

Abstract
Carbonic anhydrases constitute a group of enzymes that catalyse reversible hydration of carbon dioxide leading to the formation of bicarbonate and proton. The platelet carbonic anhydrase II (CAII) was described for the first time in the '80s of the last century. Nevertheless, its direct role in platelet physiology and pathology still remains poorly understood. The modulation of platelet CAII action as a therapeutic approach holds promise as a novel strategy to reduce the impact of cardiovascular diseases. This short review paper summarises the current knowledge regarding the role of human CAII in regulating platelet function. The potential future directions considering this enzyme as a potential drug target and important pathophysiological chain in platelet-related disorders are described.
1. Carbonic Anhydrases
Carbonic anhydrases constitute a group of zinc containing lyases, classified, according to the Enzyme Catalogue to EC 4.2.1.1., into the lyases subclass “carbon-oxygen lyases” and subclasses “hydrolyses” [1]. They catalyse reversible hydration of carbon dioxide to form bicarbonate ion and proton. Carbonic anhydrase is present in prokaryotic and eukaryotic cells. Genetically they belong to seven subgroups (α, β, γ, δ, ζ, η, θ) that are not evolutionarily related [2–4]. There are 15 carbonic anhydrase isoforms in humans (belonging to the α subgroup) [5]. They occur in various tissues in the cytoplasm, cell membrane, and mitochondria, or as extracellular enzymes (e.g., breast milk) [6]. Carbonic anhydrases (CAs) are almost ubiquitously present in human cells. The members of the CAs family play a role in pH regulation, gas exchange, and ion transport, as well as urine acidification, cerebrospinal fluid secretion, ocular fluid production, bone resorption, fatty acid metabolism, testicular fluid production, and many others [7].
Numerous diseases result from inappropriate function of carbonic anhydrases. There are scarcely several case reports regarding CAs deficiency; nonetheless they constitute a valuable source of knowledge. The erythrocyte CAI deficiency has no clinically relevant consequences [8], but CAII deficiency usually results in osteoporosis, renal tubular acidosis, and brain calcification [9, 10]. CAIII deficiency in skeletal muscles seems to play an import role in pathogenesis of myasthenia gravis [11, 12]. The carbonic anhydrase VA deficiency may present as early-onset liver failure with hyperammonemia, hyperlactatemia, and ketonuria [13]. CAXII deficiency may promote hyponatremic dehydration and rhabdomyolysis after intense physical exercises [14]. The effectiveness of carbonic anhydrase may be also altered by autoantibodies. Ignaki et al. demonstrated in 1991 that anti-carbonic anhydrase autoantibodies (aCAAs) are present in serum of 32% patients with systemic lupus erythematosus, in 21% of patients with Sjögren's syndrome, but not in healthy volunteers. They showed that aCAAs have affinity to epidermal cells, hair follicles, sweat glands, and renal tubular cells [15]. Moreover, in 2007, it was presented that also in rheumatoid arthritis there are present aCAAs in serum, showing affinity to bind to carbonic anhydrase III in synovial membranes [16]. Up to date aCAAs have been described in patients with rheumatoid disorders (rheumatoid arthritis, Behçet's disease, lupus erythematosus, polymyositis, systemic sclerosis, and Sjögren syndrome) [16–18], digestive tract disorders (idiopathic chronic pancreatitis, primary biliary cirrhosis, autoimmune cholangitis, and gastric cancer) [18, 19], endometriosis [18], Grave's disease [20], acute myeloid leukaemia [21], renal tubular acidosis [22] (significant influence on pathogenesis proved in the animal model of Sjögren's syndrome [23]), and end-stage kidney disease [24]. Moreover, autoantibodies against carbonic anhydrase II were proved to play an important role in pathogenesis of retinopathy [25] and these against CAVI seem to induce dry eye syndrome in Sjögren's syndrome [26, 27]. Interestingly, the autoantibodies against carbonic anhydrase II might be produced in humans due to cross-reactivity with carbonic anhydrase of Helicobacter pylori [28].
Carbonic anhydrases (CAs) have become an interesting enzyme for clinicians when first drugs inhibiting this enzyme established promising group of diuretics. Acetazolamide was introduced into clinical practice in 1956 as a first nonmercurial diuretic. Among “classic” carbonic anhydrase inhibitors also methazolamide, ethoxzolamide, and dichlorphenamide were applied in congestive heart failure treatment. Nowadays, more preferable are the next-generation diuretics (loop, thiazide, thiazide-like diuretic, and aldosterone antagonists), but most of them still exhibit different magnitude of carbonic anhydrase inhibition [38]. Acetazolamide remains a useful drug in case of intracranial hypertension [39], improving sleep quality (reducing the apnea-hypopnea index in both obstructive sleep apnea and healthy trekkers) [40] and reducing the risk of mountain sickness at high altitudes [41]. Topical dorzolamide and brinzolamide and in some cases systemic acetazolamide are commonly used for reduction of intraocular pressure in glaucoma [42]. Topiramate and zonisamide are anticonvulsants with a multiple mechanism of action among which is also carbonic anhydrase inhibition. Not only does this effect seem to be important in the main (antiepileptic) activity of this drug, but also it might result in weight loss. It is regarded as a side effect but may be the basis for designing the new antiobesity drugs [38]. Moreover, acetazolamide is also proved to be effective in seizure treatment [43]. Induced by hypoxia, overexpression of carbonic anhydrase IX in neoplastic cells may produce acidosis and thus reduce the effectiveness of chemotherapies [38]. Inhibition of carbonic anhydrase IX improves effects of cisplatin in small cell lung cancer [44].
2. Carbonic Anhydrase II
Carbonic anhydrase II (CAII) is a cytoplasmic enzyme with a very high affinity for sulphonamides and high catalytic activity (for structure see Figure 1.). It is one of the fastest, working enzymes in the human body, 106 cycles of enzyme per second [45]. CAII is present in numerous tissues (e.g., erythrocytes, eye, gastrointestinal tract, bone osteoclasts, kidney, lung, testis, and brain) and it constitutes a possible drug target in some diseases (glaucoma, oedema, epilepsy, and attitude sickness). However, so far it has been mostly studied in the red blood cells and has been extracted from them for the purpose of in vitro studies [5].
Figure 1.

The structure and amino acid sequence of the human carbonic anhydrase II [29].
The gene for human CAII is located on the chromosome 8q22 [46]. The diseases associated with CAII include the following: Autosomal Recessive Osteopetrosis Type 3 with Renal Tubular Acidosis (ARO3, OPTB3), which commonly manifests in early infancy with macrocephaly, feeding difficulties, evolving blindness and deafness, bone marrow failure, severe anaemia, and hepatosplenomegaly. Deafness and blindness are generally thought to represent effects of pressure on nerves. OPTB3 is associated with renal tubular acidosis and cerebral calcification (marble brain disease) and in some cases with mental retardation [47]. Among its related pathways is vitamin D-receptor mediated regulation of genes involved in osteoporosis [48].
Numerous compounds have been demonstrated to modify carbonic anhydrase activity. They are physiological substances, drugs currently used in clinical practice and other chemicals tested ex vivo in laboratories. Potential CAII activators physiologically present in human organism include biogenic amines (histamine, catecholamines, and serotonin) and amino acids (phenylalanine and histidine) [49]. Most drugs inhibiting carbonic anhydrase are sulphonamides (over 20 FDA approved drugs including diuretics like hydrochlorothiazide, indapamide, chlortalidone, and furosemide) [50, 51]. There is a wide variety of substances testes in laboratory conditions for their CA activatory and inhibitory properties [5, 52].
The aim of this review article was to collect available data regarding studies on carbonic anhydrase in platelets and concerning the influence of carbonic anhydrase regulators on platelet function. Taking into account limited number of studies directly analyzing carbonic anhydrase properties in platelets, a secondary objective was to analyze the involvement of drugs regulating CAII in platelet pathophysiology and to discuss hypothetical contribution of platelet carbonic anhydrase in these drug-platelet interactions.
3. Platelet Carbonic Anhydrase
The first statements of CAII in platelets were made 60 years ago [53] and its more accurate characterization was created in the '80s of the last century [54]. CAII, by catalysing the formation of H+ and HCO3−, reduces the cytosol pH of the platelet [55]. The reaction products can be excreted outside the plasma membrane – H+ is being exchanged for Na+ whereas HCO3− for Cl− (Figure 2).
