

Summary
Visualization of the transcriptome and the nuclear organization in situ has been challenging for single cell analysis. Here, we demonstrate a multiplexed single molecule in situ method, intron seqFISH, that allows imaging of 10,421 genes at their nascent transcription active sites in single cells, followed by mRNA and lncRNA seqFISH and immunofluorescence. This nascent transcriptome profiling method can identify different cell types and states with mouse embryonic stem cells and fibroblasts. The nascent sites of RNA synthesis tend to be localized on the surfaces of chromosome territories and their organization in individual cells is highly variable. Surprisingly, the global nascent transcription oscillated asynchronously in individual cells with a period of 2 hours in mouse embryonic stem cells as well as in fibroblasts. Together, spatial genomics of the nascent transcriptome by intron seqFISH reveals nuclear organizational principles and fast dynamics in single cells that are otherwise obscured.
Keywords: seqFISH, oscillations, nascent transcriptome, intron, chromosome, smFISH, transcription
In brief
Development of intron seqFISH allows in situ profiling and visualization of nascent transcription at the single cell level, revealing spatial and temporal features of nascent transcriptome.
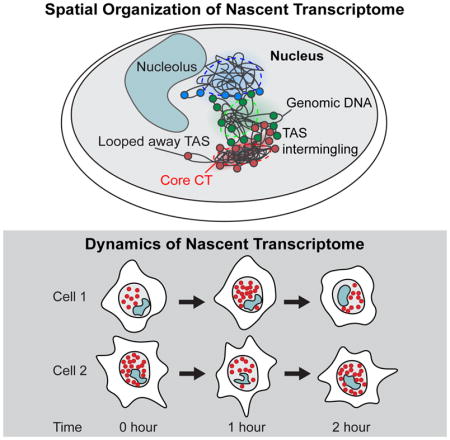
Introduction
The recent explosion of single cell sequencing technologies is leading to unprecedented insight into the structure of the nucleus and the transcriptome with Hi-C (Lieberman-Aiden et al., 2011; Nagano et al., 2013; Rao et al., 2014; Stevens et al., 2017) and single cell RNA-seq (Darmanis et al., 2015, Klein et al., 2015, Lee et al., 2014, Macosko et al., 2015, Zeisel et al., 2015) respectively. However, there exist few methods which allow direct imaging of both chromosome structure and transcriptomics information in the same cells. Furthermore, sequencing based approaches require inefficient biochemical steps to generate sequencing libraries which lower sensitivity, and are costly. Therefore, a method is needed that allows the imaging of chromosome structure and transcriptome in the same single cells in situ without sequencing.
Pioneering work on single molecule Fluorescence in situ Hybridization (smFISH) (Femino et al., 1998; Raj et al., 2006) observed that nascent mRNAs are produced in bursts at transcription active sites (TAS) in individual nuclei. In particular, these nascent sites of transcription near the genomic loci can be selectively labeled over mature transcripts, by targeting introns, which are co-transcriptionally processed out (Levesque and Raj, 2013). This intron chromosomal expression FISH (iceFISH) assay (Levesque and Raj, 2013) showed that at least 20 TAS from a single chromosome can be detected to measure their spatial positions and expression levels in individual single human cells.
We had previously developed sequential FISH (seqFISH) (Lubeck et al., 2014) to multiplex a large number of mRNA molecules in cells by single molecule imaging and sequential barcoded rounds of hybridization. seqFISH has successfully profiled hundreds of mRNAs in tissues and revealed distinct spatial structures in the mouse brain (Shah et al., 2016b) and the chick embryo (Lignell et al., 2017).
Here, we demonstrate transcriptome-scale intron seqFISH by labeling the TAS of 10,421 genes in single cells to capture the nascent transcriptome and its spatial organization with single molecule sensitivity. We also apply seqFISH in the same cells to profile mRNAs and long noncoding (lnc)RNAs along with immunofluorescence to detect pluripotency factors, cell cycle markers, and nuclear bodies. Thus, intron seqFISH provides a direct image of all the active sites within a nucleus and is complementary to ligation based sequencing methods.
Furthermore, the relatively short lifetimes of TAS compared to the longer lifetime of mRNAs (Sharova et al., 2009) mean that intron seqFISH can capture fast dynamics in the nascent transcriptome that would otherwise be obscured in mRNA measurements. Many pathways, such as NFkB, NFAT, Erk and calcium signaling can pulse on a timescale of minutes to hours (Hoffman et al., 2002; Yissachar et al., 2013; Shankaran et al., 2009; Dolmetsch et al., 1998). Indeed, a recent work showed that intron to exon ratios in single cells can provide “velocity” trajectories of cellular differentiation processes (La Manno et al., 2017).
Results
Intron seqFISH targets transcription active sites in single cells
To multiplex TAS detection for 10,421 genes, we use sequential rounds of hybridization to generate a unique temporal barcode sequence on each gene, which we then decode by aligning images from multiple round of barcoding hybridization. Specifically, we targeted the introns at the 5′ regions of genes by a set of 25 probes. A total of 260,525 primary probes were synthesized for all 10,421 genes. Each primary probe contained multiple overhang sequences that could be hybridized by fluorophore labeled readout probes in sequential round of hybridizations to impart a temporal barcode on each target (Figure 1A and S1A–B). After the primary probes were hybridized to the cells, readout probes were introduced with automated fluidics, and cells were imaged on a spinning disk confocal microscope with z-sections. After imaging, the readout probes were removed by denaturation in 55% formamide, while the primary probes remain bound on the intronic RNA due to longer probe length and higher DNA-RNA affinity. A different set of readout probes were then hybridized to the primary probes and imaged until all 60 readout probes and 20 rounds of hybridization were complete (Figure 1A–B and S1A–B). There was little decrease in signal over 20 rounds of hybridization (Figure S1C).
Figure 1. Intron seqFISH enables transcriptome profiling of nascent active sites.

(A) Schematics of intron seqFISH. Nascent RNA molecules are produced in bursts at the transcription active site at the genomic locus. Each gene is targeted by 25 primary probes. Barcodes are read out by secondary fluorescent readout probes that are complementary to the 15-nt barcode region. Detail of the primary probe design is shown in Figure S1. Each of the five barcoding rounds (I, II, III, IV and V) is based on 12 pseudocolors constructed by four serial hybridizations. In each serial hybridization, three readout probes conjugated to Alexa 647, Cy3B or Alexa 488 are hybridized to the primary probes, imaged and then stripped. Images from four serial hybridizations are collapsed into a single composite 12 pseudocolor image, which corresponds to one barcoding round. In total, five barcoding rounds were performed with 20 serial hybridizations to cover 12^4 = 20,736 barcodes with an extra round for error correction. (B) Combined seqFISH and immunofluorescence images with multiple mESCs. Barcoding round I image of 10,421 gene intron seqFISH from three z-sections (top left), and zoomed-in view of the yellow boxed region through five rounds of barcoding (top right panels). White boxes indicate identified barcodes, yellow boxes are recovered signal from error corrected barcodes, red boxes indicate false positives from mishybridizations. Bottom panels are maximum intensity projections of a z-stack of mRNA seqFISH, first internal transcribed spacer of rRNA (ITS1) probes staining nucleoli (Shishova et al., 2011), and H3S10Ph antibody staining G2/M phase cells. Dashed white lines in intron seqFISH image display nucleus boundaries (determined by z-projecting DAPI signal) while in mRNA seqFISH images display cytoplasmic boundaries of cells. Scale bars represent 5 μm in images with multiple single cells, and 0.5 μm in the zoomed-in images. (C) Two intron seqFISH replicates are highly reproducible (n1 = 314 cells; n2 = 382 cells). E14 cells were cultured under serum/LIF condition. (D) Comparison of intron seqFISH and intron smFISH with 34 genes verifies the accuracy of intron seqFISH. Error bars represent s.e.m. of intron seqFISH replicates. (E) Frequencies of on and off target barcodes in each cell. On average, 1266 ± 288 on target barcodes are typically detected per cell, while few off target barcodes (6.98 ± 1.62) are detected in any cell. (F) Frequencies of individual on target and off target barcodes detected. Introns display a wide range of expression levels. False positives are rarely detected, demonstrating the accuracy of the intron seqFISH. (G) Comparison of intron seqFISH (left) and smFISH (right) with GRO-seq. (H) Comparison of burst frequencies between E14 cells grown in serum/LIF vs. 2i conditions (left) and E14 cells grown in serum/LIF vs. NIH3T3 cells (right). GRO-seq data are from Jonkers et al. (2014).
We use a coding scheme (Eng et al., 2017) with 12 pseudocolors and 5 barcoding rounds to generate 12^5=248,832 barcodes, which allows us to uniquely identify 12^4=20,736 genes with tolerance for drops in any one round of barcoding (Figure 1A–B). The 12 pseudocolors for each barcoding round are generated with 4 serial hybridizations each with 3 readout probes labeled with three different fluorophores (Figure 1A–B and S1A–B). The advantage of using a large number of pseudocolors and a smaller number of barcoding rounds is that fewer mistakes occur in alignment and readout of barcodes when there are fewer barcoding rounds over which to accumulate errors. Furthermore, spreading out dots across many pseudocolors reduces spot density. We recently implemented this 12 color pseudocolor scheme in RNA SPOTs (Eng et al., 2017) to profile the transcriptome of 10,212 mRNAs in vitro, which showed high precision and concordance with RNA-seq measurements.
