

Abstract
Our experiences and memories define who we are, and evidence has accumulated that memory formation is dependent on functional and structural adaptations of synaptic structures in our brain. Especially dendritic spines, the postsynaptic compartments of synapses show a strong structure‐to‐function relationship and a high degree of structural plasticity. Although the molecular mechanisms are not completely understood, it is known that these modifications are highly dependent on the actin cytoskeleton, the major cytoskeletal component of the spine. Given the crucial involvement of actin in these mechanisms, dysregulations of spine actin dynamics (reflected by alterations in dendritic spine morphology) can be found in a variety of neurological disorders ranging from schizophrenia to several forms of autism spectrum disorders such as fragile X syndrome (FXS). FXS is caused by a single mutation leading to an inactivation of the X‐linked fragile X mental retardation 1 gene and loss of its gene product, the RNA‐binding protein fragile X mental retardation protein 1 (FMRP), which normally can be found both pre‐ and postsynaptically. FMRP is involved in mRNA transport as well as regulation of local translation at the synapse, and although hundreds of FMRP‐target mRNAs could be identified only a very few interactions between FMRP and actin‐regulating proteins have been reported and validated. In this review we give an overview of recent work by our lab and others providing evidence that dysregulated actin dynamics might indeed be at the very base of a deeper understanding of neurological disorders ranging from cognitive impairment to the autism spectrum.
Keywords: actin, FMRP, FXS, structural plasticity, hippocampus, spines, ASD
Introduction
The immense computational power of the central nervous system depends on the formation of functional neuronal networks, which are further refined and adapted to environmental changes by processes of neuronal plasticity throughout the entire lifespan of an individual. Synapses are considered as the single processing units of these networks, and in the cortex the majority of excitatory synapses are located on spines, dendritic protrusions containing the postsynaptic compartment. Typically, dendritic spines show a variety of shapes and sizes even on a single neuron. Although spines are tiny in size, spine shape and function are fundamental for the network performance of our brain, a fact that we are just beginning to understand. Importantly, these parameters are significant indicators of proper neuronal function, as spines are constantly adapting their structural and functional characteristics to changes in neuronal activity (reviewed in Caroni et al. 2012). Although constant changes in spine morphology occurring within a time range of seconds to minutes (spine motility, short‐term structural plasticity) are well described for more than 30 years, the underlying mechanisms remain rather elusive except for the principle involvement of the actin cytoskeleton (Fischer et al. 1998; Dunaevsky et al. 1999; Bonhoeffer & Yuste, 2002; Oertner & Matus, 2005; Michaelsen‐Preusse et al. 2016). To date, motile spines have been described both in vitro and in vivo (Fischer et al. 1998; Dunaevsky et al. 1999; Berning et al. 2012). It is speculated that besides the obvious reason of facilitating contact formation between future synaptic partners, spine motility might in addition serve to orchestrate the positioning and reorganization of macromolecular assemblies in spine nanodomains or to modulate the engagement of the spine membrane with extracellular matrix components (Halpain, 2000; Frost et al. 2010; Koleske, 2013). Besides displaying basal motility (which might reflect subtle changes in baseline synaptic transmission), spines can exhibit more pronounced changes in shape in response to stronger alterations in synaptic efficacy, namely long‐term potentiation (LTP) or long‐term depression (LTD) (Engert & Bonhoeffer, 1999; Matsuzaki et al. 2004; Nagerl et al. 2004; for reviews see Bosch & Hayashi, 2012; Sala & Segal, 2014). These long‐term structural plasticity processes can lead to substantial changes in spine volume and are considered as a key cellular correlate of learning and memory formation (as reviewed in Lamprecht & LeDoux, 2004; Caroni et al. 2012; Korte & Schmitz, 2016).
Structural plasticity and actin
These strong alterations in spine shape are directly associated with the dynamic actin cytoskeleton, which is highly enriched in dendritic spines (Fischer et al. 1998; Fukazawa et al. 2003; Chen et al. 2007; Honkura et al. 2008; Hotulainen et al. 2009; Bosch et al. 2014; Michaelsen‐Preusse et al. 2016). In fact, up to 80% of actin filaments turn over in less than 2 min in the spine head (Star et al. 2002). Results from our lab show that actin filament turnover is modulated during LTP with a reduction of the turnover time during the initial phase possibly allowing spine head expansion and a significant increase after 1 h most likely important for the stabilization and maintenance of the new spine structure (Michaelsen‐Preusse et al. 2016). Interestingly, we have recently shown that the actin‐binding protein profilin2a (PFN2a) is crucially involved in this modulation, as activity‐dependent spine head enlargement was completely abolished in PFN2a‐deficient cells along with a significant general reduction in the F‐actin turnover time, which showed no modulation during synaptic plasticity (Michaelsen‐Preusse et al. 2016). An understanding of the detailed molecular machinery and identification of key molecules which control actin polymerization in space and time will help to reveal further details about spine function and might eventually also provide a better understanding of neurological disorders characterized by defects in spinogenesis and spine maintenance (Blanpied & Ehlers, 2004; Penzes et al. 2011). The strong structure‐to‐function relationship of dendritic spines (Holtmaat & Svoboda, 2009) and the fact that alterations in their morphology and density can be found in various neurological disorders such as autism spectrum disorder (ASD), schizophrenia and several forms of mental retardation in humans (reviewed in Fiala et al. 2002) opens the possibility that defects in the modulation of dynamic actin in spines might be a key neuropathological mechanism for the aetiology of these diseases.