Figure 2.
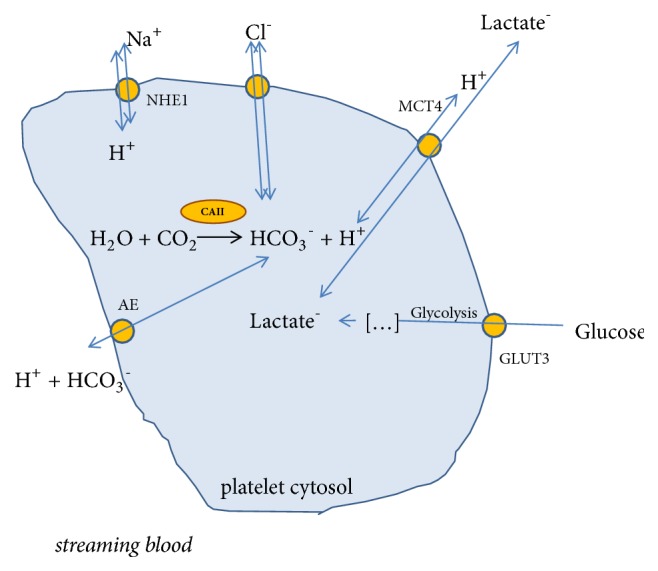
A potential role of carbonic anhydrase in the regulation of platelet metabolism [30–32]. GLUT3: glucose transporter; MCT4: H+/monocarboxylate transporter; AE: Anion exchanger; NHE1: the Na+/H+ exchanger.
The exact role of CAII in platelet physiology requires more detailed research. A question arises whether their products play a direct role in platelet physiology or enhance the transmembrane ion exchange, sodium and chloride influx. So far there has been no clinical case presented in the PubMed database that describes CAII deficiency in platelets and, consequently, the effects of this deficiency on the phenotype. The only available case is the description of a murine mutation located in the direct proximity to the CAII gene locus, which affected both platelet morphology and function [56].
The CAII activity in platelets has been hypothesized since Akkerman et al. described proton efflux from platelets following thrombin stimulation [57, 58]. Siffert et al. verified the presence of CO2 hydration in platelets, subsequently proved that this process may be inhibited by ethoxzolamide, and showed that examined enzyme kinetics strictly corresponds with carbonic anhydrase II [54]. Afterwards, ethoxzolamide was shown to reduce the thrombin stimulated aggregation by about 20%-40% [59, 60]. Similar result was achieved by the removal of CO2 from platelets environment, which additionally confirms the importance of carbon dioxide hydration in the aggregation process [59].
3.1. Effect of Carbonic Anhydrase Activators on Platelet Function
Adrenaline is CAII agonist [61] and it can sensitize platelets to thrombin by induction of CAII activity [62, 63]. Adrenaline stimulates platelet carbonic anhydrase directly or by enhancing the membrane HCO3−/Cl− exchange since such subthreshold doses of adrenaline (in concentrations which do not initiate aggregation) added to platelets are capable of increasing the chloride concentration in platelet cytosol. [55]. Not only does a CAII inhibitor, acetazolamide, reduce resting and adrenaline-induced chloride concentration in platelets, but it also abolishes the synergism of thrombin and low concentration of adrenaline [63]. The proaggregatory effect of adrenaline is also abolished by another CAII inhibitor, chlortalidone. Furthermore, the ability of adrenaline to initiate platelet aggregation has been demonstrated to be directly proportional to the activity of platelet carbonic anhydrase [64].
Serotonin is an activator of carbonic anhydrase [49], which is also a poor agonist for platelet aggregation [65]. Interestingly, it enhances adrenaline, adenosine diphosphate (ADP) and collagen induced aggregation [65]. Furthermore, while being hyperreactive to adrenaline alone or adrenaline+serotonin, platelets present increased binding affinity for serotonin [65]. This observation may be clinically important as in an in vivo dog model serotonin plasma level was increased in stress conditions and it was suspected to significantly interplay with the evolution of an occlusive coronary thrombus [66], which was verified in human observation trial where high plasma serotonin level was associated with presence of coronary artery disease and future cardiac events [67]. This effect seems to be mediated through platelet 5-HT (5-hydroxytryptamine) receptors as saprogelate (a 5-HT2 receptors antagonist) limits platelet aggregation [68–70]. To the best of our knowledge, the possible participation of direct serotonin influence on platelet carbonic anhydrase has not been evaluated.
The selective serotonin reuptake inhibitors (SSRI), fluoxetine, sertraline, and citalopram, are potent activators of carbonic anhydrase II [71] and therefore they may be suspected of proaggregatory properties. Nevertheless, another SSRI, paroxetine, decreases intraplatelet serotonin storage and at the same time lowers platelet activation [72]. Similarly, sertraline and citalopram also limit platelet aggregation [73, 74]. Fluoxetine was to decrease platelet aggregability in the case of a 49-year-old man [75]. Therefore, SSRI effect on platelet function may be the balance between their direct action on platelets and the result of decreased platelet serotonin content. Nevertheless, the impact of this group of drugs on both thrombotic [76] and bleeding events is not substantial [77].
Histamine is a poor carbonic anhydrase II activator [52] present in humans platelets and it plays an important role in modulating platelet function. Histamine does not initiate platelet aggregation by itself, but it increases platelet sensitivity to aggregation agonists like ADP, thrombin, collagen, arachidonic acid [78], and adrenaline [79]. In a clinical model of increased histamine release, patients with chronic urticaria, higher platelet activity was observed when compared to healthy controls: increased levels of soluble P-selectin in one study [80] and both enhanced aggregation in response to ADP and increased soluble P-selectin levels compared to healthy population in another paper (both parameters were strongly and positively correlated with the Urticaria Severity Score) [81]. The mechanism of histamine impact on platelet pathophysiology is not fully understood. Histamine proaggregatory properties seem to result from H1 receptor stimulation (located in platelet membrane [82]). However, involvement of histamine in intracellular metabolism is also suggested [78], which was the subject matter of some studies [79, 83, 84]. Interestingly, there are no studies verifying whether CA inhibitors attenuate histamine affect platelet aggregation and if such an inhibition may be useful, e.g., in chronic urticaria management.
Histidine, phenylalanine, and carnosine (β-alanine-histidine dipeptide) are carbonic anhydrase activators [52]. Data regarding effect of histidine and carnosine on platelet function are not consistent. In some studies, histidine was verified to be an inhibitor of ADP-induced platelet aggregation [85] or spontaneous platelet aggregation and thromboxane B2 (TxB2) formation [86], but in others it demonstrated both slight proaggregative activity (50% of the patients) and antiaggregatory activity (the other 50% of patients) [87]. Similarly, in one study carnosine slightly stimulated ADP-induced aggregation [87], but in another it enhanced platelet aggregation only in patients with a low rate of aggregation, but inhibited platelet aggregation in patients with high index of aggregation [88]. There are hardly any available data on phenylalanine influence on platelet function; in a single study phenylalanine methyl ester inhibited ADP-induced platelet aggregation. Interestingly, similar results were observed with histidine methyl ester, but not with pure histidine (no inhibition) [89].
There are no available papers analyzing histidine, phenylalanine, and carnosine influence on CAII in platelets. Nevertheless, while analyzing their action profile, they do not appear to act on platelets through CAII.
3.2. Diuretics as CAII Inhibitors
Systemic use of acetazolamide is dedicated for altitude sickness treatment and prevention. Primarily, it reverses hypocapnic alkalosis occurring due to ventilatory response to hypoxemia, but the exact mechanism of its beneficial action seems to be more complex and is not fully recognized [41]. Moreover, in people exposed to high altitudes, thrombosis is a proposed mechanism of several complications as in autopsy many megakaryocytes were present in the lungs of people diagnosed with pulmonary edema and in one case they were also accompanied by thrombi in the kidneys and liver [90]. Nevertheless, at high altitude platelets present increased adhesiveness [91], but decreased aggregation in response to ADP, adrenaline, and collagen [92, 93] so eventual benefit of acetazolamide antiaggregatory properties remains uncertain.