We followed the intron seqFISH experiment with additional rounds of mRNA and lncRNA seqFISH as well as antibody staining to label pluripotency, differentiation and cell cycle markers in addition to nuclear bodies in the same single cells (Figure 1B).
Intron seqFISH accurately measures nascent transcriptome in single cells
The intron seqFISH measurements are highly reproducible between biological replicates (R = 0.93, Figure 1C, Table S1) using mESCs cultured in serum/LIF conditions, indicating that there is little technical noise in the data. We compared the seqFISH data to 34 introns measured by single molecule FISH (smFISH), the gold standard quantification method. The average frequencies of observing TAS (burst frequency) for these 34 genes were correlated with Pearson correlation coefficient of 0.73 (Figure 1D) and a slope of 0.44, indicating a relative detection efficiency of 44%. This efficiency compares favorably with the 5–20% efficiency of single cell RNA-seq. In addition, the false positive rate is low (Figure 1E), as determined by the number of off target barcodes (6.98 ± 1.62 per cell (mean ± s.d.)) found in cells versus the number on target barcodes (1,266 ± 288 per cell (mean ± s.d.)). Across the data set, on target barcodes were on average hit with greater than 1,000 times higher frequency than off target barcodes (Figure 1F), suggesting a very low false positive rate.
We also compared intron seqFISH and smFISH results with GRO-seq (Jonkers et al., 2014) and found that they were correlated with Pearson correlation coefficient of 0.57 and 0.67, respectively (Figure 1G). As GRO-seq measures the amount of productively elongating RNA polymerase II, this correlation indicates an overall agreement between the burst frequency of active loci measured directly by intron seqFISH and the density of polymerases on gene loci measured by GRO-seq. On the other hand, the intron seqFISH for 10,421 genes and smFISH had lower correlation with bulk RNA-seq (R = 0.40 and R=0.63 respectively, Figure S1D–E) as expected, because of the difference in the lifetimes of mRNAs (on average 4–5 hours, Sharova et al., 2009) versus TAS (<30 minutes) (Femino et al., 1998; Levesque and Raj 2013). Consistent with a model of bursty and stochastic gene transcription, we find that the burst frequency of many genes are close to Poisson distributed (Figure S1F).
We further performed 10,421 gene intron seqFISH experiments on mESCs cultured under 2i condition (inhibition of MEK and GSK3β; Marks et al., 2012), as well as on NIH3T3 mouse fibroblast cells (Table S1). While biological replicates of mESCs in serum/LIF condition showed high Pearson correlation (R= 0.93) (Figure 1C), we obtained lower Pearson correlation coefficients of 0.73 (mESCs in serum/LIF vs. in 2i) and 0.33 (mESCs in serum/LIF vs. NIH3T3) (Figure 1H), consistent with previous studies showing differential gene expression in these samples (Marks et al., 2012; Kolodziejczyk et al., 2015; Eng et al., 2017). Together, these results demonstrate that intron seqFISH accurately and robustly measures nascent transcriptome in single cells.
Active transcription occurs at the surface of chromosomes territories
The spatial organization of the chromosomes and the TAS in single cells can be reconstructed from the intron seqFISH data (Figure 2A). Overall, TAS appear uniformly across the nucleus, and are excluded from the DAPI dense heterochromatic regions as well as from the interior of nucleoli (Figure 2A; Supplemental Movie S1–3). There do not appear to be major factories of active transcription in the nucleus, although smaller local foci cannot be ruled out. TAS are also not strongly colocalized with nuclear bodies (Figure S2A–C), such as the paraspeckle marked by Neat1, nuclear speckle marked by Malat1 and SC35, and lncRNA Firre. On the other hand, Locked Nucleic Acid (LNA) probes targeting polyA sequences to detect mature mRNAs colocalized with SC35 speckles (Figure S2B–C).
Figure 2. Intron seqFISH reveals nascent transcription active sites are on the surface of chromosome territories.

(A) 3D reconstruction of TAS in a single mESC nucleus, with individual chromosomes occupying distinct spatial territories (bottom). In total, 982 nascent sites were present in this cell. Nucleoli were labeled by ITS1 FISH and the nucleus was stained by DAPI. (B) Sequential intron paint (956 and 736 genes probed in chromosome 1 and 11, respectively) in a particular chromosome (red) followed by chromosome paint of the same chromosome (yellow) in mESC nuclei (blue) shows that TAS are on the surface of CTs and can loop away from the core CTs. (C) Violin plots showing the distance distributions of TAS relative to the edge of their CT in mESC nuclei for chromosome 1, 7, 11, 19 and X. In total, 913–8550 spots from 49–234 cells were analyzed per one chromosome. (D) The mean (± SEM) distance of intron FISH spots, DNA FISH spots and chromosome paints from the center of CTs,as a function of chromosome size. 49–234 cells were analyzed per chromosome (chromosome 1, 7, 11, 19 and X). (E) Representative confocal images from a single z-section showing loci looped away from their core CTs, imaged by DNA FISH (cyan) and intron FISH (red) targeting Pten along with chromosome paint (yellow) in mESC nuclei (blue). The DNA FISH spot confirms that coding genomic regions are looped away from the core CTs and are colocalized with the intron FISH spot. (F) Transcriptional statuses of loci do not affect their spatial positioning with respect to the CT boundary. DNA FISH (cyan) and intron FISH (red) targeted both allele of a gene (Adora1, as an example) along with chromosome 1 paint (yellow) in a mESC nucleus (blue). Signals outside nuclei (dashed white lines) are not shown for visual clarity (E, F). (G) Violin plots showing the distance distribution relative to their CT edge for loci with either “on” or “off” intron signals. NS, not significant with Wilcoxon’s rank sum test (P > 0.05). Results from 8 genes in chromosome 1, spanning a range of expression values (Figure S3D), are shown (n = 8–396 and 231–842 spots for “on” and “off” status introns, respectively, in 404–563 cells were analyzed per gene). Scale bars (B, E, F), 5 μm.
Two distinct sets of TAS from a given chromosome and one set from chromosome X in male mESCs (Figure 2A, lower panels) are typically observed in each cell, corresponding to the individual chromosomes. Most of the TAS from the same chromosome appear to occupy a compact region and span discrete core chromosome territories (CTs) as observed in chromosome paint studies (Bolzer et al., 2005). However, some TAS from the same chromosome do appear to be positioned away from the core CTs (Figure 2A).
To determine the relative positioning of the TAS and the core CTs, we combined intron FISH targeting the TAS in one chromosome and chromosome paint of the same chromosome to directly visualize the individual CTs. We observed that TAS are located on the surfaces of CTs with some genes positioned significantly away from the core territories (Figure 2B and S3A). On average, TAS are located 0.82 ± 1.08 μm (mean ± standard deviation) exterior relative to their CT edge (Figure 2C and S3B). We found this result to be consistent across all 6 chromosomes examined (Figure 2C and S3B). For chromosome 11, we also imaged the coding regions of the chromosome by DNA FISH in addition to intron FISH and chromosome paint (Figure S3A). We showed that the coding genomic regions, as measured by DNA FISH, are colocalized with the intron FISH signals (Figure S3A) and are also on the surface of the core CTs (Figure 2D).
These observations systematically show that actively transcribed genes are present at the exterior of core CTs, regardless of chromosome genomic size (Figure 2D). This is consistent with DNA FISH results that regions containing coding sequences are separated from the rest of the chromosome (Mahy et al., 2002b; Boyle et al., 2011).
As observed from the reconstruction from the 10,421 gene intron seqFISH (Figure 2A), we further confirmed that many TAS are indeed positioned significantly away from the core CTs (Figure 2C). Intron and DNA FISH against individual genes showed that those coding loci themselves were looped out from their chromosome territories (Figure 2E and S3C). We further investigated 8 genes that have a wide range of burst frequencies with intron FISH, DNA FISH and chromosome paint (Figure 2F). Notably, the relative spatial positions between those loci and their CTs were not influenced by their instantaneous transcriptional activities (Figure 2G and S3D), inferring that the loci positioning is not dynamically regulated according to the instantaneous transcriptional activity of the site. These results suggest that transcriptionally active regions are structurally looped out from the core domains, which likely reside inside the CTs, and are positioned at the CT surface.
Nascent transcription sites from different chromosomes are intermingled in single cells
The reconstruction of TAS in single cells shows that TAS from different chromosomes are often intertwined (Figure 3A and Supp Movie S1–3), reflecting underlying chromosome organization as well as the observation that TAS are present on the surface of CTs. To validate this observation from the intron seqFISH reconstruction, we labeled chromosomes 1, 2, 7, 11, 19 and X individually with probes targeting introns in the 10,421 gene list on each of the chromosomes with serial hybridization (Figure 3A and S3E). We observed significant overlaps between TAS of different chromosomes, suggesting the prevalence of intermingling of TAS from individual chromosomes.
Figure 3. Chromosomes intermingle and are heterogeneously organized in single cells.