The fragile X syndrome
In light of this, fragile X syndrome (FXS) can be considered as a paradigmatic disease to study spine dysfunction. FXS is the most frequent inherited single‐gene cause of ASD and mental retardation (Crawford et al. 2001) and is characterized by translational silencing of the fragile X mental retardation 1 (fmr1) gene. Caused by partial or complete loss of the fmr1‐encoded fragile X mental retardation protein 1 (FMRP; Verkerk et al. 1991), patients suffering from FXS exhibit several neurological symptoms including hyperactivity, diversely severe forms of cognitive impairments, poor motor coordination and autistic behaviour (Beckel‐Mitchener & Greenough, 2004). Surprisingly, despite having strong intellectual deficits, autopsy studies revealed that brains of FXS patients morphologically appear normal and only a subtle synaptic phenotype could be identified: a hyperabundance of long and thin dendritic spines (Rudelli et al. 1985; Hinton et al. 1991; Irwin et al. 2001).
To investigate the underlying cellular and genetic mechanism of FXS, fmr1 knockout (fmr1 KO) mice were generated (Bakker et al. 1994), which mirror some of the key behavioural phenotypes observed in humans, such as hyperactivity and increased anxiety. Most importantly, they also show deficits in cognitive function as learning in the Morris water maze is impaired (Kooy et al. 1996; D'Hooge et al. 1997). In line with this, there is evidence for an impairment in LTP both in the neocortex and in the hippocampus of fmr1 KO animals (Desai et al. 2006; Lauterborn et al. 2007; Meredith et al. 2007; Wilson & Cox, 2007; Hu et al. 2008). Hence, these mice have been intensively used to study the pre‐ and postsynaptic function of FMRP. In some neurons FMRP has been shown to be present directly at the presynapse as well as in axon‐restricted fragile X granules (FXGs), which can be observed especially during particularly plastic developmental stages (Christie et al. 2009; Akins et al. 2012). A presynaptic role has been suggested, as the absence of FMRP correlates with dysregulations in GABA release (Centonze et al. 2008; Kang et al. 2017), causing imbalances between inhibitory and excitatory neurotransmission. Additionally, several studies showed that FMRP is able to regulate presynaptic ion channel stability, trafficking, surface expression as well as sensitivity indicating a presynaptic role (Brown et al. 2010; Strumbos et al. 2010; Gross et al. 2011; Lee et al. 2011; Zhang et al. 2012; Ferron et al. 2014; Wang et al. 2014; Deng & Klyachko, 2016).
Nevertheless, most studies in fmr1 KO mice focus on the postsynapse where investigations of neuronal morphology show that the immature spine profile of human patients can be mimicked. However, reports about the detailed changes in dendritic spine density and morphology are controversial (Braun & Segal, 2000; Nimchinsky et al. 2001; Galvez & Greenough, 2005; Antar et al. 2006; Grossman et al. 2006, 2010; Bilousova et al. 2009; Cruz‐Martin et al. 2010; Pan et al. 2010; Levenga et al. 2011; Wijetunge et al. 2014). Whereas a large proportion of studies reported the classical immature spine phenotype in the hippocampus and neocortex (Nimchinsky et al. 2001; Antar et al. 2006; Grossman et al. 2006, 2010; Bilousova et al. 2009; Levenga et al. 2011), other studies analysing the same brain regions were not able to detect an impairment in spine maturation (Braun & Segal, 2000; Cruz‐Martin et al. 2010) or detected only minimal alterations (Wijetunge et al. 2014). Similar controversies are reported for spine density analyses as well, especially in the hippocampus where a decrease (Braun & Segal, 2000), no alterations (Pfeiffer & Huber, 2007; de Vrij et al. 2008; Levenga et al. 2011) or an increase in spine density (Antar et al. 2006; Grossman et al. 2006; Swanger et al. 2011) was described. In part, this controversy might be due to different labelling methods as, for instance, it is not clear if there is a specific subpopulation of cells labelled by Golgi–Cox impregnation and if so whether this population might be affected by the loss of FMRP. Despite the wealth of data, it is therefore still under debate if postsynaptic alterations in FXS are persistent or transient, which brain regions are affected most and whether some neuronal subpopulations might be specifically impaired (also reviewed extensively in He & Portera‐Cailliau, 2013). In addition, super‐resolution microscopy techniques will help in the future to reveal details of the structural phenotype at synapses which might be very subtle in some brain regions or in specific neuronal subpopulations (Wijetunge et al. 2014) but could be much more pronounced in other areas or following different experiences. In this respect it has been shown that rearing in an enriched environment either led to the enhancement of the immature spine profile or ameliorated it (Restivo et al. 2005; Lauterborn et al. 2015).
Given the fact that FMRP has both pre‐ and postsynaptic functions, it seems to be especially important in future studies to view the pre‐ and postsynapse as entities which at best should be studied together in order to obtain more conclusive results. We recently made the effort to analyse pre‐ as well as postsynaptic specializations at the same time, which allowed us to uncover a novel role of FMRP in restricting development of one of the most powerful synapses in the central nervous system, the mossy fibre synapse (Scharkowski et al. 2017). It is composed of presynaptic large mossy fibre terminals (LMTs) of granule cells connecting with postsynaptic thorny excrescences (TEs) on proximal dendrites of CA3 pyramidal neurons in the stratum lucidum. Each TE consists of multiple postsynaptic densities, rendering this synapse especially strong (Amaral & Dent, 1981), and together with their location close to the soma of CA3 neurons, these synapses are able to drive the firing of CA3 neurons just by activation of a few TEs (reviewed in Evstratova & Toth, 2014). Accordingly, the mossy fibre synapse can be described as a ‘detonator’ or ‘teacher’ directing the storage of information in the CA3 network (Urban et al. 2001). This allows single granule cells to precisely time the activity of CA3 pyramidal neurons and thereby provide the necessary depolarization needed for Hebbian plasticity at the associational/commissural inputs in the stratum radiatum and stratum oriens (see also Henze et al. 2000). Mutations in a different gene (grik2, coding for the kainate receptor subunit GluK2) are also associated with ASD and intellectual disability and have been shown to influence LMT‐TE development (Lanore et al. 2012). Moreover, a key role of the mossy fibre synapse in mediating homeostatic synaptic plasticity was reported in mature hippocampal neurons (Lee et al. 2013). Behavioural phenotypes appearing in autism spectrum disorders, as hyperactivity and hypersensitivity, especially point towards an impaired homeostatic synaptic plasticity, which therefore renders the mossy fibre synapse a key structure for investigating the cellular basis of ASD.