Diuretics commonly used in clinical practice (hydrochlorothiazide, chlortalidone, indapamide, furosemide, and bumetanide) are well-established carbonic anhydrase inhibitors [51]. Thiazide-like diuretics, indapamide and chlortalidone, are widely used in cardiovascular medicine. They are proved to prevent cardiovascular events (CVE) and reduce all-cause mortality. The CVE prophylactic effect exceeds the benefits of lowering blood pressure. The difference was revealed when this thiazide-like diuretics were compared with thiazide-type diuretics [94]. The important differences in activity between these two groups of drugs regard platelet inhibition, which is much more intense in thiazide-like than thiazide-type diuretics as shown in work by Rendu at al. comparing indapamide and hydrochlorothiazide properties [95]. Furthermore, the platelet inhibition by thiazide-like diuretics seems to be carbonic anhydrase dependent [64].
Loop diuretics, furosemide and bumetanide, are loop diuretics potent to inhibit carbonic anhydrase II activity [51]. In one clinical study, furosemide was shown to inhibit ADP-induced platelet aggregation both ex vivo and after intravenous infusion [96] and in another study furosemide inhibited in vitro both ADP- and AA-induced aggregation [97]. Bumetanide was also shown to inhibit adrenaline-induced aggregation [63]. The mechanism of influence of loop diuretics on platelets remains unclear and the involvement of carbonic anhydrase may be expected.
3.3. Physiological Nitrogen Compounds versus Platelet Carbonic Anhydrase II Function
Platelet aggregation may be inhibited by nitric oxide (NO). However there is a controversy about possible significance of this dependence since the extra-platelet NO concentration is low and it seems to be no expression of nitric oxide synthase in platelets [33, 34]. Nevertheless, the effect of NO on platelets is mediated through the activation of guanylyl cyclase to produce cyclic guanosine monophosphate (cGMP) [98, 99]. Interestingly, carbonic anhydrase II was found to catalyse generation of nitric oxide form nitrite [100], which may be an additional origination of intra-platelet NO. This reaction is significantly enhanced by both decreased pH and dorzolamide, which are inhibitors of CAII main activity [100]. On the other hand, other research groups provide evidence negating this CA activity [36].
Nitrite is a platelet inhibitor in whole blood [101], but added to platelets suspended in platelet rich plasma it does not inhibit platelet aggregation [102] or activate soluble guanylate cyclase (sCG) [36]. It does not produce increased intraplatelet nitrite concentration [103], either. Therefore it (in these conditions) cannot rich cytosolic enzymes like carbonic anhydrase II.
Platelet supplementation with S-Nitrosocysteine (SNC), a NO donor, also provides intraplatelet NO2− formation and leads to platelet sCG activation [35, 36, 103]. Interestingly, SNC aggregation inhibition is not mediated by sCG activation. SNC also presents direct cyclooxygenase (COX) inhibiting properties, but an involvement of one more mechanism is postulated as it produces complete blockage of AA-induced aggregation, when TxB2 formation is only partially decreased [103]. Therefore, SNC is supposed to be involved in one more mechanism modulating platelet function, which may be CA. This thesis of CA contribution may be supported by observation that SNC produces increased intraplatelet nitrite formation [103]. CA II is proven to catalyse opposite reaction; it can produce S-nitrosothiols from inorganic nitrite [36] (Figure 3.).
Figure 3.
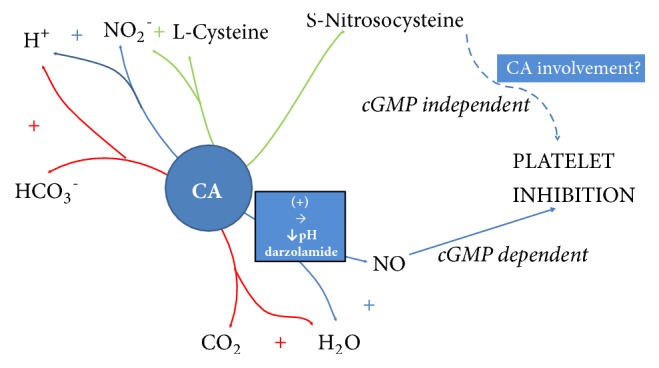
Postulated map of carbonic anhydrase II activities involving nitrosothiols, nitrate, and nitric oxide metabolism and they influence on platelet aggregation. S-Nitrosocysteine and NO are established platelet aggregation inhibitors [33, 34]. S-Nitrosocysteine is able to act independently of cGMP formation [35]. Dorzolamide and decreased pH increase nitric oxide formation [36]. CA ability to form NO from nitrite inside platelets remains controversial [36]. Nevertheless, NaHCO3− + NO2− in presence of erythrocytic CA II provides platelet cGMP formation [36] (more detailed description in the main text).
Nitrate is able to bind to carbonic anhydrase active site [104], but not it cannot be utilized to form S-nitrosothiols as nitrite can be [105]. It can inhibit platelet aggregation in whole blood, but the mechanism remains unclear [101].
3.4. Other Drugs Potentially Inhibiting Platelet Carbonic Anhydrase II
An experiment considering influence of N-hydroxyurea on platelet activity was performed by Lahiri at al. in chronic myelogenous leukaemia. There were at least three patterns of response to hydroxyurea, no response, partial aggregation inhibition, and “de-aggregation”. Interestingly, both aggregation inhibition and “de-aggregation” were only partially reversible by ODQ (1H-(1,2,4)Oxadiazolo(4,3-a)quinoxalin-1-one), which indicates involvement of other mechanism than postulated by authors NO release stimulating cGMP formation [106]. The inhibition of platelet carbonic anhydrase II by N-hydroxyurea may be possible [107].
Coumarin and its derivates are carbonic anhydrase inhibitors and substrates [108, 109]. Unfortunately, there is no information on acenocoumarol and warfarin (two coumarin derivates widely used as anticoagulant drugs [110]) regarding their individual influence on carbonic anhydrase. Nevertheless, these two drugs do not seem to inhibit platelet carbonic anhydrase in humans as both of them enhance platelet activity [111, 112].
A substantial part of nonsteroid anti-inflammatory drugs (NSAIDs) present anti-CAII activity. Celecoxib [113, 114] and valdecoxib [115], but not rofecoxib [114], meloxicam, piroxicam, and lornoxicam [116], are carbonic anhydrase II inhibitors. Diclofenac, which does not possess sulphonamide moiety, is not active against CA [116]. Flurbiprofen is a weak carbonic anhydrase II inhibitor whereas ibuprofen and indomethacin are even much less active against this enzyme [117]. In two papers by Puscas et al. indomethacin was said to be an agonist of CA, but this conclusion was reached after indomethacin limited inhibitory effect of acetazolamide on carbonic anhydrase [118, 119] and it was uncertain whether it was indeed CA agonist or just weaker than acetazolamide CA inhibitor competing for a binding site. Acetylsalicylic acid (ASA) is a noncompetitive carbonic anhydrase II inhibitor [120], but this activity may be associated with adjusting environmental pH by ASA [121]. The effect of other NSAID on platelet function does not seem to depend on CAII regulation, but exclusively on COX-1 inhibition. A nonspecific COX inhibitor naproxen, but not COX-2 selective inhibitor celecoxib inhibits both collagen and arachidonate induced platelet aggregation [122]. Nevertheless, in a recent report we provided evidence that interindividual variability in in vitro platelet responsiveness to acetylsalicylic acid may be associated with carbonic anhydrase II concentration. Briefly, ASA low-responders presented increased intraplatelet CAII concentration and more intense baseline arachidonic acid induces aggregation compared to ASA sensitive individuals [123]. Among the CAII-dependent mechanisms modifying the platelet responsiveness, the pH changes of platelet cytosol leading to impaired acetylating of cyclooxygenase by ASA are noteworthy. This in turn could affect the antiplatelet effect of ASA as well as platelet inflammatory activity and energetic metabolism (Figure 4).
Figure 4.
