

Abstract
Disrupted in schizophrenia 1 (DISC1) is an important hub protein, forming multimeric complexes by self‐association and interacting with a large number of synaptic and cytoskeletal molecules. The synaptic location of DISC1 in the adult brain suggests a role in synaptic plasticity, and indeed, a number of studies have discovered synaptic plasticity impairments in a variety of different DISC1 mutants. This review explores the possibility that DISC1 is an important molecule for organizing proteins involved in synaptic plasticity and examines why mutations in DISC1 impair plasticity. It concentrates on DISC1's role in interacting with synaptic proteins, controlling dendritic structure and cellular trafficking of mRNA, synaptic vesicles and mitochondria. N‐terminal directed mutations appear to impair synaptic plasticity through interactions with phosphodiesterase 4B (PDE4B) and hence protein kinase A (PKA)/GluA1 and PKA/cAMP response element‐binding protein (CREB) signalling pathways, and affect spine structure through interactions with kalirin 7 (Kal‐7) and Rac1. C‐terminal directed mutations also impair plasticity possibly through altered interactions with lissencephaly protein 1 (LIS1) and nuclear distribution protein nudE‐like 1 (NDEL1), thereby affecting developmental processes such as dendritic structure and spine maturation. Many of the same molecules involved in DISC1's cytoskeletal interactions are also involved in intracellular trafficking, raising the possibility that impairments in intracellular trafficking affect cytoskeletal development and vice versa. While the multiplicity of DISC1 protein interactions makes it difficult to pinpoint a single causal signalling pathway, we suggest that the immediate‐term effects of N‐terminal influences on GluA1, Rac1 and CREB, coupled with the developmental effects of C‐terminal influences on trafficking and the cytoskeleton make up the two main branches of DISC1's effect on synaptic plasticity and dendritic spine stability.
Introduction
Disrupted in schizophrenia 1 (DISC1) is a protein that when disrupted by a chromosomal t(1;11) translocation (Thomson et al. 2016) or mutated (Sachs et al. 2005) predisposes the carrier to a number of mental health disorders including schizophrenia, bipolar disorder and recurrent major depression. The synaptic location of DISC1 (Kirkpatrick et al. 2006; Carlisle et al. 2011), together with recent evidence for its role in cortical synaptic plasticity (Greenhill et al. 2015; Tropea et al. 2016) raises the question of how DISC1 affects plasticity and whether disruption of plasticity might itself lead to psychiatric symptoms. Indeed, a number of large‐scale genomics studies also implicate copy number variation and mutations in synaptic proteins involved in plasticity as risks factors for psychiatric conditions (Fromer et al. 2014; Pocklington et al. 2014; Hall et al. 2015; Marshall et al. 2017). In addition to the pressing need to understand these debilitating disorders, there is a fundamental biological question to answer about the function of DISC1 in the neuron. In vitro studies show that DISC1's pleiotropic function occurs through interactions with several proteins (Fig. 1) including phosphodiesterase 4B (PDE4B), glycogen synthase kinase 3 β (GSK3β), kalirin 7 (Kal‐7), fasciculation and elongation protein zeta‐1 (FEZ1), kendrin, lissencephaly protein 1 (LIS1) and nuclear distribution protein nudE‐like 1 (NDEL1)/nuclear distribution protein nudE homolog 1 (NDE1) (Miyoshi et al. 2003; Ozeki et al. 2003; Brandon et al. 2004; Millar et al. 2005b; Mao et al. 2009; Lipina et al. 2012). Which of these pathways are important for plasticity and what processes are altered to disrupt plasticity when DISC1 is mutated?
Figure 1. DISC1 protein–protein known interaction sites and DISC1 mouse mutants.
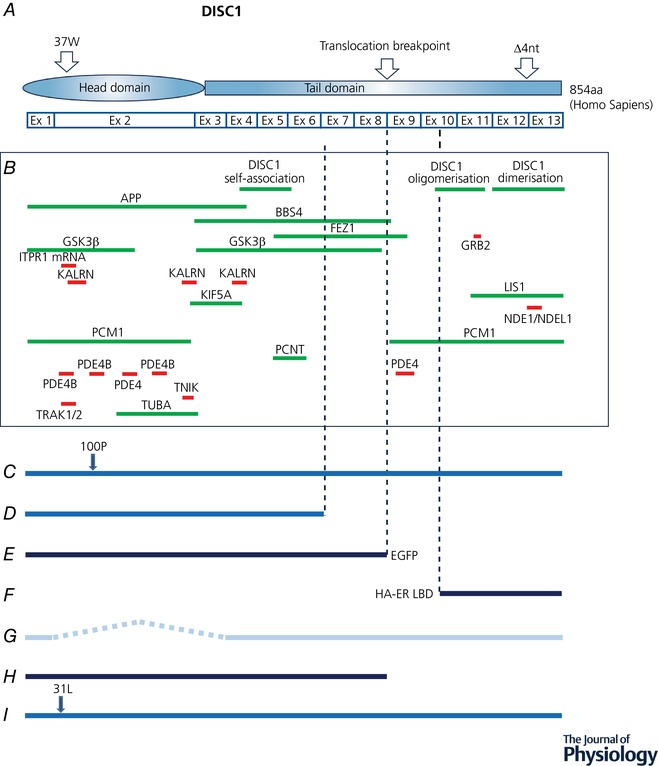
A, schematic representation of DISC1 human protein indicating naturally occurring mutation sites. 37W and the four‐nucleotide deletion in exon 12 are human mutations detected in psychiatric patients (Sachs et al. 2005; Song et al. 2008; Thomson et al. 2013). B, self‐association sites and sites required for partner binding/association. Fine‐mapped binding/association sites are indicated in red, and green indicates regions of DISC1 known to contain a site. See Table 1 for references to the original studies reporting the DISC1 protein interactions indicated here. C, endogenous mouse DISC1 carrying ethylnitrosourea‐induced point mutation (Clapcote et al. 2007). D, endogenous mouse DISC1 carrying targeted exon 8 stop codon and intron 8 polyA site plus a natural 25‐nucleotide deletion in exon 6; full‐length DISC1 expression is abolished (Koike et al. 2006). E, truncated mouse DISC1 with C‐terminal enhanced green fluorescent protein tag, expressed under native DISC1 promoter from transgenic bacterial artificial chromosome (Shen et al. 2008). F, inducible DISC1 expressed under a CAMKII promoter with N‐terminal mutant oestrogen receptor ligand binding domain fused to HA peptide tag (Li et al. 2007). G, endogenous mouse DISC1 exon 2/3 deletion; DISC1 expression is abolished (Kuroda et al. 2011). H, truncated human DISC1 cDNA inducibly expressed under cytomegalovirus (CMV) promoter with N‐terminal Myc peptide tag (Pletnikov et al. 2008) I, endogenous mouse DISC1 carrying ethylnitrosourea‐induced point mutation (Clapcote et al. 2007). J, human DISC1 cDNA carrying functional sequence variants, expressed under PrP promoter in rat. Four amino acid sequence changes are indicated; 607F and 704C are common human sequence variants (Trossbach et al., 2016). Dark blue lines indicate transgenic DISC1 expression against a background of endogenous expression; mid blue lines indicate modified endogenous expression; pale blue line indicates endogenous gene modification to abolish expression.
DISC1 is known to play a role in early development and has been implicated in neuronal proliferation, cell migration and neurite extension (Brandon et al. 2009). Several excellent reviews on DISC1's role in these processes have been published (Brandon and Sawa, 2011; Bradshaw & Porteous, 2012; Wu and Xiao, 2013; Muraki and Tanigaki, 2015) and in this review, we focus instead on synaptic plasticity in the adult as a critical process deficient in animals with mutated DISC1. However, we also consider the possibility that DISC1's action in adulthood is the result of modifications that occur at early phases of postnatal development. In particular, we discuss the hypothesis that through specific protein–protein interactions, DISC1 mediates the impairment of intracellular trafficking, dendritic growth and spine maturation during development, which ultimately lead to alterations in circuit plasticity in adulthood. We begin by considering DISC1's protein–protein interactions, since a crucial element of these plasticity defects is likely to lie in signalling pathways affected by DISC1.
Organization of DISC1 protein interactions
Secondary structure analysis using the amino acid sequence of full‐length DISC1 (854 amino acids for human DISC1) predicts that it consists of two major domains, the N‐terminal head domain and the C‐terminal tail (Fig. 1 A). The N‐terminal head domain (approximately amino acids 1–350 in human DISC1) exhibits poor evolutionary conservation, with the exception of two highly conserved motifs, the first rich in arginine, and the second rich in serine and phenylalanine (Chubb et al. 2008). The head domain is predicted to be largely disordered (Soares et al. 2012), implying a structural flexibility that could facilitate binding to multiple targets. On the contrary, the C‐terminal tail domain (approximately amino acids 350–854 in human DISC1) is well conserved across species and predicted to be highly structured, consisting of a series of α‐helices interspersed with at least four regions comprising strong coiled‐coil forming potential (Soares et al. 2012).
Tertiary structure analysis throws further light on the nature of DISC1. DISC1 is able to self‐associate to form multimers. Amino acids 403–504 of human DISC1 were the first shown to enable the protein to multimerize (Kamiya et al. 2005). This ability was further dissected by generating a series of human DISC1 fragments, with size exclusion chromatography demonstrating that several regions of DISC1, most of which are located within the tail domain, can form multimers or dimers (Leliveld et al. 2008; Leliveld et al. 2009; Yerabham et al. 2017). Size exclusion chromatography also indicates that full‐length DISC1 forms multimers with equilibrium analytical ultracentrifugation, demonstrating that it adopts predominantly dimeric and octameric forms, and that dimers likely act as the building blocks for orderly formation of octamers (Narayanan et al. 2011).
DISC1 has a large number of confirmed interactors (see Table 1 and Fig. 1 B) and many additional putative binding partners, and it appears to act as a ‘hub’ protein within interaction networks (Camargo et al. 2007). It is hypothesized to be a molecular scaffold, assembling multiple proteins into functional units. For some interactions that have been studied in detail, it has been found that there are several contact sites along the length of DISC1. For example, DISC1's interaction with the PDE4 family of cAMP phosphodiesterases was examined using peptide array mapping (Millar et al. 2005b; Murdoch et al. 2007). This demonstrated the existence of five contact sites for PDE4B (Fig. 1). Of these sites, four are located within the head domain, one encompassing the conserved arginine‐rich region, and another the conserved serine/phenylalanine‐rich region (Murdoch et al. 2007). GSK3β and kalirin also appear to contact DISC1 at more than one binding site (Morris et al. 2003; Ozeki et al. 2003; Brandon et al. 2004; Kamiya et al. 2006; Mao et al. 2009; Hayashi‐Takagi et al. 2010), and many DISC1 partner binding sites may overlap (Fig. 1). For example, the arginine‐rich region in the head domain to which PDE4B binds (Murdoch et al. 2007) is also essential for DISC1 to complex with trafficking kinesin protein 1/2 (TRAK1/2; Ogawa et al. 2014; Norkett et al. 2016) and with the 3′‐untranslated region (UTR) of inositol‐1,4,5‐trisphosphate receptor type 1 (ITPR1) mRNA (Tsuboi et al. 2015; Fig. 1). This multifunctional arginine‐rich region also acts as a nuclear localization signal (Sawamura et al. 2008; Malavasi et al. 2012).
Table 1.
DISC1 interactors: the DISC1 interactors shown in Fig. 1 are given alongside their full names and the references first establishing their interactions with DISC1
| Interaction factor | Name | Reference |
|---|---|---|
| DISC1 | Disrupted in schizophrenia 1 | Kamiya et al. (2005), Leliveld et al. (2009) |
| APP | Amyloid precursor protein | Young‐Pearse et al. (2010) |
| BBS4 | Bardet–Biedl syndrome 4 | Kamiya et al. (2008) |
| FEZ1 | Fasciculation and elongation protein zeta‐1 | Miyoshi et al. (2003) |
| GRB2 | Growth factor receptor‐bound protein 2 | Shinoda et al. (2007) |
| GSK3 | Glycogen synthase kinase‐3 | Mao et al. (2009) |
| ITPR1 mRNA | Inositol 1,4,5‐trisphosphate receptor type 1 | Tsuboi et al. (2015) |
| KALRN | Kalirin, RhoGEF kinase | Hayashi‐Takagi et al. (2010) |
| KIF5A | Kinesin family member 5A | Taya et al. (2007) |
| LIS1 | Lissencephaly 1 | Brandon et al. (2004) |
| NDE1/NDEL1 | nudE neurodevelopment protein like 1 or NDE like 1 | Burdick et al. (2008), Ozeki et al. (2003) |
| PCM1 | Pericentriolar material 1 | Kamiya et al. (2008) |
| PCNT | Pericentrin | Miyoshi et al. (2004) |
| PDE4/PDE4B | Phosphodiesterase 4/phosphodiesterase 4B | Millar et al. (2005b) |
| TNIK | TRAF2 (TNF receptor‐associated factor 2) and NCK‐interacting kinase | Wang et al. (2011) |
| TRAK1/2 | Trafficking kinesin protein 1/2 | Ogawa et al. (2014), Norkett et al. (2016) |
While it is possible that DISC1 binds only one interactor at any given time (at sites for multiple protein binding), it is also conceivable that the process of multimerization enables simultaneous binding of partners that utilize shared sequences on DISC1. Indeed, the structural information available to date suggests that the tail domain is involved in self‐association, while the head domain is essentially free and could therefore potentially interact with up to eight proteins at the same binding sites within an octameric structure. This scenario is further complicated by data indicating that some binding partners may only interact with a specific type of DISC1 multimer. This could be the case for NDEL1, which contacts a site within the tail domain (Soares et al. 2012) and has been reported to preferentially bind DISC1 octamers and oligomers, rather than dimers or higher order multimers formed from recombinant fragments (Leliveld et al. 2008; Leliveld et al. 2009). However, another study using full‐length DISC1 found that NDEL1 binding is not dependent upon the type of DISC1 oligomer formed (Narayanan et al. 2011).
In addition to the orderly formation of multimers, DISC1 has a propensity to aggregate, particularly when overexpressed. Under these conditions, a substantial proportion of DISC1 becomes detergent‐insoluble (Millar et al. 2005a; Leliveld et al. 2008, 2009; Zhou et al. 2010; Atkin et al. 2012; Eykelenboom et al. 2012) and forms aggresomes (Atkin et al. 2012). This may be relevant to psychiatry because detergent‐insoluble DISC1 has been detected in human brain tissue, and at increased levels in postmortem samples from patients diagnosed with schizophrenia or mood disorders (Leliveld et al. 2008). Moreover, there is evidence that DISC1 aggregation and insolubility is regulated by dopamine (Trossbach et al. 2016), at least in cell culture. Aggregated DISC1 does not interact with NDEL1 (Leliveld et al. 2008), indicating that this potentially pathological mechanism could disrupt DISC1 function at many levels, including DISC1 mislocalization, and lead to the loss of critical protein–protein interactions.
Having considered the basic organization of DISC1 interactions, we now consider how they influence synaptic function.
Synaptic plasticity
Synaptic plasticity is the ability of the synapse to change its transmission characteristics, either to increase its ability to depolarize the neuron by long‐term potentiation (LTP) or to decrease its depolarizing effect by long term depression (LTD). Synaptic plasticity is an important process for synaptic development, as synapses are initially weak when they are first formed, but is also critical in the adult animal for learning, memory and sensory adaptation (Bliss & Collingridge, 1993). Functional changes in synapses are closely linked to structural and molecular rearrangements, which, as characterized in this review, are often deficient in animals with DISC1 mutations.
DISC1 is localized in dendritic spines and in the adult is particularly enriched in the postsynaptic density (PSD) (Kirkpatrick et al. 2006; Hayashi‐Takagi et al. 2010; Carlisle et al. 2011). Curiously, synaptic transmission per se appears to be largely intact in most DISC1 mutants (Cui et al. 2016), although in some cases it may adopt a less mature form than normal (Greenhill et al. 2015). Instead, the main effect of DISC1 on synaptic function appears to be its effect on synaptic plasticity. Synaptic plasticity is often studied in animal models of schizophrenia by measuring LTP, and it is therefore highly pertinent that LTP has been found to be deficient in schizophrenia patients (Frantseva et al. 2008). In the following sections, we consider the impact of DISC1 mutations on synaptic plasticity, distinguishing between mutations affecting the N‐terminal or C‐terminal domains of DISC1.