Although a central connection for hippocampal network function, this synapse had not been studied in the FXS mouse model before. Somewhat surprisingly, we found TEs to be premature during development in fmr1 KO mice with their number and size being increased both in vitro and in vivo (Fig. 1; Scharkowski et al. 2017). In contrast to this, presynaptic LMTs were decreased in size leading to a significant change in the LMT/TE area ratio in fmr1 KO animals compared to WT mice. In parallel, single TEs contained more clusters of the actin‐binding protein synaptopodin, which is accumulated in the spine apparatus thereby indicating enhanced synapse maturation. In line with this premature phenotype, we found that the structure of TEs was hyperstabilized during development as short‐term structural plasticity was significantly decreased together with a reduction in actin polymerization rates. This phenotype could be rescued by overexpression of the actin binding protein profilin 1 (PFN1), a target of FMRP with reduced expression levels in the FXS mouse model (Scharkowski et al. 2017).
In addition to the structural alterations described above, we were also able to detect functional changes in the absence of FMRP as well. We used expression of Super Ecliptic pHluorin (SEP)–glutamate receptor subunit 1 (GluR1) (Kopec et al. 2007), which specifically labels surface AMPA receptors to quantify the amount of GluR1‐containing AMPA receptors at TEs of WT and fmr1 KO neurons (Scharkowski et al. 2017). The intensity of SEP–GluR1 clusters was analysed under baseline conditions and following increased activity. Interestingly, already under baseline conditions the SEP–GluR1 signal was stronger in fmr1 KO TEs compared to WT neurons, indicating an increased content of surface AMPA receptors. Synaptic activation via KCl led to a significant increase in the SEP–GluR1 signal both in WT neurons and in fmr1 KO cells compared to baseline conditions, but this increase was more pronounced in the absence of FMRP. In contrast to the premature phenotype of TEs, CA3 dendrites located in the stratum radiatum displayed the well documented immature spine profile with an overabundance of thin spines in fmr1 KO neurons (Fig. 1). It was indeed an intriguing finding that two different synapse types found at the same neuron are affected differentially, notably even in opposite directions, in the FXS mouse model, most likely with detrimental outcome for information processing in CA3 neurons.
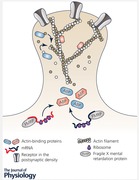
Figure 1. Two opposite synaptic phenotypes shown on a single FXS CA3 neuron.

In comparison to WT CA3 neurons (left side), FXS neurons (right side) show a hyperabundance of long and thin dendritic spines in the stratum radiatum (right upper tile). Given the fact that FMRP has an important function in mRNA transport and the regulation of local translation, our work suggests that this phenotype might be based on altered actin dynamics, caused by a dysregulation of actin‐binding proteins (ABPs) (right upper tile, here shown for the ABPs profilin 1 (PFN1) and cofilin 1 (CFL)). Surprisingly, we found mossy fibre synapses on CA3 neurons in the stratum lucidum (lower tiles) to be structurally altered as well, having increased numbers of thorny excrescences (TE), containing more surface AMPA receptors as well as more clusters of the ABP synaptopodin.
Imbalance of the actin cytoskeleton in FXS
The dysregulation of the actin cytoskeleton as observed in TEs in the FXS mouse model might emerge more and more as a central element in mediating aberrant spine phenotypes in the course of FXS and probably even in ASD in general (Chen et al. 2010). The FMRP target PFN1 with reduced levels in fmr1 KO neurons can be seen as an example where expression of recombinant PFN1 rescued actin dynamics in TEs described above as well as the immature spine phenotype of regular spines (Michaelsen‐Preusse et al. 2016; Scharkowski et al. 2017). This is in line with other studies, which also hypothesized that the rescue of spine phenotypes by manipulation of the actin cytoskeleton might represent a treatment to ameliorate ASD‐like behavioural symptoms. It was shown that a potent small molecule inhibitor of group I p21‐activated kinases (PAKs) reversed dendritic spine phenotypes in fmr1 KO mice. Moreover, this PAK inhibitor (FRAX486) rescued seizures and behavioural abnormalities such as hyperactivity and repetitive movements (Dolan et al. 2013). Also Bongmba and colleagues (2011) showed that pharmacological manipulation of overactive Rac1 partially reversed altered long‐term plasticity. Additionally, increased Rac1 activity could be also blocked by increased training time in a fear‐conditioning paradigm thereby preventing cognitive deficits (Martinez & Tejada‐Simon, 2017). Therefore, the regulation of Rac1 may indeed provide a functional link between alterations in neuronal morphology, deficits in synaptic plasticity and impaired cognition in FXS. As mentioned above, dysregulation of actin‐binding proteins and the accompanying spine phenotypes might be central not only to FXS but to ASD. Support of this comes from the Shank3 autism mouse model where pharmacological blocking of overactive cofilin 1 prevented autism‐like social deficits and repetitive behaviours, as well as significantly diminished NMDA receptor synaptic function and synaptic distribution in the prefrontal cortex (Duffney et al. 2015; for reviews see Yan et al. 2016; Joensuu et al. 2018). Normalization of excessive activity in pathways modulating actin polymerization to normalize cortical actin dynamics might indeed offer a potential therapeutic strategy to ameliorate cognitive and synaptic defects in autism. In this respect it is important to note that the expression of many actin regulators is often brain specific thereby reducing potential detrimental side effects. The identification of actin‐interacting proteins with distinct transcriptional activity in different brain regions and in the periphery would help to reveal potential therapeutic targets. Manipulation of these actin regulators (for instance of human PAK3, which is involved in X‐linked mental retardation; Allen et al. 1998) could enable the specific normalization of actin dynamics at glutamatergic synapses (see also Yan et al. 2016).