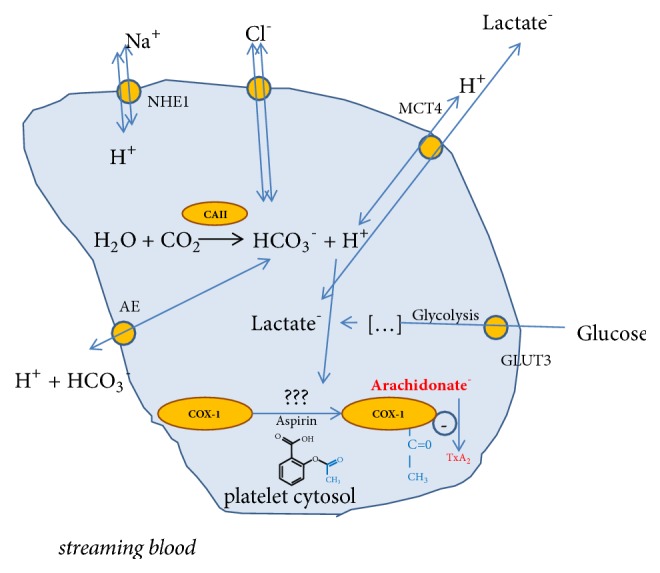
A potential role of carbonic anhydrase II (CAII) in regulating ASA response [30–32, 37]. GLUT3: glucose transporter; MCT4: H+/monocarboxylate transporter; AE: anion exchanger; NHE1: the Na+/H+ exchanger; COX-1: cyclooxygenase-1; TxA2: thromboxane A2.
To conclude, the role of carbonic anhydrase II in cardiovascular medicine is still underestimated and requires further in-depth studies.
4. Conclusions
Even though there is a well-documented rationale for an important role of carbonic anhydrase II in regulating platelet function, its exact role in platelet physiology and pathology remains poorly understood. A more frequent use of platelet CAII inhibitors holds promise as a good strategy to reduce the impact of cardiovascular diseases. However, future prospective clinical studies, supported by the evidence-based medicine principles, are needed in order to precisely elucidate the role of platelet CAII in cardiovascular medicine.
There is a need for more basic scientific investigations in order to establish the role of platelet carbonic anhydrase II in the pathogenesis of several diseases such as chronic urticaria and altitude sickness and, further, to verify the contribution of platelet CAII in metabolism of nitrites and antiaggregatory properties of S-Nitrosocysteine.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- 1.Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB) In consultation with the IUPAC-IUBMB Joint Commission on Biochemical Nomenclature (JCBN) Enzyme Nomenclature Recommendations of the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology on the Nomenclature and Classification of Enzymes by the Reactions they Catalyse IUBMB Enzyme Nomenclature EC 4.2.1.1. EC 4.2.1.1 created 1961, modified 2016, http://www.chem.qmul.ac.uk/iubmb/enzyme/EC4/2/1/1.html.
- 2.Xu Y., Feng L., Jeffrey P. D., Shi Y., Morel F. M. M. Structure and metal exchange in the cadmium carbonic anhydrase of marine diatoms. Nature. 2008;452(6):56–62. doi: 10.1038/nature06636. [DOI] [PubMed] [Google Scholar]
- 3.Del Prete S., Vullo D., Fisher G. M., et al. Discovery of a new family of carbonic anhydrases in the malaria pathogen Plasmodium falciparum - The η-carbonic anhydrases. Bioorganic & Medicinal Chemistry Letters. 2014;24(18):4389–4396. doi: 10.1016/j.bmcl.2014.08.015. [DOI] [PubMed] [Google Scholar]
- 4.Kikutani S., Nakajima K., Nagasato C., Tsuji Y., Miyatake A., Matsuda Y. Thylakoid luminal θ-carbonic anhydrase critical for growth and photosynthesis in the marine diatom. Proceedings of the National Acadamy of Sciences of the United States of America. 2016;113(35):9828–9833. doi: 10.1073/pnas.1603112113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alterio V., di Fiore A., D'Ambrosio K., Supuran C. T., de Simone G. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chemical Reviews. 2012;112(8):4421–4468. doi: 10.1021/cr200176r. [DOI] [PubMed] [Google Scholar]
- 6.Supuran C. T., Scozzafava A. Carbonic anhydrases as targets for medicinal chemistry. Bioorganic & Medicinal Chemistry. 2007;15(13):4336–4350. doi: 10.1016/j.bmc.2007.04.020. [DOI] [PubMed] [Google Scholar]
- 7.Imtaiyaz Hassan M., Shajee B., Waheed A., Ahmad F., Sly W. S. Structure, function and applications of carbonic anhydrase isozymes. Bioorganic & Medicinal Chemistry. 2013;21(6):1570–1582. doi: 10.1016/j.bmc.2012.04.044. [DOI] [PubMed] [Google Scholar]
- 8.Kendall A. G., Tashian R. E. Erythrocyte carbonic anhydrase I: Inherited deficiency in humans. Science. 1977;197(4302):471–472. doi: 10.1126/science.406674. [DOI] [PubMed] [Google Scholar]
- 9.Borthwick K. J., Kandemir N., Topaloglu R., et al. A phenocopy of CAII deficiency: A novel genetic explanation for inherited infantile osteopetrosis with distal renal tubular acidosis. Journal of Medical Genetics. 2003;40(2):115–121. doi: 10.1136/jmg.40.2.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sly W. S., Whyte M. P., Sundaram V., et al. Carbonic anhydrase II deficiency in 12 families with the autosomal recessive syndrome of osteopetrosis with renal tubular acidosis and cerebral calcification. The New England Journal of Medicine. 1985;313(3):139–145. doi: 10.1056/NEJM198507183130302. [DOI] [PubMed] [Google Scholar]
- 11.Du A.-L., Ren H.-M., Lu C.-Z., Tu J.-L., Xu C.-F., Sun Y.-A. Carbonic anhydrase III is insufficient in muscles of myasthenia gravis patients. Autoimmunity. 2009;42(3):209–215. doi: 10.1080/08916930802668610. [DOI] [PubMed] [Google Scholar]
- 12.Du A., Huang S., Zhao X., et al. Suppression of CHRN endocytosis by carbonic anhydrase CAR3 in the pathogenesis of myasthenia gravis. Autophagy. 2017;13(11):1981–1994. doi: 10.1080/15548627.2017.1375633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diez-Fernandez C., Rüfenacht V., Santra S., et al. Defective hepatic bicarbonate production due to carbonic anhydrase VA deficiency leads to early-onset life-threatening metabolic crisis. Genetics in Medicine. 2016;18(10):991–1000. doi: 10.1038/gim.2015.201. [DOI] [PubMed] [Google Scholar]
- 14.Avital D., Hershkovitz E., Loewenthal N. Exertional rhabdomyolysis in carbonic anhydrase 12 deficiency. Journal of Pediatric Endocrinology and Metabolism. 2018;31(6):697–699. doi: 10.1515/jpem-2017-0483. [DOI] [PubMed] [Google Scholar]
- 15.Inagaki Y., Jinno-Yoshida Y., Hamasaki Y., Ueki H. A novel autoantibody reactive with carbonic anhydrase in sera from patients with systemic lupus erythematosus and Sjögren's syndrome. Journal of Dermatological Science. 1991;2(3):147–154. doi: 10.1016/0923-1811(91)90060-B. [DOI] [PubMed] [Google Scholar]
- 16.Robert-Pachot M., Desbos A., Moreira A., et al. A new target for autoantibodies in patients with rheumatoid arthritis. Annals of the New York Academy of Sciences. 2007;1108:382–391. doi: 10.1196/annals.1422.039. [DOI] [PubMed] [Google Scholar]
- 17.Mentese A., Alver A., Demir S., et al. Carbonic anhydrase i and ii autoantibodies in behçet's disease. Acta Reumatólogica Portuguesa. 2017;2017(1):26–31. [PubMed] [Google Scholar]
- 18.Botrè F., Botrè C., Podestà E., Podda M., Invernizzi P. Effect of anti-carbonic anhydrase antibodies on carbonic anhydrases I and II. Clinical Chemistry. 2003;49(7):1221–1223. doi: 10.1373/49.7.1221. [DOI] [PubMed] [Google Scholar]
- 19.Mentese A., Fidan E., Alver A., et al. Detection of autoantibodies against carbonic anhydrase I and II in the plasma of patients with gastric cancer. Central European Journal of Immunology. 2017;42(1):73–77. doi: 10.5114/ceji.2017.67320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alver A., Menteşe A., Karahan S. C., et al. Increased serum anti-carbonic anhydrase II antibodies in patients with Graves' disease. Experimental and Clinical Endocrinology & Diabetes. 2007;115(5):287–291. doi: 10.1055/s-2007-960498. [DOI] [PubMed] [Google Scholar]
