

Abstract
Discovery of the STIM1 and Orai proteins as the principal components of store‐operated Ca2+ entry has drawn attention to contact sites between the endoplasmic reticulum (ER) and the plasma membrane (PM). Such contacts between adjacent membranes of different cellular organelles, primarily between the mitochondria and the ER, had already been known as the sites where Ca2+ released from the ER can be efficiently channelled to the mitochondria and also where phosphatidylserine synthesis and transfer takes place. Recent studies have identified contact sites between virtually every organelle and the ER and the functional importance of these small specialized membrane domains is increasingly recognized. Most recent developments have highlighted the role of phosphatidylinositol 4‐phosphate gradients as critical determinants of the non‐vesicular transport of various lipids from the ER to other organelles such as the Golgi or PM. As we learn more about membrane contact sites it becomes apparent that Ca2+ is not only transported at these sites but also controls both the dynamics and the lipid transfer efficiency of these processes. Conversely, lipids are critical for regulating the Ca2+ entry process. This review will summarize some of the most exciting recent developments in this rapidly expanding research field.
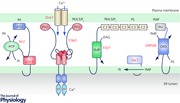
Keywords: store‐operated calcium entry, calcium entry, ER‐PM contacts
STIM and Orai proteins constitute store‐operated Ca2+ entry in endoplasmic reticulum–plasma membrane contact sites
It was over 10 years ago when the molecular basis of store‐operated Ca2+ entry (SOCE) was finally identified. SOCE has long been recognized as a ubiquitous Ca2+ entry route that is activated upon stimulation of mammalian cells through Ca2+‐mobilizing cell surface receptors, and which is ultimately controlled by the Ca2+ filling state of the endoplasmic reticulum (ER) (Putney, 1986). Based on pioneering studies from the Meyer, Stauderman, Rao, Lewis, Penner and Kinet laboratories, stromal interaction molecule 1 (STIM1) was identified as the ER luminal Ca2+ sensor (Liou et al. 2005; Roos et al. 2005) and Orai1 as the plasma membrane (PM) Ca2+ channel (Feske et al. 2006; Prakriya et al. 2006; Vig et al. 2006). These proteins work together to constitute SOCE. Because of the localization of the two proteins in the ER and PM, respectively, they can only work at sites where the two membranes are in close proximity, i.e. in ER–PM contact areas (Lewis, 2007). This simple fact has drawn significant attention to this special compartment, which turned out to hold a great amount of exciting new biology to be discovered. Great progress has since been made and it is now well established that ER luminal Ca2+ decrease causes STIM1 molecules to change their conformation such that they undergo massive clustering in the ER and interact and activate the Orai1 channels in the PM. Several important reviews have summarized these advances (Hogan et al. 2010; Soboloff et al. 2012; Hogan & Rao, 2015; Derler et al. 2016a; Putney et al. 2017; Zhou et al. 2017). More recent studies have revealed structural features of the STIM1 and Orai1 proteins that are critical for the activation process. These include the identification of the part of the STIM1 molecule that interacts with the Orai1 channel (called SOAR, Yuan et al. 2009, or CAD, Park et al. 2009) and the characterization of an intramolecular interaction between the SOAR domain and the first membrane‐adjacent coiled‐coil domain of the cytoplasmic part of STIM1 that keeps the SOAR domain from activating Orai1 in the resting state (Muik et al. 2011; Zhou et al. 2013; Fahrner et al. 2014; Ma et al. 2015). Similar structural and mutational studies reveal the key interactive motifs that form the basis of interaction between SOAR and Orai1 and the mechanism of Orai1 channel activation (Derler et al. 2013; Gudlur et al. 2014; Zhou et al. 2016; Nwokonko et al. 2017; Palty et al. 2017).
Lipid composition within special membrane domains controls STIM1/Orai1 function
While these structural studies are critically important to better understand the details of the molecular events leading to activation of STIM1/Orai1, it is equally intriguing how the lipid composition of the membrane determines the function of the STIM1/Orai1 complex. Soon after the discovery of STIM1 it was recognized that its cytoplasmic C‐terminus contains a polybasic stretch that helps the protein to maintain PM contact even in the resting state (Liou et al. 2007; Park et al. 2009). While this part of STIM1 is dispensable for activation when both STIM1 and Orai1 are overexpressed, it certainly increases the efficiency of the activation process during physiological conditions. It has been debated what the STIM1 polybasic domain binds to in the PM, but anionic phospholipids have been the prime candidates. The consensus that appears to have been reached identifies phosphatidylinositol 4,5‐bisphosphate (PI(4,5)P2) as the lipid that binds the polybasic domain of STIM1 (Walsh et al. 2010; Maleth et al. 2014; Cao et al. 2015). It should be noted, though, that our studies addressing this question could not substantiate PI(4,5)P2 as the major STIM1 PM anchor molecule, possibly because of the use of overexpressed proteins (Varnai et al. 2007; Korzeniowski et al. 2009). Yet, another inositol lipid, PI4P was suspected to control Orai1 channel activity, based on the sensitivity of SOCE and I CRAC (the current carried by Orai1) to PI‐kinase inhibitors that affected PI4P but not PI(4,5)P2 levels (Rosado & Sage, 2000; Broad et al. 2001; Korzeniowski et al. 2009). While many ion channels and transporters require phosphoinositides (mainly PI(4,5)P2 for proper function (see a list in Balla, 2013), this aspect of Orai channel regulation has not been fully explored. It will be important to see if the PI4P‐mediated regulation of Orai1 channels can be better understood with the use of newly available more specific phosphatidylinositol 4‐kinase (PI4K) inhibitors (Bojjireddy et al. 2014). In light of the new developments in the biology of contact sites and the fact that PI4P gradients shape the lipid composition of membranes, there are a number of possibilities regarding how changes in PM PI4P levels may indirectly affect the function of the Orai1 channels (see below).