Local translation of actin‐binding proteins in FXS
The altered spine morphology phenotype in FXS can be linked to a direct role of FMRP in activity‐dependent mRNA transport, docking and local translation (reviewed in Bassell & Warren, 2008; Dictenberg et al. 2008; Kao et al. 2010). Several studies showed that the RNA‐binding protein localizes together with actively translating polyribosomes in cultured neuronal and non‐neuronal cells as well as in isolated brain synaptoneurosomes (Feng et al. 1997; Khandjian et al. 2004; Stefani et al. 2004) and translational dysregulation has recently been suggested to be a major factor in ASD (Santini et al. 2013). Surprisingly, although hundreds of putative FMRP‐associated mRNAs were identified (Darnell et al. 2011; Ascano et al. 2012), little is known about the direct correlation between FMRP and actin‐binding proteins and only a very few direct interactions of actin‐binding protein mRNAs and FMRP have been validated (Reeve et al. 2005; Michaelsen‐Preusse et al. 2016). The example of MAP1B as a cytoskeletal regulatory protein described as an FMRP target indicates that there might be indeed highly conserved target mRNAs – ranging from Drosophila to mammals – which could be key molecules responsible for the most prominent phenotypes of the disease (Darnell et al. 2001; Zhang et al. 2001). However, studies on the interaction of FMRP with the mRNAs of actin regulators showed controversial results. Whereas Ascano and colleagues (2012) identified prominent candidates as profilin, cofilin, cortactin and members of the Arp2/3 complex and also work form our own lab showed an interaction of the mRNAs of PFN1 and cofilin 1 with FMRP (Michaelsen‐Preusse et al. 2016 and unpublished data), others did not detect an interaction with these mRNAs (Darnell et al. 2011). Interestingly, our results show that profilin 1 and cofilin 1 levels are altered in opposite directions in fmr1 KO animals further indicating potentially different FMRP‐dependent regulatory mechanisms. FMRP has been in general described as a negative regulator of translation for many of its targets; however, it was also shown that the protein can stabilize target mRNAs leading to a decreased expression in the absence of FMRP as is the case for PSD95 (Zalfa et al. 2007).
Conclusion
In summary, a variety of neurodevelopmental diseases such as schizophrenia, cognitive impairment and ASD share a common phenotype: alterations in dendritic spine morphology and density across different regions throughout the brain. It has to be emphasized that the reasons for impaired spine structure and function could be both pre‐ and postsynaptic. Therefore, future studies should concentrate more on a characterization of the synapse as an entity to get a deeper insight into dysregulated synaptogenesis and synaptic plasticity. The example of our own work where we found the postsynaptic site (TEs on CA3 pyramidal neurons) to be premature and the presynaptic site to be immature (LMTs of dentate granule cells) emphasizes that the phenotypes can differ extensively which will influence the way potential treatment strategies could be designed. It becomes clear that the mechanisms mediating disease phenotypes as for instance the role of FMRP for both presynaptic and postsynaptic development and function in FXS are highly complex reducing the likelihood to find a single treatment. However, whether behavioural and cognitive impairments originate from presynaptic defects, e.g. by dysregulations in ion channel composition and transmitter release or a postsynaptic defect in spinogenesis, spine maturation and spine plasticity, eventually, pathways converge on the modulation of postsynaptic actin dynamics. Thus, a deeper understanding of the spine actin cytoskeleton could provide versatile future tools and potential treatment strategies for such diverse disorders as cognitive impairment, schizophrenia and ASD.
Additional information
Competing interests
The authors declare no competing financial interests.
Author contributions
All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
K.M.P. and M.K. were supported by the Deutsche Forschungsgemeinschaft (KO 1674/8‐1 and KO 1674/16‐1).
Biography
Martin Korte, Prof. Dr, is Head of the Zoological Institute and group leader of the Division of Cellular Neurobiology at the TU Braunschweig and co‐affiliated to the Helmholtz‐Center for Infectious Disease, AG Neuroinflammation and Neurodegeneration (AG NIND). He studies cellular mechanisms of learning and memory, the physiology of APP, neurotrophins and the functional role of NogoA. In addition he is interested in structural plasticity and the functional role of actin in processes of synaptic plasticity.

Edited by: Ole Petersen & Matthew Nolan
This review was presented at the symposium ‘Neuroplasticity and Synaptic Function in Neuropsychiatric Disorders’, which took place at Cardiff University, Cardiff, UK, 27–28 April 2017.