N‐terminal effects
A number of proteins interact with the N‐terminal domain of DISC1 including PDE4B, Kal‐7 and GSK3β (Table 1 and Fig. 1 B). DISC1 interacts with PDE4B at four sites within the N‐terminal domain and one in the C‐terminal tail (Murdoch et al. 2007; Fig. 1). PDE4B works to decyclize cAMP and, as a consequence, it can antagonize protein kinase A (PKA)‐mediated effects including cAMP response element‐binding protein (CREB) activation and phosphorylation of the AMPA receptor subunit GluA1, both of which are important for plasticity.
Ethylnitrosourea‐generated mutant L100P mice have a point mutation at amino acid 100 of the DISC protein, which lies in a PDE4B binding region (Table 2 and Fig. 1 C). They have normal basic synaptic transmission and normal expression levels of synaptic proteins, but show major deficits in glutamate‐induced Ca2+ elevation. Importantly, L100P mice have impaired hippocampal LTP in slices derived from adult mice (Cui et al. 2016) (Fig. 2 H).
Table 2.
DISC1 mutants: the original and follow‐on studies using the DISC1 mutants depicted in Fig. 1 are cross‐referenced, together with a description of the mutation being tested
| Mutation | Original report | Further studies using the mutant | Nature of mutation and studied as | Fig. 1 panel |
|---|---|---|---|---|
| L100P | Clapcote et al. (2007) | Lee et al. (2011a, b ), Cui et al. (2016), Tropea et al. (2016) | Ethylnitrosourea‐induced point mutation (heterozygotes and homozygotes) | C |
| DISC1 Tm1Kara | Koike et al. (2006) | Kvajo et al. (2008, 2011), Lepagnol‐Bestel et al. (2013) | Artificial stop codon and polyadenylation signal at exon 8, combined with deletion within exon 6 (heterozygotes and homozygotes) | D |
| truncated DISC1 | Shen et al. (2008) | Booth et al. (2014) | Transgenic | E |
| DISC1cc | Li et al. (2007) | Greenhill et al. (2015) | Transgenic | F |
| DISC1 Δ2/3 | Kuroda et al. (2011) | Tsuboi et al. (2015) | knockout (homozygous null) | G |
| hDISC1 | Pletnikov et al. (2008) | Transgenic | H | |
| Q31L | Clapcote et al. (2007) | Point mutation (heterozygotes and homozygotes) | I |
Figure 2. Effects of DISC1 mutations on LTP.
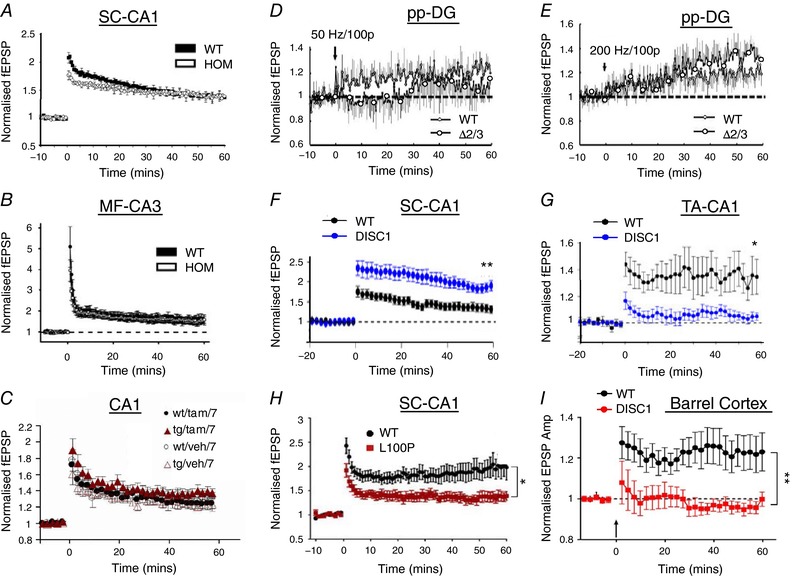
A, an early component of LTP is affected in the hippocampal CA1 (Schaffer collateral input) but LTP is still present (Kvajo et al., 2008 ). B, LTP is unaffected in the mossy fibre input to CA3 (Kvajo et al. 2011). C, LTP is unaffected in CA1 (Li et al. 2007). D and E, the early component of LTP is absent and the later component of LTP attenuated in the perforant pathway input to the dentate gyrus when induced using a 50 Hz tetanus (D), but more similar to wild‐types when induced using a 200 Hz tetanus (E) (Kuroda et al. 2011). F and G, LTP is enhanced in the Schaffer collateral pathway (F) but absent in the temperoammonic pathway of the hippocampus (G) (both, Booth et al. 2014). H, LTP is strongly reduced in the Schaffer collateral pathway (Cui et al. 2016). I, LTP is absent in the layer 4 to layer 2/3 pathway in the barrel cortex (Greenhill et al. 2015).
The impaired plasticity of L100P mice is also present in the neocortex in vivo (Tropea et al. 2016). Cortical activity and visual map organization are normal, but cortical ocular dominance plasticity is impaired in L100P mice (Tropea et al. 2016). Studies have shown that ocular dominance plasticity in the mouse visual cortex is due to a tumour necrosis factor α (TNFα)‐dependent homeostatic mechanism related to synaptic scaling (Kaneko et al. 2008; Ranson et al. 2012). Blockade of the TNFα mechanism during the critical period of development leads to a loss of potentiation of the open eye response but leaves LTP intact (Stellwagen & Malenka, 2006; Kaneko et al. 2008). The mechanism underlying this effect is not known, but phosphorylated CREB and TNFα levels are directly correlated (Datusalia & Sharma, 2014; Miao et al. 2015), and so the reduction in phosphorylated CREB by the L100P mutation may down‐regulate levels of TNFα, thereby reducing the homeostatic component of ocular dominance plasticity. Taken together with Cui et al’s findings, this implies that the L100P mutation impairs both LTP and homeostatic plasticity, suggesting that it acts at a basic mechanistic level that is common to both types of plasticity (Cui et al. 2016). Homeostatic up‐regulation of synaptic strength and LTP both involve AMPA receptor insertion, which could therefore provide a common link. However, current understanding suggests the kinase signalling pathways for LTP and homeostatic plasticity are different; while PKA influences phosphorylation of GluA1 at the S845 site to control GluA1 insertion in the membrane (Lee et al. 2000; Kopec et al. 2007; Hardingham et al. 2008), phosphoinositide‐3‐kinase (PI3K) controls GluA1 insertion through the TNFα mechanism (Stellwagen et al. 2005). Nevertheless, AMPA receptor insertion and GluA1 are important both for homeostatic up‐regulation processes (Stellwagen & Malenka, 2006; Goel et al. 2011) and ocular dominance plasticity (Ranson et al. 2013).
DISC1 interactions with PDE4B also influence plasticity through CREB's effect on gene expression. Monocular deprivation normally produces a reduction in phosphoCREB levels in the visual cortex. However, monocular deprivation fails to reduce phosphoCREB in homozygous L100P mutant mice (measured as a change in phosphoCREB immunostaining) (Tropea et al. 2016). Similarly, in neuronal culture, chemically induced LTP (cLTP) increases phosphoCREB levels in wild‐types (WT) but not in the L100P mutants (Tropea et al. 2016). Furthermore, whereas cLTP induces an increase in the expression of synaptic proteins in WTs (specifically synapsin and PSD‐95), it fails to generate significant changes in L100P‐derived cortical cultures (Tropea et al. 2016). These studies suggest that DISC1 may influence synaptogenesis and/or maturation of synapses via CREB signalling, which is defective in the L100P mutant.
The importance of the PDE4B–DISC1 interaction for the modulation of LTP is further emphasized by experiments on homozygous PDE4B mutant mice, where PDE4B was manipulated so that the affinity for cAMP was either increased or decreased (McGirr et al. 2016). Neurons in CA1 did not show LTP after high frequency stimulation in mice where the affinity between PDE4B and cAMP was increased (PDE4B+). PDE4B can influence two stages of LTP; by affecting PKA activation, PDE4B can affect the early phases of LTP (Lee et al. 2000; Kopec et al. 2007; Hardingham et al. 2008) and by affecting CREB activation, PDE4B can affect late phase LTP (Barco et al. 2002).
Kal‐7 is another important synaptic DISC1 interactor and binds DISC1 at three sites, two of which are part of the N‐terminal head domain (41–100 and 321–355) and one of which is in the C‐terminal tail domain (376–410) (see Fig. 1). Deletion of amino acids 350–394 was found to strongly affect DISC1–Kal‐7 interactions (Hayashi‐Takagi et al. 2010). Interactions between DISC1 and Kal‐7 are important for driving the NMDA‐mediated activation of Ras‐related C3 botulinum toxin substrate 1 (Rac1). Rac1 plays an important role in spine enlargement and shrinkage and, as discussed later, this is one of the mechanisms by which DISC1 can affect spine morphology. In cortical cell cultures, blockade of DISC1 has a biphasic effect on spine size with respect to time. Within 24 h of DISC1 inactivation, spines increase in size and excitatory postsynaptic currents (EPSCs) increase in frequency. In contrast, prolonged down‐regulation of DISC1 over 6 days causes a shrinkage of spines and a reduction in EPSCs (Hayashi‐Takagi et al. 2010). One note of caution, however, is that because of the limited lifetime of neuronal dissociated cultures, synapses may not reach the equivalent age at which effects are observed in vivo, though they may still give insights into the time course of DISC1 action at the cellular level. The peak of DISC1 expression in mice is P35 (Schurov et al. 2004), which is later than most conventional culture methods allow. Nevertheless, it can be seen from the above studies that the N‐terminal head of DISC1 plays an important role in shaping spine structure and plasticity through interactions with Kal‐7 and PDE4B.
C‐terminal effects
Experiments have also been performed on mice missing the C‐terminal portion of DISC1. These mutations give insight into the function of the DISC1 C‐terminal tail. In 2006, Koike and colleagues inserted a stop codon into DISC1 exon 8, followed by a polyadenylation signal, and at the same time noted that the mouse strain they had used, 129SvEv, carries a natural deletion in DISC1 exon 6 (Koike et al. 2006) (Fig. 1 D). This mutation results in an overall diminished expression of DISC1 protein, and very weak expression of a C‐terminally truncated protein corresponding to the size predicted by introduction of a premature stop codon due to the exon 6 deletion (Kvajo et al. 2008). Kvajo and colleagues transferred the modified mutated DISC1 allele into a C57BL/6 background and used this mouse model to study hippocampal circuitry. In these mice, they found that LTP can still be elicited in hippocampal CA1 (Fig. 2 A), although the initial magnitude of LTP is lower than in WT (Kvajo et al. 2008). In a follow‐up study, the authors generated a mouse strain where in addition to the DISC1 mutation, a specific population of neurons expressed green fluorescent protein (GFP) under the Thy1 promoter (Kvajo et al. 2011). They found that the axonal projections in the DISC1 mutants were disrupted, particularly those of the granule cells in the dentate gyrus (Kvajo et al. 2011), which project to CA3. In CA3, ultramicroscopic examination showed an unaltered number of synaptic vesicles but their size was reduced in mutants. Consistent with this finding, facilitation decreased more rapidly in homozygous mice as the frequency of stimulation was reduced. However, the mice again showed no deficit in LTP in this pathway (Fig. 2 B).
During the same period, another DISC1 mouse model was generated (Shen et al. 2008). This model was generated by overexpressing two copies of DISC1 exons 1–8 via an artificial chromosome (Fig. 1 E). The authors showed many anatomical and morphological alterations in these mice that mimic the neuropsychiatric phenotype, some of which are consistent with reduced neurogenesis and cell migration, such as thinning of cortical layers II/III, and others that are consistent with effects on neurite outgrowth. Booth and colleagues studied electrophysiological properties of this mouse in the hippocampal Schaffer collateral (SC) and temporo‐ammonic (TA) pathway inputs to CA1 (Booth et al. 2014). Basal synaptic transmission in mutant mice was relatively normal except that, studying the SC pathway, DISC1 slices were more variable and epileptogenic. Furthermore, the SC pathway produced higher levels of LTP in DISC1 mutants than in WTs (Fig. 2 F). In contrast, the TA pathway exhibited a profound reduction in LTP in DISC1‐mutant slices (Fig. 2 G). In both cases, theta burst stimulation (TBS) was used to induce LTP. The differences evident between TA and SC pathways in this study show that the same DISC1 mutation can differentially affect alternative inputs onto the same cells.
Booth et al.’s study and Kvajo et al.’s studies both used a DISC1 model with a C‐terminal truncation (Fig. 1 D and E), but observed different effects on LTP. There may be several reasons for the discrepancy; first, despite similarities, the animal models do differ: Booth et al. used a model with an over‐expression of the truncated form of DISC1, with maintained expression of the endogenous protein, while Kvajo and colleagues used a model with a loss of full‐length protein and an unstable expression of a C‐terminally truncated protein (Fig. 1 and Table 2). Second, Booth and colleague used TBS to induce LTP, while Kvajo et al. used high frequency stimulation. It has been shown that TBS is required to induce both nitric oxide and GluA1 components of LTP in the hippocampus, while high frequency stimulation only induces the GluA1 component (Phillips et al. 2008). It is conceivable that only one component of LTP was studied in Kvajo et al.’s experiments and the nitric oxide component of LTP in Booth et al.’s study is deficient in the TA pathway.
All the studies described so far use mutants where the expression of the mutant DISC1 is effective throughout development. However, Li et al. (2007) generated a transgenic mouse expressing the C‐terminal portion of DISC1 (Fig. 1 F) in pyramidal neurons of the forebrain where the timing of the disruption can be controlled (Li et al. 2007). The mutant protein is expressed constitutively, but continually degraded unless tamoxifen is administered, whereupon the C‐terminal fragment of DISC1 becomes active. Once the tamoxifen is metabolized, degradation of the DISC1 fusion protein resumes. This allows for a fast onset and offset of free mutant protein availability (6–48 h with a single tamoxifen injection), which then acts as a dominant negative for endogenous DISC1 by binding to the C‐terminal domain protein binding partners of DISC1, including LIS and NDEL1 (Li et al. 2007) and probably also interfering with orderly multimerization of DISC1 (Fig. 1 A). The authors disrupted DISC1 function during development or in adulthood, then ran a series of behavioural tests to identify the age at which interference of DISC1 signalling caused most functional disruption. They found that disruption of DISC1 signalling at P7 was most effective at impairing working memory and dendritic complexity in the dentate gyrus, both of which are related to human schizophrenia pathology. However, there was no effect of DISC1 disruption in adulthood, implying a developmental requirement for DISC1.
Li et al. found that LTP in the SC pathway in this heterozygous mutant was not reduced by induction of mutant DISC1 at P7, in common with Booth et al. (Fig. 2 C), but did not test the temporoammonic pathway, which has since been shown to exhibit C‐terminal‐related impairments in LTP (Booth et al. 2014).
Further studies on neocortical plasticity have thrown more light on these findings. Greenhill et al. (2015) studied the same mouse as Li et al. (2007) and found that normal DISC1 function during development is required for adult expression of LTP and experience dependent plasticity in layers 2/3 of the somatosensory cortex (Greenhill et al. 2015). Similar to Li and colleagues’ findings, the dominant negative DISC1 fragment did not affect plasticity when activated in the adult (Greenhill et al. 2015). However, a single tamoxifen injection at P7 produced a deficit in adult cortical plasticity measured months later (Fig. 2 I). Tamoxifen injections to disrupt DISC1 function at P11–13 also impaired adult plasticity, but to a lesser degree than at P7 (Greenhill et al. 2015). These findings show that DISC1 is required during a developmentally critical period in order for adult plasticity to develop in the cortex. Once again, spike timing plasticity was used to induce LTP, which affects both GluA1 and NO‐dependent aspects of plasticity (Hardingham & Fox, 2006). This early critical period coincides with the formation of synapses and dendritic branching patterns on the affected neurons (Greenhill et al. 2015), which may explain why plasticity is affected in some pathways and not others (Li et al. 2007), given that different neuronal pathways develop at different ages. The exact nature of the plasticity affected by early DISC1 dysfunction is quite specific in that it affects adult LTP and experience‐dependent potentiation, but only has a limited effect on LTD and de‐depression (Greenhill et al. 2015).