Additional data also indicated that membrane microdomains and their special lipid composition play critical roles in the control of SOCE. Cholesterol depletion was reported to increase STIM1/Orai1‐mediated Ca2+ influx or I CRAC currents, an effect that was attributed to cholesterol interaction with the N‐terminal segment of Orai1 (Derler et al. 2016b) and also with the STIM1 SOAR domain affecting its association with Orai1 (Pacheco et al. 2016). Even earlier studies had reported on the influence of cholesterol status on SOCE, showing its effect on changes in Ca2+‐dependent inactivation (Dionisio et al. 2011) and the control of STIM1 clustering and interaction with transient receptor potential channel (TrpC1) (Pani et al. 2008). Furthermore, additional studies suggested that cholesterol status also affects the PI(4,5)P2‐mediated lateral segregation of STIM1–Orai complexes (Calloway et al. 2011). This notion was extended in studies where the Ca2+‐dependent inactivation of Orai1 channels by the accessory protein SARAF (SOCE‐associated regulatory factor) (Palty et al. 2012) was found to require partitioning of the STIM1/Orai1 complex to PI(4,5)P2‐enriched PM compartments (Maleth et al. 2014).
ER–PM contacts control lipid dynamics between these organelles
The question of what molecules keep the ER in close contact with the PM has been first addressed in yeast. It was found that the synaptotagmin‐like proteins, tricarbins (Tcb1/2/3), and three other proteins, one called Ist2 and two ER‐resident proteins, Scs2 and Scs22, were responsible for maintaining the ER–PM connection. Elimination of all six proteins disrupted association of ER with the PM and caused major dysregulation of phosphoinositide signalling and induced the unfolded protein response in the ER (Manford et al. 2012). The mammalian homologues of the Tcb proteins, the extended synaptotagmins (ESyt1/2/3), were then subsequently found to be important for ER–PM tethering in mammalian cells (Chang et al. 2013; Giordano et al. 2013). An important feature of the ESyts and Tcbs is that they possess an SMP (synaptotagmin‐like mitochondrial‐lipid‐binding protein) domain, which is found in many proteins that localize to membrane contact sites (Toulmay & Prinz, 2012). The structure of the SMP domain of ESyt2 revealed a β‐barrel module similar to those found in the tubular‐lipid‐binding (TULIP) superfamily. In its dimeric state, the SMP domain of ESyt2 forms an approximately 90 Å‐long cavity encompassing a hydrophobic channel that could serve as a tunnel to pass hydrophobic molecules, including lipids, through the aqueous phase that separates the two membranes in contact sites (Schauder et al. 2014; Jeong et al. 2017). Indeed, cells that lack all three ESyts showed a defect in removing diacylglycerol (DAG) from the PM during massive Ca2+‐induced phospholipase C (PLC) activation suggesting that they can participate in DAG transfer between the PM and other membranes (Saheki et al. 2016). This latter study also showed that lipid transfer by SMP domains in vitro requires Ca2+ concentrations in the hundred micromolar range (Saheki et al. 2016). It is noteworthy, though, that ESyt depleted or knockout cells appear to have normal SOCE mediated by STIM1/Orai1 (Giordano et al. 2013) and the fact that ESyt triple knockout mice show no obvious phenotype (Sclip et al. 2016) suggests that functionality of the ER–PM contacts can be maintained by compensatory mechanisms. Another SMP domain‐containing protein, TMEM24, has been shown to play an important role in pancreatic β cells for glucose‐induced Ca2+ signals and insulin secretion (Pottekat et al. 2013), and it was suggested that it serves as a phosphatidylinositol (PI) transfer protein to supply the PM with PI for maintaining the PM PI(4,5)P2 pool necessary for exocytosis of insulin‐containing vesicles (Lees et al. 2017). ESyts also contain C‐terminal C2 domains that make contacts with PM PI(4,5)P2 and perhaps with other anionic phospholipids depending on the Ca2+ concentration (Chang et al. 2013; Giordano et al. 2013) (see below).