References
- Akins MR, Leblanc HF, Stackpole EE, Chyung E & Fallon JR (2012). Systematic mapping of fragile X granules in the mouse brain reveals a potential role for presynaptic FMRP in sensorimotor functions. J Comp Neurol 520, 3687–3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen KM, Gleeson JG, Bagrodia S, Partington MW, MacMillan JC, Cerione RA, Mulley JC & Walsh CA (1998). PAK3 mutation in nonsyndromic X‐linked mental retardation. Nat Genet 20, 25–30. [DOI] [PubMed] [Google Scholar]
- Amaral DG & Dent JA (1981). Development of the mossy fibers of the dentate gyrus: I. A light and electron microscopic study of the mossy fibers and their expansions. J Comp Neurol 195, 51–86. [DOI] [PubMed] [Google Scholar]
- Antar LN, Li C, Zhang H, Carroll RC & Bassell GJ (2006). Local functions for FMRP in axon growth cone motility and activity‐dependent regulation of filopodia and spine synapses. Mol Cell Neurosci 32, 37–48. [DOI] [PubMed] [Google Scholar]
- Ascano M Jr, Mukherjee N, Bandaru P, Miller JB, Nusbaum JD, Corcoran DL, Langlois C, Munschauer M, Dewell S, Hafner M, Williams Z, Ohler U & Tuschl T (2012). FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature 492, 382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker CE, Verheij C, Willemsen R, van der Helm R, Oerlemans F, Vermey M, Bygrave A, Hoogeveen AT, Oostra BA, Reyniers E, De Boule K, D'Hooge R, Cras P, van Velzen D, Nagels G, Martin JJ, De Deyn PP, Darby JK & Willems PJ (1994). Fmr1 knockout mice: A model to study fragile X mental retardation. Cell 78, 23–33. [PubMed] [Google Scholar]
- Bassell GJ & Warren ST (2008). Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron 60, 201–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckel‐Mitchener A & Greenough WT (2004). Correlates across the structural, functional, and molecular phenotypes of fragile X syndrome. Ment Retard Dev Disabil Res Rev 10, 53–59. [DOI] [PubMed] [Google Scholar]
- Berning S, Willig KI, Steffens H, Dibaj P & Hell SW (2012). Nanoscopy in a living mouse brain. Science 335, 551. [DOI] [PubMed] [Google Scholar]
- Bilousova TV, Dansie L, Ngo M, Aye J, Charles JR, Ethell DW & Ethell IM (2009). Minocycline promotes dendritic spine maturation and improves behavioural performance in the fragile X mouse model. J Med Genet 46, 94–102. [DOI] [PubMed] [Google Scholar]
- Blanpied TA & Ehlers MD (2004). Microanatomy of dendritic spines: emerging principles of synaptic pathology in psychiatric and neurological disease. Biol Psychiatry 55, 1121–1127. [DOI] [PubMed] [Google Scholar]
- Bongmba OY, Martinez LA, Elhardt ME, Butler K & Tejada‐Simon MV (2011). Modulation of dendritic spines and synaptic function by Rac1: A possible link to Fragile X syndrome pathology. Brain Res 1399, 79–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonhoeffer T & Yuste R (2002). Spine motility. Phenomenology, mechanisms, and function. Neuron 35, 1019–1027. [DOI] [PubMed] [Google Scholar]
- Bosch M, Castro J, Saneyoshi T, Matsuno H, Sur M & Hayashi Y (2014). Structural and molecular remodeling of dendritic spine substructures during long‐term potentiation. Neuron 82, 444–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch M & Hayashi Y (2012). Structural plasticity of dendritic spines. Curr Opin Neurobiol 22, 383–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun K & Segal M (2000). FMRP involvement in formation of synapses among cultured hippocampal neurons. Cereb Cortex 10, 1045–1052. [DOI] [PubMed] [Google Scholar]
- Brown MR, Kronengold J, Gazula VR, Chen Y, Strumbos JG, Sigworth FJ, Navaratnam D & Kaczmarek LK (2010). Fragile X mental retardation protein controls gating of the sodium‐activated potassium channel Slack. Nat Neurosci 13, 819–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caroni P, Donato F & Muller D (2012). Structural plasticity upon learning: regulation and functions. Nat Rev Neurosci 13, 478–490. [DOI] [PubMed] [Google Scholar]
- Centonze D, Rossi S, Mercaldo V, Napoli I, Ciotti MT, De Chiara V, Musella A, Prosperetti C, Calabresi P, Bernardi G & Bagni C (2008). Abnormal striatal GABA transmission in the mouse model for the fragile X syndrome. Biol Psychiatry 63, 963–973. [DOI] [PubMed] [Google Scholar]
- Chen LY, Rex CS, Babayan AH, Kramar EA, Lynch G, Gall CM & Lauterborn JC (2010). Physiological activation of synaptic Rac>PAK (p‐21 activated kinase) signaling is defective in a mouse model of fragile X syndrome. J Neurosci 30, 10977–10984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LY, Rex CS, Casale MS, Gall CM & Lynch G (2007). Changes in synaptic morphology accompany actin signaling during LTP. J Neurosci 27, 5363–5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie SB, Akins MR, Schwob JE & Fallon JR (2009). The FXG: a presynaptic fragile X granule expressed in a subset of developing brain circuits. J Neurosci 29, 1514–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford DC, Acuna JM & Sherman SL (2001). FMR1 and the fragile X syndrome: human genome epidemiology review. Genet Med 3, 359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz‐Martin A, Crespo M & Portera‐Cailliau C (2010). Delayed stabilization of dendritic spines in fragile X mice. J Neurosci 30, 7793–7803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Jensen KB, Jin P, Brown V, Warren ST & Darnell RB (2001). Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell 107, 489–499. [DOI] [PubMed] [Google Scholar]
- Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, Licatalosi DD, Richter JD & Darnell RB (2011). FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng PY & Klyachko VA (2016). Genetic upregulation of BK channel activity normalizes multiple synaptic and circuit defects in a mouse model of fragile X syndrome. J Physiol 594, 83–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai NS, Casimiro TM, Gruber SM, Vanderklish PW (2006). Early postnatal plasticity in neocortex of Fmr1 knockout mice. J Neurophysiol 96, 1734–1745. [DOI] [PubMed] [Google Scholar]
- de Vrij FM, Levenga J, van der Linde HC, Koekkoek SK, de Zeeuw CI, Nelson DL, Oostra BA & Willemsen R (2008). Rescue of behavioral phenotype and neuronal protrusion morphology in Fmr1 KO mice. Neurobiol Dis 31, 127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Hooge R, Nagels G, Franck F, Bakker CE, Reyniers E, Storm K, Kooy RF, Oostra BA, Willems PJ & De Deyn PP (1997). Mildly impaired water maze performance in male Fmr1 knockout mice. Neuroscience 76, 367–376. [DOI] [PubMed] [Google Scholar]
- Dictenberg JB, Swanger SA, Antar LN, Singer RH & Bassell GJ (2008). A direct role for FMRP in activity‐dependent dendritic mRNA transport links filopodial‐spine morphogenesis to fragile X syndrome. Dev Cell 14, 926–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolan BM, Duron SG, Campbell DA, Vollrath B, Shankaranarayana Rao BS, Ko HY, Lin GG, Govindarajan A, Choi SY & Tonegawa S (2013). Rescue of fragile X syndrome phenotypes in Fmr1 KO mice by the small‐molecule PAK inhibitor FRAX486. Proc Natl Acad Sci USA 110, 5671–5676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffney LJ, Zhong P, Wei J, Matas E, Cheng J, Qin L, Ma K, Dietz DM, Kajiwara Y, Buxbaum JD & Yan Z (2015). Autism‐like deficits in Shank3‐deficient mice are rescued by targeting actin regulators. Cell Rep 11, 1400–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunaevsky A, Tashiro A, Majewska A, Mason C & Yuste R (1999). Developmental regulation of spine motility in the mammalian central nervous system. Proc Natl Acad Sci USA 96, 13438–13443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engert F & Bonhoeffer T (1999). Dendritic spine changes associated with hippocampal long‐term synaptic plasticity. Nature 399, 66–70. [DOI] [PubMed] [Google Scholar]
- Evstratova A & Toth K (2014). Information processing and synaptic plasticity at hippocampal mossy fiber terminals. Front Cell Neurosci 8, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Gutekunst CA, Eberhart DE, Yi H, Warren ST & Hersch SM (1997). Fragile X mental retardation protein: nucleocytoplasmic shuttling and association with somatodendritic ribosomes. J Neurosci 17, 1539–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferron L, Nieto‐Rostro M, Cassidy JS & Dolphin AC (2014). Fragile X mental retardation protein controls synaptic vesicle exocytosis by modulating N‐type calcium channel density. Nat Commun 5, 3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiala JC, Spacek J & Harris KM (2002). Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res Brain Res Rev 39, 29–54. [DOI] [PubMed] [Google Scholar]
- Fischer M, Kaech S, Knutti D & Matus A (1998). Rapid actin‐based plasticity in dendritic spines. Neuron 20, 847–854. [DOI] [PubMed] [Google Scholar]
- Frost NA, Kerr JM, Lu HE & Blanpied TA (2010). A network of networks: cytoskeletal control of compartmentalized function within dendritic spines. Curr Opin Neurobiol 20, 578–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukazawa Y, Saitoh Y, Ozawa F, Ohta Y, Mizuno K & Inokuchi K (2003). Hippocampal LTP is accompanied by enhanced F‐actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron 38, 447–460. [DOI] [PubMed] [Google Scholar]
- Galvez R & Greenough WT (2005). Sequence of abnormal dendritic spine development in primary somatosensory cortex of a mouse model of the fragile X mental retardation syndrome. Am J Med Genet A 135, 155–160. [DOI] [PubMed] [Google Scholar]
- Gross C, Yao X, Pong DL, Jeromin A & Bassell GJ (2011). Fragile X mental retardation protein regulates protein expression and mRNA translation of the potassium channel Kv4.2. J Neurosci 31, 5693–5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman AW, Aldridge GM, Lee KJ, Zeman MK, Jun CS, Azam HS, Arii T, Imoto K, Greenough WT & Rhyu IJ (2010). Developmental characteristics of dendritic spines in the dentate gyrus of Fmr1 knockout mice. Brain Res 1355, 221–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman AW, Elisseou NM, McKinney BC & Greenough WT (2006). Hippocampal pyramidal cells in adult Fmr1 knockout mice exhibit an immature‐appearing profile of dendritic spines. Brain Res 1084, 158–164. [DOI] [PubMed] [Google Scholar]
- Halpain S (2000). Actin and the agile spine: how and why do dendritic spines dance? Trends Neurosci 23, 141–146. [DOI] [PubMed] [Google Scholar]
- He CX & Portera‐Cailliau C (2013). The trouble with spines in fragile X syndrome: density, maturity and plasticity. Neuroscience 251, 120–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henze DA, Urban NN & Barrionuevo G (2000). The multifarious hippocampal mossy fiber pathway: a review. Neuroscience 98, 407–427. [DOI] [PubMed] [Google Scholar]
- Hinton VJ, Brown WT, Wisniewski K & Rudelli RD (1991). Analysis of neocortex in three males with the fragile X syndrome. Am J Med Genet 41, 289–294. [DOI] [PubMed] [Google Scholar]
- Holtmaat A & Svoboda K (2009). Experience‐dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci 10, 647–658. [DOI] [PubMed] [Google Scholar]
- Honkura N, Matsuzaki M, Noguchi J, Ellis‐Davies GC & Kasai H (2008). The subspine organization of actin fibers regulates the structure and plasticity of dendritic spines. Neuron 57, 719–729. [DOI] [PubMed] [Google Scholar]