(A) Representative confocal images of a single z-section showing introns in 6 chromosomes (Chr 1, 2, 7, 11, 19 and X). Regions where introns from different chromosomes intermingled are shown with white arrows labeled with their corresponding chromosome numbers. In total, 956, 795, 624, 736, 313 and 347 genes in chromosome 1, 2, 7, 11, 19 and X were targeted respectively using the 10,421 intron seqFISH primary probe sequences. Signals outside nuclei (dashed white lines) are not shown for visual clarity. Scale bars, 5 μm. (B) Heatmap of normalized contact frequencies between pairs of loci (number of contacts within 0.5 μm normalized by burst frequencies) averaged over 420 single cells. Genes are sorted based on chromosome coordinates, and gray boxes represent individual chromosomes from chromosome 1 to X. (C–D) Concordance between the heatmaps of normalized contacts from intron seqFISH (upper right) and Hi-C (lower left) are shown for (C) 349 genes in the X chromosome and (D) a zoomed-in of 41 genes boxed in (C). Cyan boxes represent individual TADs. Mean burst frequency of each gene is visualized above the contact heatmaps and reflects the sampling of individual loci. More long range contacts are observed in the intron seqFISH contact maps compared to ligation based Hi-C maps. (E) Comparisons of mean burst frequency and burst size of genes as a function of the distance between their transcription start site and the closest TAD boundary. TAD assignments were shuffled in the randomized control. Shading shows 95% interval for bootstrapped data. On average, genes within 60 kb from the TAD boundary showed 17.8% higher burst frequency than expected, while burst size was minimally affected (1.7%). (F) Heatmaps showing normalized contact frequency between pairs of chromosomes, representing intra- and inter-chromosomal contacts. Hi-C and TAD data are from Dixon et al. (2012).
See also Figure S3.
Consistent with this observation, the pairwise contact map between loci (the frequency of two loci being within 0.5 μm with one another) showed that 82.4% of the contacts are between different chromosomes, despite the enrichment of many intrachromosomal contacts shown as blocks along the diagonal (Figure 3B and S3F). The observation that the most frequent contacts measured are interchromosomal further supports the notion that, while chromosomes mostly occupy discrete territories in the nucleus, active transcription occurs on the surfaces of these territories and likely near other chromosomes.
The contact map from seqFISH (upper right Figure 3C–D and S3G) matches the pairwise contact frequency of coding genes from Hi-C experiments (Dixon et al., 2012; lower left Figure 3C–D and S3G) for individual chromosomes. While a large number of contacts are between loci that are genomically close, representing contacts within topologically associated domains (TADs; Dixon et al., 2012), there are significant inter-TAD contacts (Figure S3F), indicating spatial proximity between loci that are genomically far apart. The total number of pairwise contacts consistently increased when the contact search radius for spatial proximity of pairwise loci was increased from 0.1 μm to 2 μm. However, the number of intra-TAD contacts saturate at 0.5 μm (Figure S3F), indicating the characteristic physical dimension of the domains and chromosomes in the cells.
At the boundaries of TAD, the burst frequencies of genes were on average 21.1% higher than those of genes in the interior of TADs as well as 17.8% higher than those of genes with randomized TAD assignments (Figure 3E), consistent with observations with smFISH (Arjun Raj, personal communication). At the same time, the burst sizes of genes do not change across different distances from TAD boundaries (Figure 3E). These results suggest a potential link between local chromosome structures as defined by TADs and nascent transcriptional activity.
Single cell chromosome conformations are heterogeneous
At the single cell level, contacts appear to be mostly interchromosomal and stochastic, and vary significantly from cell-to-cell (Figure 3F). A histogram of the ratio of intrachromosomal contacts to the total contacts in single cell shows a distribution of values, indicating that different cells have differing amounts of interchromosomal vs intrachromosomal contacts (Figure S3H). Recent single cell Hi-C measurements (Nagano et al., 2013; Stevens et al., 2017) showed similar high interchromosomal interactions in single cells. Together, these data indicate that while TAS are patterned on the surface of CTs, the CTs themselves are randomly distributed in individual nuclei and spatially overlap with other chromosomes stochastically. These random spatial organizations are averaged out in ensemble experiments.
Taken together, intron seqFISH directly images transcriptionally active genes in single cells, and provides complementary information about the spatial organization of the nucleus compared to ensemble Hi-C (Lieberman-Aiden et al., 2009; Dixon et al., 2012; Rao et al., 2014), which captures highly consistent features amongst cells such as CTCF mediated loops and TADs.
Intron seqFISH profiles can identify cell types and cell states
To investigate the similarities and differences of the nascent transcriptome, we clustered cells based on their 10,421 gene intron profiles obtained by intron seqFISH. Principal component analysis (PCA) showed that the mESCs grown in serum/LIF conditions clustered separately from mESCs grown in 2i conditions and NIH3T3 fibroblasts (Figure 4A). At the same time, the two biological replicates of mESCs under serum/LIF condition overlapped in PCA space. To determine the robustness of the clustering analysis, we downsampled the number of genes used and measured the cell type assignment accuracy (i.e., serum mESCs vs. 2i mESCs vs. NIH3T3 cells) as a function of the number of genes sampled. The two replicates of mESCs in serum were found to be indistinguishable across all range of downsampling of genes, suggesting minimal experimental variability (Figure 4B), while the mESCs grown in serum and NIH3T3 cells were distinguishable with high accuracy with just 700–1,000 genes (Figure 4B). mESCs grown in serum/LIF and 2i conditions are more subtly different and require between 2,500–3,000 genes to be clearly separated (Figure 4B). Thus, the nascent transcriptome can differentiate cells in different states as well as cell types.
Figure 4. The nascent transcriptomes can distinguish cell types and cell states.

(A) Principal Component Analysis (PCA) of the nascent transcriptome separates NIH3T3 cells and mESCs grown in different conditions (serum/LIF and 2i). All cells obtained from four intron seqFISH experiments (n = 1158 in total; n = 314 in E14 serum rep. 1; n = 382 in E14 serum rep. 2; n = 347 in E14 2i; n = 115 in NIH3T3) are projected onto the first two principal components. PCA analysis was performed on the correlation matrix of all cells after normalizing individual intron counts in each cell by total number of introns of the cell. Note that biological replicates of mESCs grown in serum/LIF are clustered together indicating little batch effect. (B) Cells from the serum replicates clustered together even when the number of genes used is downsampled (left). Serum vs. NIH3T3 cells (right panel, yellow) and serum vs. 2i cells (right panel, red) are well separated when 700 and 2,000 genes are used respectively. Separation index is the overlap between the cluster assignments between the cell types subtracted from unity. Shaded regions shows the 95% confidence interval of the separation index with 100 trials of downsampling. (C) PCA of the genes differentially expressed in each cell line. Nascent transcriptomes for NIH3T3 and, mESCs in serum and 2i were clustered and the genes differentially expressed were further analyzed by PCA. (D) Heatmaps of differentially expressed genes for cell cycle (left) and pluripotency (right) with mESCs in serum replicate 1 (n = 314 cells). In the left panel, cells were sorted by G2/M marker gene mRNA levels (Aurka and Plk1) with cell cycle phases assigned by H3S10Ph immunofluorescence. Introns differentially expressed in the G2/M phase are shown in the heatmap and found by Pearson correlation analysis with Aurka mRNA levels. In the right panel, cells were sorted by pluripotency associated gene Zfp42 mRNA levels. Other pluripotency associated marker mRNAs are shown as well as the differentially expressed introns in the Zfp42 high and low states.
See also Figure S4 and Supplementary Table S1–3.
We further examined the nascent transcripts that are differentially expressed in different cell types and states (Figure 4C and S4A–C). Using hierarchical clustering (Figure S4A), we found distinct sets of genes are differentially expressed in the three conditions that can be visualized in PCA space (Figure 4C and Table S2). The nascent transcripts upregulated in mESCs grown in serum/LIF include pluripotency associated stem cell markers such as Zfp42 (Rex1), Tet1 and Pou5f1 and genes involved in embryogenesis such as Rbpj and Dppa4, and the genes upregulated in 2i condition contain Wdr5 and Ash2l, regulators of ESC self-renewal via maintenance of H3K4me3 (Ang et al., 2011), and Tfcp2l1, a naive pluripotency marker. On the other hand, the genes upregulated in NIH3T3 cells correspond to factors related to cytoskeleton (Myh9, Acta2) and extracellular matrix (Emp1,Grasp), reflecting the signature of the differences of the different cell types. Most differentially expressed genes are regulated by both burst frequency and size, with the changes correlated between the two parameters (Figure S4D).
Furthermore, the intron profiles are informative of differences in cellular states, such as cell cycle phases and metastable pluripotent states of mESCs grown in serum/LIF (Marks et al., 2012). For example, in G2/M cells identified using Aurka and Plk1 mRNA and phospho-Histone H3 (Ser10) antibody as markers, we observed upregulation of a panel of cell cycle related introns (i.e. Cks2, Arl6ip1, Cenpa, Mis18bp1) (Figure 4D and Table S3). Similarly, using pluripotency gene Zfp42 mRNA as a marker, we observed a panel of significantly upregulated introns in the Zfp42 high cells, including known pluripotency related genes (i.e. Zfp42, Fbxo15, Smarcad1, Tet1, Tdh; Figure 4D and Table S3). On the other hand, another set of introns (i.e. Rap1gds1, Esd, Podxl, Mfap3l) are upregulated in the Zfp42 low cells similar to the more differentiated states (Figure 4D and Table S3). These results demonstrate that intron seqFISH can identify differentially regulated genes in different dynamic states of cells.
