperature and incubated with mouse anti‐CD56 (aka: NCAM) (IgG1, diluted 1:40; Invitrogen MA5‐11563), with serial sections incubated with monoclonal mouse anti‐MHC I IgG2b (BA‐F8, DSHB) and rabbit anti‐laminin (L9393, Sigma Aldrich) overnight at 4°C. Alexa Fluor 647 (1:100) and Alexa Fluor 488 (1:500) conjugated goat anti‐mouse secondary antibodies were used for visualization.
Image analyses for MHC fibre typing were performed using the open source image analysis software program Fiji on images obtained using a Zeiss Axio Imager M2 upright microscope. Atrophied fibres were classified as having an area ≥ 2 standard deviations below the mean fibre area of age‐matched controls. Fibre shape was classified using a shape factor (4π × area × perimeter−2), where values <0.6 were defined as angular. Muscle fibres surrounded entirely by fibres of their same type were classified as grouped (Lexell & Downham, 1991). Imaging for NCAM immunofluorescence was done with a Leica SP8 confocal microscope with a 20× objective and a pinhole size of 0.81 airy units, and HyVolution deconvolution.
Smoking mouse and neuromuscular junction morphological analyses
To facilitate understanding the direct impact of chronic TS on the neuromuscular junction, we used a smoking mouse model where we could perform detailed neuromuscular junction morphological analyses in the endplate zone of muscle. Male C57BL/6 mice aged 8 months were obtained from our in‐house colony and exposed to either room air or TS for 16 weeks, with TS exposures as described previously (de Souza et al. 2014). Briefly, TS exposures used research cigarettes (3R4F; University of Kentucky, Lexington, KY, USA) and were smoked according to the protocol approved by the Federal Trade Commission (1 puff per minute per cigarette, where each puff was 2 s in duration and 35 ml in volume) in a SCIREQ InExpose Exposure System (SCIREQ, Montreal, QC, Canada). Mice in the TS group were exposed for 1 h, twice per day, 5 days per week for 16 weeks, such that animals were 12 months of age when they were killed.
On the day of terminal experiments mice were killed by CO2 asphyxiation followed by cervical dislocation. The right tibialis anterior muscle from five animals per group (air, TS) was gently removed, and then the deep oxidative region containing a mixture of slow and fast oxidative fibre types was dissected out and subsequently prepared for neuromuscular junction labelling, as done previously (Spendiff et al. 2016). Briefly, dissected muscle portions were washed (3 × 5 min) in PBS and then fixed overnight at 4°C in 2% formaldehyde. The fixed muscle portions were separated into small bundles by gentle dissection using forceps and blocked overnight at 4°C in 5% NGS, 5% BSA, 2% Triton X100 in PBS, and then incubated with mouse anti‐synaptophysin (1:25 dilution; ab8049, Abcam, Cambridge, MA, USA) overnight at 4°C to label the pre‐synpatic motor neuron terminals. Muscle bundles were washed in PBS and incubated overnight at 4°C with AF594‐conjugated goat anti‐mouse secondary antibody and Alexa488‐conjugated α‐bungarotoxin [to identify post‐junctional acetylcholine receptor (AChR) clusters; dilution 1:500, B‐13422, Life Technologies]. Muscle bundles were subsequently mounted on slides with Prolong Gold. Neuromuscular junction image stacks were obtained with a Zeiss LSM880 confocal microscope with a 63× objective, and analysed using Fiji. Neuromuscular junction morphology was characterized for en face endplates based upon the following categories: (1) fraction of fragmented junctions (AChR clusters with > 4 segments); (2) endplate area; (3) fraction of endplate area occupied by AChRs; (4) fraction of endplate area occupied by synaptophysin; and (5) fraction of abandoned endplates (no detectable synaptophysin signal at endplate), as previously described (Spendiff et al. 2016). An average of 46 neuromuscular junctions per animal were analysed (range: 30–59). All analyses were done by a single observer blinded to the identity of the samples.
Data analysis
Results are expressed as means ± SEM. All statistical analysis was performed using GraphPad Prism Software, with significance set at P < 0.05. Comparisons for human data amongst the three groups involving a single parameter, such as proportion of grouped fibres of a given type, average fibre size, proportion of atrophied fibres, proportions of angular fibres and all mRNA data, were made using one‐Way ANOVA. A Tukey post‐hoc test was used for normally distributed data and Kruskal–Wallis with Dunn's multiple comparison test was used for non‐normally distributed data. Comparisons involving fibre type and a single parameter were made using a two‐Way ANOVA with Sidak correction for multiple comparisons. Comparisons between COPD patients with normal versus low FFMI were done using Student's t‐test. Comparisons of neuromuscular junction morphology in air versus TS‐exposed mice were done using Student's t‐test.
Results
Subject characteristics
Descriptive data for our subjects are shown in Table 2. All COPD patients who responded to the question about smoking history were former or current smokers, with no difference in number of pack‐years between subjects with normal versus low FFMI (noting that three COPD patients abstained from answering about their smoking history). BMI, FFMI and mid‐thigh circumference were similar between COPD patients with normal FFMI and controls; however, these were all significantly lower in COPD patients with low FFMI. Although all COPD patients had severe airflow obstruction, those patients with low FFMI had even greater obstruction, lower diffusion capacity and greater residual volume. COPD patients had lower and higher at rest than control subjects but this did not differ between COPD patients based on FFMI. COPD patients had lower peak work capacity than control subjects, and COPD patients with low FFMI had lower peak work capacity than COPD patients with normal FFMI (P = 0.06). COPD patients with low FFMI also tended to have lower isokinetic strength than COPD patients with normal FFMI (P = 0.05). Within the COPD group, only 1 patient within the low FFMI group was on statins, whereas there were 4 patients within the normal FFMI group on both statins and corticosteroids and 3 patients on statins alone.
Table 2.
Subject characteristics
| Variable | Control (n = 11) | COPD normal FFM (n = 15) | COPD low FFM (n = 10) |
|---|---|---|---|
| Age, years | 67 ± 2 | 65 ± 1 | 65 ± 2 |
| BMI, kg m2 | 26 ± 1 | 28 ± 1 | 20 ± 1* , ** |
| FFMI, kg m2 | 18.5 ± 0.4 | 18.5 ± 0.4 | 15.7 ± 0.3* , ** |
| Mid‐thigh circumference, cm | 52 ± 1 | 50 ± 2 | 43 ± 1* , ** |
| Smoking pack years | NS | 46.2 ± 5.4 | 55.6 ± 14.1 |
| FEV1 (litres) | 3.0 ± 0.1 | 1.2 ± 0.1* | 0.7 ± 0.1* |
| FEV1 (% pred) | 101 ± 5 | 42 ± 3* | 29 ± 4* |
| FEV1/FVC (%) | 76 ± 2 | 39 ± 2* | 30 ± 3* , ** |
| D LCO (% pred) | 116 ± 7 | 58 ± 5* | 41 ± 4* , ** |
| RV (% pred) | 120 ± 9 | 171 ± 11 | 207 ± 11* , ** |
| , mmHg | 85 ± 5 | 72 ± 3 | 68 ± 3* |
| , mmHg | 34 ± 1 | 41 ± 1* | 42 ± 1* |
| Peak work, W | 136 ± 12 | 62 ± 7* | 38 ± 9* , ** |
| Peak work, % pred | 79 ± 2 | 36 ± 4* | 22 ± 5* |
| Peak isokinetic torque, Nm | 130 ± 10 | 120 ± 12 | 84 ± 9P = 0.05 vs . other |
BMI = body mass index; FFMI = fat‐free mass index, FEV1 = forced expiratory volume in 1 s; FVC = forced vital capacity; D LCO = diffusing capacity for carbon monoxide; RV = residual volume; = arterial partial pressure of oxygen at rest; = arterial partial pressure of carbon dioxide at rest; NS = non‐smoker; NM = not measured. Values are means ± SEM. *P < 0.05 versus Control, **P < 0.05 versus COPD with normal FFMI.