Hippocampal versus cortical effects
A number of studies have been performed on LTP in the hippocampus using a variety of DISC1 mutants. As shown in Fig. 2 A, B and C, three studies have found little or no effect of DISC1 mutation on LTP in the SC pathway (Li et al. 2007; Kvajo et al. 2008, 2011) and one actually showed an increase in LTP (Booth et al. 2014), possibly due to reduced inhibition in these mutants (Fig. 2 F). In these cases the C‐terminal domain of DISC1 was truncated for investigation. The only case where a partial reduction was seen in the level of LTP in the SC pathway was due to an N‐terminal DISC1 mutation (Fig. 2 H) (Cui et al. 2016).
Plasticity has been studied in other hippocampal pathways and found either to be abolished, as with the TA pathway, or the threshold for plasticity induction affected, as with the perforant path input to the dentate gyrus (Kuroda et al. 2011). In the latter case, an effective knockout of both the N‐ and C‐terminal domains of DISC1 was produced by deletion of exons 2 and 3 (Fig. 1 G). While it was still possible to trigger LTP using a frequency generally used to investigate LTP (50–100 Hz), LTP was reduced but not eliminated in DISC1 mutant mice (Fig. 2 D), while at 200 Hz LTP was greater in the DISC1 mutants than in WTs (Fig. 2 E).
These partial and pathway‐specific effects in the hippocampus are for the most part in contrast to the far starker effects of DISC1 mutations on plasticity in the neocortex. Greenhill et al. (2015) saw a complete abolition of LTP in layer 2/3 neurons (Fig. 2 I) and a loss of whisker deprivation induced experience‐dependent potentiation with a C‐terminally directed DISC1 manipulation, while Tropea et al. (2016) saw a clear abolition of ocular dominance plasticity in the visual cortex and a lack of cLTP in cortical cell cultures with an N‐terminally directed DISC1 mutation. Similarly, studies on spine morphology and structural plasticity in the cortex have consistently demonstrated effects of DISC1 mutations (Hayashi‐Takagi et al. 2010). It remains to be determined whether these differences are due to intrinsic properties of the neocortex and hippocampus, or whether some of the differences arise due to the different DISC1 mutations used in each study. It is also important to note that ocular dominance plasticity involves LTD and homeostatic plasticity, which are generally not investigated in the hippocampus of DISC1 mutants (LTP is normally measured). One of the major differences between the neocortex and hippocampus is the longevity and stability of synapses. Synapses can last for a period of several months in the neocortex, while most hippocampal synapses turn over within a period of a month (Yang et al. 2009a; Attardo et al. 2015). If DISC1 does have a different role in the hippocampus and cortex, it could indicate a role for DISC1 in structural plasticity and the long‐term stability of synapses, particularly given the demonstrated role of DISC1 in cytoskeletal interactions and intracellular trafficking, as will be discussed in the following sections.
Development and control of neuronal morphology
Schizophrenic patients show a deficit in spine density in deep layer 3 cortical pyramidal neurons in the prefrontal cortex (Glantz & Lewis, 2000), and a number of mental disorders have been linked to deficits in dendritic spines (Penzes et al. 2011). Dendritic spines contain the postsynaptic compartment and receive the majority of the excitatory synaptic input in the brain and thus changes in dendritic spine morphology are closely linked to synaptic plasticity (Kasai et al. 2010). The link between DISC1 and mental disorders has thus prompted research into the effects of DISC1 at the structural level
An initial study on human frontal and parietal cortex showed that a pool of DISC1 is located at the synapse (Kirkpatrick et al. 2006) with some 40% of synapses, particularly excitatory synapses, labelled for DISC1. A similar localization of DISC1 at the PSD was shown both in rat cortical and hippocampal cell cultures (Hayashi‐Takagi et al. 2010; Wang et al. 2011). However, as many studies have shown, the effects of DISC1 are not only imposed at the synaptic level (Fig. 3 A). The DISC1 interactome points towards multiple functions of DISC1 during development: effects on neuronal proliferation, migration, neurite outgrowth and the formation and maintenance of synapses (Brandon, 2007; Camargo et al. 2007). Whilst changes in spine morphology are most closely linked to synaptic plasticity, it is apparent that any change in neuronal morphology, from incorrect neuronal location to retarded or reduced neurite outgrowth, could lead to incorrect connections being formed between neurons and synaptic plasticity subsequently being affected in the mature brain. In this sense, plasticity could be impaired by malformation of a neuronal circuit. Alternatively, by affecting the dendritic composition during development, DISC1 could permanently affect the properties of the spines that constantly form and retract on the dendrites and thereby affect their ability to undergo synaptic plasticity. Consideration of these hypotheses requires a discussion of DISC1's cytoskeletal interactions.
Figure 3. Cellular locations and protein interactions of DISC1.
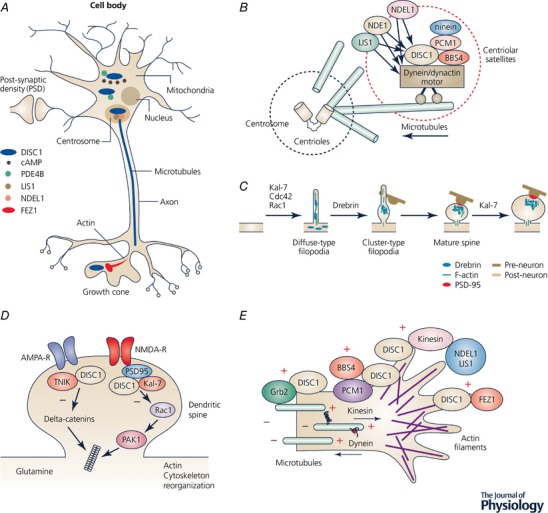
A, a schematic representation of a neuronal cell body and dendritic processes, axon‐aligned microtubules and actin‐rich growth cones. DISC1 complexes are found at the centrosome (cream) and mitochondria (black) in the cell body and at the growth cones (invagination at tip of axon). DISC1 interacts with PDE4B at mitochondria and in the cytosol and also localizes to the centrosome where it complexes with NDEL1 and LIS1. DISC1 is also found at the postsynaptic density (expanded in figure) and also colocalizes with FEZ1 at growth cones (also expanded in figure). B, DISC1 at the centrosome. DISC1 is known to bind to a number of proteins localized to the centrosome including NDEL1, LIS1, PCM1 and Bardet–Biedl syndrome proteins (BBSs). DISC1 plays a role in anchoring these molecules in association with the dynein motor complex and the centrosome, regulating microtubule organization. C, schematic representation of spine formation. During synaptogenesis, dendrites are covered with many diffuse‐type filopodia in which drebrin is diffusely distributed. After an axon terminal makes contact with a filopodium, drebrin clusters with F‐actin at the postsynaptic site and forms a cluster‐type filopodium. PSD‐95 cluster formation follows drebrin–actin cluster formation. The drebrin–actin complex tethers the postsynaptic machinery and is crucial to the maturation of the dendritic spines. Molecules implicated in each of the developmental processes are drawn above the adjoining arrows. Kal‐7, Cdc42 and Rac1 are all implicated in filopodium formation, drebrin in the conversion of filopodia to spines and Kal‐7 is also implicated in spine growth. D, DISC1 at the synapse. Multiple lines of evidence show that DISC1 is a component of the postsynaptic density (PSD) of excitatory synapses and regulates their form and function. The effects of DISC1 are mediated through a range of protein interaction partners. Two of these interactors that have been studied considerably are PSD proteins Kal‐7 and TNIK, which both regulate the actin cytoskeleton, most probably via separate pathways. E, organization of the axonal growth cone. Microtubules distributed along the axonal shaft extend into the growth cone. The growth cone is enriched with F‐actin bundles that form filopodia or lamellipodia. Cytoplasmic dynein moves along the microtubules towards the minus end, while kinesin moves along the microtubules towards the plus end. Various DISC1 interactors have been implicated in the activation of kinesin, including FEZ1, Grb2, PCM1 and the NDE1/NDEL‐1‐LIS1 complex.
Effects on dendrites
Multiple laboratories have shown that DISC1 interacts with proteins that bind to microtubules and associated complexes (Fig. 3 B); these proteins include NDE1, NDEL1 and LIS1 (Morris et al. 2003; Ozeki et al. 2003; Miyoshi et al. 2004; Brandon et al. 2005; Bradshaw et al. 2009; Wang and Brandon, 2011). The role of DISC1 disruption in attenuating neurite outgrowth is well established (Miyoshi et al. 2003; Ozeki et al. 2003; Pletnikov et al. 2008; Shen et al. 2008), as is the involvement of NDEL1 and LIS1 in this process, (Kamiya et al. 2006; Taya et al. 2007; Shim et al. 2008). As discussed in more detail in intracellular trafficking (see below), DISC1 is a component of the microtubule‐associated dynein motor complex (Fig. 3 E). Disruption of DISC1 early in development has been shown to affect microtubule dynamics and produce a disorganized microtubular network, subsequently impairing neurite outgrowth (Kamiya et al. 2005). In these experiments a C‐terminally truncated form of DISC1, which functions as a dominant negative protein, was used to block accumulation of DISC1, NDEL1 and their dynein‐associated proteins at the centrosome (Fig. 3 B). This disruption caused a marked retardation in both neuronal migration and neurite outgrowth in the developing cortex (Kamiya et al. 2005). The Disc1‐L100P mutant mouse has an N‐terminal DISC1 mutation (Fig. 1 C) that also produces a change in dendrite and spine organization. Homozygous L100P mice exhibit deficits in both dendrite length and spine density in frontal cortex neurons (Lee et al. 2011b). A key mechanism in this effect was shown to be the loss of the interaction between DISC1 and GSK3β, as inactivation of GSK3α rescue spine density in L100P mutant mice but does not rescue spine density (Lee et al. 2011b).
Abnormalities in dendritic complexity in C‐terminally truncated DISC1 mutant mice have also been found, with dentate gyrus granule cells from homozygous DISC1Tm1Kara mice showing reduced dendritic length (Kvajo et al. 2008, 2011); dendritic length was also reduced in the frontal cortex but not in CA1 hippocampus (Lee et al. 2011a). There is evidence that effects of DISC1 mutations are sometimes cell type specific; cultured hippocampal neurons exhibited deficits in both axonal and dendritic length and branching and also at the spine level, but in cultured cortical neurons, reductions in spine density were observed in both heterozygous and homozygous mutant mice (Lepagnol‐Bestel et al. 2013). DISC1 has also been demonstrated to regulate neurite outgrowth by controlling cell–cell adhesion and overexpression of DISC1 increased the expression of N‐cadherin and β‐1‐integrin protein as well as enhancing neurite outgrowth (Hattori et al. 2010).
Three further studies have examined the role of DISC1 in controlling dendritic morphology at particular stages of development. Pletnikov et al. (2008) used the Tet‐off system to express a C‐terminally truncated form of DISC1 (Fig. 1 H). This led to decreased neurite outgrowth in primary cortical neurons and reduced levels of LIS1 and SNAP‐25 in forebrain areas (Pletnikov et al. 2008). Li et al. used a mouse with an inducible and transient (6–48 h) disruption of DISC1 signalling (see ‘Synaptic plasticity’ for details) (Li et al. 2007). They found that a single period of disruption at P7 produced chronic reductions in dendritic complexity in the adult hippocampus in transgenic heterozygous mice. Using the same mouse with inducible disruption of DISC1 signalling at P7, Greenhill et al. showed that dendritic growth was significantly attenuated in layer 2/3 cortical neurons at both P11 and P14, but returned to normal levels in adulthood, presumably due to the transient expression of the dominant negative protein (Greenhill et al. 2015).
Effects on dendritic spines
Given the initial observation that schizophrenic patients exhibited a reduced spine density and the link between DISC1 and neurological disorders, it is pertinent that one of the most consistent observations made so far in many DISC1 mutants is a reduction in spine density. The Q31L (Fig. 1 I) and L100P (Fig. 1 C) mutant mice, which both have mutations in the PDE4B binding sites, show reduced spine density on pyramidal neurons in layers III and V of the frontal cortex and CA1 neurons of the hippocampus in homozygous mutants (Lee et al. 2011a). While the DISC1 mutation in this mouse does not occur in human patients, it nevertheless demonstrates the role of DISC1 in spine regulation. The homozygous DISC1TM1Kara, carrying a targeted exon 8 stop codon and creating a mouse with truncated DISC1 and no full length DISC1 expression (Fig. 1 D), also shows a decrease in synaptic spines in the dentate gyrus and in layer 2/3 prefrontal cortex (or PFC) neurons, but not in layer 5 neurons (Kvajo et al. 2008; Juan et al. 2014; Crabtree et al. 2017). Cultured hippocampal neurons from these mice showed reductions in mushroom and stubby spines but not reductions in filopodia, whilst cultured cortical neurons showed reductions in all classes of spines in both heterozygous and homozygous mutant mice (Lepagnol‐Bestel et al. 2013). Long‐term suppression of DISC1 has also been shown to produce changes at the spine level in cultured cortical neurons, with reductions in spine density and effects on spine morphology, specifically reductions in spine size and spine length (Hayashi‐Takagi et al. 2010).
In concert with this finding, a disruption of DISC1 signalling for 6–48 h (dominant negative, Fig. 1 F) starting at P7 produced almost immediate deficits in spine density just 24 h later at P8. Furthermore, even though the DISC1 disruption only lasted 48 h, the deficit in spine density lasted into adulthood and was accompanied by lasting deficits in synaptic plasticity (see previous section) (Greenhill et al. 2015). At P7, layer 2/3 neurons show immature morphology with short dendrites and limited dendritic branching (Greenhill et al. 2015). Only primary neurites are developed at this time and higher order dendrites are in the midst of developing (Greenhill et al. 2015). The reductions in spine density caused by DISC1 disruption only occurred on second and third order dendrites, i.e. those dendrites developing at the time that DISC1 function was impaired. Among the spines that did develop following DISC1 disruption, a higher proportion than normal were thin spines and a lower proportion were mushroom spines (Greenhill et al. 2015). Since spine morphology is representative of maturation state, with thin spines often associated with an early stage of synapse stabilization and maturation, while mushroom‐type spines are associated with stabilized synapses important for synaptic transmission (Tropea et al. 2010), one can infer that DISC1 has an effect both on synaptic density and on spine maturation.
How does DISC1 affect spine density and maturation? Regulation of the actin cytoskeleton is one of the cellular processes enriched in the DISC1 interactome (Camargo et al. 2007). The actin cytoskeleton is critical in regulating growth cone motility (Fig. 3 E), spine formation (Fig. 3 C) and underlying synaptic plasticity (Ramakers 2002; Sekino et al. 2007; Cingolani & Goda, 2008). An increasing number of intracellular signalling pathways have been shown to regulate the actin cytoskeleton and to be critical for spine structure (Calabrese et al. 2006). The RhoGTPases Cdc42 and Rac1, which regulate the outgrowth of the actin cytoskeleton, are involved in the formation of filopodia (Kozma et al. 1997) and are also implicated in the maintenance of dendritic spines and thus in the regulation of spine density (Nakayama et al. 2000). Rho proteins regulate cell adhesion through proteins linked to the actin cytoskeleton, such as cadherins and integrins, which are present at synapses and are also involved in synaptic plasticity (Ramakers, 2002). As we have already noted (see ‘Synaptic plasticity’), DISC1 binds to Kal‐7 and prevents access of Kal‐7 to Rac1 (Fig. 3 D), controlling the duration and intensity of Rac1 activation in response to NMDA activation, when the DISC1 anchor to Kal‐7 is weakened, leading to subsequent spine enlargement (Hayashi‐Takagi et al. 2010). Kal‐7 also increases spine formation when overexpressed (Hayashi‐Takagi et al. 2010) and there is a strong correlation found between spine density and Duo (the human version of Kal‐7) in schizophrenic patients (Hill et al. 2006).
DISC1 also interacts with another kinase, Traf2 and Nck interacting kinase (TNIK), in the dendritic spine (Fig. 1 B), and inhibits the kinase activity of TNIK (Wang et al. 2011). TNIK is specifically expressed in the brain and highly enriched in the PSD, while inhibition of TNIK leads to loss of PSD95, surface GluA1 and reductions in both mEPSC frequency and mEPSC amplitude (Hussain et al. 2010; Wang et al. 2011; Burette et al. 2015). Furthermore, knockdown of TNIK signalling through RNAi leads to reductions in hippocampal dendritic branching and spine density (Hussain et al. 2010). Recent studies suggest these effects may be mediated by δ‐catenin family proteins, part of the classic cadherin adhesion complexes at synapses required for synaptogenesis, spine growth and synaptic plasticity (Wang et al. 2016).