Heterogeneity is present in the global instantaneous transcriptional activity
Surprisingly, there is large variability in the global nascent transcriptional states of cells, as observed from the total number of active transcription sites in each nucleus in the 10,421 gene intron experiments (Figure 5A) even after considering differences in cell cycle phase and cell size (Figure S4E–I). In G1/S cells of a given size, there are on average 1,361 ± 169 TAS per cell (mean ± s.d.), indicating that some cells are globally transcriptionally active while other cells are globally quiescent. In contrast, simulation assuming that each gene fires randomly and independently produce a much narrower distribution of TAS (1,264 ± 32 TAS per cell (mean ± standard deviation)) (Figure 5A).
Figure 5. Single cell pulse chase experiments reveal 2 hour oscillations in global nascent transcription.

(A) Histograms showing wide distributions of TAS per cell from the decoded 10,421 gene intron seqFISH (n = 188 cells). Cells in G1/S cell cycle phase and a small cell size window were used. In comparison, simulations assuming Poisson bursting of each gene (bottom, n = 188 simulated cells) yield a narrower distribution of TAS per cell, showing that the global nascent transcription states are more heterogeneous than expected in individual cell. (B) Cytoplasmic polyA intensity per cell by poly-dT FISH probes shows lower correlation with the 1,000 gene intron seqFISH total counts (n = 763 cells) than expected if global intron levels were static. Intensity and counts are normalized by nuclear volume (pL, picoliter). (C) Schematic of the pulse-chase experiment (top). 5-EU was pulsed for 30 min to globally label nascent transcripts, then chased for different periods of time in growth medium lacking 5-EU, followed by fixation and intron hybridization. Confocal images with maximum intensity z projection of 5-EU signals detected by click linkage to an azide-dye and 1,000 gene introns in single cells shown for three time points (middle). The correlations between 5-EU levels and intron numbers examined after different chase time are shown as scatterplots (bottom) (n = 747, 570 and 901 cells). Dashed yellow lines in the images display nuclear boundaries determined by DAPI. Scale bars, 10 μm. (D) Pulse-chase correlation measurements show oscillatory dynamics on the time scale of two hours. Oscillations are observed in two different mESCs (E14 and wild-type mESCs from Hu et al., 2014) and mouse fibroblast cell line (left and right panels). Additional replicates are shown in Figure S5C. In contrast to mESCs in serum+LIF condition (blue), mESCs in 2i condition do not show oscillations (red). At each time point, 329–901, 435–1725, 311–658, 1035–1556, and 444–618 cells were analyzed with E14 cells in serum/LIF and 2i, wild-type mESCs in serum/LIF and 2i, and NIH3T3 cells, respectively. Shaded regions represent 95% confidence intervals. R, Pearson correlation coefficient.
See also Figure S5.
Therefore, the large variability in global transcriptional states raises the question of whether these global states are static in time or interconvert dynamically. The correlation (R = 0.28) between the total TAS number in the nucleus and the amount of mature mRNA in the cytoplasm measured by LNA probes targeting polyA (Figure 5B and S5A) is lower than expected if global nascent transcriptional activities are static in time. However, as the dynamics are unlikely to be synchronized amongst cells, we cannot measure the interconversion rate between active and inactive global transcriptional states by population averaged experiments. At the same time, it is also difficult to perform direct live cell experiments with reporter based assays to measure the transcriptional activities across thousands of genes.
Global oscillations are seen in transcriptional activities
To measure the dynamics of TAS globally, we developed a single cell pulse chase experiment that records the nascent transcriptional activities at two time points in a cell’s history (Figure 5C). We first fed cells with a modified Uridine (5-EU) (Jao and Salic, 2008) to record the global transcriptional activity during a short 30 minute pulse. Then we washed out the 5-EU and let the cells grow in the original medium for different amounts of time from 0 to 3 hours. We fixed the cells, measured the 5-EU incorporation levels with a clickable fluorescent dye and counted the total number of TAS seen from 1,000 gene intron probes in the same cells. Probes for 1,000 intronic genes were used because they could be quantified accurately in a single fluorescence channel without optical crowding. The variability in the 5-EU signal in individual cells (Figure S5B) is similar to the intron variability observed (Figure 5A), confirming nascent transcriptional heterogeneity in single cells.
We then determined whether transcriptional activities change over time or are static by comparing the global instantaneous transcriptional activity at defined time points in the past, as labeled by 5-EU incorporation, with the nascent activity at the time of fixation, as measured by intron levels in the same cells (Figure 5C–D). At early time points, the 5-EU and intron levels are correlated in single cells (Figure 5C, bottom left panel), confirming that the heterogeneities observed in both measurements are consistent. The correlation coefficient decayed within 1 hour, with little correspondence between the 5-EU signal and intron levels in single cells (Figure 5C, bottom middle panel). Surprisingly, the correlation is restored at around 2 hours (Figure 5C and D, left blue lines). This result suggests that mESCs oscillate between low and high transcriptional states with a roughly 2 hour time period. Our data at each time point consist of hundreds of cells and the 2 hour oscillation is observed with the biological replicate of mESCs (Figure S5C), as well as with a different mESC cell line (Figure 5D middle). The same oscillation is observed with a different method of analyzing the pulse-chase data by binning the 5-EU levels at each time point (Figure S5D–E).
The observation of 2 hour oscillations from single cell pulse chase experiments implies that oscillation periods are relatively consistent in different cells. The oscillation amplitude in single cells may indeed even be higher, but are dampened in the population based pulse chase experiments due to slight differences in the period of oscillations. If the global transcriptional activities were fluctuating stochastically without a defined period, or if different cells had completely different oscillation periods, then the correlation coefficients would simply decay without re-cohering at 2 hours.
Interestingly, the 2 hour fast dynamics in global nascent transcription can be abolished in 2i condition where Wnt and Mek pathways are perturbed (Figure 5D, red lines in left and middle panels; Figure S5D–E). Furthermore, the 2 hour global transcription oscillation is also observed in NIH3T3 fibroblast cells using the single cell pulse chase experiment (Figure 5D, right). We found similar heterogeneity of the nascent transcription states with NIH3T3 cells (Figure S5F) as observed in mESCs (Figure 5C). These results suggest that global nascent transcriptional oscillations may be present in many cell types and are not limited to the pluripotent state of mESCs.
Global nascent transcriptional oscillations are linked to Hes1 oscillations
The 2i results suggest that perturbation of signaling pathways can change the dynamics of the global transcriptional states. To further investigate the molecular origins of this 2 hour oscillation, we measured the mRNA expression levels of 48 genes, including transcription factors and signaling pathway components, along with the total intron levels for 1,000 genes. This data helped determine whether any mRNAs are correlated with global transcriptional activities in the same cells (Figure 6A and S6A–C).
Figure 6. Global nascent transcription links to Hes1 dynamics in mESCs.

(A) Heatmap showing Pearson gene-to-gene correlation coefficients between total introns counts for 1,000 genes and 47 mRNAs involved in pluripotency, signaling pathway and other processes by non-barcoded seqFISH (n = 605 cells cultured under serum/LIF condition). Red and black boxes show the correlated clusters, and blue boxes show clusters of genes that are anticorrelated. (B) Confocal images of mESCs (serum/LIF condition) with Hes1 immunofluorescence (magenta) and 5-EU staining (green) used in the Hes1 protein pulse chase experiment for the initial time point (top) and 30 minute chase time point (bottom). Images are shown as a maximum intensity projection of z stacks of the fluorescence images. Scale bars, 10 μm. (C) Pulse-chase correlation measurements between detected 5-EU signals and Hes1 immunofluorescence signals. Similar 2 hour oscillatory dynamics are observed as intron pulse chase experiments. The data at each time point consist of 328–510, or 272–1305 cells in biological replicates 1 and 2, respectively. Shaded regions represent 95% confidence intervals.
See also Figure S6.
We found that total intron number clustered most closely with Hes1, Stat3, Socs3 and Fgf4 (Figure 6A), suggesting that global transcription activity closely follows the pattern of the aforementioned genes. Hes1 mRNA and protein have been shown to have short lifetimes and oscillate with 2–4 hour periods in many mouse cell lines, including mESCs and fibroblasts (Kobayashi et al., 2009, Yoshiura et al., 2007), as well as in vivo (Zhang et al., 2014). It has also been shown that Socs3 mRNAs and proteins, and phosphorylated Stat3 protein, oscillate with Hes1 transcripts with a periodicity of ~2 hours in mouse fibroblast cells (Yoshiura et al., 2007). This gene cluster (consisting of 1,000 gene total introns, Hes1, Stat3, and Socs3) is observed even after taking into account the cell cycle effect (Figure S6D–E). Furthermore, Hes1 has been shown to negatively regulate Delta 1 (Dll1) (Shimojo et al., 2008) and Gadd45g (Kobayashi et al., 2009). Indeed, Dll1 and Gadd45g form a distinct cluster that are negatively correlated with the Hes1-intron cluster (Figure 6A). Our results also recapitulate the metastable pluripotent mESC subpopulations (Figure S6F–G), consistent with single-cell RNA-seq measurement (Kolodziejczyk et al., 2015). Interestingly, gene-to-gene hierarchical clustering results showed differences between serum/LIF and 2i conditions of mESCs, showing more distinct clusters in serum/LIF condition (Figure S6H).