Other lipid transfer proteins are also linked to ER–PM contact sites. The Nir2 protein, which belongs to the family of PI transfer proteins (PITPs), was shown to translocate to ER–PM contact sites during activation of PLC‐coupled receptors to support PI transfer from the ER to the PM during PLC‐mediated consumption of PM PI(4,5)P2 (Chang et al. 2013; Kim et al. 2013). Subsequent studies showed that the Nir2 protein and its fly orthologue, RdgBα, can also transfer phosphatidic acid (PA) from the PM to the ER at these contact sites when PLC is activated (Kim et al. 2015; Yadav et al. 2015). Recruitment of the Nir2 proteins to the contact sites is provided by their interaction with the ER‐localized VAP‐A and VAP‐B proteins via the Nir2 FFAT (double phenylalanine in an acidic tract) domain (Peretti et al. 2008) on the one hand, and with the PA that is generated by PLC activation in the PM on the other (Chang et al. 2013; Kim et al. 2013, 2015). Although Nir2 may also function in other contexts at different contact sites, such as the ER and Golgi (Litvak et al. 2005), its dynamic recruitment to ER–PM contacts, together with its PI and PA transport functions, supports the reutilization of PA and maintenance of PI(4,5)P2 during PLC action.
Another lipid transfer mechanism at ER–PM contacts has been recently described that links non‐vesicular phosphatidylserine (PS) transport to the PM with utilization of PI4P gradients that exist between the PM and ER. It has been known for a while that the PI4P pool made by the yeast Stt4 PI4K is kept under control by the Sac1 phosphatase located in the ER (Foti et al. 2001). More recent yeast studies showed that the ER‐localized Sac1 can access PI4P produced in the PM with the aid of some yeast oxysterol binding protein homologues, such as Osh3 (Stefan et al. 2011). Curiously, in earlier studies, Sac1‐deleted yeast strains showed a large accumulation of PI4P that did not translate to increased PI(4,5)P2 levels (Guo et al. 1999; Hughes et al. 2000; Foti et al. 2001). These findings suggested that the PI4P accumulated in a compartment where the PIP 5‐kinase was unable to convert it to PI(4,5)P2, possibly in the ER. This apparent conundrum can be explained by the recent discovery that PI4P gradients can be utilized to support non‐vesicular transport of cholesterol from the ER to the Golgi both in yeast and mammalian cells (de Saint‐Jean et al. 2011; Mesmin et al. 2013) and, in a similar fashion, to control transport of PS from the ER to the PM (Maeda et al. 2013; Chung et al. 2015; Moser von Filseck et al. 2015). In the mammalian cell, the latter transport is mediated by the oxysterol binding protein related proteins (ORP5) and ORP8. Both of these molecules are anchored to the ER by their C‐terminal hydrophobic domains and make contact with the PM via their PH domains that bind PI4P (Chung et al. 2015). Their lipid transfer domain is able to bind either PI4P or PS but not both at the same time. The PI4P gradients between the ER and the PM are maintained by the action of the PI4KA enzyme in the PM (Balla et al. 2007; Nakatsu et al. 2012; Bojjireddy et al. 2014) and the Sac1 phosphatase in the ER to which PI4P is delivered by the ORP proteins. This arrangement raises the question of how PM PI4P flux is divided between conversion to PI(4,5)P2 and consumption for PI4P/PS exchange. Curiously, PI(4,5)P2 levels can be maintained even when PI4P levels fall in the PM, as long as PLC is not activated, suggesting a functional dissociation between these two lipid pools in spite of their known substrate–product relationship (Hammond et al. 2012; Nakatsu et al. 2012; Bojjireddy et al. 2014). Another important conclusion of these findings is that PI4P metabolism has a great impact on PS levels and, conversely, PS overproduction has a major effect on PI4P levels both in the PM and other PI4P‐rich compartments, such as the Golgi (Sohn et al. 2016).
Lastly, a new family of proteins called Lams (Elbaz‐Alon et al. 2015; Gatta et al. 2015) or Ltcs (Murley et al. 2015) were identified in yeast as sterol transport proteins that function between contact sites of various organelles, including the ER and PM. Mammalian homologues of these proteins have been identified (Gatta et al. 2015; Murley et al. 2015) and it will be interesting to identify which if any of them works in ER–PM contacts and has an impact on lipid composition and Ca2+ regulation.
Ca2+ has important roles in the control of ER–PM contacts
It is hard not to note the close interrelationship between Ca2+ signals generated at the PM in ER–PM contact sites and the tethering molecules that maintain the structure and the lipid composition on either side of the contact region. First, the interaction of ESyt1 with the PM is regulated by Ca2+ (Chang et al. 2013; Giordano et al. 2013), while ESyt2 and ESyt3 use one of their C2 domains to interact with PM PI(4,5)P2. It is not entirely clear what lipid is the main binding partner of ESyt1 and it may depend on the Ca2+ concentration. ESyt1 localization during Gq‐mediated PLC activation suggests that moderate cytoplasmic Ca2+ elevations enhance binding to PI(4,5)P2, which then becomes a limiting factor when high PLC activity induces PI(4,5)P2 depletion. However, further Ca2+ increases (probably reaching over 10 μm) again increase ESyt1 PM association even when PI(4,5)P2 is no longer available (Saheki et al. 2016). This dual PM recruitment depending on the Ca2+ may reflect the participation of more than one of the five C2 domains found in this molecule and more than one lipid in the PM (Idevall‐Hagren et al. 2015).