- Hotulainen P, Llano O, Smirnov S, Tanhuanpaa K, Faix J, Rivera C & Lappalainen P (2009). Defining mechanisms of actin polymerization and depolymerization during dendritic spine morphogenesis. J Cell Biol 185, 323–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Qin Y, Bochorishvili G, Zhu Y, van AL & Zhu JJ (2008). Ras signaling mechanisms underlying impaired GluR1‐dependent plasticity associated with fragile X syndrome. J Neurosci 28, 7847–7862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin SA, Patel B, Idupulapati M, Harris JB, Crisostomo RA, Larsen BP, Kooy F, Willems PJ, Cras P, Kozlowski PB, Swain RA, Weiler IJ & Greenough WT (2001). Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile‐X syndrome: a quantitative examination. Am J Med Genet 98, 161–167. [DOI] [PubMed] [Google Scholar]
- Joensuu M, Lanoue V & Hotulainen P (2018). Dendritic spine actin cytoskeleton in autism spectrum disorder. Prog Neuropsychopharmacol Biol Psychiatry 84, 362–381. [DOI] [PubMed] [Google Scholar]
- Kang JY, Chadchankar J, Vien TN, Mighdoll MI, Hyde TM, Mather RJ, Deeb TZ, Pangalos MN, Brandon NJ, Dunlop J & Moss SJ (2017). Deficits in the activity of presynaptic gamma‐aminobutyric acid type B receptors contribute to altered neuronal excitability in fragile X syndrome. J Biol Chem 292, 6621–6632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao DI, Aldridge GM, Weiler IJ & Greenough WT (2010). Altered mRNA transport, docking, and protein translation in neurons lacking fragile X mental retardation protein. Proc Natl Acad Sci USA 107, 15601–15606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandjian EW, Huot ME, Tremblay S, Davidovic L, Mazroui R & Bardoni B (2004). Biochemical evidence for the association of fragile X mental retardation protein with brain polyribosomal ribonucleoparticles. Proc Natl Acad Sci USA 101, 13357–13362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koleske AJ (2013). Molecular mechanisms of dendrite stability. Nat Rev Neurosci 14, 536–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooy RF, D'Hooge R, Reyniers E, Bakker CE, Nagels G, De Boulle K, Storm K, Clincke G, De Deyn PP, Oostra BA & Willems PJ (1996). Transgenic mouse model for the fragile X syndrome. Am J Med Genet 64, 241–245. [DOI] [PubMed] [Google Scholar]
- Kopec CD, Real E, Kessels HW & Malinow R (2007). GluR1 links structural and functional plasticity at excitatory synapses. J Neurosci 27, 13706–13718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte M & Schmitz D (2016). Cellular and system biology of memory: Timing, molecules, and beyond. Physiol Rev 96, 647–693. [DOI] [PubMed] [Google Scholar]
- Lamprecht R & LeDoux J (2004). Structural plasticity and memory. Nat Rev Neurosci 5, 45–54. [DOI] [PubMed] [Google Scholar]
- Lanore F, Labrousse VF, Szabo Z, Normand E, Blanchet C & Mulle C (2012). Deficits in morphofunctional maturation of hippocampal mossy fiber synapses in a mouse model of intellectual disability. J Neurosci 32, 17882–17893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauterborn JC, Jafari M, Babayan AH & Gall CM (2015). Environmental enrichment reveals effects of genotype on hippocampal spine morphologies in the mouse model of Fragile X Syndrome. Cereb Cortex 25, 516–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauterborn JC, Rex CS, Kramar E, Chen LY, Pandyarajan V, Lynch G & Gall CM (2007). Brain‐derived neurotrophic factor rescues synaptic plasticity in a mouse model of fragile X syndrome. J Neurosci 27, 10685–10694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HY, Ge WP, Huang W, He Y, Wang GX, Rowson‐Baldwin A, Smith SJ, Jan YN & Jan LY (2011). Bidirectional regulation of dendritic voltage‐gated potassium channels by the fragile X mental retardation protein. Neuron 72, 630–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KJ, Queenan BN, Rozeboom AM, Bellmore R, Lim ST, Vicini S & Pak DT (2013). Mossy fiber‐CA3 synapses mediate homeostatic plasticity in mature hippocampal neurons. Neuron 77, 99–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenga J, de Vrij FM, Buijsen RA, Li T, Nieuwenhuizen IM, Pop A, Oostra BA & Willemsen R (2011). Subregion‐specific dendritic spine abnormalities in the hippocampus of Fmr1 KO mice. Neurobiol Learn Mem 95, 467–472. [DOI] [PubMed] [Google Scholar]
- Martinez LA & Tejada‐Simon MV (2017). Increased training intensity induces proper membrane localization of actin remodeling proteins in the hippocampus preventing cognitive deficits: implications for Fragile X syndrome. Mol Neurobiol, 10.1007/s12035-017-0666-4. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis‐Davies GC & Kasai H (2004). Structural basis of long‐term potentiation in single dendritic spines. Nature 429, 761–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith RM, Holmgren CD, Weidum M, Burnashev N & Mansvelder HD (2007). Increased threshold for spike‐timing‐dependent plasticity is caused by unreliable calcium signaling in mice lacking fragile X gene FMR1. Neuron 54, 627–638. [DOI] [PubMed] [Google Scholar]
- Michaelsen‐Preusse K, Zessin S, Grigoryan G, Scharkowski F, Feuge J, Remus A & Korte M (2016). Neuronal profilins in health and disease: Relevance for spine plasticity and Fragile X syndrome. Proc Natl Acad Sci USA 113, 3365–3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagerl UV, Eberhorn N, Cambridge SB & Bonhoeffer T (2004). Bidirectional activity‐dependent morphological plasticity in hippocampal neurons. Neuron 44, 759–767. [DOI] [PubMed] [Google Scholar]