- 21.Menteşe A., Erkut N., Demir S., et al. Autoantibodies against Carbonic Anhydrase I and II in Patients with Acute Myeloid Leukemia. Turkish Journal of Hematology. 2017 doi: 10.4274/tjh.2016.0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pertovaara M., Bootorabi F., Kuuslahti M., Pasternack A., Parkkila S. Novel carbonic anhydrase autoantibodies and renal manifestations in patients with primary Sjogren's syndrome. Rheumatology. 2011;50(8):1453–1457. doi: 10.1093/rheumatology/ker118. [DOI] [PubMed] [Google Scholar]
- 23.Takemoto F., Katori H., Sawa N., et al. Induction of anti-carbonic-anhydrase-II antibody causes renal tubular acidosis in a mouse model of Sjögren's syndrome. Nephron Physiology. 2007;106(4):p63–p68. doi: 10.1159/000104873. [DOI] [PubMed] [Google Scholar]
- 24.Alver A., Menteşe A., Menteşe Ü., Sümer A., Uçar F., Us Altay D. Anti-carbonic anhydrase ii antibodies in end-stage renal disease patients. Medical Principles and Practice. 2014;23(4):331–335. doi: 10.1159/000362667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adamus G., Karren L. Autoimmunity against carbonic anhydrase II affects retinal cell functions in autoimmune retinopathy. Journal of Autoimmunity. 2009;32(2):133–139. doi: 10.1016/j.jaut.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karakus S., Baer A. N., Agrawal D., Gurakar M., Massof R. W., Akpek E. K. Utility of Novel Autoantibodies in the Diagnosis of Sjögrenʼs Syndrome Among Patients With Dry Eye. Cornea. 2018;37(4):405–411. doi: 10.1097/ICO.0000000000001471. [DOI] [PubMed] [Google Scholar]
- 27.Phung L., Lollett I. V., Goldhardt R., et al. Parallel ocular and serologic course in a patient with early Sjogren's syndrome markers. American Journal of Ophthalmology Case Reports. 2017;8:48–52. doi: 10.1016/j.ajoc.2017.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guarneri F., Guarneri C., Benvenga S. Helicobacter pylori and autoimmune pancreatitis: role of carbonic anhydrase via molecular mimicry? Journal of Cellular and Molecular Medicine. 2005;9(3):741–744. doi: 10.1111/j.1582-4934.2005.tb00506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Author: Fvasconcellos Source. Public Domain, 2011, https://commons.wikimedia.org/wiki/File:Carbonic_anhydrase_1CA2.png.
- 30.Merezhinskaya N., Ogunwuyi S. A., Fishbein W. N. Expression of monocarboxylate transporter 4 in human platelets, leukocytes, and tissues assessed by antibodies raised against terminal versus pre-terminal peptides. Molecular Genetics and Metabolism. 2006;87(2):152–161. doi: 10.1016/j.ymgme.2005.09.029. [DOI] [PubMed] [Google Scholar]
- 31.Ferreira I. A., Mocking A. I. M., Urbanus R. T., Varlack S., Wnuk M., Akkerman J.-W. N. Glucose uptake via glucose transporter 3 by human platelets is regulated by protein kinase B. The Journal of Biological Chemistry. 2005;280(38):32625–32633. doi: 10.1074/jbc.M507221200. [DOI] [PubMed] [Google Scholar]
- 32.Rosskopf D. Sodium-hydrogen exchange and platelet function. Journal of Thrombosis and Thrombolysis. 1999;8(1):15–23. doi: 10.1023/A:1008986329267. [DOI] [PubMed] [Google Scholar]
- 33.Gambaryan S., Tsikas D. A review and discussion of platelet nitric oxide and nitric oxide synthase: do blood platelets produce nitric oxide from l-arginine or nitrite? Amino Acids. 2015;47(9, article no. 1986):1779–1793. doi: 10.1007/s00726-015-1986-1. [DOI] [PubMed] [Google Scholar]
- 34.Broos K., Feys H. B., De Meyer S. F., Vanhoorelbeke K., Deckmyn H. Platelets at work in primary hemostasis. Blood Reviews. 2011;25(4):155–167. doi: 10.1016/j.blre.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 35.Lieberman E. H., O'Neill S., Mendelsohn M. E. S-nitrosocysteine inhibition of human platelet secretion is correlated with increases in platelet cGMP levels. Circulation Research. 1991;68(6):1722–1728. doi: 10.1161/01.RES.68.6.1722. [DOI] [PubMed] [Google Scholar]
- 36.Hanff E., Böhmer A., Zinke M., et al. Carbonic anhydrases are producers of S-nitrosothiols from inorganic nitrite and modulators of soluble guanylyl cyclase in human platelets. Amino Acids. 2016;48(7):1695–1706. doi: 10.1007/s00726-016-2234-z. [DOI] [PubMed] [Google Scholar]
- 37.Smyth S. S., Whiteheart S., Joseph E. I., Barry S. Platelet morphology, biochemistry and function; In: Williams Hematology by Marshall A. Lichtman, Thomas J. Kipps, Uri Seligsohn, Kenneth Kaushansky, Josef T. Prchal. 2010. [Google Scholar]
- 38.Supuran C. T. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nature Reviews Drug Discovery. 2008;7(2):168–181. doi: 10.1038/nrd2467. [DOI] [PubMed] [Google Scholar]
- 39.Supuran C. T. Acetazolamide for the treatment of idiopathic intracranial hypertension. Expert Review of Neurotherapeutics. 2015;15(8):851–856. doi: 10.1586/14737175.2015.1066675. [DOI] [PubMed] [Google Scholar]
- 40.Liu H., Chiang I., Kuo K. N., Liou C., Chen C. The effect of acetazolamide on sleep apnea at high altitude: a systematic review and meta-analysis. Therapeutic Advances in Respiratory Disease. 2016;11(1):20–29. doi: 10.1177/1753465816677006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Swenson E. R. Carbonic Anhydrase: Mechanism, Regulation, Links to Disease, and Industrial Applications. Vol. 75. Dordrecht: Springer Netherlands; 2014. Carbonic Anhydrase Inhibitors and High Altitude Illnesses; pp. 361–386. (Subcellular Biochemistry). [DOI] [PubMed] [Google Scholar]
- 42.Schmidl D., Schmetterer L., Garhöfer G., Popa-Cherecheanu A. Pharmacotherapy of Glaucoma. Journal of Ocular Pharmacology and Therapeutics. 2015;31(2):63–77. doi: 10.1089/jop.2014.0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reiss W. G., Oles K. S. Acetazolamide in the Treatment of Seizures. Annals of Pharmacotherapy. 2016;30(5):514–519. doi: 10.1177/106002809603000515. [DOI] [PubMed] [Google Scholar]
- 44.Bryant J. L., Gieling R. G., Meredith S. L., et al. Novel carbonic anhydrase IX-targeted therapy enhances the anti-tumour effects of cisplatin in small cell lung cancer. International Journal of Cancer. 2018;142(1):191–201. doi: 10.1002/ijc.31042. [DOI] [PubMed] [Google Scholar]
- 45.Lindskog S. Structure and mechanism of Carbonic Anhydrase. Pharmacology & Therapeutics. 1997;74(1):1–20. doi: 10.1016/S0163-7258(96)00198-2. [DOI] [PubMed] [Google Scholar]
- 46.Nakai H., Byers M. G., Venta P. J., Tashian R. E., Shows T. B. The gene for human carbonic anhydrase ii (Ca2) is located at chromosome 8q22. Cytogenetic and Genome Research. 1987;44(4):234–235. doi: 10.1159/000132378. [DOI] [PubMed] [Google Scholar]
- 47.Sobacchi C., Schulz A., Coxon F. P., Villa A., Helfrich M. H. Osteopetrosis: genetics, treatment and new insights into osteoclast function. Nature Reviews Endocrinology. 2013;9(9):522–536. doi: 10.1038/nrendo.2013.137. [DOI] [PubMed] [Google Scholar]