Muscle fibre type and type‐grouped fibres
Representative images of MHC‐labelled muscle cross‐sections are shown in Fig. 1 A. In calculating fibre type proportions and fibre size and shape, an average of 411 ± 34 fibres were analysed per biopsy (range: 83–955 fibres). Type I fibre proportion was lower in COPD muscle and was accompanied by a proportional increase in fibres co‐expressing two MHC isoforms, particularly those expressing IIa/x (Fig. 1 B). Interestingly, there was a marked increase in fibres completely enclosed by fibres of the same type (so‐called type‐grouped fibres that are indicative of recurring cycles of denervation–reinnervation) in COPD that was restricted to the fast/type II fibres, and this was further exacerbated in the COPD patients with low FFMI (Fig. 1 C).
Figure 1. Muscle fibre type in COPD versus Control.
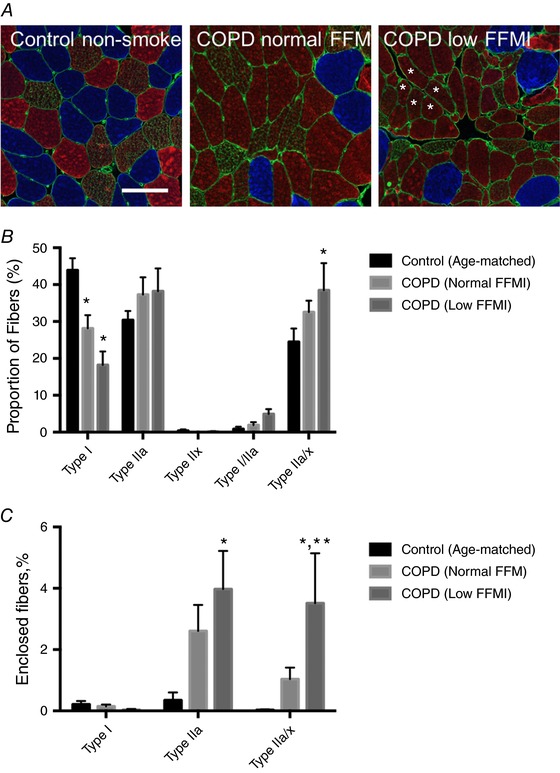
A, representative immunoflorescence images labelled for type I (blue), type IIa (red), type IIx (green) and laminin (green), where grouped fast fibres are indicated (*). Fibres co‐expressing type IIa and IIx were seen in all groups and these appear reddish/green/brown in the images (bar = 100 μm). B, fibre type proportions. C, frequency of fibres completely surrounded by fibres of the same type (Grouped Fibres). *P < 0.05 versus Control; **P < 0.05 versus COPD patients with normal FFMI. [Color figure can be viewed at http://wileyonlinelibrary.com]
Muscle fibre size and shape
Whereas type I fibre size was not different across groups, type IIa and IIa/x fibre size was lower in COPD patients with low FFMI (Fig. 2 A). In interpreting this result it is important to understand that many of the type II fibres in COPD subjects were formerly type I fibres (as reflected by the large shift away from type I fibres seen in Fig. 1 A and B), and thus the lack of change in size of pure type I fibres is misleading with respect to type I atrophy susceptibility, as discussed elsewhere (Purves‐Smith et al. 2012). The fibre size distribution for all fibres revealed a general flattening of the curve in COPD patients (Fig. 2 B), with a striking increase in the number of very small fibres (1495 μm2; a size ≥2 standard deviations below the mean of the age‐matched controls: denoted by the vertical dashed line in Fig. 2 B) in COPD patients with low FFMI (Fig. 2 C). Similarly, there was a large increase in fibres with angular shape [≤0.6; a shape associated with persistent denervation (Baloh et al. 2007; Rowan et al. 2012)] in COPD patients that was exacerbated in those with low FFMI (Control: 1.3 ± 0.4%; COPD normal FFMI: 6.4 ± 1.3%; COPD low FFMI: 18.3 ± 3.4% of fibres).
Figure 2. Muscle morphology in COPD versus Control.
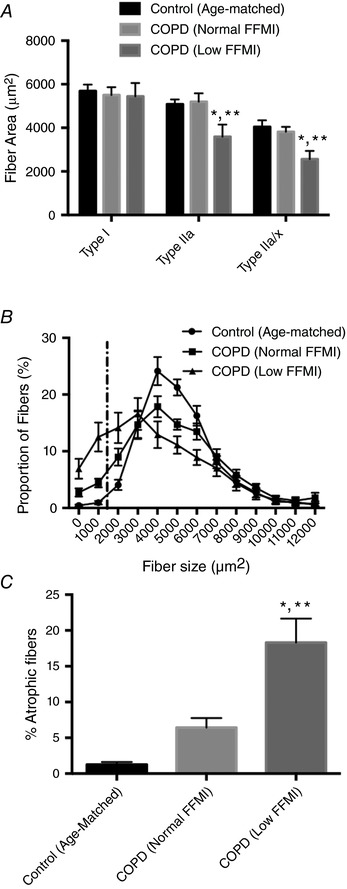
A, mean fibre cross‐sectional area by fibre type. B, fibre size distribution, with dashed vertical line representing fibres that were atrophied, defined as a size ≥2 standard deviations less than the mean for Controls. C, fraction of atrophied fibres in each group. *P < 0.05 versus Control; **P < 0.05 versus COPD patients with normal FFMI.
Morphological and transcriptional markers of denervation
Non‐specific esterase labelling is a clinical histological stain used to identify neurogenic atrophies associated with accumulation of denervated muscle fibres (Goebel et al. 2013). As shown in Fig. 3 A, COPD muscle was characterized by an accumulation of small angular fibres harbouring intense non‐specific esterase staining indicative of persistently denervated muscle fibres, and this was further exacerbated in patients with low FFMI. Further to this, we also examined immunolabelling for NCAM, which is a cytokine produced in neurons and muscle fibres that is key not only to producing stable neuromuscular junctions during development, but is also essential for restoring innervation by promoting axonal sprouting to reinnervate denervated muscle fibres (Walsh et al. 2000; Chipman et al. 2010). As shown in Fig. 3 B, NCAM labelling was greater in COPD patients with low FFMI and affected both type I and type II (unlabelled) fibres, and as such, corroborates the non‐specific esterase assay, indicating that there is an exacerbated accumulation of severely atrophied persistently denervated muscle fibres in COPD patients with low FFMI.
Figure 3. Evidence for denervation in COPD locomotor muscle.
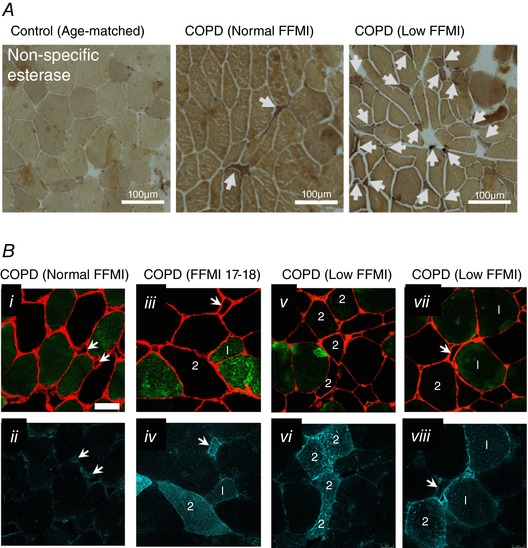
A, non‐specific esterase histochemically stained sections showing the presence of darkly staining small angular fibres in COPD and this was exacerbated in patients with low FFMI (bar = 100 μm). B, NCAM immunofluorescence (dark green in ii, iv, vi and viii) evident in COPD muscle in both type I (green labelled fibres in panels i, iii, v and vii denoted by ‘1’) and type II (unlabelled fibres in panels i, iii, v and vii denoted by ‘2’) fibres, and this was exacerbated in COPD patients with low FFMI (laminin is in red in panels i, iii, v and vii). Arrows denote very small fibres with positive NCAM signal. [Color figure can be viewed at http://wileyonlinelibrary.com]
Analysis of denervation‐responsive transcripts (Fig. 4) revealed a marked up‐regulation of Agrin (>50‐fold) and MuSK (8‐fold) in COPD patients with normal FFMI but not low FFMI relative to controls, whereas Lrp4 was modestly upregulated in COPD patients as a whole versus age‐matched controls and rapsyn was unchanged. Furthermore, transcript levels of the reinnervation‐promoting FGFBP1 were elevated >70‐fold in COPD patients with normal FFMI versus controls but unchanged in patients with low FFMI. Similarly, whereas AChRα was elevated in COPD patients as a whole, AChRγ was lower in COPD patients as a whole, and the reinnervation‐responsive AChRε was elevated only in patients with normal FFMI but not low FFMI relative to controls, despite histological evidence for exacerbated persistent denervation in the patients with low FFMI.