To understand differences between mESCs in serum/LIF versus 2i conditions that may affect their distinct oscillation behaviors, we compared their mRNA expressions profiles, and found that Hes1, Dll1, Gadd45g, and Dnmt3a/b are strongly differentially expressed (Figure S6I). These results are consistent with the intron profile differences between 2i and serum showing certain signaling pathway genes are differentially expressed (Table S2).
Using the 10,421 intron seqFISH data, we further found that most genes are oscillating in synchrony with the global dynamics (Figure S6J). TAS for most genes occur at a linearly higher frequency in cells with high total number of introns than cells with lower total number of introns, suggesting that there is a global mechanism that upregulate the nascent production of most genes in the TAS high state and vice versa in the low states.
To further investigate the relationship between the known Hes1 oscillation and the nascent transcription oscillations, we performed single cell pulse chase experiments with the 5-EU labeling and Hes1 antibody staining using mESCs grown in the serum/LIF condition (Figure 6B–C). Hes1 protein oscillation is time delayed about 20 minutes compared to Hes1 mRNA oscillation but both have the same period (Hirata et al., 2002). We observed the similar 2 hour oscillation period, showing that Hes1 protein oscillations and global transcriptional oscillations follow a similar period (Figure 6C). Together, our results provide multiple lines of evidence suggesting that the global nascent transcription states oscillate with a 2 hour period, which is potentially related to the known 2 hour oscillation of Hes1 and other components of signaling pathways.
Discussion
In this study, we showed that the “spatial genomics” approach with intron seqFISH can scale to the transcriptome level and capture both the nascent transcriptome and the spatial architecture in the nucleus of single cells. The sensitivity and spatial imaging nature of the single molecule based seqFISH methods allow us to obtain insights that are unavailable with existing methods. First, we are able to explore the nascent transcriptome of single cells, which is highly informative of cell types and cell states, with specific introns upregulated dynamically in different cell cycle phases and metastable pluripotent states in mESCs. Second, by imaging the spatial organization of the nucleus in situ, we showed systematically that transcription active regions occur at the surface of chromosome territories, and are not dynamically positioned according to the instantaneous transcriptional activity, providing an actual picture of the nuclei beyond the pairwise interactions measured by Hi-C experiments. We further showed that gene burst frequencies, but not burst size, measurements uniquely possible with FISH, is higher in genes near TAD boundaries. Lastly, we observed surprisingly fast dynamics with 2 hour oscillations in mESCs and fibroblasts with single cell pulse chase measurements using intron FISH. Such global oscillations would otherwise be lost in population average measurements because cells are not synchronized, and would also be missed in single gene live cell experiments which are dominated by stochastic bursting at each active site.
The high dimensional spatial genomics data allow us to generate new models of chromosome organization combining insights from Hi-C data and multi-color imaging data. Specifically, our systematic observation that active genes are positioned away from the core chromosome territories explains the high interchromosomal contacts observed in the single cell Hi-C data (Nagano et al., 2013; Stevens et al., 2017) and the enrichment of transcriptionally active phosphorylated RNAPII at the inter-chromosome contact regions by immunofluorescence (Maharana et al., 2016). Combined with observations that burst frequencies of genes are higher near TAD boundaries and that many domain boundaries are invariant across cell types and species (Dixon et al., 2012; Rao et al., 2014), it is possible that there are structural elements in the genome that loop out potential active genes while the inactive domains remain in the interior of chromosome territories. Further investigation of dynamics can take advantage of intron seqFISH together with multiplexed live cell imaging of genomic loci (Takei et al., 2017).
The measurement of the nascent transcriptome combined with many mRNAs in the same cells by seqFISH allowed us to link the fast 2 hour global nascent dynamics with other molecular pathways. We found that global intron levels varied with Hes1, Stat3, Socs3 transcripts, and were anticorrelated with Delta1 and Gadd45g transcripts, which were downregulated by Hes1. Hes1 has been shown to oscillate in many cell lines including mESCs and fibroblasts (Kobayashi et al., 2009, Yoshiura et al., 2007). Many of these genes were studied previously in the context of a somitogenesis clock in embryos, which also oscillates in 2 hour periods in mouse (Dequeant et al., 2006).
Thus, it is possible that a common 2 hour oscillation gates global transcriptional activity in many cell types, but is unsynchronized amongst cells and therefore previously unrecognized. This 2 hour global nascent transcription dynamics could be also be related to a 2 hour methylation oscillation observed in mESCs released from 2i to serum/LIF conditions (personal communications, Reik and Simon).
Using pulsatile and oscillatory dynamics, cells can achieve states not accessible with amplitude based regulation schemes (Letsou and Cai, 2016). For example, cells can use fluctuations in global transcriptional activity to coordinate the stoichiometry of many transcripts in a mechanisms akin to the frequency modulated signaling observed in yeast and mammalian pathways (Cai et al., 2008; Yissachar et al., 2013).
Finally, an exciting recent work showed that intron-to-exon ratios across the transcriptome can be used to determine the direction of of cells on the developmental trajectory (La Manno et al., 2017). As we showed, the nascent transcriptome profiles can not only distinguish cell types and cell states, but also detect fast dynamics in single cells. Applications of intron seqFISH with signal amplification (Shah et al., 2016a) along with mRNA seqFISH (Shah et al., 2016b, Lignell et al., 2017), can enable simultaneous profiling of nascent and mature RNAs in tissues, with spatial information preserved. It will be fascinating to explore the nascent transcriptome in single cells in many tissue settings and developmental contexts.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Long Cai (lcai@caltech.edu).
Experimental Model and Subject Details
Cell Lines
E14 mESCs (E14Tg2a.4) from Mutant Mouse Regional Resource Centers were maintained under serum/LIF or serum/LIF/2i conditions as described previously (Singer et al., 2014; Takei et al., 2017). Wild-type mESCs (Hu et al., 2014) were kindly provided by Wolf Reik and maintained on gelatin-coated dishes at 37°C with 5% CO2 in DMEM (Thermo Fisher Scientific), 15% FBS (Hyclone serum SH30070.02), 2 mM L-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin (Thermo Fisher Scientific), 1000 units/ml Leukemia Inhibitory Factor (LIF, Millipore), 1× Minimum Essential Medium Non-Essential Amino Acids (Thermo Fisher Scientific) and 100 μM β-Mercaptoethanol (Thermo Fisher Scientific). NIH/3T3 cells (ATCC) were cultured at 37°C with 5% CO2 in DMEM (Thermo Fisher Scientific) supplemented with 10% FBS (Atlanta biologicals S11150) and 100 units/ml penicillin, 100 μg/ml streptomycin (Thermo Fisher Scientific).
Method Details
Intron FISH Probe Design
Oligoarray 2.1 (Rouillard et al., 2003) was run on all constitutive introns present in the masked mm10 mouse genome with parameters min/max length 35-nucleotide (nt), max TM 100°C, min TM 74°C, secondary structure temp 76°C, cross hyb temp 72°C, max distance 1,000-nt and max oligos 100. Genes with more than 48 probes designed were used for secondary filtering. All probes were blasted against the mouse transcriptome and expected copy numbers of off-target probe hits were calculated using predicted RNA counts in the ENCODE database for 11.5 day old murine embryos.
Probe optimization was initially run to minimize expected off target hits for any given probe. An outer loop was run until at least 25 probes were designed per gene initially permitting a predicted 2,000 off-target hits for any probe, increasing every round of optimization by 2,000 off-target hits until a maximum of 10,000 predicted off-target hits were permitted or the target number of probes was designed. For every cycle of probe optimization an inner loop was run iteratively choosing probes until no off-target RNA was hit more than 7 times for any genes probeset. If more than 25 probes were found for any given intron, the 25 probes with the predicted GC range closest to 55% was chosen.
A second round of optimization was performed on the entire probeset to minimize the combined off-target hits. If any RNA was predicted to be hit more than 7 times by all of the combined probesets, probes were iteratively dropped from the probe sets with the largest number of genes until no more than 7 off-target hits existed for any predicted off-target RNA. If less than 25 probes could be designed for any gene it was dropped from the probeset.
Probe pools were assigned with a validated primer and assembled according to the following template with 60 readout sequences used in RNA SPOTs (Eng et al., 2017):
Primary probes: 5′-[Primer 1] - [readout 1] - [readout 2] - [probe] - [readout 3] - [readout 4] - [Primer 2] - 3′
Intron Probes Generation
The intron probes for 10,421 gene experiment were generated through oligoarray pools. The oligoarray pools of probes were purchased from Twist Biosciences. Probes were amplified from array-synthesized oligopool as previously described (Beliveau et al., 2012; Shah et al., 2016; Takei et al., 2017; Eng et al., 2017) with the following modifications:
The template oligo for each encoding primary probe contains: (i) a 35-nt intron-targeting sequence for in situ hybridization, (ii) Four 18-nt gene specific readout sequence combinations (readout+spacer), (iii) two hybridization specific flanking primer sequences to allow PCR amplification of the probe set.