Calcium, of course, will have an impact on phosphoinositides, especially during receptor activation of PLC, and through those changes can affect both ESyt PM interactions (see above), they can also affect the ORP5 and ORP6‐mediated PI4P/PS exchange process. Since these ORPs maintain PM contact through PI4P interaction, their PM engagement and hence lipid transfer function is limited by the available PI4P in the PM as PI4P levels in the PM usually follow the same kinetics as PI(4,5)P2 during strong PLC activation. Turning off ORPs this way will affect PS transport and perhaps that of other lipids that may impact the STIM1/Orai1 function. Another recent study identified RASFF4, one of the Ras association domain family proteins, as regulators of SOCE via controlling PI(4,5)P2 production thus maintaining ESyt2 and ESyt3 PM contacts (Chen et al. 2017). Higher cytoplasmic Ca2+ increases that are evoked by Ca2+ ionophores, or during PM damage and repair can activate PLC without receptor stimulation, but also initiate more profound changes in the distribution of other lipids, such as DAG (see above) and PS. PS externalization occurs via Ca2+ activation of scramblases such as the TMEM16F (Suzuki et al. 2010; Bevers & Williamson, 2016; Brunner et al. 2016; Gyobu et al. 2017). Incidentally, TMEM16F is a member of the anoctamin family of 10 proteins (hence also called ANO6). Several members of this family are chloride channels (Kunzelmann et al. 2016) and, notably, they are the mammalian orthologues of the yeast Ist2 that was identified as one of six yeast proteins important for maintaining ER–PM contacts (see above). Anoctamins are Ca2+‐regulated proteins that have been linked to a variety of diseases and it is hotly debated whether their Cl− channel or scramblase activities are more important. It is noteworthy, though, that yeast Ist2 is primarily ER localized contacting the PM with its C‐terminal polybasic domains, whereas the cellular distribution of anoctamins and their role(s) in stabilizing ER–PM contacts is still poorly understood (Kunzelmann et al. 2016).
Morphological data indicate that ER–PM contact sites are heterogeneous: some of the cortical ER juxtaposed to the PM has a very narrow lumen excluding even ER luminally targeted proteins, such as green fluorescent protein (GFP) (Orci et al. 2009; Giordano et al. 2013; Fernandez‐Busnadiego et al. 2015; Wu et al. 2017). Overexpression of STIM1 causes enlargement of the ER–PM contact areas (even without expression of Orai1) and can generate stacks of ER beneath the PM (Orci et al. 2009). It is not clear if the morphological differences reflect functionally distinct contact areas and what signalling modalities are associated with them.
Concluding remarks
The physiology and pathology of ER–PM contacts has become one of the most exciting research areas since the discovery of STIM1/Orai1 proteins. More and more proteins are discovered that work at these contact sites and it is a recurring question whether they are simply recruited to these sites to fulfil their function or they contribute to the structural stability of this compartment. ER–PM contacts are critically important for non‐vesicular lipid transfer and also for SOCE, but the possible presence of anoctamine Cl− channels and even potassium channels (Fox et al. 2015; Fu et al. 2017) in these sites suggest that other less explored functions may add to their complexities. Even from this limited overview one theme has to emerge: that is the close interrelationship between Ca2+ and lipid signals that influence one another in profound ways. While most attention is focused on the lipid composition of the PM at these contact zones, relatively little is known about the lipid composition of the ER side of the contacts and how those lipids affect ER‐related processes, including Ca2+ release and uptake pathways. Lastly, these new developments have made it clear that the imaging tools we have been using to determine lipid distribution are too crude to assess the local enrichment of these lipids around specific structures or molecular clusters. Another important point to realize is that in vitro lipid transfer assays based on transport between lipid vesicles may not be sufficient to understand how these processes are organized in the intact cell. We have to find ways to monitor lipid transfer in the intact cell where the fine structure is undisturbed. In summary, ER–PM contacts represent an exciting research field that not only shapes our current concepts regarding lipid distribution and metabolism, but also poses methodological challenges that we have to meet to fully understand the impact of this membrane organization on specific cellular functions.
Additional information
Competing interests
None declared.
Funding
The research of T.B. is supported by the intramural research program of the Eunice Kennedy Shriver National Institutes of Child Health and Human Development of the National Institutes of Health (HD000196‐20).
Biography
Tamas Balla is a tenured Senior Investigator at the National Institute of Child Health and Human Development, NIH, Bethesda, MD. His research is concerned with lipid homeostasis, membrane biology and signalling in eukaryotic cells. He has been working on various aspects of inositol lipid signalling for almost 30 years mainly focusing on phosphatidylinositol 4‐kinases. In the last decade, his laboratory has developed a number of fluorescence‐based tools to monitor and alter cellular phosphoinositide levels and kinetics in living cells. The overall goal of his research is to understand how phosphoinositide signals regulate cellular functions working both at the molecular, cellular and organismal level.

Edited by: Ole Petersen & Reinhold Penner
This review was presented at the symposium “Intracellular Calcium Signals: Generation, Function and Therapeutic Intervention” which took place at Gordon Research Conferences 2017, Lucca, Italy, 18‐23 June 2017.