- Nimchinsky EA, Oberlander AM & Svoboda K (2001). Abnormal development of dendritic spines in FMR1 knock‐out mice. J Neurosci 21, 5139–5146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oertner TG & Matus A (2005). Calcium regulation of actin dynamics in dendritic spines. Cell Calcium 37, 477–482. [DOI] [PubMed] [Google Scholar]
- Pan F, Aldridge GM, Greenough WT & Gan WB (2010). Dendritic spine instability and insensitivity to modulation by sensory experience in a mouse model of fragile X syndrome. Proc Natl Acad Sci USA 107, 17768–17773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P, Cahill ME, Jones KA, VanLeeuwen JE & Woolfrey KM (2011). Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci 14, 285–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer BE & Huber KM (2007). Fragile X mental retardation protein induces synapse loss through acute postsynaptic translational regulation. J Neurosci 27, 3120–3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve SP, Bassetto L, Genova GK, Kleyner Y, Leyssen M, Jackson FR & Hassan BA (2005). The Drosophila fragile X mental retardation protein controls actin dynamics by directly regulating profilin in the brain. Curr Biol 15, 1156–1163. [DOI] [PubMed] [Google Scholar]
- Restivo L, Ferrari F, Passino E, Sgobio C, Bock J, Oostra BA, Bagni C & Ammassari‐Teule M (2005). Enriched environment promotes behavioral and morphological recovery in a mouse model for the fragile X syndrome. Proc Natl Acad Sci USA 102, 11557–11562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudelli RD, Brown WT, Wisniewski K, Jenkins EC, Laure‐Kamionowska M, Connell F & Wisniewski HM (1985). Adult fragile X syndrome. Clinico‐neuropathologic findings. Acta Neuropathol 67, 289–295. [DOI] [PubMed] [Google Scholar]
- Sala C & Segal M (2014). Dendritic spines: the locus of structural and functional plasticity. Physiol Rev 94, 141–188. [DOI] [PubMed] [Google Scholar]
- Santini E, Huynh TN, MacAskill AF, Carter AG, Pierre P, Ruggero D, Kaphzan H & Klann E (2013). Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature 493, 411–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharkowski F, Frotscher M, Lutz D, Korte M & Michaelsen‐Preusse K (2017). Altered connectivity and synapse maturation of the hippocampal mossy fiber pathway in a mouse model of the Fragile X syndrome. Cereb Cortex, 10.1093/cercor/bhw408. [DOI] [PubMed] [Google Scholar]
- Star EN, Kwiatkowski DJ & Murthy VN (2002). Rapid turnover of actin in dendritic spines and its regulation by activity. Nat Neurosci 5, 239–246. [DOI] [PubMed] [Google Scholar]
- Stefani G, Fraser CE, Darnell JC & Darnell RB (2004). Fragile X mental retardation protein is associated with translating polyribosomes in neuronal cells. J Neurosci 24, 7272–7276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strumbos JG, Brown MR, Kronengold J, Polley DB & Kaczmarek LK (2010). Fragile X mental retardation protein is required for rapid experience‐dependent regulation of the potassium channel Kv3.1b. J Neurosci 30, 10263–10271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanger SA, Yao X, Gross C & Bassell GJ (2011). Automated 4D analysis of dendritic spine morphology: applications to stimulus‐induced spine remodeling and pharmacological rescue in a disease model. Mol Brain 4, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban NN, Henze DA & Barrionuevo G (2001). Revisiting the role of the hippocampal mossy fiber synapse. Hippocampus 11, 408–417. [DOI] [PubMed] [Google Scholar]
- Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP, Eussen BE, van Ommen G, Bionden LAJ, Riggins GJ, Chastain JL, Kunst C, Gaijaard H, Caskey CT, Nelson DL, Oostra BA & Warren ST (1991). Identification of a gene (FMR‐1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914. [DOI] [PubMed] [Google Scholar]
- Wang XS, Peng CZ, Cai WJ, Xia J, Jin D, Dai Y, Luo XG, Klyachko VA & Deng PY (2014). Activity‐dependent regulation of release probability at excitatory hippocampal synapses: a crucial role of fragile X mental retardation protein in neurotransmission. Eur J Neurosci 39, 1602–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijetunge LS, Angibaud J, Frick A, Kind PC & Nagerl UV (2014). Stimulated emission depletion (STED) microscopy reveals nanoscale defects in the developmental trajectory of dendritic spine morphogenesis in a mouse model of fragile X syndrome. J Neurosci 34, 6405–6412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson BM & Cox CL (2007). Absence of metabotropic glutamate receptor‐mediated plasticity in the neocortex of fragile X mice. Proc Natl Acad Sci USA 104, 2454–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Kim E, Datta D, Lewis DA & Soderling SH (2016). Synaptic actin dysregulation, a convergent mechanism of mental disorders? J Neurosci 36, 11411–11417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalfa F, Eleuteri B, Dickson KS, Mercaldo V, De Rubeis S, di Penta A, Tabolacci E, Chiurazzi P, Neri G, Grant SG & Bagni C (2007). A new function for the fragile X mental retardation protein in regulation of PSD‐95 mRNA stability. Nat Neurosci 10, 578–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Brown MR, Hyland C, Chen Y, Kronengold J, Fleming MR, Kohn AB, Moroz LL & Kaczmarek LK (2012). Regulation of neuronal excitability by interaction of fragile X mental retardation protein with slack potassium channels. J Neurosci 32, 15318–15327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YQ, Bailey AM, Matthies HJ, Renden RB, Smith MA, Speese SD, Rubin GM & Broadie K (2001). Drosophila fragile X‐related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell 107, 591–603. [DOI] [PubMed] [Google Scholar]