Intron seqFISH, mRNA seqFISH, lncRNA seqFISH, and Immunofluorescence
E14 mESCs were were plated on poly-D-lysine (Sigma P6407) and human laminin (BioLamina LN511) coated coverslips (3421; Thermo Scientific), and incubated for 2–3 hours. Then cells were fixed using 4% Formaldehyde (Thermo Scientific 28908) in 1× PBS (Invitrogen AM9624; diluted in Ultrapure water (Invitrogen 10977-015)) for 15 minutes at 20°C, washed with 1× PBS for a few times, and stored in 70% ethanol for more than overnight at −20°C or for a few hours at room temperature. NIH/3T3 cells were prepared similarly using poly-D-lysine coated coverslips. The coverslips were air dried, attached with flow cell (Grace Bio-Labs RD478685-M), and incubated with 0.2 μm blue fluorescent (365/415) beads (Thermo Scientific F8805) with 2000-fold dilution in 2× SSC (Invitrogen 15557-044 diluted in Ultrapure water (Invitrogen 10977-015)) at room temperature for 5 minutes for the alignment of images. The coverslips were then washed twice with 2× SSC.
For hybridization of the probes, samples were 1) hybridized for 30 hours at 37°C with primary intron probes and mRNA/lncRNA probes at 1 nM each oligo concentration in 50% Hybridization Buffer (50% HB: 2× SSC, 50% Formamide (v/v) (Invitrogen AM9344), 10% Dextran Sulfate (Sigma D8906) in Ultrapure water), then 2) washed in 55% Wash Buffer (55% WB: 2× SSC, 55% Formamide (v/v), 0.1% Triton-X 100 (Sigma 93443)) for 30 minutes at room temperature, followed by 2× SSC wash. The fluorophore-coupled 15-nt readout probes (Alexa 488, 647 (Thermo Fisher Scientific) and Cy3B (GE Healthcare)) for first round of hybridization were incubated for 20 minutes at 50 nM each at room temperature in 10% EC buffer (10% Ethylene carbonate (Sigma E26258), 2× SSC, 0.1 g/ml Dextran sulfate (Sigma D4911) and 0.02 U/μL SUPERase In RNase Inhibitor (Invitrogen AM2694)), which is optimized for 15-nt readout probe hybridization from the EC buffer (Matthiesen et al., 2012), and washed for 5 minute at room temperature in 10% Wash Buffer (10% WB: 2× SSC, 10% Formamide (v/v), 0.1% Triton-X 100) followed by 1 minute wash in 2× SSC. 3) Once the first hybridization was complete, the flow cell was connected to an automated fluidics delivery system made from three multichannel fluidics valves (EZ1213-820-4; IDEX Health & Science) and a Hamilton syringe pump (63133-01, Hamilton Company). The integration of the fluidics valves, peristaltic pump through homemade connectors, and microscope imaging were controlled through a custom script written in Micromanager software. 4) Imaging positions were then registered using nuclei signals stained by 5 μg/mL DAPI (Sigma D8417). Then the sample was 5) proceed to imaging as described below. After image acquisition, 6) the samples were incubated with 55% WB at room temperature for 5 minutes to strip off readout probes, followed by 2× SSC wash for 1 minutes each round. 7) Then, the fluorophore-coupled readout probes were incubated at 50 nM each concentration at room temperature for 20 minutes in 10% EC buffer followed by 8) 5 minute wash in 10% WB, 1 minute wash in 2× SSC and DAPI staining. The procedures 5)–8) were repeated with the next round of readout hybridization until the completion of all rounds of seqFISH.
Following intron seqFISH, the mRNA seqFISH was performed. The mRNA seqFISH primary probes were hybridized at the same time as the intron seqFISH primary probes, and then read out without barcoding using addition readout probes, with the same procedures as described above 5)–8).
After mRNA seqFISH, nucleolus and lncRNA probes (ITS1, Malat1, Neat1, Firre) and and a 25-nt polyT LNA probe (Exiqon: 300510-04) were imaged similar to non-barcoded mRNA seqFISH.
Following the lncRNA seqFISH, one round of immunofluorescence was carried out. The samples were blocked with blocking buffer (1× PBS, 1% UltraPure BSA (Thermo Scientific AM2616), 0.3% Triton-X 100) at room temperature for 30 minutes. The samples were then incubated with 100-fold diluted primary antibody (anti-Phospho-Histone H3 (Ser10) (Thermo Fisher Scientific PA5-17869)) in blocking buffer at room temperature for 1 hour, followed by washes with 1× PBS for a few times, incubation with 500-fold diluted secondary antibody (Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 (Thermo Scientific A-31573)) in blocking buffer at room temperature for 1 hour, and washes with 1× PBS for a few times, and imaged as described below.
Samples were imaged in an anti-bleaching buffer (50 mM Tris-HCl pH 8.0, 300 mM NaCl, 2× SSC, 3 mM Trolox (Sigma 238813), 0.8% D-glucose (Sigma G7528), 100-fold diluted Catalase (Sigma C3155), 0.5 mg/mL Glucose oxidase (Sigma G2133) and and 0.02 U/μL SUPERase In RNase Inhibitor (Invitrogen AM2694)) with the microscope (Leica, DMi8) equipped with a confocal scanner unit (Yokogawa CSU-W1), a sCMOS camera (Andor Zyla 4.2 Plus), 63× oil objective lens (Leica 1.40 NA), and a motorized stage (ASI MS2000). Lasers from CNI and filter sets from Semrock were used. Snapshots were acquired with 0.35 μm z steps for more than 10 positions per sample.
DNA FISH Probe Design and Synthesis
DNA FISH probes were designed and synthesized by following the previous protocol (Takei et al., 2017) with minor modifications. For DNA FISH paint of chromosome 11, 380 genes were selected from the 10,421 intron seqFISH gene list. To label genomic regions of selected genes, regions from transcription start sites to 20 kb downstream of each gene were selected according to mm10 RefGene database (UCSC Genome Bioinformatics). Across those regions, a set of non-overlapping 35-nt probes were designed using the masked mm10 mouse genome with several constraints including 40–60% GC content, no more than 5 contiguous identical nucleotides, at least 2-nt spaces between adjacent probes and the same off target evaluation as previously done. At the 5′ end of the 35-nt probe sets, 20-nt adapter sequences, which are identical in each gene probe set but orthogonal among different probe sets, are attached with a 4-nt spacer in-between. For the array-based oligo library synthesis, universal primer binding sequences were attached at 5′ and 3′ ends. The oligonucleotide probe pools were purchased from Twist Bioscience (141–398 probes per gene). DNA FISH probes were generated in the same way as intron probe generation, without restriction enzyme digestion at the final step.
Intron FISH, DNA FISH and Chromosome Paint
Intron paint experiments using 10,421 gene probe set were carried out using oligoarray pool (Twist Bioscience) based probes, generated without cutting the primer binding sites. Probe sequences of the genes in the same chromosome were amplified from one primer pairs, and those primer binding sequences (5′ and 3′ end) were targeted with 20-nt dye-conjugated readout probes to paint introns in particular chromosomes. Images were taken with the microscope (Leica DMi8 automated) equipped with a confocal scanner unit (Yokogawa CSU-W1), a sCMOS camera (Andor Zyla 4.2 PLUS), 63× oil objective lens (Leica NA 1.40), and a motorized stage (ASI MS2000). Lasers from CNI and filter sets from Semrock were used. Snapshots were acquired with 0.35 μm z steps
Following the intron FISH, DNA FISH experiments were performed as described (Takei et al., 2017). Briefly, after intron FISH imaging, cells were incubated in 55% formamide and 2× SSC at room temperature for 30 minutes, and then washed three times with 2× SSC to strip off fluorophore-coupled readout probes. Samples were treated with a prechilled solution of methanol and acetic acid at a 4:1 ratio at room temperature for 1 hour, and then with 0.1 mg/ml RNase A (Thermo Fisher Scientific EN0531) at 37°C for 1 hour. Then samples were washed and dried with 1× PBS, 70% ethanol and 100% ethanol. The samples were then heated at 95°C for 10 minutes in 70% formamide and 2× SSC. Cells were hybridized with DNA FISH probe pool at 37°C overnight, where the final concentration of each probe was estimated as 10 nM in nuclease free water with 50% formamide, 2× SSC and 0.1 g/ml dextran sulfate (Sigma D8906). After incubation with the probes, samples were washed three times in 50% formamide, 0.1% Triton-X 100 and 2× SSC at room temperature, and hybridized with 20-nt readout probes coupled to Alexa 488, 647 or Cy3B at 10 nM final concentration at room temperature for at least 1 hour in nuclease free water with 30% formamide, 2× SSC and 0.1 g/ml dextran sulfate (Sigma D8906). Samples were then washed three times in 30% formamide, 0.1% Triton-X 100 and 2× SSC at room temperature, stained with DAPI and imaged under the same condition as intron FISH.
Following the DNA FISH, chromosome paint experiments were performed. Samples were heated at 95°C for 10 minutes in 70% formamide and 2× SSC, and then washed three times with 2× SSC for DNA FISH probe stripping. Chromosome paint probes (MetaSystems, XMP X Green or Orange) for chromosome 1, 2, 7, 11 19 or X were incubated with samples at 95°C for 10 minutes followed by incubation at 37°C overnight. Afterwards, samples were washed with 30% formamide, 0.1% Triton-X 100 and 2× SSC at room temperature for 15 minutes. Then samples were stained with DAPI and imaged under the same condition as intron FISH. In case multiple chromosomes were painted, sequential rounds of chromosome paint were performed.