Figure 4. Denervation and reinnervation responsive transcript responses in COPD locomotor muscle.
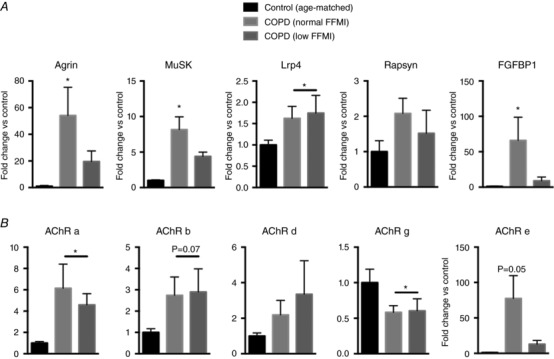
A, transcripts that are up‐regulated with denervation to promote reinnervation (agrin, MuSK, Lrp4, rapsyn, FGFBP1); B, transcripts for AChR subunits. *P < 0.05 versus Control.
Smoking mouse neuromuscular junction morphology
Representative images of the neuromuscular junctions in air‐ versus TS‐exposed mice is shown in Fig. 5. Whereas the fraction of fragmented endplates did not differ between groups (Air: 26 ± 6%; TS: 25 ± 5%), there was an increase in fraction of endplates having an area <500 μm2, a reduced endplate area occupied by AChRs (Air: 39.6 ± 1.1%; TS: 35.9 ± 0.6%), a reduced endplate area occupied by synaptophysin (Air: 26.3 ± 1.0%; TS: 20.5 ± 2.1%) and a large increase in fraction of abandoned endplates (Air: 0.4 ± 0.4%; TS: 6.4 ± 1.5%) in TS‐exposed mice (Fig. 5 B).
Figure 5. Neuromuscular junction morphology in mice following 16 weeks of tobacco smoke exposure.
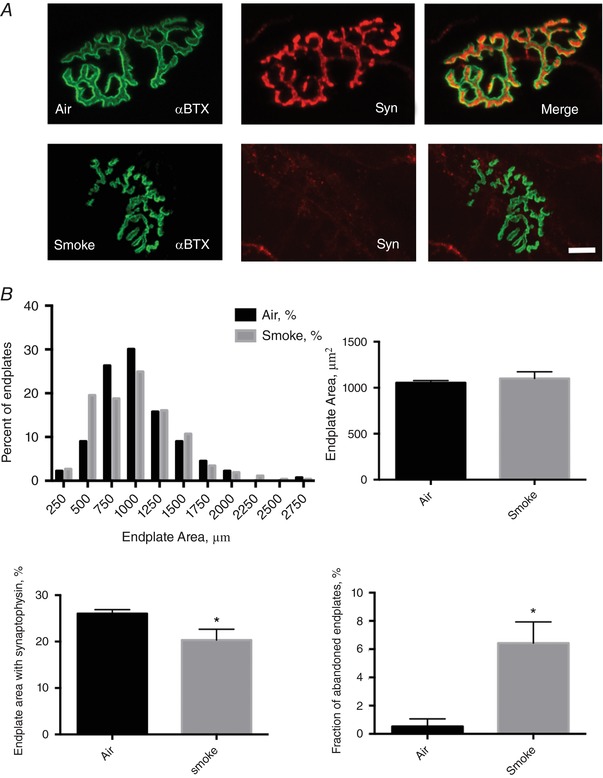
A, confocal images of the deep region of tibialis anterior muscle showing the post‐synaptic acetylcholine receptor cluster (α‐bungarotoxin, green) and pre‐synaptic motor neuron terminals (synaptophysin, red) at the neuromuscular junction in air‐exposed versus tobacco smoke‐exposed mice (bar = 10 μm). B, endplate morphology results, where the fraction of abandoned endplates = endplates with no detectable synaptophysin (bottom right). *P < 0.05 versus Air. [Color figure can be viewed at http://wileyonlinelibrary.com]
Discussion
Our objectives were to evaluate the contribution of recurring cycles of denervation–reinnervation (motor unit remodelling) to the fast fibre shift and muscle atrophy seen in COPD locomotor muscle, and evaluate the potential role of chronic TS exposure in causing denervation. Consistent with our hypothesis, COPD patients exhibited significant motor unit remodelling characterized by a reduced proportion of slow twitch fibres and a large abundance of grouped fast fibres, suggesting that repeating cycles of denervation of slow fibres followed by reinnervation by fast twitch motor units is driving the fast fibre shift in COPD locomotor muscle. Consistent with this notion, COPD muscle exhibited a significant accumulation of small angular fibres wherein the smallest of these were positive for a marker of persistent denervation (non‐specific esterase), and this was further exacerbated in patients with low FFMI. Furthermore, NCAM immunolabelling revealed denervated type I and type II fibres in COPD patients and this was exacerbated in patients with low FFMI. Interestingly, whereas COPD patients with normal FFMI exhibited strong induction of denervation‐responsive transcripts associated with promoting reinnervation, this was muted in patients with low FFMI, suggesting a failure of reinnervation signalling is associated with the exacerbated accumulation of persistently denervated fibres in COPD patients who develop muscle atrophy. In addition to these observations and consistent with our hypothesis, in adult mice following 16 weeks of passive TS exposure there was significant neuromuscular junction degeneration. Thus, our results provide the first evidence that TS‐induced neuromuscular junction degeneration is likely to be an important initiator of denervation and subsequent motor unit remodelling in COPD and other smoking‐related diseases. The significance of our results is therefore that they provide for the first time compelling evidence that denervation is a major contributor to both the fast fibre shift and the muscle atrophy seen in COPD locomotor muscle, and that chronic TS exposure probably plays a key role in initiating this effect by causing chronic destabilization of the neuromuscular junction.
Muscle fibre innervation, fibre type and fibre size
Although many factors are posited to contribute to the fibre type shift and atrophy seen in COPD locomotor muscle, including systemic hypoxia (Turan et al. 2011), physical inactivity (Wagner, 2006) and systemic inflammation (Gayan‐Ramirez & Decramer, 2013), amongst many others, the relative contribution of any of the previously considered factors remains unclear. The complexity of this issue has been nicely reviewed by The American Thoracic Society and European Respiratory Society Ad Hoc Committee on Limb Muscle Dysfunction in COPD (Maltais et al. 2014). Notably, no previous study has considered the potential involvement of denervation in these changes in COPD muscle. In view of the well‐known occurrence of neuromuscular junction degeneration with normal ageing (Hepple & Rice, 2016), the potential exacerbation of this impact in COPD is worthy of consideration.