References
- Balla A, Kim YJ, Varnai P, Szentpetery Z, Knight Z, Shokat KM & Balla T (2007). Maintenance of hormone‐sensitive phosphoinositide pools in the plasma membrane requires phosphatidylinositol 4‐kinase IIIα. Mol Biol Cell 19, 711–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balla T (2013). Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol Rev 93, 1019–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevers EM & Williamson PL (2016). Getting to the outer leaflet: physiology of phosphatidylserine exposure at the plasma membrane. Physiol Rev 96, 605–645. [DOI] [PubMed] [Google Scholar]
- Bojjireddy N, Botyanszki J, Hammond G, Creech D, Peterson R, Kemp DC, Snead M, Brown R, Morrison A, Wilson S, Harrison S, Moore C & Balla T (2014). Pharmacological and genetic targeting of the PI4KA enzyme reveals its important role in maintaining plasma membrane phosphatidylinositol 4‐phosphate and phosphatidylinositol 4,5‐bisphosphate levels. J Biol Chem 289, 6120–6132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broad LM, Braun FJ, Lievremont JP, Bird GS, Kurosaki T & Putney JW Jr (2001). Role of the phospholipase C‐inositol 1,4,5‐trisphosphate pathway in calcium release‐activated calcium current and capacitative calcium entry. J Biol Chem 276, 15945–15952. [DOI] [PubMed] [Google Scholar]
- Brunner JD, Schenck S & Dutzler R (2016). Structural basis for phospholipid scrambling in the TMEM16 family. Curr Opin Struct Biol 39, 61–70. [DOI] [PubMed] [Google Scholar]
- Calloway N, Owens T, Corwith K, Rodgers W, Holowka D & Baird B (2011). Stimulated association of STIM1 and Orai1 is regulated by the balance of PtdIns(4,5)P 2 between distinct membrane pools. J Cell Sci 124, 2602–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Choi S, Maleth JJ, Park S, Ahuja M & Muallem S (2015). The ER/PM microdomain, PI(4,5)P2 and the regulation of STIM1‐Orai1 channel function. Cell Calcium 58, 342–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CL, Hsieh TS, Yang TT, Rothberg KG, Azizoglu DB, Volk E, Liao JC & Liou J (2013). Feedback regulation of receptor‐induced Ca2+ signaling mediated by E‐Syt1 and Nir2 at endoplasmic reticulum‐plasma membrane junctions. Cell Rep 5, 813–825. [DOI] [PubMed] [Google Scholar]
- Chen YJ, Chang CL, Lee WR & Liou J (2017). RASSF4 controls SOCE and ER‐PM junctions through regulation of PI(4,5)P2 . J Cell Biol 216, 2011–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung J, Torta F, Masai K, Lucast L, Czapla H, Tanner LB, Narayanaswamy P, Wenk MR, Nakatsu F & De Camilli P (2015). PI4P/phosphatidylserine countertransport at ORP5‐ and ORP8‐mediated ER–plasma membrane contacts. Science 349, 428–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derler I, Jardin I & Romanin C (2016a). Molecular mechanisms of STIM/Orai communication. Am J Physiol Cell Physiol 310, C643–C662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derler I, Jardin I, Stathopulos PB, Muik M, Fahrner M, Zayats V, Pandey SK, Poteser M, Lackner B, Absolonova M, Schindl R, Groschner K, Ettrich R, Ikura M & Romanin C (2016b). Cholesterol modulates Orai1 channel function. Sci Signal 9, ra10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derler I, Plenk P, Fahrner M, Muik M, Jardin I, Schindl R, Gruber HJ, Groschner K & Romanin C (2013). The extended transmembrane Orai1 N‐terminal (ETON) region combines binding interface and gate for Orai1 activation by STIM1. J Biol Chem 288, 29025–29034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Saint‐Jean M, Delfosse V, Douguet D, Chicanne G, Payrastre B, Bourguet W, Antonny B & Drin G (2011). Osh4p exchanges sterols for phosphatidylinositol 4‐phosphate between lipid bilayers. J Cell Biol 195, 965–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dionisio N, Galan C, Jardin I, Salido GM & Rosado JA (2011). Lipid rafts are essential for the regulation of SOCE by plasma membrane resident STIM1 in human platelets. Biochim Biophys Acta 1813, 431–437. [DOI] [PubMed] [Google Scholar]
- Elbaz‐Alon Y, Eisenberg‐Bord M, Shinder V, Stiller SB, Shimoni E, Wiedemann N, Geiger T & Schuldiner M (2015). Lam6 regulates the extent of contacts between organelles. Cell Rep 12, 7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahrner M, Muik M, Schindl R, Butorac C, Stathopulos P, Zheng L, Jardin I, Ikura M & Romanin C (2014). A coiled‐coil clamp controls both conformation and clustering of stromal interaction molecule 1 (STIM1). J Biol Chem 289, 33231–33244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez‐Busnadiego R, Saheki Y & De Camilli P (2015). Three‐dimensional architecture of extended synaptotagmin‐mediated endoplasmic reticulum‐plasma membrane contact sites. Proc Natl Acad Sci USA 112, E2004–E2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M & Rao A (2006). A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 441, 179–185. [DOI] [PubMed] [Google Scholar]