Intron FISH and Non-Barcoded mRNA seqFISH
For the 1,000 gene intron imaging without decoding, 1,000 intron seqFISH probe set was generated for 48 probes per intron as described above. Both primer binding sequences (5′ and 3′ end) were targeted with 20-nt Alexa 488-conjugated readout probes. For the non-barcoded mRNA seqFISH, probes with primary probe sequences and readout sequences were used, which were either generated by oligoarray pool (Twist Bioscience) synthesis described above or purchased by IDT.
E14 samples were prepared as described above, and 18 rounds of sequential imaging were performed to cover 50 genes mRNAs and introns of 1,000 gene. For the first 12 rounds, after imaging, fluorophores (Alexa 647, Alexa 594 or Cy3B) in dye-conjugated readout probes were removed by TCEP cleavage (Sigma-Aldrich 646547; Eng et al., 2017). For the next 6 rounds, readout probes were stripped off using 70% formamide as described above. After cleaving or stripping off the probes at each round, one position was imaged to confirm the loss of signals. Two genes (Sfrp2 and Dnmt3l) were excluded from the analysis due to the poor signal.
Samples were imaged in the anti-bleaching buffer with the microscope (Leica DMi8 automated) equipped with a confocal scanner unit (Yokogawa CSU-W1), a sCMOS camera (Andor Zyla 4.2 PLUS), 40× oil objective lens (Leica NA 1.30), and a motorized stage (ASI MS2000). Lasers from CNI and filter sets from Semrock were used. Snapshots were acquired with 0.5 μm z steps.
Single-Cell Pulse Chase
Cells were plated on coated coverslips at about 50% confluency, and incubated for two hours before any treatment. Then cells were treated with 100 ng/ml Actinomycin D (Thermo Fisher Scientific 11805017) at 37°C for 30 minutes, followed by 30 minutes incubation for the “pulse” with final concentration of 2mM 5-ethynyl uridine (5-EU) (Thermo Fisher Scientific E10345) and 100 ng/ml Actinomycin D to prevent the 5-EU incorporation into transcripts from RNA polymerase I. The cells were then incubated with fresh culture medium, and incubated for the “chase” for the following time: 0, 0.5, 1, 1.5, 2, 3 and 4 hours. Note that samples for the 0 time point chase were immediately proceeded to the cell fixation step. After the particular chase time, the cells were fixed with 4% formaldehyde in 1× PBS at room temperature for 10 minutes, and permeabilized in 70% ethanol at −20°C more than overnight or at room temperature for several hours.
Intron FISH experiments were firstly performed with the 1000 gene intron probes by targeting 5′ and 3′ end of the primer binding sites with fluorophore-coupled (Alexa 488) readout oligos to image 1000 gene introns in a single channel. Note that intron probes were not cut by the restriction enzymes for these experiments to preserve the common PCR primer sequences, which were targeted by readout oligos for imaging. The sample preparation conditions are described in the intron seqFISH section.
Following the intron FISH, 5-EU labeling was performed to the same samples in order to visualize global transcripts during the pulse time by using click chemistry (Jao and Salic, 2008). The Click iT RNA Alexa 594 Imaging Kit (Thermo Fisher Scientific C10330) was used according to the manufacturer’s instruction, and the samples were incubated with the reaction mixture for one hour at room temperature in dark. The reaction mixture was then removed and the samples were washed once with a reaction rinse buffer, followed by 1× PBS wash for a few times. Afterwards, the samples were stained with DAPI and imaged under the condition below.
The Hes1 immunofluorescence and 5-EU single-cell pulse chase experiments were performed similarly. Immunofluorescence preparation and imaging were firstly performed with the immunofluorescence method described above. Primary antibodies and the dilution used were anti-HES1 (E-5) (Santa Cruz sc-166410) (1:100) and anti-Phospho-Histone H3 (Ser10) (Thermo Fisher Scientific PA5-17869) (1:100). Secondary antibodies and the dilution used were Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 (Thermo Fisher Scientific A-21206) (1:500), and Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 (Thermo Fisher Scientific A-31571) (1:500). After immunofluorescence labeling, samples were post-fixed with 4% formaldehyde in 1× PBS at room temperature for 5 minutes, and washed several times with 1xPBS. Following this step, 5-EU labeling and imaging were performed as described above.
Samples were imaged in the same setup as described in Intron FISH and non-barcoded mRNA seqFISH section. Snapshots were acquired with 0.5 μm z steps for more than 10 positions per sample of each time point. Samples from multiple time points were imaged on the same day and those samples were imaged with random orders.
Quantification and Statistical Analysis
Image Processing
To remove the effects of chromatic aberration, 0.1 μm TetraSpeck beads’ (Thermo Scientific T7279) images were first used to create geometric transforms to align all fluorescence channels. Next, the background illumination profile of every fluorescence channel was mapped using a morphological image opening with a large structuring element. These illumination profile maps were used to flatten the illumination in post-processing resulting in relatively uniform background intensity and preservation of the intensity profile of fluorescent points. The background signal was then subtracted using the imagej rolling ball background subtraction algorithm with a radius of 3 pixels. Finally, the calculated geometric transforms were applied to each channel respectively.
Image Registration
Each round of imaging included imaging with the 405 channel which included the DAPI stain of the cell along with 200 nm blue fluorescent (365/415) beads (Thermo Scientific F8805). Rounds of hybridization that belonged to a single barcoding round were first aligned by 3D phase correlation. Once all 5 barcoding rounds were all internally aligned, each barcoding round was aligned to round 1 using the same method.
Cell Segmentation
For nuclear segmentation, the DAPI image was first blurred using a 2D gaussian blur with a sigma of 1 pixel. The ImageJ built in Li thresholding algorithm was then used to separate out nuclear regions from background. Finally, to demarcate individual nuclei, the thresholded image was run through a watershed algorithm. The subsequent segmentation results were manually curated and corrected to obtain a final accurate segmentation of images. For cytoplasmic segmentation, the segmentation was performed manually using ImageJ’s ROI tool.
Barcode Calling
The potential intron signals were then found by finding local maxima in the image above a predetermined pixel threshold in the registered images. Once all potential points in all channels of all hybridizations were obtained, dots were matched to potential barcode partners in all other channels of all other hybridizations using a 2.45 pixel search radius to find symmetric nearest neighbors in 3D. Point combinations that constructed only a single barcode were immediately matched to the on-target barcode set. For points that matched to construct multiple barcodes, first the point sets were filtered by calculating the residual spatial distance of each potential barcode point set and only the point sets giving the minimum residuals were used to match to a barcode. If multiple barcodes were still possible, the point was matched to its closest on-target barcode with a hamming distance of 1. If multiple on target barcodes were still possible, then the point was dropped from the analysis as an ambiguous barcode. This procedure was repeated using each hybridization as a seed for barcode finding and only barcodes that were called similarly in at least 4 out of 5 rounds were used in the analysis. The number of each barcode was then counted in each of the assigned cell volumes and transcript numbers were assigned based on the number of on-target barcodes present in the cell volume. All image processing and image analysis code can be obtained upon request.
Image Analysis for Distance from Chromosome Surface
The images were initially processed similarly to the intron barcoding images. Multiple rounds of imaging were aligned by cross-correlation of the DAPI counterstain image of the cells taken with each round of imaging. The edges of the chromosome were found by thresholding the chromosome paint signal to remove all background signal. The perimeter pixel of each thresholded object was then determined. Next the intron dots were identified by LOG filtering and picking local maxima in the LOG filtered image above a specified threshold as a true positive point. The distance between these putative intron points and the perimeter points of the chromosome were determined first in pixels and then converted to micron distance values based on pixel size calibration. Intron points from chromosome 1, 2 and 11 were also assigned to the nearest chromosome 19 surface in the same single cells, shown as shuffle controls (Figure S3B).
Image Analysis for Single Cell Pulse Chase
The images were initially processed similarly to the intron barcoding images. Once the corrected images were obtained, the background intensity in the pulse (click signal) experiments was removed to isolate click dye specific signal. The click images were then maximum intensity projected in the Z axis to remove the effects of 3 dimensional variation in nuclear shape. The mean intensity per voxel was then calculated to obtain a representative numerical value for instantaneous transcription as measured by the pulse phase of the experiment. The intron number per nuclei was determined by finding dots using the same algorithm outlined in the “Image Analysis for Distance from Chromosome Surface” and then counting the number of true positives found. This number was then normalized by the volume of the nuclei of the cell. Immunofluorescence analysis was performed the same as the click signal analysis above. The Pearson correlation value and 95% confidence intervals were calculated for each time point using these two normalized values per cell. Two time points (t = 90 min in replicate 1 and t = 30 min in replicate 2) were dropped due to the misalignment of the Hes1 and 5-EU images. For the binning analysis of the pulse chase experiments, cells were divided into quarters based on nuclear volume normalized 5-EU signals, and then normalized intron counts in individual subpopulations were compared at each time point.
Image Analysis for Colocalization Quantification
Image correlation analysis was performed using custom ImageJ macros using Matlab codes with Miji function. Nuclei were segmented using the ImageJ Auto Threshold and ROI tool. The background of each channel image except DAPI channel was subtracted using ImageJ’s rolling ball background subtraction algorithm with a radius of 3 pixels. Images were then z projected with maximum intensities. Intensities in each channel and nucleus were converted to 1D array of sum intensities of 2×2 pixels with removal of nucleolus pixels. Then Pearson correlation coefficient per nucleus between two images was computed based on the arrays.