Haploinsufficiency in LIS1, one of the major binding partners at the C‐terminal domain of DISC1 (Fig. 1 B), has also been shown to reduce spine density both in hippocampal CA1 and barrel cortex and also filopodial length in CA1, with a lower spine turnover and elimination in vivo, while down‐regulation of RhoA rescued spine motility. Therefore, this is likely to be a pathway involved in DISC1's effects on spines (Sudarov et al. 2013). GSK3α is known to be a specific modulator of spine density in L100P mice, because its genetic inactivation rescues spine density in L100P/GSK3α mutants, while all the other morphological alterations remain unchanged (Lee et al. 2011b). Interestingly, Dixdc1 also affects dendritic spines and interacts with DISC1 to control GSK3 and Wnt signalling (Martin et al. 2018), though unlike the other interactors mentioned in this section, it is not yet known to be located synaptically. In conclusion, there is overwhelming evidence that DISC1 is important for dendritic spine structure and function. Interestingly, DISC1 can also influence development of the presynaptic side of the synapse, as will be discussed in the following section.
Effects on axons
In addition to its established role in dendritic development, DISC1 is also involved in axonal development, most notably in granule cells in the dentate gyrus projecting to their targets in CA3 (Kvajo et al. 2011) but also in cultured hippocampal neurons (Lepgagnol‐Bestel et al. 2013). FEZ1 and pericentriolar material (PCM1) are two microtubule interacting proteins that bind to DISC1 (Fig. 1 B) and are linked to axonal development. Centrosomal PCM1 is required for correct development of axonal morphology and is recruited co‐operatively by interacting proteins DISC1 and Bardet–Biedl syndrome 4 (BBS4) (Kamiya et al. 2008; de Anda et al. 2010), while alterations in axonal targeting may well involve abnormal elevations in cAMP, as correction of cAMP levels was found to be a means of reversing the deficits (Kvajo et al. 2011).
FEZ1 has been identified as an important DISC1 interacting partner that participates in neurite outgrowth and co‐localizes with DISC1 at axonal growth cones (Fig. 1 B), whilst interactions are associated with F‐actin (Fig. 3 E) (Fujita et al. 2007). FEZ1 is known to be involved in the activation of the kinesin‐1 motor protein and in the transport of mitochondria (Blasius et al. 2007; Fujita et al. 2007) (see ‘Mitochondrial trafficking’ below). The expression of FEZ1 is again developmentally controlled and regulated synchronously with DISC1 (Honda et al. 2004). FEZ1, and by implication DISC1, are thus thought to function in axon growth and guidance, possibly involving cytoskeletal remodelling at the axonal growth cone (Miyoshi et al. 2003). Consistent with this, up‐regulation of the DISC1/FEZ1 interaction enhances the extension of neurites while silencing of DISC1 inhibits neurite production (Miyoshi et al. 2003; Kamiya et al. 2005). DISC1 also plays a role in axon elongation through an interaction with growth factor receptor‐bound protein 2 (GRB2), resulting in activation of extracellular signal‐regulated kinase (ERK) (Fig. 3 E) (Shinoda et al. 2007), or through kinesin and the NDEL1–LIS1 complex in axonal growth cones (Shinoda et al. 2007; Taya et al. 2007), while knockdown of DISC1, kinesin1, NDEL1 or LIS1 inhibited axon elongation (Taya et al. 2007).
Thus, while it is clear that DISC1 perturbation affects various aspects of neurogenesis, neural migration, neurite growth and both spinogenesis and spine maintenance, questions remain about how these processes are controlled. At a superficial level, it is possible to imagine that many of the processes involved in cytoskeletal organization during migration and neurite outgrowth are also involved in the maintenance and plasticity of dendrites and dendritic spines, as discussed above. Given the role of DISC1 in synaptic plasticity and spine morphology, particularly in the neocortex, understanding this process is likely to help in understanding spine plasticity and stability as well as the consequences of disrupting it. One further process that also involves DISC1, which may affect both the development and maintenance of neuronal structure, is neuronal trafficking, and this topic is discussed in the next section.
Neuronal intracellular trafficking
Due to their highly elongated morphology, neurons are reliant upon efficient microtubule‐based transport of various cargoes between soma and synapses, which can be a considerable distance in some neurons. This process utilizes the molecular motors kinesin and dynein, which transport cargo in the anterograde (away from the soma) or retrograde (towards the soma) directions, respectively, along the polarized microtubules in axons (Fig. 3 E). As discussed below, both kinesin and dynein are DISC1 interactors. Dendrites contain microtubule bundles of mixed polarity and thus cargo transport is more complex in this neuronal compartment (Franker & Hoogenraad, 2013). In axons and dendrites, many cargoes undergo bidirectional transport, often exhibiting saltatory movements in both directions, but with overall movement in a single direction eventually achieved (Franker & Hoogenraad, 2013). The mechanism underlying this pattern of movement is unknown, but cargoes appear to be simultaneously complexed with both dynein and kinesin, and it has been proposed that a ‘tug‐of‐war’ occurs between the opposing motors, with one ultimately predominating to determine the overall direction of travel (Bryantseva & Zhapparova, 2012). This hypothesis is, however, complicated by the observation that disruption of the activity of either kinesin or dynein affects movement in both directions (Franker & Hoogenraad, 2013).
A general role is emerging for DISC1 in microtubule‐based cargo transport because it complexes with both kinesin and dynein. DISC1 affinity chromatography using rat brain lysates demonstrated that DISC1 interacts or associates with kinesin family member (KIF) 1B, KIF5A, KIF5B, KIF5C and kinesin light chains 1 and 2 (KLC1 and KLC2) (Shinoda et al. 2007; Taya et al. 2007; Tsuboi et al. 2015). Dynein intermediate chain (DIC) was also found to complex with DISC1 by co‐immunoprecipitation from PC12 cells (Kamiya et al. 2005) and by DISC1 affinity chromatography (Taya et al. 2007). DISC1 is therefore well placed to regulate both kinesin and dynein, possibly co‐ordinately, and/or to connect cargo with these motors to facilitate their anterograde and retrograde trafficking within axons and dendrites.
There are currently few mechanistic clues as to how DISC1 affects kinesin activity and anterograde cargo transport, apart from a possible role in facilitating cargo recruitment to kinesin adaptors (Flores et al. 2011), or in kinesin activation through an interaction with FEZ1 (Blasius et al. 2007). More is known, however, about its role in dynein regulation. Dynein activity is controlled by the multiprotein dynactin complex with which DISC1 associates (Kamiya et al. 2005), and by the DISC1 binding partner LIS1 (Ozeki et al. 2003; Brandon et al. 2004)(Burdick et al. 2008; Bradshaw et al. 2009) and its accessory proteins NDE1 and NDEL1 (Cianfrocco et al. 2015; Bradshaw & Hayashi, 2017). The DISC1 interactor and cAMP phosphodiesterase PDE4 also interacts with LIS1 (Murdoch et al. 2011; Houslay et al. 2017), NDE1 (Bradshaw et al. 2008) and NDEL1 (Collins et al. 2008), and DISC1 and PDE4 control the composition of LIS1–NDE1–NDEL1 complexes via cAMP‐dependent phosphorylation (Collins et al. 2008; Bradshaw et al. 2011; Murdoch et al. 2011). It follows that DISC1 is likely to regulate retrograde cargo transport through interactions with LIS1, NDE1 and NDEL1 (Bradshaw et al. 2011; Murdoch et al. 2011), and that this mechanism may involve the second messenger cAMP, although the latter has yet to be proven.
Consistent with the above, DISC1 is now known to modulate motility of diverse cargoes in neurons. It is suggested to be involved in endocytosis of amyloid precursor protein (APP) (Shahani et al. 2015) and GABAA receptor trafficking (Wei et al. 2015), but the best studied DISC1‐modulated cargoes to date are mitochondria, messenger RNAs and synaptic vesicles. In the following sections we will discuss how defective trafficking of these three cargoes is likely to affect synapse function and plasticity.
Mitochondrial trafficking
The brain uses approximately 20% of all the energy utilized by the body at rest, and most of this energy is used by neurons (Harris et al. 2012). Mitochondria provide the majority of energy utilized by neurons, in the form of ATP (Harris et al. 2012). Much of this ATP consumption is due to neurotransmission, for example driving synaptic vesicle cycling in dendrites and powering ion pumps at the post‐synapse (Harris et al. 2012). Mitochondria also power all aspects of neuron development. Of particular relevance to this review, mitochondria are abundant at axonal growth cones and are required for dendritic spine remodelling (Li et al. 2004). Neurons are therefore extremely sensitive to mitochondrial defects and/or mitochondrial trafficking dysfunction.
DISC1 is directly involved in bidirectional mitochondrial motility through interaction with the adaptor proteins trafficking kinesin‐binding protein (TRAK) 1 and TRAK2 (Schwarz, 2013; Ogawa et al. 2014; Norkett et al. 2016). Both TRAK1 and TRAK2 also bind mitochondrial Rho GTPase (Ras homolog family member T1/2 (RHOT1/2, also known as MIRO1/2)), which is embedded in the outer mitochondrial membrane (Schwarz, 2013), where they therefore help link mitochondria to the molecular motors that drive their bidirectional transport along neuronal processes. TRAK1 and TRAK2 interact with DISC1 through a highly conserved arginine‐rich motif within the DISC1 head domain that is essential for both TRAK1 and TRAK2 binding (Ogawa et al. 2014; Norkett et al. 2016) and mitochondrial localization of DISC1 (Ogawa et al. 2014), indicating that the TRAK proteins recruit DISC1 to mitochondria.
Time‐lapse imaging of live neurons containing fluorescently labelled mitochondria has demonstrated that DISC1 regulates mitochondrial motility. DISC1 RNA interference and overexpression reduce and increase the proportion of moving mitochondria in axons, respectively (Atkin et al. 2012), while DISC1 overexpression also promotes axonal movement in the anterograde direction at the expense of retrograde movement (Ogawa et al. 2014). In contrast, a common DISC1 sequence variant, involving a change of leucine to phenylalanine at position 607 (607F, Fig. 1 A) that influences brain structure and function (Thomson et al. 2013), abolishes the ability of exogenous DISC1 to increase axonal mitochondrial motility (Atkin et al. 2012). Moreover, a rare sequence change of arginine to tryptophan at position 37 (37W), found only in four psychiatric patients to date (Song et al. 2008; Thomson et al. 2014), blocks the ability of exogenous DISC1 to promote anterograde axonal mitochondrial movement (Ogawa et al. 2014). Leucine 607 is located within a putative leucine zipper (Thomson et al. 2013), while arginine 37 is located within the conserved arginine‐rich motif that is required for DISC1–TRAK interaction (Ogawa et al. 2014; Norkett et al. 2016). Indeed, the 37W variant increases DISC1–TRAK1 binding and decreases TRAK1–MIRO binding (Ogawa et al. 2014), which may contribute to its effect on axonal mitochondrial transport regulation by DISC1. As well as these sequence variants, an aberrant C‐terminally truncated chimeric form of DISC1, which may arise from the t(1;11) translocation, has been investigated in relation to mitochondrial motility. This DISC1 species is strongly targeted to mitochondria and causes extreme mitochondrial dysfunction when artificially overexpressed (Eykelenboom et al. 2012). It also specifically inhibits axonal mitochondrial motility when overexpressed (Norkett et al. 2016), although it is not clear whether this is a direct effect, or whether it is due to neuronal pathology as a result of mitochondrial dysfunction. In addition to these axonal effects, DISC1 also regulates mitochondrial motility within dendrites (Norkett et al. 2016).
The DISC1 interactor NDE1 (Burdick et al. 2008; Bradshaw et al. 2009) also complexes with TRAK1 (Ogawa et al. 2016) and controls mitochondrial motility. As predicted from its role as a dynein activator (Liang et al. 2004; Li et al. 2005; McKenney et al. 2010), NDE1 promotes retrograde axonal mitochondrial transport (Ogawa et al. 2016). The NDE1 interactors LIS1 and NDEL1 also regulate axonal mitochondrial motility (Shao et al. 2013). Although LIS1 and NDEL1 have not yet been directly linked to the mitochondrial trafficking machinery, since NDE1 is a component, it is probable that LIS1 and NDEL1 are also components of the transportation complex that are required for dynein regulation.
GSK3β is another TRAK1‐associated DISC1 interactor (Mao et al. 2011; Ogawa et al. 2016). Its role in mitochondrial trafficking has been examined in several studies (Murphy & Millar, 2017) and, while the results are somewhat conflicting, there is broad agreement that GSK3β regulates the number of motile mitochondria, as well as their velocity, primarily in the anterograde direction (Murphy & Millar, 2017). Moreover, the DISC1 607F variant interferes with DISC1/GSK3β binding (Singh et al. 2011) and may therefore disrupt DISC1's ability to promote mitochondrial motility via a loss of GSK3β function in the trafficking complex.
Altogether, these observations indicate that DISC1 co‐ordinates directional mitochondrial movement through its binding partners. Another DISC1 interactor, FEZ1 (Miyoshi et al. 2003), may also be involved because it is known to specifically regulate mitochondrial trafficking (Butkevich et al. 2016), although its role in this process has not yet been examined with respect to its interaction with DISC1.
As well as being transported along neuronal processes, mitochondria must also respond to stop signals at specific sites, where functions such as mitochondrial ATP production can take place. NMDA receptor‐mediated calcium influx is one of the major signals that causes mitochondria to stop, and this occurs via the calcium‐sensing activity of MIRO (Macaskill et al. 2009; Wang & Schwarz, 2009). Another mechanism arresting mitochondrial movement involves syntaphilin (SNPH) which has been described as a docking receptor for axonal mitochondria, since it associates with both mitochondria and the microtubules to immobilize mitochondria (Kang et al. 2008). DISC1 may associate with SNPH to arrest axonal mitochondrial movement in response to neuronal activation (Park et al. 2016). DISC1 and its binding partners therefore regulate multiple aspects of mitochondrial trafficking, and neurons are thus critically dependent upon these proteins for their energy requirements to be met for proper development and synaptic transmission.
Dendritic messenger RNA transport
Synaptic activity is partly regulated by localized protein expression, achieved through control of dendritic distribution and translation of mRNAs (Klein et al. 2016). At least 2550 mRNAs are known to be present in dendrites, and localization of many of these is dependent upon structural elements within their 3′‐UTR (Klein et al. 2016).
A role for DISC1 in dendritic mRNA targeting was revealed by DISC1 affinity chromatography, which demonstrated its association with the RNA binding proteins heterogeneous nuclear ribonucleoproteins (hnRNP), synaptotagmin‐binding cytoplasmic RNA interacting protein (SYNCRIP), hematopoietic zinc finger (HZF), PURα and receptor for activated C kinase 1 (RACK1), as well as LIS1 and KIF5A (Tsuboi et al. 2015). Of these proteins, SYNCRIP and HZF are components of RNA granules that transport and regulate translation of mRNA in dendrites (Bannai et al. 2004; Iijima et al. 2005; Duning et al. 2008). Consistent with this, DISC1 was found to co‐localize with markers of RNA granules within axons and dendrites (Tsuboi et al. 2015). HZF binds the 3′‐UTR of mRNA encoding the intracellular calcium release channel ITPR1 (Iijima et al. 2005), while live neuron time‐lapse imaging demonstrated that DISC1 is co‐transported bidirectionally with fluorescently tagged ITPR1 mRNA 3′‐UTR within dendrites (Tsuboi et al. 2015), suggesting a role for DISC1 in dendritic ITPR1 mRNA transport. To examine this possibility further, a mutant mouse was studied in which DISC1 exons 2 and 3 were knocked out (Fig. 1 G) (Kuroda et al. 2011). Cultured neurons from this mutant mouse, which lacks any full‐length DISC1 expression, exhibit decreased ITPR1 3′‐UTR transport (Tsuboi et al. 2015), confirming a role for DISC1 in dendritic ITPR1 mRNA trafficking. RNA binding assays were then used to demonstrate that DISC1 binds directly to the 3′‐UTR of ITPR1 mRNA in an HZF‐dependent manner, and this was found to involve the conserved multifunctional arginine‐rich motif within the head domain of DISC1 (Tsuboi et al. 2015) that is also required for mitochondrial localization and trafficking (Ogawa et al. 2014).