There is a rich history of using muscle morphology to infer the causes of muscle atrophy in neuromuscular disease (Carpenter & Karpati, 2001; O'Ferrall & Sinnreich, 2009), wherein morphological features are used to distinguish between neurogenic atrophies (those involving the neuromuscular junction and/or motor neuron) and dystrophic atrophies (Baloh et al. 2007; Saez et al. 2013). In the context of neurogenic atrophies, the distinguishing morphological traits are a direct consequence of the well‐established influence of innervation/denervation on the characteristics of the motor unit. For example, innervation of a muscle fibre by a motor neuron, and the associated stimulation pattern, is a primary determinant of muscle fibre type and muscle contractile characteristics (Buller et al. 1960; Ausoni et al. 1990), driven by stimulation frequency‐dependent regulation of specific nuclear factor of activated T‐cells (NFAT) isoforms (Ehlers et al. 2014). Indeed, mouse studies have shown that during development, prototypical slow and fast muscles begin with similar levels of troponin I fast expression, but shortly after birth and initiation of locomotor muscle activity, the distinct motor neuron activation patterns in fast versus slow muscles cause a dramatic divergence such that troponin I fast expression increases in fast muscle and decreases in slow muscle (Rana et al. 2009). This divergent adaptation in the fibre type programme regresses to embryonic patterns with denervation (Rana et al. 2009) and is characterized by expression of multiple MHC isoforms within a single muscle fibre (so‐called co‐expressing or hybrid muscle fibres) (Patterson et al. 2006). In the context of normal ageing and neuromuscular diseases having a neurogenic involvement (e.g. ALS and spinal muscular atrophy), repeating cycles of denervation–reinnervation cause an increased frequency of MHC co‐expressing fibres and fibre type grouping, illustrating how these features can be used in identifying the occurrence of denervation–reinnervation events in muscle under various conditions (Baloh et al. 2007; Hepple & Rice, 2016). Innervation status also plays a key role in fibre size, with long‐term denervation causing marked fibre atrophy (Dow et al. 2005). Indeed, a sporadic pattern of muscle fibre atrophy (i.e. severely atrophied and frequently angular fibres interspersed amongst normal sized and shaped fibres) is typical of neurogenic atrophies (Baloh et al. 2007) and advanced ageing (Rowan et al. 2012).
In our current study, we used the aforementioned information as a means of gauging the involvement of denervation in the shift in fibre type and atrophy seen in COPD locomotor muscle. Our analysis showed that the reduced slow fibre abundance and commensurate increase in fast fibre abundance (specifically IIa/x hybrid fibres) was associated with a large fraction of grouped fast fibres, and these features were more pronounced in patients with low FFMI. Whereas the fast fibre type shift in COPD is well established (Whittom et al. 1998; Gosker et al. 2002), and a previous study also noted an abundance of type‐grouped fibres in skeletal muscle in patients with chronic emphysema (Sato et al. 1997), the collective interpretive value of these changes in the context of a neurological process in COPD has not been considered previously. Furthermore, no prior study has compared these features in patients with normal versus low muscle mass. In addition to these fibre type changes, we also observed a striking accumulation of sporadically distributed small and small angular fibres that exhibited high levels of non‐specific esterase activity, a widely used clinical marker of denervation (Goebel et al. 2013). Notably, we also observed in COPD patient muscle that subsets of both type I and type II fibres exhibited positive labelling for the denervation‐responsive cytokine, NCAM, that is essential for restoring stable neuromuscular junctions following denervation (Chipman et al. 2010), and this was aggravated in patients with low FFMI. Finally, we also observed a marked up‐regulation of several denervation‐responsive transcripts in COPD muscle (agrin, MuSK, Lrp4, FGFBP1, AChRα and AChRγ). Viewed as a whole, the presence of these morphological features, combined with the high NCAM expression seen in some fibres and marked denervation‐responsive transcriptional profile seen in COPD muscle, provides strong evidence that a neurogenic process is involved. Importantly, this conclusion does not rule out involvement of other factors, such as prolonged physical inactivity and systemic inflammation, that have also been implicated in the fibre type shift seen in COPD patients. For example, because several denervation‐responsive transcripts remain elevated during muscle reloading following hindlimb unloading in aged muscle (e.g. HDAC4, myogenin, AChRγ) (Baehr et al. 2016), it is possible that prolonged inactivity in older COPD patients would exacerbate denervation which is already occurring in the context of normal ageing (and TS exposure, see below), and thus contribute to the fibre type shift observed in COPD patient muscle. Furthermore, because inflammation associated with sepsis induces neuromuscular junction degeneration (Liu et al. 2016), perhaps the chronic systemic inflammation seen with COPD could have a similar impact and thus also exacerbate denervation already occurring with ageing (and TS exposure, see below). On the other hand, although systemic hypoxia has been implicated in fibre type shift in COPD (Turan et al. 2011) and our COPD patients exhibited mild systemic hypoxemia ( = 68 mmHg in the group with low FFMI), we do not believe this played a significant role in driving their fast fibre shift. This is based upon the minimal changes in fibre type/MHC composition seen with much more severe systemic hypoxia ( < 45 mmHg) in both rodent models (Itoh et al. 1990; Slot et al. 2016) and muscle cell culture (Slot et al. 2014). Furthermore, a meta‐analysis has shown that there is no relationship between fibre type shift and systemic hypoxia in COPD patients when is > 55 mmHg (7.3 kPa) (Gosker et al. 2007).
Mechanisms of denervation and reinnervation in skeletal muscle
The neuromuscular junction is the interface between the motor neuron and muscle fibre and is a point of vulnerability in various neuromuscular diseases (Comley et al. 2016; Boido & Vercelli, 2016) and normal ageing (Hepple & Rice, 2016). Importantly, previous studies have established that factors intrinsic to motor neurons, perisynaptic Schwann cells and muscle fibres may each contribute to neuromuscular junction degeneration in different contexts (Lieberman et al. 2014; Sakellariou et al. 2014; Arbour et al. 2017). For example, in the context of muscle‐specific retrograde signalling to the neuromuscular junction, recent studies have shown that muscle‐specific disturbances in (1) autophagy (Carnio et al. 2014), (2) the G93A mutation of superoxide dismutase 1 (SOD1) seen in a familial form of ALS (Dobrowolny et al. 2018) and (3) FGFBP1 secretion (Taetzsch et al. 2017) can each cause neuromuscular junction degeneration. On the other hand, although muscle‐specific knockout of SOD1 (Zhang et al. 2013) or SOD2 (Kuwahara et al. 2010) can cause muscle contractile defects with no muscle atrophy or neuromuscular junction degeneration, both neuron‐specific (Sataranatarajan et al. 2015) and whole body knockout (Jang et al. 2010) of SOD1 cause neuromuscular junction degeneration, but muscle atrophy is seen only in the whole body SOD1 knockout. These findings illustrate the complex tissue‐specific nature by which varied mechanisms can impair the neuromuscular junction and possibly result in muscle atrophy.
Amongst the pathways identified as important to regulating the integrity of the neuromuscular junction, the agrin–MuSK signalling axis is essential not only to the development of the neuromuscular junction but also to its maintenance in adulthood. Specifically, neural agrin (also known as z‐agrin) released from the terminal axons of the motor neuron binds to low‐density lipoprotein receptor‐related protein 4 (Lrp4) on the endplate region of the muscle fibre, which in turn activates MuSK (Kim et al. 2008) to recruit rapsyn to the muscle fibre subsynaptic microdomain, where it facilitates anchoring of the post‐synaptic acetylcholine receptors (AChRs) to the actin cytoskeleton (Choi et al. 2012; Ghazanfari et al. 2014). Experimental denervation (e.g. sciatic nerve transection) causes marked increases in signals related to maintaining/restoring the neuromuscular junction, including agrin (Eusebio et al. 2003) and MuSK (Valenzuela et al. 1995; Ip et al. 2000). Thus, the fact that we observed marked increases in both agrin and MuSK in muscle from COPD subjects with normal but not low FFMI suggests an active response to denervation (in an attempt to promote reinnervation) in the former but a blunted response in the latter. Further to this point, FGFBP1 is upregulated in denervated muscle to facilitate reinnervation (Williams et al. 2009; Valsecchi et al. 2015) and during reinnervation of muscle the AChRε subunit is induced in response to neural agrin signalling at the neuromuscular junction (Rimer et al. 1997). Thus, the induction of FGFBP1 and AChRε that we observed in COPD patients with normal FFMI but not low FFMI further suggests an active reinnervation process that is impaired in COPD patients who exhibit muscle atrophy. On the basis of these observations, we surmise that atrophy at the whole muscle level occurs in COPD when the capacity for reinnervation fails (e.g. when the surviving motor units cannot expand further, such as occurs with post‐Polio syndrome; Tiffreau et al. 2010), leading to an accelerated accumulation of persistently denervated myofibres that become severely atrophied. Because smoking is by far the primary cause of COPD, and our cohort of COPD patients exhibited on average 56 pack years of smoking history, in seeking to understand what might precipitate the motor unit remodelling seen in COPD patient muscle, we examined the impact of chronic TS exposure on the neuromuscular junction, as discussed below.