- Foti M, Audhya A & Emr SD (2001). Sac1 lipid phosphatase and Stt4 phosphatidylinositol 4‐kinase regulate a pool of phosphatidylinositol 4‐phosphate that functions in the control of the actin cytoskeleton and vacuole morphology. Mol Biol Cell 128, 2396–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox PD, Haberkorn CJ, Akin EJ, Seel PJ, Krapf D & Tamkun MM (2015). Induction of stable ER‐plasma‐membrane junctions by Kv2.1 potassium channels. J Cell Sci 128, 2096–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Dai X, Plummer G, Suzuki K, Bautista A, Githaka JM, Senior L, Jensen M, Greitzer‐Antes D, Manning Fox JE, Gaisano HY, Newgard CB, Touret N & MacDonald PE (2017). Kv2.1 clustering contributes to insulin exocytosis and rescues human β‐cell dysfunction. Diabetes 66, 1890–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatta AT, Wong LH, Sere YY, Calderon‐Norena DM, Cockcroft S, Menon AK & Levine TP (2015). A new family of StART domain proteins at membrane contact sites has a role in ER‐PM sterol transport. Elife 4; 10.7554/eLife.07253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano F, Saheki Y, Idevall‐Hagren O, Colombo SF, Pirruccello M, Milosevic I, Gracheva EO, Bagriantsev SN, Borgese N & De Camilli P (2013). PI(4,5)P2‐dependent and Ca2+‐regulated ER‐PM interactions mediated by the extended synaptotagmins. Cell 153, 1494–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudlur A, Quintana A, Zhou Y, Hirve N, Mahapatra S & Hogan PG (2014). STIM1 triggers a gating rearrangement at the extracellular mouth of the ORAI1 channel. Nat Commun 5, 5164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S, Stolz LE, Lemrow SM & York JD (1999). SAC1‐like domains of yeast SAC1, INP52, and INP53 and of human synaptojanin encode polyphosphoinositide phosphatases. J Biol Chem 274, 12990–12995. [DOI] [PubMed] [Google Scholar]
- Gyobu S, Ishihara K, Suzuki J, Segawa K & Nagata S (2017). Characterization of the scrambling domain of the TMEM16 family. Proc Natl Acad Sci USA 114, 6274–6279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond GR, Fischer MJ, Anderson KE, Holdich J, Koteci A, Balla T & Irvine RF (2012). PI4P and PI(4,5)P2 are essential but independent lipid determinants of membrane identity. Science 337, 727–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan PG, Lewis RS & Rao A (2010). Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu Rev Immunol 28, 491–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan PG & Rao A (2015). Store‐operated calcium entry: mechanisms and modulation. Biochem Biophys Res Commun 460, 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes WE, Woscholski R, Cooke FT, Patrick RS, Dove SK, McDonald NQ & Parker PJ (2000). SAC1 encodes a regulated lipid phosphoinositide phosphatase, defects in which can be suppressed by the homologous Inp52p and Inp53p phosphatases. J Biol Chem 275, 801–808. [DOI] [PubMed] [Google Scholar]
- Idevall‐Hagren O, Lu A, Xie B & De Camilli P (2015). Triggered Ca2+ influx is required for extended synaptotagmin 1‐induced ER‐plasma membrane tethering. EMBO J 34, 2291–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong H, Park J, Jun Y & Lee C (2017). Crystal structures of Mmm1 and Mdm12‐Mmm1 reveal mechanistic insight into phospholipid trafficking at ER‐mitochondria contact sites. Proc Natl Acad Sci USA 114, E9502–E9511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Kedan A, Marom M, Gavert N, Keinan O, Selitrennik M, Laufman O & Lev S (2013). The phosphatidylinositol‐transfer protein Nir2 binds phosphatidic acid and positively regulates phosphoinositide signalling. EMBO Rep 14, 891–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YJ, Guzman‐Hernandez ML, Wisniewski E & Balla T (2015). Phosphatidylinositol‐phosphatidic acid exchange by Nir2 at ER‐PM contact sites maintains phosphoinositide signaling competence. Dev Cell 33, 549–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzeniowski MK, Popovic MA, Szentpetery Z, Varnai P, Stojilkovic SS & Balla T (2009). Dependence of STIM1/Orai1‐mediated calcium entry on plasma membrane phosphoinositides. J Biol Chem 284, 21027–21035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunzelmann K, Cabrita I, Wanitchakool P, Ousingsawat J, Sirianant L, Benedetto R & Schreiber R (2016). Modulating Ca2+ signals: a common theme for TMEM16, Ist2, and TMC. Pflugers Arch 468, 475–490. [DOI] [PubMed] [Google Scholar]
- Lees JA, Messa M, Sun EW, Wheeler H, Torta F, Wenk MR, De Camilli P & Reinisch KM (2017). Lipid transport by TMEM24 at ER‐plasma membrane contacts regulates pulsatile insulin secretion. Science 355, eaah6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis RS (2007). The molecular choreography of a store‐operated calcium channel. Nature 446, 284–287. [DOI] [PubMed] [Google Scholar]