ITPR1 transcripts encode an inositol 1,4,5,‐trisphosphate (IP3) receptor that mediates release of intracellular calcium to control several cellular functions including neurotransmission and synaptic plasticity (Fedorenko et al. 2014), indicating a novel route by which DISC1 can control neuronal function. Importantly, however, DISC1 was also found to bind to the 3′‐UTRs from a number of other mRNAs that are trafficked within dendrites. Of the 40 dendritic transcripts tested for DISC1 binding, mRNAs encoding the DISC1 interactor kalirin, which regulates neuronal morphology and plasticity (see ‘Effects on dendritic spines’) (Hayashi‐Takagi et al. 2010), and the voltage‐gated potassium channels KCNC1 and KCNC4 were identified as being DISC1‐associated (Tsuboi et al. 2015). Transcripts from the CACNA1C and CACNA2D1 genes encoding voltage‐gated calcium channel subunits were also found to be DISC1‐associated. Taking each in turn, Cav1.2 subunits encoded by the CACNA1C gene operate postsynaptically at glutamatergic synapses, where they regulate synaptic plasticity through the CREB signalling pathway and synaptic expression of calcium‐permeable AMPA receptors (Moosmang et al. 2005), while at GABAergic synapses they regulate short term GABAergic plasticity (Yang et al. 2009b). Second, the CACNA2D1 gene encodes auxiliary α2/δ1 subunits that regulate presynaptic voltage‐gated calcium channel trafficking, plasma membrane expression and biophysical properties, as well as synaptogenesis (Yang et al. 2009b).
The mRNA transport mechanism involves KIF5A (Tsuboi et al. 2015), and since the RNA granules mediating mRNA trafficking move bidirectionally (Tsuboi et al. 2015), dynein, regulated by LIS1, may also participate in RNA granule transport. Moreover, since DISC1‐associated RNA granules were detected in axons as well as dendrites (albeit to a lesser extent) (Tsuboi et al. 2015), it is possible that DISC1 also modulates axonal mRNA trafficking, consistent with the postsynaptic location of some of the encoded proteins. This role in mRNA trafficking implicates DISC1 in regulation of the localized expression of a specific set of proteins that regulate neurotransmission and is likely related to effects on late phase LTP in the DISC1 knockout mouse (Fig. 1 G, see also ‘Synaptic plasticity’) (Tsuboi et al. 2015). Indeed, disruption of DISC1–mRNA binding blocks the maintenance of LTP (Tsuboi et al. 2015). Notably, however, the strategy used to disrupt DISC1–mRNA binding, a peptide encompassing the DISC1 arginine‐rich region, is also likely to disrupt the DISC1 interaction with TRAK1/2 and therefore affect mitochondrial trafficking (Fig. 1 B). It may also affect the binding of PDE4B and kalirin to DISC1 (Fig. 1 B). In conclusion, there is good evidence that DISC1 can affect mRNA trafficking of several factors involved in synaptic structure and function. In the final section on trafficking we consider the evidence regarding the role of DISC1 in trafficking proteins in synaptic vesicles.
Synaptic vesicle transport
Synaptic vesicles store various neurotransmitters that are released into the synaptic cleft in response to presynaptic depolarization‐induced calcium ion influx through voltage‐gated calcium channels. Following neurotransmitter release, the synaptic vesicles are endocytosed, recycled and reloaded with neurotransmitter. These processes require large amounts of energy in the form of ATP.
Regulation of synaptic vesicle movement along neuronal processes by DISC1 was first demonstrated by tagging vesicles with a fluorescently labelled synaptic vesicle marker, vesicle‐associated membrane protein 2 (VAMP2), in cultured mouse neurons (Flores et al. 2011). DISC1 knockdown by RNA interference was found to cause aberrant vesicle distribution in neuronal processes, and live neuron time‐lapse imaging demonstrated diminished synaptic vesicle movement. Overexpression of a C‐terminally truncated form of DISC1 also decreased synaptic vesicle motility, and intriguingly, this effect was reversed by treatment with lithium, which is in clinical use as a mood stabiliser (Flores et al. 2011). This mechanism involves the DISC1 interactor FEZ1 (Miyoshi et al. 2003), which binds to and facilitates activation of kinesin (Blasius et al. 2007). FEZ1 regulates synaptic vesicle transport and binds to the synaptic vesicle membrane protein synaptotagmin‐1 (SYT1) in Drosophila (Gindhart et al. 2003; Toda et al. 2008). In mammalian cells DISC1 enhances FEZ1–SYT1 interaction, with the enhancing effect blocked by the C‐terminally truncated DISC1 species and rescued by lithium treatment (Flores et al. 2011). DISC1 therefore promotes synaptic vesicle movement by linking synaptic vesicles to kinesin through its interactor FEZ1.
The next study to demonstrate involvement of DISC1 in the synaptic vesicle cycle utilized induced pluripotent stem cell (IPSC)‐derived neurons (Wen et al. 2014) from a family carrying a 4 bp deletion in DISC1 exon 12 (Sachs et al. 2005). This mutation causes a shift of frame and introduces a premature stop codon (Sachs et al. 2005) and is likely to affect DISC1 binding to NDE1 and NDEL1, as well as potentially affecting DISC1 binding to LIS1 and the orderly assembly of DISC1 multimers (Fig. 1 A and B; see also ‘Organization of DISC1 protein interactions’). Several members of this family have a diagnosis of psychiatric illness, and of those examined, two mutation carriers have schizophrenia and one carrier has schizoaffective disorder, while two non‐carriers have major depression and one non‐carrier has a schizotypal personality disorder (Sachs et al. 2005). It has been suggested that the DISC1 mutation is linked to mental illness in this family, although co‐segregation of the mutation with psychiatric illness is incomplete. In IPSC‐derived neurons, mutant DISC1 is expressed normally at the transcript level, but the protein expression is substantially reduced (Wen et al. 2014). Mutant neurons express lower levels of the synaptic vesicle marker SV2 and, consistent with this, mutant neurons also exhibit a lower frequency of excitatory spontaneous synaptic currents, implying a defect in presynaptic neurotransmitter release (Wen et al. 2014). This was further investigated using the dye FM1‐43, which is intensely fluorescent when taken up by synaptic vesicles. Potassium chloride‐induced synaptic vesicle release was quantified using this dye, and fluorescence reduction over time was found to be lower in mutant neurons, confirming a deficit in synaptic vesicle release (Wen et al. 2014). The involvement of DISC1 in these effects was confirmed by correcting the DISC1 expression in mutant lines, and introducing the DISC1 mutation into control lines. Expression of a number of synaptic proteins was altered in the mutant neurons, including increased expression of synapsin (SYN) 2 and SYN3 (Wen et al. 2014). Upregulation of SYN1 has previously been shown to suppress neurotransmitter release (Hackett et al. 1990; Rosahl et al. 1993), and therefore it is possible that increased SYN2 and SYN3 expression acts similarly. Expression of the transcription factor myocyte‐specific enhancer factor 2C (MEF2C) was decreased in the mutant neurons (Wen et al. 2014); this is a change which has previously been shown to decrease the frequency of spontaneous synaptic currents (Barbosa et al. 2008). This DISC1 mutation therefore dysregulates synaptic vesicle release.
The influence of DISC1 on synaptic vesicle cycling has also been examined using pHluorin fused to the synaptic vesicle protein vesicular glutamate transporter 1 (VGLUT1; Tang et al. 2016). pHluorin is a pH‐sensitive form of GFP that fluoresces when released from acidic synaptic vesicles, and that is quenched following reuptake during synaptic vesicle recycling. Release of vesicles labelled with VGLUT1–pHluorin was triggered using trains of action potentials in neurons with DISC1 knocked down using RNA interference. DISC1 knockdown revealed a decreased rate and amplitude of vesicle exocytosis and a trend towards slower endocytosis (Tang et al. 2016). When this experiment was repeated with neurons cultured from the homozygous mutant mouse lacking DISC1 exons 2 and 3, which lacks full‐length DISC1 expression (Fig. 1 G; Kuroda et al. 2011), similar effects were observed on vesicle exocytosis (Tang et al. 2016). As calcium ion influx through voltage‐gated calcium channels triggers synaptic vesicle release, the presynaptic marker synaptophysin, fused to the calcium indicator GCamp3, was used to examine presynaptic calcium currents, and action potential trains were used to stimulate calcium transients. DISC1 knockdown or knockout of DISC1 exons 2 and 3 produced a reduction in calcium transients (Kuroda et al. 2011; Tang et al. 2016). In neurons with DISC1 expression knocked down, this effect was reversed by increasing the extracellular calcium concentration, indicating that in wild‐type neurons DISC1 may facilitate presynaptic calcium ion influx (Tang et al. 2016). Calcium channel‐specific blockers were then used to demonstrate that the measured calcium transients are largely mediated by N‐type voltage‐gated calcium channels (CAV2.2), although presynaptic CAV2.2 protein expression is not significantly influenced by DISC1 (Tang et al. 2016). The authors concluded that DISC1 enhances synaptic vesicle release mediated by N‐type voltage‐gated calcium channels and that this may be due to altered calcium channel function rather than differential expression of channel proteins (Tang et al. 2016).
Taken together, these three studies demonstrate that (1) DISC1 promotes synaptic vesicle transport along neuronal processes, which may, in turn, affect the size of the presynaptic vesicle pools; (2) DISC1 promotes the depolarization‐induced presynaptic calcium influx that drives vesicle exocytosis and neurotransmitter release into the synaptic cleft; and (3) DISC1 promotes neurotransmitter release into the synaptic cleft. The latter may therefore be due to combined effects on vesicle trafficking and calcium influx. Moreover, since mitochondrial ATP is believed to drive both the synaptic vesicle cycle and the ion pumps that restore calcium gradients following presynaptic depolarization (Zenisek & Matthews, 2000; Harris et al. 2012), it is possible that DISC1's role in mitochondrial trafficking also contributes to its regulation of the presynaptic calcium current and subsequent neurotransmitter release. Deficiencies in any of these processes are likely to alter the amount of neurotransmitter released into the synaptic cleft and will affect the postsynaptic responses.
In summary, DISC1 has been shown to regulate trafficking of three important neuronal cargoes, mitochondria, mRNA and synaptic vesicles, whose co‐ordinated action is required for neurotransmission. It is possible that future work will extend these observations to additional cargoes by extending the preliminary findings on a role for DISC1 in APP (Shahani et al. 2015) or GABAA receptor trafficking (Wei et al. 2015). In addition, the DISC1‐associated adaptor TRAK1 is involved in endosome to lysosome trafficking (Webber et al. 2008) as well as mitochondrial motility, while TRAK2 is required for movement of the voltage‐gated potassium channel Kir2.1 (Grishin et al. 2006) and, like DISC1, is implicated in GABAA receptor transport (Stephenson, 2014). Moreover the DISC1 interactor and kinesin adaptor FEZ1 potentially traffics a large number of proteins required for synaptic transmission and neural development (Butkevich et al. 2016). DISC1 may therefore have a general role in transportation of cargo along axons and dendrites to facilitate neurotransmission. Available data indicate that the motility defects observed in response to DISC1 overexpression or knockdown, mutation or knockout may be due to deficits in cargo recruitment to the motors, and/or to effects upon the activity of either dynein, kinesin or both motor proteins.
Conclusions
Irrespective of its role in schizophrenia, studies on DISC1 have revealed this molecule to be of fundamental importance in many neuronal processes including synaptic plasticity, dendritic growth and intracellular trafficking. These three aspects of DISC1 function are likely to be interrelated and in some cases causal. For example, by altering cellular trafficking, DISC1 can affect dendritic growth, axonal growth and synapse formation, which can then subsequently affect circuit formation and plasticity within the system. However, DISC1 function does not just affect plasticity by affecting the development of neuronal circuits. N‐terminal DISC1 mutations affect plasticity in the adult through their signalling interactions with PDE4B and Kal‐7. These studies imply a continuous role for DISC1 in synaptic organization and hence in synaptic plasticity. It is therefore important to note that among the great variety of DISC1 mutations that have been studied, impairment of normal DISC1 function consistently leads to decreases in synapse density.
However, there are still a number of areas that remain to be understood, most notably how synaptic DISC1 is involved in the stability and plasticity of adult synapses. Due to the variety of DISC1 signalling interactions, there are potentially many ways in which this could happen. Does DISC1 act via PDE4B and hence PKA‐related signalling pathways to control AMPA receptor phosphorylation and CREB activation? Similar considerations could be given to TNIK and Kal‐7 pathways. Does DISC1 act via trafficking of important molecules required for synaptic function in the adult such as synaptic vesicles, messenger RNA and mitochondria? Finally, how does DISC1 alter development of the neuron in such a way as to affect synaptic plasticity in the adult? Answers to these questions should lead not only to a greater understanding of normal synaptic function and plasticity, but also to an understanding of the aberrant synaptic function and plasticity encountered in mental health conditions such as schizophrenia.
Additional information
Competing interests
None declared.
Acknowledgements
We would like to thank the sponsors of the Cardiff workshop on ‘Neuroplasticity and synaptic function in neuropsychiatric disorders’ for instigating this review by bringing the authors together in discussion (The Journal of Physiology, Academia Europaea, The Learned Society for Wales, The Academy of Medical Sciences, The Neuroscience and Mental Health Research Institute and Cardiff College of Health and Life Sciences). We should also like to acknowledge the MRC and Wellcome Trust for the author's studies cited here.
Biographies
Daniela Tropea studies the biology of genes involved in neurodevelopmental disorders. In vivo studies uncovered novel molecular mechanisms of cortical plasticity with application to diagnostics and clinic. Her current work focuses on the role of specific mutations and environmental factors on the biology and function of genes involved in neurodevelopmental disorders.

Neil Hardingham studies synaptic transmission in the cerebral cortex and how it can be modulated by development, sensory activity or synaptic plasticity: how are neurones compromised by neurological disorders and how are structural deficits manifested functionally? His recent work on DISC1 has revealed a critical period where DISC1 signalling is required for the correct development of plasticity, electrical and structural properties in the mature cortex.
This review was presented at the symposium ‘Neuroplasticity and Synaptic Function in Neuropsychiatric Disorders’, which took place at Cardiff University, Cardiff, UK, 27‐28 April 2017.