To gain insight into whether chronic TS exposure can induce motor unit remodelling, we examined the impact of 16 weeks of TS exposure on neuromuscular junction morphology in adult mice. Although many other studies have examined the impact of chronic TS exposure on skeletal muscle in a smoking mouse model (Gosker et al. 2009; Caron et al. 2013; Kruger et al. 2015), to our knowledge this is the first study to examine the TS impact on neuromuscular junction morphology. Our analysis revealed significant neuromuscular junction degeneration in TS‐exposed mice, characterized by a loss of motor neuron terminals (synaptophysin depletion) from endplates. As such, these results suggest that the denervation induced by chronic TS exposure is probably a key factor driving the motor unit remodelling (fast fibre shift) and eventual depletion of reinnervation potential that causes muscle atrophy in COPD locomotor muscle and perhaps other TS‐related diseases. Note that based upon the lifespan of mouse versus human, we estimate that 16 weeks of TS exposure in a mouse models 10 years of smoking in humans. Thus, given that our patients exhibited a more than 5‐fold greater relative duration of TS exposure than the smoking mice, and that denervation‐induced changes in muscle fibre type are cumulative (Butikofer et al. 2011), we suggest that the smoking history in these patients played a significant role in the fast fibre shift observed. Although we are not aware of any previous studies evaluating a neuromuscular junction impact in response to chronic TS smoke exposure, cigarette smoking is a risk factor for the neurological disease ALS (de Jong et al. 2012), and is also a triggering factor in Leber's hereditary optic neuropathy (Giordano et al. 2015). Furthermore, chronic TS exposure is an established risk factor for exacerbating the normal loss of muscle seen with ageing (van den Borst et al. 2011; Curtis et al. 2015), a process characterized by neuromuscular junction degeneration and motor unit remodelling (Hepple & Rice, 2016). Finally, it is noteworthy that patients with other smoking‐related diseases such as cardiovascular disease and cancers also exhibit a fast fibre shift (Kitzman et al. 2014; Toth et al. 2016), underlining the potentially broad clinical significance of our observations showing chronic TS exposure causes neuromuscular junction degeneration.
There are two key caveats with respect to using the smoking mouse model to infer the impact of chronic TS exposure in COPD patients. Firstly, the smoking mouse model utilizes a standardized cigarette smoking protocol developed by the Federal Trade Commission (USA) nearly 30 years ago. Based upon the high degree of inter‐individual variability in smoking behaviour (puff duration/volume/frequency, smoke‐holding in the mouth, etc.), and the variations in type of cigarettes smoked (e.g. filtered versus non‐filtered), it has been suggested that there is no representative norm for human smoking behaviour (Marian et al. 2009). This variability in smoking behaviour is relevant because the chemical composition of sidestream versus mainstream smoke is different, it can be impacted by the presence of a filter and it can vary by brand of cigarette. Thus, the chemical composition of inhaled smoke (which is a mixture of sidestream and mainstream smoke) varies as a function of differences in smoking behaviour, as well as the characteristics of the cigarettes smoked (U.S. Department of Health and Human Service, 2010). As such, it is clear that individual impact from chronic TS exposure will be much more variable than represented in the smoking mouse model. The second caveat concerns the muscle studied for TS‐induced neuromuscular junction impact. The muscle studied in our COPD patients was the vastus lateralis muscle, which in healthy young adults typically exhibits 45% of each of type I (slow oxidative) and IIa (fast oxidative) fibres, with the balance being type IIx and hybrid fibres co‐expressing multiple MHC isoforms (Gouspillou et al. 2014), whereas mouse vastus lateralis comprises ≥70% fast glycolytic fibre types (Burkholder et al. 1994). As such, to obtain a more representative match to human vastus lateralis muscle, we evaluated the impact of chronic TS exposure on the neuromuscular junction in the deep region of the mouse tibialis anterior muscle, which consists of oxidative fibre types, albeit largely fast (Burkholder et al. 1994).
In conclusion, and putting our results into the context of the TS‐related disease patient who develops muscle atrophy, our results from the preclinical smoking mouse experiment suggest that TS‐induced neuromuscular degeneration could play a key initiating role in the adverse muscle affect by creating a point of vulnerability that is preferentially affected by the systemic components of the disease. For example, in the context of pre‐existing neuromuscular junction degeneration induced by TS exposure the systemic milieu of TS‐related disease (inflammation, hypoxaemia, hypercapnia, etc.), combined with the advancing age of the patients, could accelerate the occurrence of denervation events to precipitate an exhaustion of reinnervation capacity that would then result in muscle atrophy. This is consistent with the greater denervation impact in our patients with low FFMI where there was greater COPD disease burden (lower forced expiratory volume in 1 s/forced vital capacity as a percentage of vital capacity, lower diffusing capacity of the lung for carbon monoxide, lower , etc.).
Additional information
Conflicts of interest
Jean Bourbeau receives grant funding from the (1) Research Chair COPD McGill University; (2) Research Institute of the McGill University Health Centre; (3) Research Chair COPD from GlaxoSmithKline; (4) CanCOLD consortium grant by Aerocrine, Almiral, AstraZeneca, Boehringer‐Ingleheim, GlaxoSmithKline, Novartis; and (v) Canadian Respiratory Research Network (CRRN) – Canadian Institutes of Health Research. R. Thomas Jagoe is a consultant for Immunotec Inc and related to this he has a patent for Compositions and Methods for Treatment of Muscle Wasting [US Patent Application 10/050,686 filed January 16 2003 (Harvard case 1829)]. None of the remaining authors declare any conflict of interest.
Author contributions
Conception and design: C.J.B., T.T., R.T.H. Acquisition of data: S.K., M.V., K.M., D.E., V.S., S.A., N.M., J.Baril, P.R., C.P., M.A‐L., J.A.M., R.T.J., R.T.H. Analysis and interpretation of data: S.K., M.V., D.E., S.A., T.T., R.T.H. Drafting of manuscript: S.K., T.T., R.T.H. Review and final approval of manuscript: all authors.
Acknowledgements
Funding was provided by an operating grant from the Canadian Institutes of Health Research (MOP 119583 to R.T.H.). S. Spendiff was supported in part by a Postdoctoral Fellowship from the Research Institute of the McGill University Health Centre. M. Vuda was supported by a Postdoctoral Fellowship for Prospective Researchers’ Award from the Swiss National Science Foundation. K. Miguez was supported by a Canada Graduate Scholarship from the Canadian Institutes of Health Research. S. Kapchinsky was supported by a PhD Fellowship from the Canadian Institutes of Health Research. V. Sonjak was supported by a Bloomberg‐Manulife PhD Scholarship. M Aubertin‐Leheudre was supported by a Chercheur Boursier Junior II Award from the Fond de Recherche du Québec en Santé (FRQS).
Biographies
Sophia Kapchinsky was a PhD student in the Department of Kinesiology & Physical Education at McGill University during the conduct of these studies as part of her dissertation.2

Madhusarano Vuda was a Postdoctoral Fellow at the Meakins Christie Institute at McGill University during conduct of these studies and is currently a Postdoctoral Fellow at the University of Minnesota.
Kayla Miguez was an MSc student in the Department of Kinesiology & Physical Education at McGill University during the conduct of these studies as part of her MSc.
Russell Hepple, formerly of McGill University, is a Professor of Muscle Biology in the Department of Physical Therapy at the University of Florida. Dr Hepple's research focuses upon the mechanisms of skeletal muscle deterioration with ageing and age‐related disease.