- 48.Imai Y., Youn M.-Y., Inoue K., Takada I., Kouzmenko A., Kato S. Nuclear receptors in bone physiology and diseases. Physiological Reviews. 2013;93(2):481–523. doi: 10.1152/physrev.00008.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Supuran C. T., Scozzafava A. Activation of carbonic anhydrase isozymes. In: Chegwidden W. R., Carter N. D., Edwards Y. H., editors. The Carbonic Anhydrases. 90, EXS 90. Birkhäuser, Basel: 2000. [DOI] [PubMed] [Google Scholar]
- 50.Supuran C. T. How many carbonic anhydrase inhibition mechanisms exist? Journal of Enzyme Inhibition and Medicinal Chemistry. 2015;31(3):345–360. doi: 10.3109/14756366.2015.1122001. [DOI] [PubMed] [Google Scholar]
- 51.Temperini C., Cecchi A., Scozzafava A., Supuran C. T. Carbonic anhydrase inhibitors. Interaction of indapamide and related diuretics with 12 mammalian isozymes and X-ray crystallographic studies for the indapamide-isozyme II adduct. Bioorganic & Medicinal Chemistry Letters. 2008;18(8):2567–2573. doi: 10.1016/j.bmcl.2008.03.051. [DOI] [PubMed] [Google Scholar]
- 52.Saada M.-C., Vullo D., Montero J.-L., Scozzafava A., Supuran C. T., Winum J.-Y. Mono- and di-halogenated histamine, histidine and carnosine derivatives are potent carbonic anhydrase I, II, VII, XII and XIV activators. Bioorganic & Medicinal Chemistry. 2014;22(17):4752–4758. doi: 10.1016/j.bmc.2014.07.005. [DOI] [PubMed] [Google Scholar]
- 53.Belloni F., Turpini R. Presence of carbonic anhydrase in human platelets. Haematologica. 1957;42(3):217–225. [PubMed] [Google Scholar]
- 54.Siffert W., Gros G. Carbonic anhydrase in human platelets. Biochemical Journal. 1984;217(3):727–730. doi: 10.1042/bj2170727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vaitkevicius H., Turner I., Spalding A., Lockette W. Chloride increases adrenergic receptor-mediated platelet and vascular responses. American Journal of Hypertension. 2002;15(6):492–498. doi: 10.1016/S0895-7061(02)02276-8. [DOI] [PubMed] [Google Scholar]
- 56.Novak E. K., Sweet H. O., Prochazka M., et al. Cocoa: A new mouse model for platelet storage pool deficiency. British Journal of Haematology. 1988;69(3):371–378. doi: 10.1111/j.1365-2141.1988.tb02376.x. [DOI] [PubMed] [Google Scholar]
- 57.Akkerman J. W. N., Holmsen H., Loughnane M. Simultaneous measurement of aggregation, secretion, oxygen uptake, proton production, and intracellular metabolites in the same platelet suspension. Analytical Biochemistry. 1979;97(2):387–393. doi: 10.1016/0003-2697(79)90090-3. [DOI] [PubMed] [Google Scholar]
- 58.Akkerman J. W. N., Holmsen H. Interrelationships among platelet responses: Studies on the burst in proton liberation, lactate production, and oxygen uptake during platelet aggregation and Ca2+ secretion. Blood. 1981;57(5):956–966. [PubMed] [Google Scholar]
- 59.Siffert W., Fox G., Gros G. Carbonic Anhydrase in Human Platelets: Effects of Carbonic Anhydrase Inhibition on Platelet Aggregation. Annals of the New York Academy of Sciences. 1984;429(1):207–209. doi: 10.1111/j.1749-6632.1984.tb12334.x. [DOI] [PubMed] [Google Scholar]
- 60.Siffert W., Fox G., Gros G. The effect of carbonic anhydrase inhibition on the velocity of thrombin-stimulated platelet aggregation under physiological conditions. Biochemical and Biophysical Research Communications. 1984;121(1):266–270. doi: 10.1016/0006-291X(84)90717-4. [DOI] [PubMed] [Google Scholar]
- 61.Temperini C., Innocenti A., Scozzafava A., Mastrolorenzo A., Supuran C. T. Carbonic anhydrase activators: l-Adrenaline plugs the active site entrance of isozyme II, activating better isoforms I, IV, VA, VII, and XIV. Bioorganic & Medicinal Chemistry Letters. 2007;17(3):628–635. doi: 10.1016/j.bmcl.2006.11.027. [DOI] [PubMed] [Google Scholar]
- 62.Temperini C., Scozzafava A., Supuran C. T. Carbonic anhydrase activation and the drug design. Current Pharmaceutical Design. 2008;14(7):708–715. doi: 10.2174/138161208783877857. [DOI] [PubMed] [Google Scholar]
- 63.Spalding A., Vaitkevicius H., Dill S., MacKenzie S., Schmaier A., Lockette W. Mechanism of epinephrine-induced platelet aggregation. Hypertension. 1998;31(2):603–607. doi: 10.1161/01.HYP.31.2.603. [DOI] [PubMed] [Google Scholar]
- 64.Woodman R., Brown C., Lockette W. Chlorthalidone decreases platelet aggregation and vascular permeability and promotes angiogenesis. Hypertension. 2010;56(3):463–470. doi: 10.1161/HYPERTENSIONAHA.110.154476. [DOI] [PubMed] [Google Scholar]
- 65.Berger J. S., Becker R. C., Kuhn C., Helms M. J., Ortel T. L., Williams R. Hyperreactive platelet phenotypes: Relationship to altered serotonin transporter number, transport kinetics and intrinsic response to adrenergic co-stimulation. Thrombosis and Haemostasis. 2013;109(1):85–92. doi: 10.1160/TH12-03-0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Benedict C. R., Mathew B., Rex K. A., Cartwright J., Jr., Sordahl L. A. Correlation of plasma serotonin changes with platelet aggregation in an in vivo dog model of spontaneous occlusive coronary thrombus formation. Circulation Research. 1986;58(1):58–67. doi: 10.1161/01.RES.58.1.58. [DOI] [PubMed] [Google Scholar]
- 67.Vikenes K., Farstad M., Nordrehaug J. E. Serotonin is associated with coronary artery disease and cardiac events. Circulation. 1999;100(5):483–489. doi: 10.1161/01.CIR.100.5.483. [DOI] [PubMed] [Google Scholar]
- 68.Kajiwara I., Soejima H., Miyamoto S., Ogawa H. Effects of additional treatment of sarpogrelate to aspirin therapy on platelet aggregation and plasma plasminogen activator inhibitor activity in patients with stable effort angina. Thrombosis Research. 2011;128(6):547–551. doi: 10.1016/j.thromres.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 69.Uchiyama S., Ozaki Y., Satoh K., Kondo K., Nishimaru K. Effect of sarpogrelate, a 5-HT2A antagonist, on platelet aggregation in patients with ischemic stroke: Clinical-pharmacological dose-response study. Cerebrovascular Disease. 2007;24(2-3):264–270. doi: 10.1159/000105135. [DOI] [PubMed] [Google Scholar]
- 70.Nishihira K., Yamashita A., Tanaka N., et al. Inhibition of 5-hydroxytryptamine2A receptor prevents occlusive thrombus formation on neointima of the rabbit femoral artery. Journal of Thrombosis and Haemostasis. 2006;4(1):247–255. doi: 10.1111/j.1538-7836.2005.01702.x. [DOI] [PubMed] [Google Scholar]
- 71.Casini A., Caccia S., Scozzafava A., Supuran C. T. Carbonic anhydrase activators. The selective serotonin reuptake inhibitors fluoxetine, sertraline and citalopram are strong activators of isozymes I and II. Bioorganic & Medicinal Chemistry Letters. 2003;13(16):2765–2768. doi: 10.1016/S0960-894X(03)00507-9. [DOI] [PubMed] [Google Scholar]
- 72.Hergovich N., Aigner M., Eichler H.-G., Entlicher J., Drucker C., Jilma B. Paroxetine decreases platelet serotonin storage and platelet function in human beings. Clinical Pharmacology & Therapeutics. 2000;68(4):435–442. doi: 10.1067/mcp.2000.110456. [DOI] [PubMed] [Google Scholar]