Edited by: Ole Paulsen and Jesper Sjöström
References
- Atkin TA, Brandon NJ & Kittler JT (2012). Disrupted in Schizophrenia 1 forms pathological aggresomes that disrupt its function in intracellular transport. Hum Mol Genet 21, 2017–2028. [DOI] [PubMed] [Google Scholar]
- Attardo A, Fitzgerald JE & Schnitzer MJ (2015). Impermanence of dendritic spines in live adult CA1 hippocampus. Nature 523, 592–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannai H, Fukatsu K, Mizutani A, Natsume T, Iemura S, Ikegami T, Inoue T & Mikoshiba K (2004). An RNA‐interacting protein, SYNCRIP (heterogeneous nuclear ribonuclear protein Q1/NSAP1) is a component of mRNA granule transported with inositol 1,4,5‐trisphosphate receptor type 1 mRNA in neuronal dendrites. J Biol Chem 279, 53427–53434. [DOI] [PubMed] [Google Scholar]
- Barbosa AC, Kim MS, Ertunc M, Adachi M, Nelson ED, McAnally J, Richardson JA, Kavalali ET, Monteggia LM, Bassel‐Duby R & Olson EN (2008). MEF2C, a transcription factor that facilitates learning and memory by negative regulation of synapse numbers and function. Proc Natl Acad Sci U S A 105, 9391–9396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barco A, Alarcon JM & Kandel ER (2002). Expression of constitutively active CREB protein facilitates the late phase of long‐term potentiation by enhancing synaptic capture. Cell 108, 689–703. [DOI] [PubMed] [Google Scholar]
- Blasius TL, Cai D, Jih GT, Toret CP & Verhey KJ (2007). Two binding partners cooperate to activate the molecular motor Kinesin‐1. J Cell Biol 176, 11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV & Collingridge GL (1993). A synaptic model of memory: long‐term potentiation in the hippocampus. Nature 361, 31–39. [DOI] [PubMed] [Google Scholar]
- Booth CA, Brown JT & Randall AD (2014). Neurophysiological modification of CA1 pyramidal neurons in a transgenic mouse expressing a truncated form of disrupted‐in‐schizophrenia 1. Eur J Neurosci 39, 1074–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw NJ, Christie S, Soares DC, Carlyle BC, Porteous DJ & Millar JK (2009). NDE1 and NDEL1: multimerisation, alternate splicing and DISC1 interaction. Neurosci Lett 449, 228–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw NJ & Hayashi MA (2017). NDE1 and NDEL1 from genes to (mal)functions: parallel but distinct roles impacting on neurodevelopmental disorders and psychiatric illness. Cell Mol Life Sci 74, 1191–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw NJ, Ogawa F, Antolin‐Fontes B, Chubb JE, Carlyle BC, Christie S, Claessens A, Porteous DJ & Millar JK (2008). DISC1, PDE4B, and NDE1 at the centrosome and synapse. Biochem Biophys Res Commun 377, 1091–1096. [DOI] [PubMed] [Google Scholar]
- Bradshaw NJ & Porteous DJ (2012). DISC1‐binding proteins in neural development, signalling and schizophrenia. Neuropharmacology 62, 1230–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw NJ, Soares DC, Carlyle BC, Ogawa F, Davidson‐Smith H, Christie S, Mackie S, Thomson PA, Porteous DJ & Millar JK (2011). PKA phosphorylation of NDE1 is DISC1/PDE4 dependent and modulates its interaction with LIS1 and NDEL1. J Neurosci 31, 9043–9054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon NJ ( 2007). Dissecting DISC1 function through protein‐protein interactions. Biochem Soc Trans 35, 1283–1286. [DOI] [PubMed] [Google Scholar]
- Brandon NJ, Handford EJ, Schurov I, Rain JC, Pelling M, Duran‐Jimeniz B, Camargo LM, Oliver KR, Beher D, Shearman MS & Whiting PJ (2004). Disrupted in Schizophrenia 1 and Nudel form a neurodevelopmentally regulated protein complex: implications for schizophrenia and other major neurological disorders. Mol Cell Neurosci 25, 42–55. [DOI] [PubMed] [Google Scholar]
- Brandon NJ, Millar JK, Korth C, Sive H, Singh KK & Sawa A (2009). Understanding the role of DISC1 in psychiatric disease and during normal development. J Neurosci 29, 12768–12775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon NJ & Sawa A (2011). Linking neurodevelopmental and synaptic theories of mental illness through DISC1. Nat Rev Neurosci 12, 707–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon NJ, Schurov I, Camargo LM, Handford EJ, Duran‐Jimeniz B, Hunt P, Millar JK, Porteous DJ, Shearman MS & Whiting PJ (2005). Subcellular targeting of DISC1 is dependent on a domain independent from the Nudel binding site. Mol Cell Neurosci 28, 613–624. [DOI] [PubMed] [Google Scholar]
- Bryantseva SA & Zhapparova ON (2012). Bidirectional transport of organelles: unity and struggle of opposing motors. Cell Biol Int 36, 1–6. [DOI] [PubMed] [Google Scholar]
- Burdick KE, Kamiya A, Hodgkinson CA, Lencz T, DeRosse P, Ishizuka K, Elashvili S, Arai H, Goldman D, Sawa A & Malhotra AK (2008). Elucidating the relationship between DISC1, NDEL1 and NDE1 and the risk for schizophrenia: evidence of epistasis and competitive binding. Hum Mol Genet 17, 2462–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burette AC, Phend KD, Burette S, Lin Q, Liang M, Foltz G, Taylor N, Wang Q, Brandon NJ, Bates B, Ehlers MD & Weinberg RJ (2015). Organization of TNIK in dendritic spines. J Comp Neurol 523, 1913–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butkevich E, Hartig W, Nikolov M, Erck C, Grosche J, Urlaub H, Schmidt CF, Klopfenstein DR & Chua JJ (2016). Phosphorylation of FEZ1 by Microtubule Affinity Regulating Kinases regulates its function in presynaptic protein trafficking. Sci Rep 6, 26965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese B, Wilson MS & Halpain S (2006). Development and regulation of dendritic spine synapses. Physiology 21, 38–47. [DOI] [PubMed] [Google Scholar]
- Camargo LM, Collura V, Rain JC, Mizuguchi K, Hermjakob H, Kerrien S, Bonnert TP, Whiting PJ & Brandon NJ (2007). Disrupted in Schizophrenia 1 Interactome: evidence for the close connectivity of risk genes and a potential synaptic basis for schizophrenia. Mol Psychiatry 12, 74–86. [DOI] [PubMed] [Google Scholar]
- Carlisle HJ, Luong TN, Medina‐Marino A, Schenker L, Khorosheva E, Indersmitten T, Gunapala KM, Steele AD, O'Dell TJ, Patterson PH & Kennedy MB (2011). Deletion of densin‐180 results in abnormal behaviors associated with mental illness and reduces mGluR5 and DISC1 in the postsynaptic density fraction. J Neurosci 31, 16194–16207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chubb JE, Bradshaw NJ, Soares DC, Porteous DJ & Millar JK (2008). The DISC locus in psychiatric illness. Mol Psychiatry 13, 36–64. [DOI] [PubMed] [Google Scholar]
- Cianfrocco MA, DeSantis ME, Leschziner AE & Reck‐Peterson SL (2015). Mechanism and regulation of cytoplasmic dynein. Annu Rev Cell Dev Biol 31, 83–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani LA & Goda Y (2008). Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat Rev Neurosci 9, 344–356. [DOI] [PubMed] [Google Scholar]
- Clapcote SJ, Lipina TV, Millar JK, Mackie S, Christie S, Ogawa F, Lerch JP, Trimble K, Uchiyama M, Sakuraba Y, Kaneda H, Shiroishi T, Houslay MD, Henkelman RM, Sled JG, Gondo Y, Porteous DJ & Roder JC (2007). Behavioral phenotypes of Disc1 missense mutations in mice. Neuron 54, 387–402. [DOI] [PubMed] [Google Scholar]
- Collins DM, Murdoch H, Dunlop AJ, Charych E, Baillie GS, Wang Q, Brandon N, Prinz A & Houslay MD (2008). Ndel1 alters its conformation by sequestering cAMP‐specific phosphodiesterase‐4D3 (PDE4D3) in a manner that is dynamically regulated through Protein Kinase A (PKA). Cell Signal 20, 2356–2369. [DOI] [PubMed] [Google Scholar]
- Crabtree GW, Sun Z, Kvajo M, Broek JA, Fenelon K, McKellar H, Xiao L, Xu B, Bahn S, O'Donnell JM & Gogos JA (2017). Alteration of neuronal excitability and short‐term synaptic plasticity in the prefrontal cortex of a mouse model of mental illness. J Neurosci 37, 4158–4180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui L, Sun W, Yu M, Li N, Guo L, Gu H & Zhou Y (2016). Disrupted‐in‐schizophrenia1 (DISC1) L100P mutation alters synaptic transmission and plasticity in the hippocampus and causes recognition memory deficits. Mol Brain 9, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datusalia AK & Sharma SS (2014). Amelioration of diabetes‐induced cognitive deficits by GSK‐3β inhibition is attributed to modulation of neurotransmitters and neuroinflammation. Mol Neurobiol 50, 390–405. [DOI] [PubMed] [Google Scholar]
- de Anda FC, Meletis K, Ge X, Rei D & Tsai LH (2010). Centrosome motility is essential for initial axon formation in the neocortex. J Neurosci 30, 10391–10406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duning K, Buck F, Barnekow A & Kremerskothen J (2008). SYNCRIP, a component of dendritically localized mRNPs, binds to the translation regulator BC200 RNA. J Neurochem 105, 351–359. [DOI] [PubMed] [Google Scholar]
- Eykelenboom JE, Briggs GJ, Bradshaw NJ, Soares DC, Ogawa F, Christie S, Malavasi EL, Makedonopoulou P, Mackie S, Malloy MP, Wear MA, Blackburn EA, Bramham J, McIntosh AM, Blackwood DH, Muir WJ, Porteous DJ & Millar JK (2012). A t(1;11) translocation linked to schizophrenia and affective disorders gives rise to aberrant chimeric DISC1 transcripts that encode structurally altered, deleterious mitochondrial proteins. Hum Mol Genet 21, 3374–3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorenko OA, Popugaeva E, Enomoto M, Stathopulos PB, Ikura M & Bezprozvanny I (2014). Intracellular calcium channels: inositol‐1,4,5‐trisphosphate receptors. Eur J Pharmacol 739, 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores R 3rd, Hirota Y, Armstrong B, Sawa A & Tomoda T (2011). DISC1 regulates synaptic vesicle transport via a lithium‐sensitive pathway. Neurosci Res 71, 71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franker MA & Hoogenraad CC (2013). Microtubule‐based transport – basic mechanisms, traffic rules and role in neurological pathogenesis. J Cell Sci 126, 2319–2329. [DOI] [PubMed] [Google Scholar]
- Frantseva MV, Fitzgerald PB, Chen R, Moller B, Daigle M & Daskalakis ZJ (2008). Evidence for impaired long‐term potentiation in schizophrenia and its relationship to motor skill learning. Cereb Cortex 18, 990–996. [DOI] [PubMed] [Google Scholar]
- Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, Georgieva L, Rees E, Palta P, Ruderfer DM, Carrera N, Humphreys I, Johnson JS, Roussos P, Barker DD, Banks E, Milanova V, Grant SG, Hannon E, Rose SA, Chambert K, Mahajan M, Scolnick EM, Moran JL, Kirov G, Palotie A, McCarroll SA, Holmans P, Sklar P, Owen MJ, Purcell SM & O'Donovan MC (2014). De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita T, Maturana AD, Ikuta J, Hamada J, Walchli S, Suzuki T, Sawa H, Wooten MW, Okajima T, Tatematsu K, Tanizawa K & Kuroda S (2007). Axonal guidance protein FEZ1 associates with tubulin and kinesin motor protein to transport mitochondria in neurites of NGF‐stimulated PC12 cells. Biochem Biophys Res Commun 361, 605–610. [DOI] [PubMed] [Google Scholar]
- Gindhart JG, Chen J, Faulkner M, Gandhi R, Doerner K, Wisniewski T & Nandlestadt A (2003). The kinesin‐associated protein UNC‐76 is required for axonal transport in the Drosophila nervous system. Mol Biol Cell 14, 3356–3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glantz LA & Lewis DA (2000). Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry 57, 65–73. [DOI] [PubMed] [Google Scholar]
- Goel A, Xu LW, Snyder KP, Song L, Goenaga‐Vazquez Y, Megill A, Takamiya K, Huganir RL & Lee HK (2011). Phosphorylation of AMPA receptors is required for sensory deprivation‐induced homeostatic synaptic plasticity. PLoS One 6 , e18264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenhill SD, Juczewski K, de Haan AM, Seaton G, Fox K & Hardingham NR (2015). Adult cortical plasticity depends on an early postnatal critical period. Science 349, 424–427. [DOI] [PubMed] [Google Scholar]
- Grishin A, Li H, Levitan ES & Zaks‐Makhina E (2006). Identification of gamma‐aminobutyric acid receptor‐interacting factor 1 (TRAK2) as a trafficking factor for the K+ channel Kir2.1. J Biol Chem 281, 30104–30111. [DOI] [PubMed] [Google Scholar]
- Hackett JT, Cochran SL, Greenfield LJ Jr, Brosius DC & Ueda T (1990). Synapsin I injected presynaptically into goldfish mauthner axons reduces quantal synaptic transmission. J Neurophysiol 63, 701–706. [DOI] [PubMed] [Google Scholar]
- Hall J, Trent S, Thomas KL, O'Donovan MC & Owen MJ (2015). Genetic risk for schizophrenia: convergence on synaptic pathways involved in plasticity. Biol Psychiatry 77, 52–58. [DOI] [PubMed] [Google Scholar]
- Hardingham N & Fox K (2006). The role of nitric oxide and GluR1 in presynaptic and postsynaptic components of neocortical potentiation. J Neurosci 26, 7395–7404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham N, Wright N, Dachtler J & Fox K (2008). Sensory deprivation unmasks a PKA‐dependent synaptic plasticity mechanism that operates in parallel with CaMKII. Neuron 60, 861–874. [DOI] [PubMed] [Google Scholar]
- Harris JJ, Jolivet R & Attwell D (2012). Synaptic energy use and supply. Neuron 75, 762–777. [DOI] [PubMed] [Google Scholar]
- Hattori T, Shimizu S, Koyama Y, Yamada K, Kuwahara R, Kumamoto N, Matsuzaki S, Ito A, Katayama T & Tohyama M (2010). DISC1 regulates cell‐cell adhesion, cell‐matrix adhesion and neurite outgrowth. Mol Psychiatry 15, 778, 798–809. [DOI] [PubMed] [Google Scholar]
- Hayashi‐Takagi A, Takaki M, Graziane N, Seshadri S, Murdoch H, Dunlop AJ, Makino Y, Seshadri AJ, Ishizuka K, Srivastava DP, Xie Z, Baraban JM, Houslay MD, Tomoda T, Brandon NJ, Kamiya A, Yan Z, Penzes P & Sawa A (2010). Disrupted‐in‐Schizophrenia 1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nat Neurosci 13, 327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JJ, Hashimoto T & Lewis DA (2006). Molecular mechanisms contributing to dendritic spine alterations in the prefrontal cortex of subjects with schizophrenia. Mol Psychiatry 11, 557–566. [DOI] [PubMed] [Google Scholar]
- Honda A, Miyoshi K, Baba K, Taniguchi M, Koyama Y, Kuroda S, Katayama T & Tohyama M (2004). Expression of fasciculation and elongation protein zeta‐1 (FEZ1) in the developing rat brain. Brain Res Mol Brain Res 122, 89–92. [DOI] [PubMed] [Google Scholar]
- Houslay KF, Christian F, MacLeod R, Adams DR, Houslay MD & Baillie GS (2017). Identification of a multifunctional docking site on the catalytic unit of phosphodiesterase‐4 (PDE4) that is utilised by multiple interaction partners. Biochem J 474, 597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain NK, Hsin H, Huganir RL & Sheng M (2010). MINK and TNIK differentially act on Rap2‐mediated signal transduction to regulate neuronal structure and AMPA receptor function. J Neurosci 30, 14786–14794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima T, Imai T, Kimura Y, Bernstein A, Okano HJ, Yuzaki M & Okano H (2005). Hzf protein regulates dendritic localization and BDNF‐induced translation of type 1 inositol 1,4,5‐trisphosphate receptor mRNA. Proc Natl Acad Sci U S A 102, 17190–17195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juan LW, Liao CC, Lai WS, Chang CY, Pei JC, Wong WR, Liu CM, Hwu HG & Lee LJ (2014). Phenotypic characterization of C57BL/6J mice carrying the Disc1 gene from the 129S6/SvEv strain. Brain Struct Funct 219, 1417–1431. [DOI] [PubMed] [Google Scholar]