Edited by: Michael Hogan, Karyn Hamilton & Scott Powers
References
- http://American Thoracic Society; http://American College of Chest Physicians. (2003). ATS/ACCP statement on cardiopulmonary exercise testing. Am J Respir Crit Care Med 167, 211–277. [DOI] [PubMed] [Google Scholar]
- Arbour D, Vande Velde C & Robitaille R (2017). New perspectives on amyotrophic lateral sclerosis: the role of glial cells at the neuromuscular junction. J Physiol 595, 647–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausoni S, Gorza L, Schiaffino S, Gundersen K & Lomo T (1990). Expression of myosin heavy chain isoforms in stimulated fast and slow rat muscles. J Neurosci 10, 153–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baehr LM, West DW, Marcotte G, Marshall AG, De Sousa LG, Baar K & Bodine SC (2016). Age‐related deficits in skeletal muscle recovery following disuse are associated with neuromuscular junction instability and ER stress, not impaired protein synthesis. Aging (Albany NY) 8, 127–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baloh RH, Rakowicz W, Gardner R & Pestronk A (2007). Frequent atrophic groups with mixed‐type myofibers is distinctive to motor neuron syndromes. Muscle Nerve 36, 107–110. [DOI] [PubMed] [Google Scholar]
- Boido M & Vercelli A (2016). Neuromuscular junctions as key contributors and therapeutic targets in spinal muscular atrophy. Front Neuroanat 10, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buller AJ, Eccles JC & Eccles RM (1960). Interactions between motoneurones and muscles in respect of the characteristic speeds of their responses. J Physiol 150, 417–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkholder TJ, Fingado B, Baron S & Lieber RL (1994). Relationship between muscle fiber types and sizes and muscle architectural properties in the mouse hindlimb. J Morphol 221, 177–190. [DOI] [PubMed] [Google Scholar]
- Butikofer L, Zurlinden A, Bolliger MF, Kunz B & Sonderegger P (2011). Destabilization of the neuromuscular junction by proteolytic cleavage of agrin results in precocious sarcopenia. FASEB J 25, 4378–4393. [DOI] [PubMed] [Google Scholar]
- Carnio S, LoVerso F, Baraibar MA, Longa E, Khan MM, Maffei M, Reischl M, Canepari M, Loefler S, Kern H, Blaauw B, Friguet B, Bottinelli R, Rudolf R & Sandri M (2014). Autophagy impairment in muscle induces neuromuscular junction degeneration and precocious aging. Cell Rep 8, 1509–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron MA, Morissette MC, Theriault ME, Nikota JK, Stampfli MR & Debigare R (2013). Alterations in skeletal muscle cell homeostasis in a mouse model of cigarette smoke exposure. PLoS One 8, e66433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter S & Karpati G (2001). Pathology of Skeletal Muscle, 2nd edn Oxford University Press, Oxford. [Google Scholar]
- Chipman PH, Franz CK, Nelson A, Schachner M & Rafuse VF (2010). Neural cell adhesion molecule is required for stability of reinnervated neuromuscular junctions. Eur J Neurosci 31, 238–249. [DOI] [PubMed] [Google Scholar]
- Choi KR, Berrera M, Reischl M, Strack S, Albrizio M, Roder IV, Wagner A, Petersen Y, Hafner M, Zaccolo M & Rudolf R (2012). Rapsyn mediates subsynaptic anchoring of PKA type I and stabilisation of acetylcholine receptor in vivo . J Cell Sci 125, 714–723. [DOI] [PubMed] [Google Scholar]
- Comley LH, Nijssen J, Frost‐Nylen J & Hedlund E (2016). Cross‐disease comparison of amyotrophic lateral sclerosis and spinal muscular atrophy reveals conservation of selective vulnerability but differential neuromuscular junction pathology. J Comp Neurol 52, 1424–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis E, Litwic A, Cooper C & Dennison E (2015). Determinants of muscle and bone aging. J Cell Physiol 230, 2618–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong SW, Huisman MH, Sutedja NA, van der Kooi AJ, de Visser M, Schelhaas HJ, Fischer K, Veldink JH & van den Berg LH (2012). Smoking, alcohol consumption, and the risk of amyotrophic lateral sclerosis: a population‐based study. Am J Epidemiol 176, 233–239. [DOI] [PubMed] [Google Scholar]
- de Souza AR, Zago M, Eidelman DH, Hamid Q & Baglole CJ (2014). Aryl hydrocarbon receptor (AhR) attenuation of subchronic cigarette smoke‐induced pulmonary neutrophilia is associated with retention of nuclear RelB and suppression of intercellular adhesion molecule‐1 (ICAM‐1). Toxicol Sci 140, 204–223. [DOI] [PubMed] [Google Scholar]
- Degens H, Gayan‐Ramirez G & van Hees HW (2015). Smoking‐induced skeletal muscle dysfunction: from evidence to mechanisms. Am J Respir Crit Care Med 191, 620–625. [DOI] [PubMed] [Google Scholar]
- Dobrowolny G, Martini M, Scicchitano BM, Romanello V, Boncompagni S, Nicoletti C, Pietrangelo L, De Panfilis S, Catizone A, Bouche M, Sandri M, Rudolf R, Protasi F & Musaro A (2018). Muscle expression of SOD1G93A triggers the dismantlement of neuromuscular junction via PKC‐Theta. Antioxid Redox Signal 28, 1105–1119. [DOI] [PubMed] [Google Scholar]
- Dow DE, Dennis RG & Faulkner JA (2005). Electrical stimulation attenuates denervation and age‐related atrophy in extensor digitorum longus muscles of old rats. J Gerontol A Biol Sci Med Sci 60, 416–424. [DOI] [PubMed] [Google Scholar]
- Ehlers ML, Celona B & Black BL (2014). NFATc1 controls skeletal muscle fiber type and is a negative regulator of MyoD activity. Cell Rep 8, 1639–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eusebio A, Oliveri F, Barzaghi P & Ruegg MA (2003). Expression of mouse agrin in normal, denervated and dystrophic muscle. Neuromuscul Disord 13, 408–415. [DOI] [PubMed] [Google Scholar]
- Gayan‐Ramirez G & Decramer M (2013). Mechanisms of striated muscle dysfunction during acute exacerbations of COPD. J Appl Physiol (1985) 114, 1291–1299. [DOI] [PubMed] [Google Scholar]
- Ghazanfari N, Morsch M, Reddel SW, Liang SX & Phillips WD (2014). Muscle‐specific kinase (MuSK) autoantibodies suppress the MuSK pathway and ACh receptor retention at the mouse neuromuscular junction. J Physiol 592, 2881–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano L, Deceglie S, d'Adamo P, Valentino ML, La Morgia C, Fracasso F, Roberti M, Cappellari M, Petrosillo G, Ciaravolo S, Parente D, Giordano C, Maresca A, Iommarini L, Del Dotto V, Ghelli AM, Salomao SR, Berezovsky A, Belfort R Jr, Sadun AA, Carelli V, Loguercio Polosa P & Cantatore P (2015). Cigarette toxicity triggers Leber's hereditary optic neuropathy by affecting mtDNA copy number, oxidative phosphorylation and ROS detoxification pathways. Cell Death Dis 6, e2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebel HH, Sewry CA & Weller RO (2013). Muscle Disease: Pathology and Genetics. John Wiley & Sons, Chichcester. [Google Scholar]
- Goodchild M, Nargis N & Tursan d'Espaignet E (2018). Global economic cost of smoking‐attributable diseases. Tob Control 7276, 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosker HR, Engelen MP, van Mameren H, van Dijk PJ, van der Vusse GJ, Wouters EF & Schols AM (2002). Muscle fiber type IIX atrophy is involved in the loss of fat‐free mass in chronic obstructive pulmonary disease. Am J Clin Nutr 76, 113–119. [DOI] [PubMed] [Google Scholar]