- 73.Serebruany V. L., Gurbel P. A., O'Connor C. M. Platelet inhibition by sertraline and N-desmethylsertraline: A possible missing link between depression, coronary events, and mortality benefits of selective serotonin reuptake inhibitors. Pharmacological Research. 2001;43(5):453–461. doi: 10.1006/phrs.2001.0817. [DOI] [PubMed] [Google Scholar]
- 74.van Holten T. C., Roest M., Riphagen J., et al. Citalopram is a more potent platelet function inhibitor than paroxetine in a case-control study. Journal of Thrombosis and Haemostasis. 2012;10(6):1177–1179. doi: 10.1111/j.1538-7836.2012.04703.x. [DOI] [PubMed] [Google Scholar]
- 75.Alderman C. P., Moritz C. K., Ben-Tovim D. I. Abnormal platelet aggregation associated with fluoxetine therapy. Annals of Pharmacotherapy. 1992;26(12):1517–1519. doi: 10.1177/106002809202601205. [DOI] [PubMed] [Google Scholar]
- 76.Meier C. R., Schlienger R. G., Jick H. Use of selective serotonin reuptake inhibitors and risk of developing first-time acute myocardial infarction. British Journal of Clinical Pharmacology. 2001;52(2):179–184. doi: 10.1046/j.0306-5251.2001.01426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Smith M. M., Smith B. B., Lahr B. D., et al. Selective Serotonin Reuptake Inhibitors and Serotonin–Norepinephrine Reuptake Inhibitors Are Not Associated With Bleeding or Transfusion in Cardiac Surgical Patients. Anesthesia & Analgesia. 2018;126(6):1859–1866. doi: 10.1213/ANE.0000000000002668. [DOI] [PubMed] [Google Scholar]
- 78.Masini E., Di Bello M. G., Raspanti S., et al. The role of histamine in platelet aggregation by physiological and immunological stimuli. Inflammation Research. 1998;47(5):211–220. doi: 10.1007/s000110050319. [DOI] [PubMed] [Google Scholar]
- 79.Shah B. H., Lashari I., Rana S., et al. Synergistic interaction of adrenaline and histamine in human platelet aggregation is mediated through activation of phospholipase, map kinase and cyclo-oxygenase pathways. Pharmacological Research. 2000;42(5):479–483. doi: 10.1006/phrs.2000.0721. [DOI] [PubMed] [Google Scholar]
- 80.Zuberbier T., Schadendorf D., Haas N., Hartmann K., Henz B. M. Enhanced P-Selectin Expression in Chronic and Dermographic Urticaria. International Archives of Allergy and Immunology. 1997;114(1):86–89. doi: 10.1159/000237648. [DOI] [PubMed] [Google Scholar]
- 81.Chandrashekar L., Rajappa M., Sundar I., et al. Platelet activation in chronic urticaria and its correlation with disease severity. Platelets. 2014;25(3):162–165. doi: 10.3109/09537104.2013.786822. [DOI] [PubMed] [Google Scholar]
- 82.Hallberg T., Dohlsten M., Baldetorp B. Demonstration of Histamine Receptors on Human Platelets by Flow Cytometry. European Journal of Haematology. 1984;32(2):113–118. doi: 10.1111/j.1600-0609.1984.tb02165.x. [DOI] [PubMed] [Google Scholar]
- 83.Saxena S. P., Brandes L. J., Becker A. B., Simons K. J., Labella F. S., Gerrard J. M. Histamine is an intracellular messenger mediating platelet aggregation. Science. 1989;243(4898):1596–1599. doi: 10.1126/science.2928797. [DOI] [PubMed] [Google Scholar]
- 84.Saxena S. P., McNicol A., Brandes L. J., Becker A. B., Gerrard J. M. A role for intracellular histamine in collagen-induced platelet aggregation. Blood. 1990;75(2):407–414. [PubMed] [Google Scholar]
- 85.Li S. Q., Zhao G., Li J., Qian W. Effect of histidine on myocardial mitochondria and platelet aggregation during thrombotic cerebral ischemia in rats. Zhongguo Yao Li Xue Bao. 1998;19(5):493–496. [PubMed] [Google Scholar]
- 86.Steinhauer H. B., Kluthe R., Lubrich I., Schollmeyer P. Effect of L-histidine in vivo on human platelet function and arachidonic acid metabolism. Prostaglandins, Leukotrienes and Medicine. 1985;18(2):245–254. doi: 10.1016/0262-1746(85)90024-1. [DOI] [PubMed] [Google Scholar]
- 87.Boldyrev A., Abe H. Metabolic transformation of neuropeptide carnosine modifies its biological activity. Cellular and Molecular Neurobiology. 1999;19(1):163–75. doi: 10.1023/A:1006914028195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nikitenko N, Shavratskii V, Boldyrev A., Suslina Z., Ionova V. Effect of carnosine and its derivatives on ADP-induced human platelet aggregation (only abstract available) Voprosy Meditsinskoi Khimii. 1995;41(1):41–43. [PubMed] [Google Scholar]
- 89.Salzman E. W., Chambers D. A. Inhibition of ADP-induced platelet aggregation by substituted amino-acids [32] Nature. 1964;204(4959):698–700. doi: 10.1038/204698b0. [DOI] [PubMed] [Google Scholar]
- 90.Nayak N. C., Roy S., Narayanan T. K. Pathologic features of altitude sickness. American Journal of Pathology. 1964;45:381–391. [PMC free article] [PubMed] [Google Scholar]
- 91.Sharma S. C., Hoon R. S. Platelet adhesiveness on acute induction to high altitude. Thrombosis Research. 1978;13(5):725–732. doi: 10.1016/0049-3848(78)90178-0. [DOI] [PubMed] [Google Scholar]
- 92.Chatterji J. C., Ohri V. C., Das B. K., et al. Platelet count, platelet aggregation and fibrinogen levels following acute induction to high altitude (3200 and 3771 metres) Thrombosis Research. 1982;26(3):177–182. doi: 10.1016/0049-3848(82)90138-4. [DOI] [PubMed] [Google Scholar]
- 93.Vij A. G. Effect of prolonged stay at high altitude on platelet aggregation and fibrinogen levels. Platelets. 2009;20(6):421–427. doi: 10.1080/09537100903116516. [DOI] [PubMed] [Google Scholar]
- 94.Engberink R. H. G. O., Frenkel W. J., Van Den Bogaard B., Brewster L. M., Vogt L., Van Den Born B.-J. H. Effects of thiazide-type and thiazide-like diuretics on cardiovascular events and mortality: Systematic review and meta-analysis. Hypertension. 2015;65(5):1033–1040. doi: 10.1161/HYPERTENSIONAHA.114.05122. [DOI] [PubMed] [Google Scholar]
- 95.Rendu F., Bachelot C., Molle D., Caen J., Guez D. Indapamide inhibits human platelet aggregation in vitro: Comparison with hydrochlorothiazide. Journal of Cardiovascular Pharmacology. 1993;22(12):S57–S63. doi: 10.1097/00005344-199322008-00017. [DOI] [PubMed] [Google Scholar]
- 96.Chohan I. S., Singh I., Vermylen J. Furosemide: its role on certain platelet functions. Acta clinica Belgica. 1977;32(1):39–43. doi: 10.1080/17843286.1977.11717837. [DOI] [PubMed] [Google Scholar]
- 97.Ingerman C. M., Bryan Smith J., Silver M. J. Inhibition of the platelet release reaction and platelet prostagland in synthesis by furosemide. Thrombosis Research. 1976;8(3):417–419. doi: 10.1016/0049-3848(76)90035-9. [DOI] [PubMed] [Google Scholar]
- 98.Radomski M. W., Palmer R. M. J., Moncada S. The role of nitric oxide and cGMP in platelet adhesion to vascular endothelium. Biochemical and Biophysical Research Communications. 1987;148(3):1482–1489. doi: 10.1016/S0006-291X(87)80299-1. [DOI] [PubMed] [Google Scholar]