- Kamiya A, Kubo K, Tomoda T, Takaki M, Youn R, Ozeki Y, Sawamura N, Park U, Kudo C, Okawa M, Ross CA, Hatten ME, Nakajima K & Sawa A (2005). A schizophrenia‐associated mutation of DISC1 perturbs cerebral cortex development. Nat Cell Biol 7, 1167–1178. [DOI] [PubMed] [Google Scholar]
- Kamiya A, Tan PL, Kubo K, Engelhard C, Ishizuka K, Kubo A, Tsukita S, Pulver AE, Nakajima K, Cascella NG, Katsanis N & Sawa A (2008). Recruitment of PCM1 to the centrosome by the cooperative action of DISC1 and BBS4: a candidate for psychiatric illnesses. Arch Gen Psychiatry 65, 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya A, Tomoda T, Chang J, Takaki M, Zhan C, Morita M, Cascio MB, Elashvili S, Koizumi H, Takanezawa Y, Dickerson F, Yolken R, Arai H & Sawa A (2006). DISC1‐NDEL1/NUDEL protein interaction, an essential component for neurite outgrowth, is modulated by genetic variations of DISC1. Hum Mol Genet 15, 3313–3323. [DOI] [PubMed] [Google Scholar]
- Kaneko M, Stellwagen D, Malenka RC & Stryker MP (2008). Tumor necrosis factor‐α mediates one component of competitive, experience‐dependent plasticity in developing visual cortex. Neuron 58, 673–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JS, Tian JH, Pan PY, Zald P, Li C, Deng C & Sheng ZH (2008). Docking of axonal mitochondria by syntaphilin controls their mobility and affects short‐term facilitation. Cell 132, 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai H, Fukuda M, Watanabe S, Hayashi‐Takagi A & Noguchi J (2010). Structural dynamics of dendritic spines in memory and cognition. Trends Neurosci 33, 121–129. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick B, Xu L, Cascella N, Ozeki Y, Sawa A & Roberts RC (2006). DISC1 immunoreactivity at the light and ultrastructural level in the human neocortex. J Comp Neurol 497, 436–450. [DOI] [PubMed] [Google Scholar]
- Klein ME, Monday H & Jordan BA (2016). Proteostasis and RNA binding proteins in synaptic plasticity and in the pathogenesis of neuropsychiatric disorders. Neural Plast 2016, 3857934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike H, Arguello PA, Kvajo M, Karayiorgou M & Gogos JA (2006). Disc1 is mutated in the 129S6/SvEv strain and modulates working memory in mice. Proc Natl Acad Sci U S A 103, 3693–3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopec CD, Real E, Kessels HW & Malinow R (2007). GluR1 links structural and functional plasticity at excitatory synapses. J Neurosci 27, 13706–13718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozma R, Sarner S, Ahmed S & Lim L (1997). Rho family GTPases and neuronal growth cone remodelling: relationship between increased complexity induced by Cdc42Hs, Rac1, and acetylcholine and collapse induced by RhoA and lysophosphatidic acid. Mol Cell Biol 17, 1201–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda K, Yamada S, Tanaka M, Iizuka M, Yano H, Mori D, Tsuboi D, Nishioka T, Namba T, Iizuka Y, Kubota S, Nagai T, Ibi D, Wang R, Enomoto A, Isotani‐Sakakibara M, Asai N, Kimura K, Kiyonari H, Abe T, Mizoguchi A, Sokabe M, Takahashi M, Yamada K & Kaibuchi K (2011). Behavioral alterations associated with targeted disruption of exons 2 and 3 of the Disc1 gene in the mouse. Hum Mol Genet 20, 4666–4683. [DOI] [PubMed] [Google Scholar]
- Kvajo M, McKellar H, Arguello PA, Drew LJ, Moore H, MacDermott AB, Karayiorgou M & Gogos JA (2008). A mutation in mouse Disc1 that models a schizophrenia risk allele leads to specific alterations in neuronal architecture and cognition. Proc Natl Acad Sci U S A 105, 7076–7081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvajo M, McKellar H, Drew LJ, Lepagnol‐Bestel AM, Xiao L, Levy RJ, Blazeski R, Arguello PA, Lacefield CO, Mason CA, Simonneau M, O'Donnell JM, MacDermott AB, Karayiorgou M & Gogos JA (2011). Altered axonal targeting and short‐term plasticity in the hippocampus of Disc1 mutant mice. Proc Natl Acad Sci U S A 108, E1349–E1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee FH, Fadel MP, Preston‐Maher K, Cordes SP, Clapcote SJ, Price DJ, Roder JC & Wong AH (2011a). Disc1 point mutations in mice affect development of the cerebral cortex. J Neurosci 31, 3197–3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee FH, Kaidanovich‐Beilin O, Roder JC, Woodgett JR & Wong AH (2011b). Genetic inactivation of GSK3α rescues spine deficits in Disc1‐L100P mutant mice. Schizophr Res 129, 74–79. [DOI] [PubMed] [Google Scholar]
- Lee HK, Barbarosie M, Kameyama K, Bear MF & Huganir RL (2000). Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature 405, 955–959. [DOI] [PubMed] [Google Scholar]
- Leliveld SR, Bader V, Hendriks P, Prikulis I, Sajnani G, Requena JR & Korth C (2008). Insolubility of disrupted‐in‐schizophrenia 1 disrupts oligomer‐dependent interactions with nuclear distribution element 1 and is associated with sporadic mental disease. J Neurosci 28, 3839–3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leliveld SR, Hendriks P, Michel M, Sajnani G, Bader V, Trossbach S, Prikulis I, Hartmann R, Jonas E, Willbold D, Requena JR & Korth C (2009). Oligomer assembly of the C‐terminal DISC1 domain (640‐854) is controlled by self‐association motifs and disease‐associated polymorphism S704C. Biochemistry 48, 7746–7755. [DOI] [PubMed] [Google Scholar]
- Lepagnol‐Bestel AM, Kvajo M, Karayiorgou M, Simonneau M & Gogos JA (2013). A Disc1 mutation differentially affects neurites and spines in hippocampal and cortical neurons. Mol Cell Neurosci 54, 84–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Lee WL & Cooper JA (2005). NudEL targets dynein to microtubule ends through LIS1. Nat Cell Biol 7, 686–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Zhou Y, Jentsch JD, Brown RA, Tian X, Ehninger D, Hennah W, Peltonen L, Lonnqvist J, Huttunen MO, Kaprio J, Trachtenberg JT, Silva AJ & Cannon TD (2007). Specific developmental disruption of disrupted‐in‐schizophrenia‐1 function results in schizophrenia‐related phenotypes in mice. Proc Natl Acad Sci U S A 104, 18280–18285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Okamoto K, Hayashi Y & Sheng M (2004). The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 119, 873–887. [DOI] [PubMed] [Google Scholar]
- Liang Y, Yu W, Li Y, Yang Z, Yan X, Huang Q & Zhu X (2004). Nudel functions in membrane traffic mainly through association with Lis1 and cytoplasmic dynein. J Cell Biol 164, 557–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipina TV, Wang M, Liu F & Roder JC (2012). Synergistic interactions between PDE4B and GSK‐3: DISC1 mutant mice. Neuropharmacology 62, 1252–1262. [DOI] [PubMed] [Google Scholar]
- Macaskill AF, Rinholm JE, Twelvetrees AE, Arancibia‐Carcamo IL, Muir J, Fransson A, Aspenstrom P, Attwell D & Kittler JT (2009). Miro1 is a calcium sensor for glutamate receptor‐dependent localization of mitochondria at synapses. Neuron 61, 541–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malavasi EL, Ogawa F, Porteous DJ & Millar JK (2012). DISC1 variants 37W and 607F disrupt its nuclear targeting and regulatory role in ATF4‐mediated transcription. Hum Mol Genet 21, 2779–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao T, Kusefoglu D, Hooks BM, Huber D, Petreanu L & Svoboda K (2011). Long‐range neuronal circuits underlying the interaction between sensory and motor cortex. Neuron 72, 111–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Y, Ge X, Frank CL, Madison JM, Koehler AN, Doud MK, Tassa C, Berry EM, Soda T, Singh KK, Biechele T, Petryshen TL, Moon RT, Haggarty SJ & Tsai LH (2009). Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta‐catenin signaling. Cell 136, 1017–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall CR, Howrigan DP, Merico D, Thiruvahindrapuram B, Wu W, Greer DS, Antaki D, Shetty A, Holmans PA, Pinto D, Gujral M, Brandler WM, Malhotra D, Wang Z, Fajarado KVF, Maile MS, Ripke S, Agartz I, Albus M, Alexander M, Amin F, Atkins J, Bacanu SA, Belliveau RA Jr, Bergen SE, Bertalan M, Bevilacqua E, Bigdeli TB, Black DW, Buccola NG, Buckner RL, Bulik‐Sullivan B, Byerley W, Cahn W, Cai G, Cairns MJ, Campion D, Cantor RM, Carr VJ, Carrera N, Catts SV, Chambert KD, Cheng W, Cloninger CR, Cohen D, Cormican P, Craddock N, Crespo‐Facorro B, Crowley JJ, Curtis D, Davidson M, Davis KL, Degenhardt F, Del Favero J, DeLisi LE, Dikeos D, Dinan T, Djurovic S, Donohoe G, Drapeau E, Duan J, Eichhammer P, Eriksson J, Escott‐Price V, Essioux L, Fanous AH, Farrell MS, Frank J, Franke L, Freedman R, Freimer NB, Friedman JI, Forstner AJ, Fromer M, Genovese G, Georgieva L, Gershon ES, Giegling I, Giusti‐Rodriguez P, Godard S, Goldstein JI, Gratten J, de Haan L, Hamshere ML, Hansen M, Hansen T, Haroutunian V, Hartmann AM, Henskens FA, Herms S, Hirschhorn JN, Hoffmann P, Hofman A, Huang H, Ikeda M, Joa I, Kahler AK, Kahn RS, Kalaydjieva L, Kavanagh D, Keller MC, Kelly BJ, Kennedy JL, Kim Y, Knowles JA, Konte B, Laurent C, Lee P, Lee SH, Legge SE, Lerer B, Levy DL, Liang KY, Lieberman J, Lonnqvist J, Loughland CM, Maher BS, Maier W, Mallet J, Mattheisen M, Mattingsdal M, McCarley RW, McDonald C, McIntosh AM, Meier S, Meijer CJ, Melle I, Mesholam‐Gately RI, Metspalu A, Michie PT, Milani L, Milanova V, Mokrab Y, Morris DW, Muller‐Myhsok B, Murphy KC, Murray RM, Myin‐Germeys I, Nenadic I, Nertney DA, Nestadt G, Nicodemus KK, Nisenbaum L, Nordin A, O'Callaghan E, O'Dushlaine C, Oh SY, Olincy A, Olsen L, O'Neill FA, Van Os J, Pantelis C, Papadimitriou GN, Parkhomenko E, Pato MT, Paunio T, Psychosis Endophenotypes International Consortium , Perkins DO, Pers TH, Pietilainen O, Pimm J, Pocklington AJ, Powell J, Price A, Pulver AE, Purcell SM, Quested D, Rasmussen HB, Reichenberg A, Reimers MA, Richards AL, Roffman JL, Roussos P, Ruderfer DM, Salomaa V, Sanders AR, Savitz A, Schall U, Schulze TG, Schwab SG, Scolnick EM, Scott RJ, Seidman LJ, Shi J, Silverman JM, Smoller JW, Soderman E, Spencer CCA, Stahl EA, Strengman E, Strohmaier J, Stroup TS, Suvisaari J, Svrakic DM, Szatkiewicz JP, Thirumalai S, Tooney PA, Veijola J, Visscher PM, Waddington J, Walsh D, Webb BT, Weiser M, Wildenauer DB, Williams NM, Williams S, Witt SH, Wormley BK, Wray NR, Wu JQ, Zai CC, Adolfsson R, Andreassen OA, Bramon E, Buxbaum JD, Cichon S, Collier DA, Corvin A, Daly MJ, Darvasi A, Domenici E, Esko T, Gejman PV, Gill M, Gurling H, Hultman CM, Iwata N, Jablensky AV, Jonsson EG, Kendler KS, Kirov G, Knight J, Levinson DF, Li QS, McCarroll SA, McQuillin A, Moran JL, Mowry BJ, Nothen MM, Ophoff RA, Owen MJ, Palotie A, Pato CN, Petryshen TL, Posthuma D, Rietschel M, Riley BP, Rujescu D, Sklar P, St Clair D, Walters JTR, Werge T, Sullivan PF, O'Donovan MC, Scherer SW, Neale BM, Sebat J; CNV and Schizophrenia Working Groups of the Psychiatric Genomics Consortium (2017). Contribution of copy number variants to schizophrenia from a genome‐wide study of 41,321 subjects. Nat Genet 49, 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin PM, Stanley RE, Ross AP, Freitas AE, Moyer CE, Brumback AC, Iafrati J, Stapornwongkul KS, Dominguez S, Kivimae S, Mulligan KA, Pirooznia M, McCombie WR, Potash JB, Zandi PP, Purcell SM, Sanders SJ, Zuo Y, Sohal VS & Cheyette BN (2018). DIXDC1 contributes to psychiatric susceptibility by regulating dendritic spine and glutamatergic synapse density via GSK3 and Wnt/β‐catenin signaling. Mol Psychiatry 23, 467–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGirr A, Lipina TV, Mun HS, Georgiou J, Al‐Amri AH, Ng E, Zhai D, Elliott C, Cameron RT, Mullins JG, Liu F, Baillie GS, Clapcote SJ & Roder JC (2016). Specific inhibition of phosphodiesterase‐4b results in anxiolysis and facilitates memory acquisition. Neuropsychopharmacology 41, 1080–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenney RJ, Vershinin M, Kunwar A, Vallee RB & Gross SP (2010). LIS1 and NudE induce a persistent dynein force‐producing state. Cell 141, 304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Y, He T, Zhu Y, Li W, Wang B & Zhong Y (2015). Activation of hippocampal CREB by rolipram partially recovers balance between TNF‐α and IL‐10 levels and improves cognitive deficits in diabetic rats. Cell Mol Neurobiol 35, 1157–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar JK, James R, Christie S & Porteous DJ (2005a). Disrupted in schizophrenia 1 (DISC1): subcellular targeting and induction of ring mitochondria. Mol Cell Neurosci 30, 477–484. [DOI] [PubMed] [Google Scholar]
- Millar JK, Pickard BS, Mackie S, James R, Christie S, Buchanan SR, Malloy MP, Chubb JE, Huston E, Baillie GS, Thomson PA, Hill EV, Brandon NJ, Rain JC, Camargo LM, Whiting PJ, Houslay MD, Blackwood DH, Muir WJ & Porteous DJ (2005b). DISC1 and PDE4B are interacting genetic factors in schizophrenia that regulate cAMP signaling. Science 310, 1187–1191. [DOI] [PubMed] [Google Scholar]
- Miyoshi K, Asanuma M, Miyazaki I, Diaz‐Corrales FJ, Katayama T, Tohyama M & Ogawa N (2004). DISC1 localizes to the centrosome by binding to kendrin. Biochem Biophys Res Commun 317, 1195–1199. [DOI] [PubMed] [Google Scholar]
- Miyoshi K, Honda A, Baba K, Taniguchi M, Oono K, Fujita T, Kuroda S, Katayama T & Tohyama M (2003). Disrupted‐In‐Schizophrenia 1, a candidate gene for schizophrenia, participates in neurite outgrowth. Mol Psychiatry 8, 685–694. [DOI] [PubMed] [Google Scholar]
- Moosmang S, Haider N, Klugbauer N, Adelsberger H, Langwieser N, Muller J, Stiess M, Marais E, Schulla V, Lacinova L, Goebbels S, Nave KA, Storm DR, Hofmann F & Kleppisch T (2005). Role of hippocampal Cav1.2 Ca2+ channels in NMDA receptor‐independent synaptic plasticity and spatial memory. J Neurosci 25, 9883–9892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JA, Kandpal G, Ma L & Austin CP (2003). DISC1 (Disrupted‐In‐Schizophrenia 1) is a centrosome‐associated protein that interacts with MAP1A, MIPT3, ATF4/5 and NUDEL: regulation and loss of interaction with mutation. Hum Mol Genet 12, 1591–1608. [DOI] [PubMed] [Google Scholar]
- Muraki K & Tanigaki K (2015). Neuronal migration abnormalities and its possible implications for schizophrenia. Front Neurosci 9, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murdoch H, Mackie S, Collins DM, Hill EV, Bolger GB, Klussmann E, Porteous DJ, Millar JK & Houslay MD (2007). Isoform‐selective susceptibility of DISC1/phosphodiesterase‐4 complexes to dissociation by elevated intracellular cAMP levels. J Neurosci 27, 9513–9524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murdoch H, Vadrevu S, Prinz A, Dunlop AJ, Klussmann E, Bolger GB, Norman JC & Houslay MD (2011). Interaction between LIS1 and PDE4, and its role in cytoplasmic dynein function. J Cell Sci 124, 2253–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy LC & Millar JK (2017). Regulation of mitochondrial dynamics by DISC1, a putative risk factor for major mental illness. Schizophr Res 187, 55–61. [DOI] [PubMed] [Google Scholar]