- Liou J, Fivaz M, Inoue T & Meyer T (2007). Live‐cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc Natl Acad Sci USA 104, 9301–9306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE Jr & Meyer T (2005). STIM is a Ca2+ sensor essential for Ca2+‐store‐depletion‐triggered Ca2+ influx. Curr Biol 15, 1235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litvak V, Dahan N, Ramachandran S, Sabanay H & Lev S (2005). Maintenance of the diacylglycerol level in the Golgi apparatus by the Nir2 protein is critical for Golgi secretory function. Nat Cell Biol 7, 225–234. [DOI] [PubMed] [Google Scholar]
- Ma G, Wei M, He L, Liu C, Wu B, Zhang SL, Jing J, Liang X, Senes A, Tan P, Li S, Sun A, Bi Y, Zhong L, Si H, Shen Y, Li M, Lee MS, Zhou W, Wang J, Wang Y & Zhou Y (2015). Inside‐out Ca2+ signalling prompted by STIM1 conformational switch. Nat Commun 6, 7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda K, Anand K, Chiapparino A, Kumar A, Poletto M, Kaksonen M & Gavin AC (2013). Interactome map uncovers phosphatidylserine transport by oxysterol‐binding proteins. Nature 501, 257–261. [DOI] [PubMed] [Google Scholar]
- Maleth J, Choi S, Muallem S & Ahuja M (2014). Translocation between PI(4,5)P2‐poor and PI(4,5)P2‐rich microdomains during store depletion determines STIM1 conformation and Orai1 gating. Nat Commun 5, 5843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manford AG, Stefan CJ, Yuan HL, MacGurn JA & Emr SD (2012). ER‐to‐plasma membrane tethering proteins regulate cell signaling and ER morphology. Dev Cell 23, 1129–1140. [DOI] [PubMed] [Google Scholar]
- Mesmin B, Bigay J, Moser von Filseck J, Lacas‐Gervais S, Drin G & Antonny B (2013). A four‐step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER‐Golgi tether OSBP. Cell 155, 830–843. [DOI] [PubMed] [Google Scholar]
- Moser von Filseck J, Copic A, Delfosse V, Vanni S, Jackson CL, Bourguet W & Drin G (2015). Phosphatidylserine transport by ORP/Osh proteins is driven by phosphatidylinositol 4‐phosphate. Science 349, 432–436. [DOI] [PubMed] [Google Scholar]
- Muik M, Fahrner M, Schindl R, Stathopulos P, Frischauf I, Derler I, Plenk P, Lackner B, Groschner K, Ikura M & Romanin C (2011). STIM1 couples to ORAI1 via an intramolecular transition into an extended conformation. EMBO J 30, 1678–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murley A, Sarsam RD, Toulmay A, Yamada J, Prinz WA & Nunnari J (2015). Ltc1 is an ER‐localized sterol transporter and a component of ER‐mitochondria and ER‐vacuole contacts. J Cell Biol 209, 539–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsu F, Baskin JM, Chung J, Tanner LB, Shui G, Lee SY, Pirruccello M, Haio M, Ingolia NT, Wenk MR & De Camilli P (2012). PtdIns4P synthesis by PI4KIIIα at the plasma membrane and its impact on plasma membrane identity. J Cell Biol 199, 1003–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nwokonko RM, Cai X, Loktionova NA, Wang Y, Zhou Y & Gill DL (2017). The STIM‐Orai pathway: conformational coupling between STIM and Orai in the activation of store‐operated Ca2+ entry. Adv Exp Med Biol 993, 83–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orci L, Ravazzola M, Le Coadic M, Shen WW, Demaurex N & Cosson P (2009). STIM1‐induced precortical and cortical subdomains of the endoplasmic reticulum. Proc Natl Acad Sci USA 106, 19358–19362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco J, Dominguez L, Bohorquez‐Hernandez A, Asanov A & Vaca L (2016). A cholesterol‐binding domain in STIM1 modulates STIM1‐Orai1 physical and functional interactions. Sci Rep 6, 29634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palty R, Fu Z & Isacoff EY (2017). Sequential steps of CRAC channel activation. Cell Rep 19, 1929–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palty R, Raveh A, Kaminsky I, Meller R & Reuveny E (2012). SARAF inactivates the store operated calcium entry machinery to prevent excess calcium refilling. Cell 149, 425–438. [DOI] [PubMed] [Google Scholar]
- Pani B, Ong HL, Liu X, Rauser K, Ambudkar IS & Singh BB (2008). Lipid rafts determine clustering of STIM1 in endoplasmic reticulum‐plasma membrane junctions and regulation of store‐operated Ca2+ entry (SOCE). J Biol Chem 283, 17333–17340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CY, Hoover PJ, Mullins FM, Bachhawat P, Covington ED, Raunser S, Walz T, Garcia KC, Dolmetsch RE & Lewis RS (2009). STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 136, 876–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peretti D, Dahan N, Shimoni E, Hirschberg K & Lev S (2008). Coordinated lipid transfer between the endoplasmic reticulum and the Golgi complex requires the VAP proteins and is essential for Golgi‐mediated transport. Mol Biol Cell 19, 3871–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pottekat A, Becker S, Spencer KR, Yates JR 3rd, Manning G, Itkin‐Ansari P & Balch WE (2013). Insulin biosynthetic interaction network component, TMEM24, facilitates insulin reserve pool release. Cell Rep 4, 921–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A & Hogan PG (2006). Orai1 is an essential pore subunit of the CRAC channel. Nature 443, 230–233. [DOI] [PubMed] [Google Scholar]