- 99.Dangel O., Mergia E., Karlisch K., Groneberg D., Koesling D., Friebe A. Nitric oxide-sensitive guanylyl cyclase is the only nitric oxide receptor mediating platelet inhibition. Journal of Thrombosis and Haemostasis. 2010;8(6):1343–1352. doi: 10.1111/j.1538-7836.2010.03806.x. [DOI] [PubMed] [Google Scholar]
- 100.Aamand R., Dalsgaard T., Jensen F. B., Simonsen U., Roepstorff A., Fago A. Generation of nitric oxide from nitrite by carbonic anhydrase: A possible link between metabolic activity and vasodilation. American Journal of Physiology-Heart and Circulatory Physiology. 2009;297(6):H2068–H2074. doi: 10.1152/ajpheart.00525.2009. [DOI] [PubMed] [Google Scholar]
- 101.Park J. W., Piknova B., Huang P. L., Noguchi C. T., Schechter A. N. Effect of Blood Nitrite and Nitrate Levels on Murine Platelet Function. PLoS ONE. 2013;8(2) doi: 10.1371/journal.pone.0055699.e55699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Srihirun S., Sriwantana T., Unchern S., et al. Platelet inhibition by nitrite is dependent on erythrocytes and deoxygenation. PLoS ONE. 2012;7(1) doi: 10.1371/journal.pone.0030380.e30380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tsikas D., Ikic M., Tewes K. S., Raida M., Frölich J. C. Inhibition of platelet aggregation by S-nitroso-cysteine via cGMP-independent mechanisms: Evidence of inhibition of thromboxane A2 synthesis in human blood platelets. FEBS Letters. 1999;442(2-3):162–166. doi: 10.1016/S0014-5793(98)01633-0. [DOI] [PubMed] [Google Scholar]
- 104.Mangani S., Håkansson K. Crystallographic studies of the binding of protonated and unprotonated inhibitors to carbonic anhydrase using hydrogen sulphide and nitrate anions. European Journal of Biochemistry. 1992;210(3):867–871. doi: 10.1111/j.1432-1033.1992.tb17490.x. [DOI] [PubMed] [Google Scholar]
- 105.Zinke M., Hanff E., Böhmer A., Supuran C. T., Tsikas D. Discovery and microassay of a nitrite-dependent carbonic anhydrase activity by stable-isotope dilution gas chromatography-mass spectrometry. Amino Acids. 2016;48(1):245–255. doi: 10.1007/s00726-015-2081-3. [DOI] [PubMed] [Google Scholar]
- 106.Lahiri P., Chaudhuri U., Chattopadhyay A., Dasgupta A. K. R. Platelet aggregation profile as a marker of hydroxyurea bioavailability through nitric oxide generation in chronic myelogenous leukemia. Leukemia & Lymphoma. 2006;47(4):741–746. doi: 10.1080/10428190500375854. [DOI] [PubMed] [Google Scholar]
- 107.Scozzafava A., Supuran C. T. Hydroxyurea is a carbonic anhydrase inhibitor. Bioorganic & Medicinal Chemistry. 2003;11(10):2241–2246. doi: 10.1016/S0968-0896(03)00112-3. [DOI] [PubMed] [Google Scholar]
- 108.Poulsen S. A. Targeting Carbonic Anhydrases. 2014. Coumarins that inhibit carbonic anhydrase. [Google Scholar]
- 109.Lomelino C., Supuran C., McKenna R. Non-Classical Inhibition of Carbonic Anhydrase. International Journal of Molecular Sciences. 2016;17(7):p. 1150. doi: 10.3390/ijms17071150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Barcellona D., Vannini M. L., Fenu L., Balestrieri C., Marongiu F. Warfarin or acenocoumarol: Which is better in the management of oral anticoagulants? Thrombosis and Haemostasis. 1998;80(6):899–902. doi: 10.1055/s-0037-1615385. [DOI] [PubMed] [Google Scholar]
- 111.Mieszczak C., Winther K. Does warfarin enhance platelet activity? Thrombosis Research. 1996;84(4):285–287. doi: 10.1016/S0049-3848(96)00188-0. [DOI] [PubMed] [Google Scholar]
- 112.Dewilde W. J. M., Janssen P. W. A., Bergmeijer T. O., Kelder J. C., Hackeng C. M., ten Berg J. M. The effect of acenocoumarol on the antiplatelet effect of clopidogrel. Thrombosis and Haemostasis. 2015;114(4):708–716. doi: 10.1160/TH15-02-0130. [DOI] [PubMed] [Google Scholar]
- 113.Weber A., Casini A., Heine A., et al. Unexpected Nanomolar Inhibition of Carbonic Anhydrase by COX-2-Selective Celecoxib: New Pharmacological Opportunities Due to Related Binding Site Recognition. Journal of Medicinal Chemistry. 2004;47(3):550–557. doi: 10.1021/jm030912m. [DOI] [PubMed] [Google Scholar]
- 114.Katagiri M., Ogasawara T., Hoshi K., et al. Suppression of adjuvant-induced arthritic bone destruction by cyclooxygenase-2 selective agents with and without inhibitory potency against carbonic anhydrase II. Journal of Bone and Mineral Research. 2006;21(2):219–227. doi: 10.1359/JBMR.051025. [DOI] [PubMed] [Google Scholar]
- 115.Di Fiore A., Pedone C., D'Ambrosio K., Scozzafava A., De Simone G., Supuran C. T. Carbonic anhydrase inhibitors: Valdecoxib binds to a different active site region of the human isoform II as compared to the structurally related cyclooxygenase II 'selective' inhibitor celecoxib. Bioorganic & Medicinal Chemistry Letters. 2006;16(2):437–442. doi: 10.1016/j.bmcl.2005.09.040. [DOI] [PubMed] [Google Scholar]
- 116.De Monte C., Carradori S., Gentili A., Mollica A., Trisciuoglio D., Supuran C. T. Dual cyclooxygenase and carbonic anhydrase inhibition by nonsteroidal anti-inflammatory drugs for the treatment of cancer. Current Medicinal Chemistry. 2015;22(24):2812–2818. doi: 10.2174/0929867322666150716113501. [DOI] [PubMed] [Google Scholar]
- 117.Greene I. M., Arifullah M., Kenny A. D. Carbonic anhydrase inhibition by flurbiprofen and related agents. Pharmacology. 1992;45(5):278–284. doi: 10.1159/000139011. [DOI] [PubMed] [Google Scholar]
- 118.Puscas I., Coltau M., Pasca R. Nonsteroidal anti-inflammatory drugs activate carbonic anhydrase by a direct mechanism of action. The Journal of Pharmacology and Experimental Therapeutics. 1996;277(3):1464–1466. [PubMed] [Google Scholar]
- 119.Puscas I., Ifrim M., Maghiar T., et al. Indomethacin activates carbonic anhydrase and antagonizes the effect of the specific carbonic anhydrase inhibitor acetazolamide, by a direct mechanism of action. International Journal of Clinical Pharmacology and Therapeutics. 2001;39(6):265–270. [PubMed] [Google Scholar]
- 120.Bayram E., Senturk M., Irfan Kufrevioglu O., Supuran C. T. In vitro inhibition of salicylic acid derivatives on human cytosolic carbonic anhydrase isozymes I and II. Bioorganic & Medicinal Chemistry. 2008;16(20):9101–9105. doi: 10.1016/j.bmc.2008.09.028. [DOI] [PubMed] [Google Scholar]
- 121.Cummings A. J., Martin B. K. The effect of acetylsalicylic acid on carbonic anhydrase in vitro. Biochemical Pharmacology. 1963;12(9):1011–1016. doi: 10.1016/0006-2952(63)90024-8. [DOI] [PubMed] [Google Scholar]
- 122.Leese P. T., Hubbard R. C., Karim A., Isakson P. C., Yu S. S., Geis G. S. Effects of celecoxib, a novel cyclooxygenase-2 inhibitor, on platelet function in healthy adults: A randomized, controlled trial. Clinical Pharmacology and Therapeutics. 2000;40(2):124–132. doi: 10.1177/00912700022008766. [DOI] [PubMed] [Google Scholar]
- 123.Jakubowski M., Dȩbski J., Szahidewicz-Krupska E., et al. Platelet Carbonic Anhydrase II, a Forgotten Enzyme, May Be Responsible for Aspirin Resistance. Oxidative Medicine and Cellular Longevity. 2017;2017 doi: 10.1155/2017/3132063. [DOI] [PMC free article] [PubMed] [Google Scholar]