- Nakayama AY, Harms MB & Luo L (2000). Small GTPases Rac and Rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. J Neurosci 20, 5329–5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan S, Arthanari H, Wolfe MS & Wagner G (2011). Molecular characterization of disrupted in schizophrenia‐1 risk variant S704C reveals the formation of altered oligomeric assembly. J Biol Chem 286, 44266–44276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norkett R, Modi S, Birsa N, Atkin TA, Ivankovic D, Pathania M, Trossbach SV, Korth C, Hirst WD & Kittler JT (2016). DISC1‐dependent regulation of mitochondrial dynamics controls the morphogenesis of complex neuronal dendrites. J Biol Chem 291, 613–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa F, Malavasi EL, Crummie DK, Eykelenboom JE, Soares DC, Mackie S, Porteous DJ & Millar JK (2014). DISC1 complexes with TRAK1 and Miro1 to modulate anterograde axonal mitochondrial trafficking. Hum Mol Genet 23, 906–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa F, Murphy LC, Malavasi EL, O'Sullivan ST, Torrance HS, Porteous DJ & Millar JK (2016). NDE1 and GSK3β associate with TRAK1 and regulate axonal mitochondrial motility: identification of cyclic AMP as a novel modulator of axonal mitochondrial trafficking. ACS Chem Neurosci 7, 553–564. [DOI] [PubMed] [Google Scholar]
- Ozeki Y, Tomoda T, Kleiderlein J, Kamiya A, Bord L, Fujii K, Okawa M, Yamada N, Hatten ME, Snyder SH, Ross CA & Sawa A (2003). Disrupted‐in‐Schizophrenia‐1 (DISC‐1): mutant truncation prevents binding to NudE‐like (NUDEL) and inhibits neurite outgrowth. Proc Natl Acad Sci U S A 100, 289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C, Lee SA, Hong JH, Suh Y, Park SJ, Suh BK, Woo Y, Choi J, Huh JW, Kim YM & Park SK (2016). Disrupted‐in‐schizophrenia 1 (DISC1) and Syntaphilin collaborate to modulate axonal mitochondrial anchoring. Mol Brain 9, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P, Cahill ME, Jones KA, VanLeeuwen JE & Woolfrey KM (2011). Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci 14, 285–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips KG, Hardingham NR & Fox K (2008). Postsynaptic action potentials are required for nitric‐oxide‐dependent long‐term potentiation in CA1 neurons of adult GluR1 knock‐out and wild‐type mice. J Neurosci 28, 14031–14041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pletnikov MV, Ayhan Y, Nikolskaia O, Xu Y, Ovanesov MV, Huang H, Mori S, Moran TH & Ross CA (2008). Inducible expression of mutant human DISC1 in mice is associated with brain and behavioral abnormalities reminiscent of schizophrenia. Mol Psychiatry 13, 173–186, 115. [DOI] [PubMed] [Google Scholar]
- Pocklington AJ, O'Donovan M & Owen MJ (2014). The synapse in schizophrenia. Eur J Neurosci 39, 1059–1067. [DOI] [PubMed] [Google Scholar]
- Ramakers GJ ( 2002). Rho proteins, mental retardation and the cellular basis of cognition. Trends Neurosci 25, 191–199. [DOI] [PubMed] [Google Scholar]
- Ranson A, Cheetham CE, Fox K & Sengpiel F (2012). Homeostatic plasticity mechanisms are required for juvenile, but not adult, ocular dominance plasticity. Proc Natl Acad Sci U S A 109, 1311–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranson A, Sengpiel F & Fox K (2013). The role of GluA1 in ocular dominance plasticity in the mouse visual cortex. J Neurosci 33, 15220–15225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosahl TW, Geppert M, Spillane D, Herz J, Hammer RE, Malenka RC & Sudhof TC (1993). Short‐term synaptic plasticity is altered in mice lacking synapsin I. Cell 75, 661–670. [DOI] [PubMed] [Google Scholar]
- Sachs NA, Sawa A, Holmes SE, Ross CA, DeLisi LE & Margolis RL (2005). A frameshift mutation in Disrupted in Schizophrenia 1 in an American family with schizophrenia and schizoaffective disorder. Mol Psychiatry 10, 758–764. [DOI] [PubMed] [Google Scholar]
- Sawamura N, Ando T, Maruyama Y, Fujimuro M, Mochizuki H, Honjo K, Shimoda M, Toda H, Sawamura‐Yamamoto T, Makuch LA, Hayashi A, Ishizuka K, Cascella NG, Kamiya A, Ishida N, Tomoda T, Hai T, Furukubo‐Tokunaga K & Sawa A (2008). Nuclear DISC1 regulates CRE‐mediated gene transcription and sleep homeostasis in the fruit fly. Mol Psychiatry 13, 1138–1148, 1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurov IL, Handford EJ, Brandon NJ & Whiting PJ (2004). Expression of disrupted in schizophrenia 1 (DISC1) protein in the adult and developing mouse brain indicates its role in neurodevelopment. Mol Psychiatry 9, 1100–1110. [DOI] [PubMed] [Google Scholar]
- Schwarz TL ( 2013). Mitochondrial trafficking in neurons. Cold Spring Harb Perspect Biol 5, a011304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekino Y, Kojima N & Shirao T (2007). Role of actin cytoskeleton in dendritic spine morphogenesis. Neurocehm Int 51, 92–104. [DOI] [PubMed] [Google Scholar]
- Shahani N, Seshadri S, Jaaro‐Peled H, Ishizuka K, Hirota‐Tsuyada Y, Wang Q, Koga M, Sedlak TW, Korth C, Brandon NJ, Kamiya A, Subramaniam S, Tomoda T & Sawa A (2015). DISC1 regulates trafficking and processing of APP and Aβ generation. Mol Psychiatry 20, 874–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao CY, Zhu J, Xie YJ, Wang Z, Wang YN, Wang Y, Su LD, Zhou L, Zhou TH & Shen Y (2013). Distinct functions of nuclear distribution proteins LIS1, Ndel1 and NudCL in regulating axonal mitochondrial transport. Traffic 14, 785–797. [DOI] [PubMed] [Google Scholar]
- Shen S, Lang B, Nakamoto C, Zhang F, Pu J, Kuan SL, Chatzi C, He S, Mackie I, Brandon NJ, Marquis KL, Day M, Hurko O, McCaig CD, Riedel G & St Clair D (2008). Schizophrenia‐related neural and behavioral phenotypes in transgenic mice expressing truncated Disc1. J Neurosci 28, 10893–10904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim SY, Samuels BA, Wang J, Neumayer G, Belzil C, Ayala R, Shi Y, Shi Y, Tsai LH & Nguyen MD (2008). Ndel1 controls the dynein‐mediated transport of vimentin during neurite outgrowth. J Biol Chem 283, 12232–12240. [DOI] [PubMed] [Google Scholar]
- Shinoda T, Taya S, Tsuboi D, Hikita T, Matsuzawa R, Kuroda S, Iwamatsu A & Kaibuchi K (2007). DISC1 regulates neurotrophin‐induced axon elongation via interaction with Grb2. J Neurosci 27, 4–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh KK, De Rienzo G, Drane L, Mao Y, Flood Z, Madison J, Ferreira M, Bergen S, King C, Sklar P, Sive H & Tsai LH (2011). Common DISC1 polymorphisms disrupt Wnt/GSK3β signaling and brain development. Neuron 72, 545–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares DC, Bradshaw NJ, Zou J, Kennaway CK, Hamilton RS, Chen ZA, Wear MA, Blackburn EA, Bramham J, Bottcher B, Millar JK, Barlow PN, Walkinshaw MD, Rappsilber J & Porteous DJ (2012). The mitosis and neurodevelopment proteins NDE1 and NDEL1 form dimers, tetramers, and polymers with a folded back structure in solution. J Biol Chem 287, 32381–32393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Li W, Feng J, Heston LL, Scaringe WA & Sommer SS (2008). Identification of high risk DISC1 structural variants with a 2% attributable risk for schizophrenia. Biochem Biophys Res Commun 367, 700–706. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Beattie EC, Seo JY & Malenka RC (2005). Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor‐α. J Neurosci 25, 3219–3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen D & Malenka RC (2006). Synaptic scaling mediated by glial TNF‐α. Nature 440, 1054–1059. [DOI] [PubMed] [Google Scholar]
- Stephenson FA ( 2014). Revisiting the TRAK family of proteins as mediators of GABAA receptor trafficking. Neurochem Res 39, 992–996. [DOI] [PubMed] [Google Scholar]
- Sudarov A, Gooden F, Tseng D, Gan WB & Ross ME (2013). Lis1 controls dynamics of neuronal filopodia and spines to impact synaptogenesis and social behaviour. EMBO Mol Med 5, 591–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, Thevathasan JV, Lin Q, Lim KB, Kuroda K, Kaibuchi K, Bilger M, Soong TW & Fivaz M (2016). Stimulation of synaptic vesicle exocytosis by the mental disease gene DISC1 is mediated by N‐Type voltage‐gated calcium channels. Front Synaptic Neurosci 8, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taya S, Shinoda T, Tsuboi D, Asaki J, Hikita T, Kuroda S, Kuroda K, Hirotsune S & Kaibuchi K (2007). DISC1 regulates the transport of the NUDEL/LIS1/14‐3‐3epsilon complex through kinesin‐1. J Neurosci 27, 15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson PA, Duff B, Romaniuk L, Watson A, Whalley HC, Li X, Dauvermann MR, Moorhead TW, Bois C, Ryan NM, Redpath H, Hall L, Morris SW, van Beek EJ, Roberts N, St Clair , Whitcher B, Dunlop J, Brandon NJ, Hughes ZA, Hall J, McIntosh A & Lawrie SM (2016). Balanced translocation linked to psychiatric disorder, glutamate, and cortical structure/function. NPJ Schizophr 2, 16024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson PA, Malavasi EL, Grunewald E, Soares DC, Borkowska M & Millar JK (2013). DISC1 genetics, biology and psychiatric illness. Front Biol 8, 1–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson PA, Parla JS, McRae AF, Kramer M, Ramakrishnan K, Yao J, Soares DC, McCarthy S, Morris SW, Cardone L, Cass S, Ghiban E, Hennah W, Evans KL, Rebolini D, Millar JK, Harris SE, Starr JM, MacIntyre DJ, Generation S, McIntosh AM, Watson JD, Deary IJ, Visscher PM, Blackwood DH, McCombie WR & Porteous DJ (2014). 708 Common and 2010 rare DISC1 locus variants identified in 1542 subjects: analysis for association with psychiatric disorder and cognitive traits. Mol Psychiatry 19, 668–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda H, Mochizuki H, Flores R 3rd, Josowitz R, Krasieva TB, Lamorte VJ, Suzuki E, Gindhart JG, Furukubo‐Tokunaga K & Tomoda T (2008). UNC‐51/ATG1 kinase regulates axonal transport by mediating motor‐cargo assembly. Genes Dev 22, 3292–3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tropea D, Majewska AK, Garcia R & Sur M (2010). Structural dynamics of synapses in vivo correlate with functional changes during experience‐dependent plasticity in visual cortex. J Neurosci 30, 11086–11095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tropea D, Molinos I, Petit E, Bellini S, Nagakura I, O'Tuathaigh C, Schorova L, Mitchell KJ, Waddington J, Sur M, Gill M & Corvin AP (2016). Disrupted in schizophrenia 1 (DISC1) L100P mutants have impaired activity‐dependent plasticity in vivo and in vitro. Transl Psychiatry 6, e712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trossbach SV, Bader V, Hecher L, Pum ME, Masoud ST, Prikulis I, Schable S, de Souza Silva MA, Su P, Boulat B, Chwiesko C, Poschmann G, Stuhler K, Lohr KM, Stout KA, Oskamp A, Godsave SF, Muller‐Schiffmann A, Bilzer T, Steiner H, Peters PJ, Bauer A, Sauvage M, Ramsey AJ, Miller GW, Liu F, Seeman P, Brandon NJ, Huston JP & Korth C (2016). Misassembly of full‐length Disrupted‐in‐Schizophrenia 1 protein is linked to altered dopamine homeostasis and behavioral deficits. Mol Psychiatry 21, 1561–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuboi D, Kuroda K, Tanaka M, Namba T, Iizuka Y, Taya S, Shinoda T, Hikita T, Muraoka S, Iizuka M, Nimura A, Mizoguchi A, Shiina N, Sokabe M, Okano H, Mikoshiba K & Kaibuchi K (2015). Disrupted‐in‐schizophrenia 1 regulates transport of ITPR1 mRNA for synaptic plasticity. Nat Neurosci 18, 698–707. [DOI] [PubMed] [Google Scholar]
- Wang Q, Amato SP, Rubitski DM, Hayward MM, Kormos BL, Verhoest PR, Xu L, Brandon NJ & Ehlers MD (2016). Identification of phosphorylation consensus sequences and endogenous neuronal substrates of the psychiatric risk kinase TNIK. J Pharmacol Exp Ther 356, 410–423. [DOI] [PubMed] [Google Scholar]
- Wang Q & Brandon NJ (2011). Regulation of the cytoskeleton by Disrupted‐in‐schizophrenia 1 (DISC1). Mol Cell Neurosci 48, 359–364. [DOI] [PubMed] [Google Scholar]
- Wang Q, Charych EI, Pulito VL, Lee JB, Graziane NM, Crozier RA, Revilla‐Sanchez R, Kelly MP, Dunlop AJ, Murdoch H, Taylor N, Xie Y, Pausch M, Hayashi‐Takagi A, Ishizuka K, Seshadri S, Bates B, Kariya K, Sawa A, Weinberg RJ, Moss SJ, Houslay MD, Yan Z & Brandon NJ (2011). The psychiatric disease risk factors DISC1 and TNIK interact to regulate synapse composition and function. Mol Psychiatry 16, 1006–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X & Schwarz TL (2009). The mechanism of Ca2+‐dependent regulation of kinesin‐mediated mitochondrial motility. Cell 136, 163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webber E, Li L & Chin LS (2008). Hypertonia‐associated protein Trak1 is a novel regulator of endosome‐to‐lysosome trafficking. J Mol Biol 382, 638–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Graziane NM, Gu Z & Yan Z (2015). DISC1 protein regulates gamma‐aminobutyric acid, type A (GABAA) receptor trafficking and inhibitory synaptic transmission in cortical neurons. J Biol Chem 290, 27680–27687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Z, Nguyen HN, Guo Z, Lalli MA, Wang X, Su Y, Kim NS, Yoon KJ, Shin J, Zhang C, Makri G, Nauen D, Yu H, Guzman E, Chiang CH, Yoritomo N, Kaibuchi K, Zou J, Christian KM, Cheng L, Ross CA, Margolis RL, Chen G, Kosik KS, Song H & Ming GL (2014). Synaptic dysregulation in a human iPS cell model of mental disorders. Nature 515,414‐418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Li Y & Xiao B (2013). DISC1‐related signaling pathways in adult neurogenesis of the hippocampus. Gene 518, 223–230. [DOI] [PubMed] [Google Scholar]
- Yang G, Pan F & Gan WB (2009a). Stably maintained dendritic spines are associated with lifelong memories. Nature 462, 920–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Wang Z, Wang B, Justice NJ & Zheng H (2009b). Amyloid precursor protein regulates Cav1.2 L‐type calcium channel levels and function to influence GABAergic short‐term plasticity. J Neurosci 29, 15660–15668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yerabham ASK, Mas PJ, Decker C, Soares DC, Weiergraber OH, Nagel‐Steger L, Willbold D, Hart DJ, Bradshaw NJ & Korth C (2017). A structural organization for the Disrupted in Schizophrenia 1 protein, identified by high‐throughput screening, reveals distinctly folded regions, which are bisected by mental illness‐related mutations. J Biol Chem 292, 6468–6477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young‐Pearse TL, Suth S, Luth ES, Sawa A & Selkoe DJ, Nagel‐Steger L, Willbold D, Hart DJ, Bradshaw NJ & Korth C (2010). Biochemical and functional interaction of DISC1 and APP regulates neuronal migration during mammalian cortical development. J Neurosci 30, 10431–10440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenisek D & Matthews G (2000). The role of mitochondria in presynaptic calcium handling at a ribbon synapse. Neuron 25, 229–237. [DOI] [PubMed] [Google Scholar]
- Zhou X, Chen Q, Schaukowitch K, Kelsoe JR & Geyer MA (2010). Insoluble DISC1‐Boymaw fusion proteins generated by DISC1 translocation. Mol Psychiatry 15, 669–672. [DOI] [PMC free article] [PubMed] [Google Scholar]