- Putney JW Jr (1986). A model for receptor‐regulated calcium entry. Cell Calcium 7, 1–12. [DOI] [PubMed] [Google Scholar]
- Putney JW, Steinckwich‐Besancon N, Numaga‐Tomita T, Davis FM, Desai PN, D'Agostin DM, Wu S & Bird GS (2017). The functions of store‐operated calcium channels. Biochim Biophys Acta 1864, 900–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Veliçelebi G & Stauderman KA (2005). STIM1, an essential and conserved component of store‐operated Ca2+ channel function. J Cell Biol 169, 435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosado JA & Sage SO (2000). Phosphoinositides are required for store‐mediated calcium entry in human platelets. J Biol Chem 275, 9110–9113. [DOI] [PubMed] [Google Scholar]
- Saheki Y, Bian X, Schauder CM, Sawaki Y, Surma MA, Klose C, Pincet F, Reinisch KM & De Camilli P (2016). Control of plasma membrane lipid homeostasis by the extended synaptotagmins. Nat Cell Biol 18, 504–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauder CM, Wu X, Saheki Y, Narayanaswamy P, Torta F, Wenk MR, De Camilli P & Reinisch KM (2014). Structure of a lipid‐bound extended synaptotagmin indicates a role in lipid transfer. Nature 510, 552–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sclip A, Bacaj T, Giam LR & Sudhof TC (2016). Extended synaptotagmin (ESyt) triple knock‐out mice are viable and fertile without obvious endoplasmic reticulum dysfunction. PLoS One 11, e0158295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soboloff J, Rothberg BS, Madesh M & Gill DL (2012). STIM proteins: dynamic calcium signal transducers. Nat Rev Mol Cell Biol 13, 549–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn M, Ivanova P, Brown HA, Toth DJ, Varnai P, Kim YJ & Balla T (2016). Lenz‐Majewski mutations in PTDSS1 affect phosphatidylinositol 4‐phosphate metabolism at ER‐PM and ER‐Golgi junctions. Proc Natl Acad Sci USA 113, 4314–4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefan CJ, Manford AG, Baird D, Yamada‐Hanff J, Mao Y & Emr SD (2011). Osh proteins regulate phosphoinositide metabolism at ER‐plasma membrane contact sites. Cell 144, 389–401. [DOI] [PubMed] [Google Scholar]
- Suzuki J, Umeda M, Sims PJ & Nagata S (2010). Calcium‐dependent phospholipid scrambling by TMEM16F. Nature 468, 834–838. [DOI] [PubMed] [Google Scholar]
- Toulmay A & Prinz WA (2012). A conserved membrane‐binding domain targets proteins to organelle contact sites. J Cell Sci 125, 49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnai P, Toth B, Toth DJ, Hunyady L & Balla T (2007). Visualization and manipulation of plasma membrane‐endoplasmic reticulum contact sites indicates the presence of additional molecular components within the STIM1‐Orai1 Complex. J Biol Chem 282, 29678–29690. [DOI] [PubMed] [Google Scholar]
- Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, Koblan‐Huberson M, Kraft S, Turner H, Fleig A, Penner R & Kinet JP (2006). CRACM1 is a plasma membrane protein essential for store‐operated Ca2+ entry. Science 312, 1220–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh CM, Chvanov M, Haynes LP, Petersen OH, Tepikin AV & Burgoyne RD (2010). Role of phosphoinositides in STIM1 dynamics and store‐operated calcium entry. Biochem J 425, 159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Whiteus C, Xu CS, Hayworth KJ, Weinberg RJ, Hess HF & De Camilli P (2017). Contacts between the endoplasmic reticulum and other membranes in neurons. Proc Natl Acad Sci USA 114, E4859–E4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav S, Garner K, Georgiev P, Li M, Gomez‐Espinosa E, Panda A, Mathre S, Okkenhaug H, Cockcroft S & Raghu P (2015). RDGBα, a PtdIns‐PtdOH transfer protein, regulates G‐protein‐coupled PtdIns(4,5)P 2 signalling during Drosophila phototransduction. J Cell Sci 128, 3330–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan JP, Zeng W, Dorwart MR, Choi YJ, Worley PF & Muallem S (2009). SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat Cell Biol 11, 337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Cai X, Loktionova NA, Wang X, Nwokonko RM, Wang X, Wang Y, Rothberg BS, Trebak M & Gill DL (2016). The STIM1‐binding site nexus remotely controls Orai1 channel gating. Nat Commun 7, 13725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Cai X, Nwokonko RM, Loktionova NA, Wang Y & Gill DL (2017). The STIM‐Orai coupling interface and gating of the Orai1 channel. Cell Calcium 63, 8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Srinivasan P, Razavi S, Seymour S, Meraner P, Gudlur A, Stathopulos PB, Ikura M, Rao A & Hogan PG (2013). Initial activation of STIM1, the regulator of store‐operated calcium entry. Nat Struct Mol Biol 20, 973–981. [DOI] [PMC free article] [PubMed] [Google Scholar]