- Gosker HR, Langen RC, Bracke KR, Joos GF, Brusselle GG, Steele C, Ward KA, Wouters EF & Schols AM (2009). Extrapulmonary manifestations of chronic obstructive pulmonary disease in a mouse model of chronic cigarette smoke exposure. Am J Respir Cell Mol Biol 40, 710–716. [DOI] [PubMed] [Google Scholar]
- Gosker HR, Zeegers MP, Wouters EF & Schols AM (2007). Muscle fibre type shifting in the vastus lateralis of patients with COPD is associated with disease severity: a systematic review and meta‐analysis. Thorax 62, 944–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouspillou G, Sgarioto N, Kapchinsky S, Purves‐Smith F, Norris B, Pion CH, Barbat‐Artigas S, Lemieux F, Taivassalo T, Morais JA, Aubertin‐Leheudre M & Hepple RT (2014). Increased sensitivity to mitochondrial permeability transition and myonuclear translocation of endonuclease G in atrophied muscle of physically active older humans. FASEB J 28, 1621–1633. [DOI] [PubMed] [Google Scholar]
- Hata K, Maeno‐Hikichi Y, Yumoto N, Burden SJ & Landmesser LT (2018). Distinct roles of different presynaptic and postsynaptic NCAM isoforms in early motoneuron‐myotube interactions required for functional synapse formation. J Neurosci 38, 498–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepple RT & Rice CL (2016). Innervation and neuromuscular control in ageing skeletal muscle. J Physiol 594, 1965–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip FC, Glass DG, Gies DR, Cheung J, Lai KO, Fu AK, Yancopoulos GD & Ip NY (2000). Cloning and characterization of muscle‐specific kinase in chicken. Mol Cell Neurosci 16, 661–673. [DOI] [PubMed] [Google Scholar]
- Itoh K, Moritani T, Ishida K, Hirofuji C, Taguchi S & Itoh M (1990). Hypoxia‐induced fibre type transformation in rat hindlimb muscles. Histochemical and electro‐mechanical changes. Eur J Appl Physiol Occup Physiol 60, 331–336. [DOI] [PubMed] [Google Scholar]
- Jang YC, Lustgarten MS, Liu Y, Muller FL, Bhattacharya A, Liang H, Salmon AB, Brooks SV, Larkin L, Hayworth CR, Richardson A & Van Remmen H (2010). Increased superoxide in vivo accelerates age‐associated muscle atrophy through mitochondrial dysfunction and neuromuscular junction degeneration. FASEB J 24, 1376–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SE, Maddocks M, Kon SS, Canavan JL, Nolan CM, Clark AL, Polkey MI & Man WD (2015). Sarcopenia in COPD: prevalence, clinical correlates and response to pulmonary rehabilitation. Thorax 70, 213–218. [DOI] [PubMed] [Google Scholar]
- Kim N, Stiegler AL, Cameron TO, Hallock PT, Gomez AM, Huang JH, Hubbard SR, Dustin ML & Burden SJ (2008). Lrp4 is a receptor for Agrin and forms a complex with MuSK. Cell 135, 334–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitzman DW, Nicklas B, Kraus WE, Lyles MF, Eggebeen J, Morgan TM & Haykowsky M (2014). Skeletal muscle abnormalities and exercise intolerance in older patients with heart failure and preserved ejection fraction. Am J Physiol Heart Circ Physiol 306, H1364–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger K, Dischereit G, Seimetz M, Wilhelm J, Weissmann N & Mooren FC (2015). Time course of cigarette smoke‐induced changes of systemic inflammation and muscle structure. Am J Physiol Lung Cell Mol Physiol 309, L119–128. [DOI] [PubMed] [Google Scholar]
- Kuwahara H, Horie T, Ishikawa S, Tsuda C, Kawakami S, Noda Y, Kaneko T, Tahara S, Tachibana T, Okabe M, Melki J, Takano R, Toda T, Morikawa D, Nojiri H, Kurosawa H, Shirasawa T & Shimizu T (2010). Oxidative stress in skeletal muscle causes severe disturbance of exercise activity without muscle atrophy. Free Radic Biol Med 48, 1252–1262. [DOI] [PubMed] [Google Scholar]
- Larsson L & Orlander J (1984). Skeletal muscle morphology, metabolism and function in smokers and non‐smokers. A study on smoking‐discordant monozygous twins. Acta Physiol Scand 120, 343–352. [DOI] [PubMed] [Google Scholar]
- Lexell J & Downham DY (1991). The occurrence of fibre‐type grouping in healthy human muscle: a quantitative study of cross‐sections of whole vastus lateralis from men between 15 and 83 years. Acta Neuropathol (Berl) 81, 377–381. [DOI] [PubMed] [Google Scholar]
- Lieberman AP, Yu Z, Murray S, Peralta R, Low A, Guo S, Yu XX, Cortes CJ, Bennett CF, Monia BP, La Spada AR & Hung G (2014). Peripheral androgen receptor gene suppression rescues disease in mouse models of spinal and bulbar muscular atrophy. Cell Rep 7, 774–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Xie F, Wei K, Hao XC, Li P, Cao J & Min S (2016). Sepsis induced denervation‐like changes at the neuromuscular junction. J Surg Res 200, 523–532. [DOI] [PubMed] [Google Scholar]
- Malicdan MCV, Noguchi S & Nishino I (2009). Monitoring autophagy in muscle diseases. Methods Enzymol 453, 379–396. [DOI] [PubMed] [Google Scholar]
- Maltais F, Decramer M, Casaburi R, Barreiro E, Burelle Y, Debigare R, Dekhuijzen PN, Franssen F, Gayan‐Ramirez G, Gea J, Gosker HR, Gosselink R, Hayot M, Hussain SN, Janssens W, Polkey MI, Roca J, Saey D, Schols AM, Spruit MA, Steiner M, Taivassalo T, Troosters T, Vogiatzis I, Wagner PD & COPD AEAHCoLMDi (2014). An official American Thoracic Society/European Respiratory Society statement: update on limb muscle dysfunction in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 189, e15–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marian C, O'Connor RJ, Djordjevic MV, Rees VW, Hatsukami DK & Shields PG (2009). Reconciling human smoking behavior and machine smoking patterns: implications for understanding smoking behavior and the impact on laboratory studies. Cancer Epidemiol Biomarkers Prev 18, 3305–3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquis K, Debigare R, Lacasse Y, LeBlanc P, Jobin J, Carrier G & Maltais F (2002). Midthigh muscle cross‐sectional area is a better predictor of mortality than body mass index in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 166, 809–813. [DOI] [PubMed] [Google Scholar]
- Mollard P, Bourdillon N, Letournel M, Herman H, Gibert S, Pichon A, Woorons X & Richalet JP (2010). Validity of arterialized earlobe blood gases at rest and exercise in normoxia and hypoxia. Respir Physiol Neurobiol 172, 179–183. [DOI] [PubMed] [Google Scholar]
- O'Ferrall EK & Sinnreich M (2009). The role of muscle biopsy in the age of genetic testing. Curr Opin Neurol 22, 543–553. [DOI] [PubMed] [Google Scholar]
- Orlander J, Kiessling KH & Larsson L (1979). Skeletal muscle metabolism, morphology and function in sedentary smokers and nonsmokers. Acta Physiol Scand 107, 39–46. [DOI] [PubMed] [Google Scholar]
- Patterson MF, Stephenson GMM & Stephenson DG (2006). Denervation produces different single fiber phenotypes in fast‐ and slow‐twitch hindlimb muscles of the rat. Am J Physiol Cell Physiol 291, C518–528. [DOI] [PubMed] [Google Scholar]
- U.S. Department of Health and Human Service (2010). How Tobacco Smoke Causes Disease: The Biology and Behavioral Basis for Smoking‐Attributable Disease: A Report of the Surgeon General. U.S. Department of Health and Human Service, Atlanta: (GA: ). [PubMed] [Google Scholar]
- Purves‐Smith FM, Solbak NM, Rowan SL & Hepple RT (2012). Severe atrophy of slow myofibers in aging muscle is concealed by myosin heavy chain co‐expression. Exp Gerontol 47, 913–918. [DOI] [PubMed] [Google Scholar]
- Rana ZA, Gundersen K & Buonanno A (2009). The ups and downs of gene regulation by electrical activity in skeletal muscles. J Muscle Res Cell Motil 30, 255–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimer M, Mathiesen I, Lomo T & McMahan UJ (1997). γ‐AChR/ε‐AChR switch at agrin‐induced postsynaptic‐like apparatus in skeletal muscle. Mol Cell Neurosci 9, 254–263. [DOI] [PubMed] [Google Scholar]
- Rowan SL, Rygiel K, Purves‐Smith FM, Solbak NM, Turnbull DM & Hepple RT (2012). Denervation causes fiber atrophy and myosin heavy chain co‐expression in senescent skeletal muscle. PLoS One 7, e29082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saez A, Rivas E, Montero‐Sanchez A, Paradas C, Acha B, Pascual A, Serrano C & Escudero LM (2013). Quantifiable diagnosis of muscular dystrophies and neurogenic atrophies through network analysis. BMC Med 11, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakellariou GK, Davis CS, Shi Y, Ivannikov MV, Zhang Y, Vasilaki A, Macleod GT, Richardson A, Van Remmen H, Jackson MJ, McArdle A & Brooks SV (2014). Neuron‐specific expression of CuZnSOD prevents the loss of muscle mass and function that occurs in homozygous CuZnSOD‐knockout mice. FASEB J 28, 1666–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sataranatarajan K, Qaisar R, Davis C, Sakellariou GK, Vasilaki A, Zhang Y, Liu Y, Bhaskaran S, McArdle A, Jackson M, Brooks SV, Richardson A & Van Remmen H (2015). Neuron specific reduction in CuZnSOD is not sufficient to initiate a full sarcopenia phenotype. Redox Biology 5, 140–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Asoh T, Honda Y, Fujimatsu Y, Higuchi I & Oizumi K (1997). Morphologic and histochemical evaluation of muscle in patients with chronic pulmonary emphysema manifesting generalized emaciation. Eur Neurol 37, 116–121. [DOI] [PubMed] [Google Scholar]
- Schutz Y, Kyle UU & Pichard C (2002). Fat‐free mass index and fat mass index percentiles in Caucasians aged 18–98 y. Int J Obes Relat Metab Disord 26, 953–960. [DOI] [PubMed] [Google Scholar]
- Shanely RA, Zwetsloot KA, Triplett NT, Meaney MP, Farris GE & Nieman DC (2014). Human skeletal muscle biopsy procedures using the modified Bergstrom technique. J Vis Exp 10, 51812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slot IG, Schols AM, de Theije CC, Snepvangers FJ & Gosker HR (2016). Alterations in skeletal muscle oxidative phenotype in mice exposed to 3 weeks of normobaric hypoxia. J Cell Physiol 231, 377–392. [DOI] [PubMed] [Google Scholar]
- Slot IG, Schols AM, Vosse BA, Kelders MC & Gosker HR (2014). Hypoxia differentially regulates muscle oxidative fiber type and metabolism in a HIF‐1α‐dependent manner. Cell Signal 26, 1837–1845. [DOI] [PubMed] [Google Scholar]
- Spendiff S, Vuda M, Gouspillou G, Aare S, Perez A, Morais JA, Jagoe RT, Filion ME, Glicksman R, Kapchinsky S, MacMillan NJ, Pion CH, Aubertin‐Leheudre M, Hettwer S, Correa JA, Taivassalo T & Hepple RT (2016). Denervation drives mitochondrial dysfunction in skeletal muscle of octogenarians. J Physiol 594, 7361–7379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swallow EB, Reyes D, Hopkinson NS, Man WD, Porcher R, Cetti EJ, Moore AJ, Moxham J & Polkey MI (2007). Quadriceps strength predicts mortality in patients with moderate to severe chronic obstructive pulmonary disease. Thorax 62, 115–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taetzsch T, Tenga MJ & Valdez G (2017). Muscle fibers secrete FGFBP1 to slow degeneration of neuromuscular synapses during aging and progression of ALS. J Neurosci 37, 70–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiffreau V, Rapin A, Serafi R, Percebois‐Macadre L, Supper C, Jolly D & Boyer FC (2010). Post‐polio syndrome and rehabilitation. Ann Phys Rehabil Med 53, 42–50. [DOI] [PubMed] [Google Scholar]
- Toth MJ, Callahan DM, Miller MS, Tourville TW, Hackett SB, Couch ME & Dittus K (2016). Skeletal muscle fiber size and fiber type distribution in human cancer: Effects of weight loss and relationship to physical function. Clin Nutr 35, 1359–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turan N, Kalko S, Stincone A, Clarke K, Sabah A, Howlett K, Curnow SJ, Rodriguez DA, Cascante M, O'Neill L, Egginton S, Roca J & Falciani F (2011). A systems biology approach identifies molecular networks defining skeletal muscle abnormalities in chronic obstructive pulmonary disease. PLoS Comput Biol 7, e1002129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela DM, Stitt TN, DiStefano PS, Rojas E, Mattsson K, Compton DL, Nunez L, Park JS, Stark JL, Gies DR, Thomas S, Le Beau MM, Fernald AA, Copeland NG, Jenkins NA, Burden SJ, Glass DJ & Yancopoulos GD (1995). Receptor tyrosine kinase specific for the skeletal muscle lineage: expression in embryonic muscle, at the neuromuscular junction, and after injury. Neuron 15, 573–584. [DOI] [PubMed] [Google Scholar]
- Valsecchi V, Boido M, De Amicis E, Piras A & Vercelli A (2015). Expression of muscle‐specific MiRNA 206 in the progression of disease in a murine SMA model. PLoS One 10, e0128560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Borst B, Koster A, Yu B, Gosker HR, Meibohm B, Bauer DC, Kritchevsky SB, Liu Y, Newman AB, Harris TB & Schols AM (2011). Is age‐related decline in lean mass and physical function accelerated by obstructive lung disease or smoking? Thorax 66, 961–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner PD ( 2006). Skeletal muscles in chronic obstructive pulmonary disease: deconditioning, or myopathy? Respirology 11, 681–686. [DOI] [PubMed] [Google Scholar]
- Walsh FS, Hobbs C, Wells DJ, Slater CR & Fazeli S (2000). Ectopic expression of NCAM in skeletal muscle of transgenic mice results in terminal sprouting at the neuromuscular junction and altered structure but not function. Mol Cell Neurosci 15, 244–261. [DOI] [PubMed] [Google Scholar]
- Wanger J, Clausen JL, Coates A, Pedersen OF, Brusasco V, Burgos F, Casaburi R, Crapo R, Enright P, van der Grinten CP, Gustafsson P, Hankinson J, Jensen R, Johnson D, Macintyre N, McKay R, Miller MR, Navajas D, Pellegrino R & Viegi G (2005). Standardisation of the measurement of lung volumes. Eur Respir J 26, 511–522. [DOI] [PubMed] [Google Scholar]
- Warren GW, Alberg AJ, Kraft AS & Cummings KM (2014). The 2014 Surgeon General's report: “The health consequences of smoking–50 years of progress”: a paradigm shift in cancer care. Cancer 120, 1914–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittom F, Jobin J, Simard PM, Leblanc P, Simard C, Bernard S, Belleau R & Maltais F (1998). Histochemical and morphological characteristics of the vastus lateralis muscle in patients with chronic obstructive pulmonary disease. Med Sci Sports Exerc 30, 1467–1474. [DOI] [PubMed] [Google Scholar]
- Williams AH, Valdez G, Moresi V, Qi X, McAnally J, Elliott JL, Bassel‐Duby R, Sanes JR & Olson EN (2009). MicroRNA‐206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science 326, 1549–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Davis C, Sakellariou GK, Shi Y, Kayani AC, Pulliam D, Bhattacharya A, Richardson A, Jackson MJ, McArdle A, Brooks SV & Van Remmen H (2013). CuZnSOD gene deletion targeted to skeletal muscle leads to loss of contractile force but does not cause muscle atrophy in adult mice. FASEB J 27, 3536–3548. [DOI] [PMC free article] [PubMed] [Google Scholar]