Chromosome contact frequency analysis
First, distances between all pairs of the TAS in single cells were calculated. The pairwise contact frequency is determined by the number of times two active loci are found within 0.1, 0.25, 0.5, 1 and 2 um of each other. Each pixel in xy corresponded to 103 nm in the 10,421 gene experiment, as it was performed with a 63× objective and a camera with pixel size of 6.5 μm. Each pixel in Z corresponded to 350 nm steps in the z-sectioning on a confocal microscope. To calculate the interchromosomal contact frequency (Figure 3F), for a given pair of chromosomes, interactions between all gene pairs belonging to the two chromosomes were summed and then normalized by the expected frequency of appearance of the gene pairs, and summed over all gene pairs for each pair of chromosome interactions, shown in (Figure 3B). For the heatmap of pairwise gene interaction (Figure 3B), normalized contact frequencies were shown for all cells in G1/S phase.
Hi-C data was taken from Dixon et al., 2012 at 40 kb resolution and selecting only the positions that corresponded to the first intron probe. To compute the burst size and burst frequency distribution as a function of distance to TAD boundary, we take the nearest distance from the first intron probe of a gene to a TAD boundary (defined from Dixon et al., 2012), and averaged the burst size and frequency over all the genes for a particular distance bin. For the randomize data, we reshuffled the distance to TAD boundary and the burst statistics for all genes for 100 trials.
Simulations
Using the experimentally determined burst frequencies in the 10,421 seqFISH experiment, we simulated the intron expression profiles of 188 cells assuming that each gene was bursting independently and randomly. We sampled from Poisson distribution for each gene for all the cells. The total intron per cell distribution obtained from this simulation is plotted in Figure 5A, as a comparison to the experimentally determined distribution for 188 cells in G1/S phase and within a cell size window (12,000–16,000 pixel^2).
Principal component analysis (PCA)
For Figure 4A, we first calculated the cell to cell correlation matrix using the 10,421 gene intron seqFISH data. Each cell’s intron levels were normalized by the total intron numbers per cell. The PCA analysis was performed on the cell-to-cell correlation matrix. All cells from 4 datasets were used in this analysis regardless of cell cycle phases.
For Figure 4B, we use bootstrap to downsample the number of genes from the 10,421 intron experiment for mESC serum replicates, mESC 2i and NIH3T3 cells. We draw a given number of genes (100, 200 to 10,000 genes) randomly for 100 trials in each datapoint for all the cells. We then computed the cell-to-cell correlation matrices at each datapoint as above and hierarchically clustered the correlation matrices into 8 clusters (twice the number of datasets). The number of cells that falls into the 8 clusters from each of the 4 datasets was tabulated. To determine how much cells in different datasets fall into the same 8 clusters, we calculated the correlation coefficient of the cell clustering vector between pairs of datasets. The separation index is 1 minus this correlation coefficient. The separation indices are then computed for all bootstrap datasets for Figure 4B.
For Figure 4C, we first combined the mESC serum (replicate 1), 2i and NIH3T3 dataset, and Z-score normalized the combined dataset. We selected G1/S cells and within a FOV window to minimize variation due to cell cycle or illumination differences. We then clustered the genes with hierarchical clustering (Figure S4A) and selected the clusters showing introns upregulated in one of the three cell types/states, but downregulated in the other 2 cell types. PCA is performed on the gene-to-gene correlation matrix of these differentially expressed genes.
Data and Software Availability
MATLAB code for barcode calling and all image analysis steps: https://github.com/CaiGroup/Intron-SeqFish
Raw data of TAS counts and mRNA counts used in the analysis presented in this article can be found in Supplementary Table 1.
Supplementary Material
Reconstruction of all introns detected in individual mESCs grown in serum/LIF. Introns on individual chromosomes are shown. Introns are colored with respect to the chromosome to which it belongs. The color legend for each chromosome is shown in Figure 2A.
10,421 gene intron seqFISH and mRNA seqFISH data for 4 conditions (E14 cells grown in serum/LIF with replicate 1, 2, grown in 2i and NIH3T3 cells). Rows are cells and columns are genes. Values correspond to the number of TAS or mRNAs detected for each of the gene in the given cell.
Lists of differentially expressed genes enriched in one of the conditions of 10,421 gene intron seqFISH (E14 cells grown in serum/LIF, 2i and NIH3T3 cells).
Lists of gene pairs (introns and mRNAs), showing statistically significant Pearson correlation coefficient (p value < 0.01) in E14 cells grown in serum/LIF across two biological replicates.
(A) Detail primary probe design schematics for intron seqFISH experiments. Each gene is targeted by 25 primary probes with 35-nt gene specific sequence complementary to the intron region, four 15-nt barcode sites (a, b, c, d), 20-nt PCR primer binding sites and nucleotide spacers. Each barcode site (a, b, c, d) corresponds to one of the five barcoding rounds (I, II, III, IV and V). The 5 rounds of barcodes are distributed over 25 primary probes for each gene, such that each probe contains 4 barcode sites. (B) Schematic illustration of hybridization, stripping and re-hybridization of readout probes per one gene over 5 rounds of barcoding rounds. In each barcoding round, barcode sites (a, b, c, d) of the barcoding round (I, II, III, IV or V), are read out by a readout probe conjugated with one of the fluorophores (Alexa 647, Cy3B or Alexa 488). After imaging, readout probes are stripped off by 55% formamide solution, while primary probes remain bound to intron sequences due to longer probe length and higher DNA-RNA affinity. (C) Representative image of one of the channels (hyb1 channel 1; left) and its repeat after 20 rounds of hybridizations (hyb21 channel 1; middle) using the same readout probes as hyb1 channel 1. Merged image (right) shows many colocalized spots (white) between those two images (green and magenta), showing the robustness of the intron seqFISH protocol over 20 rounds of hybridizations without significant decrease of the signals. (D) Comparison of 10,421 gene intron seqFISH (n = 314 cells) and RNA-seq FPKM values with Pearson correlation coefficient of 0.40. (E) Comparison of 34 gene intron smFISH (n = 446–480 cells) and RNA-seq FPKM values with Pearson correlation coefficient of 0.63. Following 34 genes were used for this validation (Akt1s1, Fam120c, Pou5f1, Igf1r, Ap1s2, Lmx1a, Dlg2, Dock11, Scamp1, Wnt11, Mbtps2, Dnmt3b, Pdha1, Acsl4, Pgk1, Echdc3, Chm, Mras, Esrrb, Prrg1, Ric3, Sall4, Zfp42, Sox6, Src, Fgf1, Dusp8, Il6st, Dennd4c, 4933407K13Rik, Tet1, Zfp516, Eef2). Note that Dlg2 intron spots were not detected in our mESC population measured. (F) Fano factors as a function of mean burst frequency plotted for each gene in the 10,421 gene intron seqFISH using G1/S phase E14 cells grown in serum/LIF (n = 257 cells). Most genes have Fano factors close to unity. RNA-seq data from Antebi et al., (2017).
(A) Representative images showing intron spots from the Alexa 647 channel in the first hybridization of the 10,421 gene intron seqFISH (green), lncRNAs by lncRNA seqFISH (magenta) and nuclear stain by DAPI (blue) in mESCs. Images are a single confocal section. Introns are not necessarily colocalized with lncRNAs investigated here. (B) Representative images showing intron spots, polyA FISH, SC35 immunofluorescence, and nuclear stain by DAPI. Scale bars (A, B), 5 μm. (C) Distributions of localization correlation scores (Pearson correlation coefficient) in single cells (n = 437 nuclei). Solid lines display density plots and dashed lines indicate median correlation scores from our data. Note that Rex1 (mRNA FISH) & SC35 correlation score represents baseline correlation.
(A) Representative confocal images of a single z-section showing intron FISH targeting genes from individual chromosomes, DNA FISH targeting corresponding coding regions and chromosome paints in mESC nuclei stained by DAPI. Intron FISH probes targeting 736 genes, and DNA FISH probes targeting 380 genes in chromosome 11 are used. White arrow represents introns looped away from their core CT boundaries. Panel on the right displays quantified fluorescence intensity of the intron FISH, DNA FISH and chromosome paint along the yellow line within a single z section. Solid black arrow shows overlapped pattern between intron and DNA FISH peaks while dashed black arrow shows chromosome paint peak. (B) Violin plots showing the distance distribution of TAS relative to their chromosomal territory (CT) edge in mESC nuclei (n = 2880, 4609, 7372 and 1296 spots from 312, 301, 330 and 217 cells respectively) for chromosome 1, 2, 11 and 19 (intron probes targeting 77, 86, 79, and 30 genes on their chromosomes, respectively). Much larger displacements are observed if the introns and chromosome territories are scrambled (i.e. calculating distance from chromosome 1 introns to chromosome 19 CT). ****, significant with Wilcoxon’s rank sum test (P < 0.0001). (C) Representative confocal images from a single z-section showing loci looped away from their core CTs, imaged by DNA FISH (cyan) and intron FISH (red) targeting Ppp3r1 along with chromosome paint (yellow) in mESC nuclei (blue). DNA FISH spot confirms that coding genomic regions are looped away from the core CTs and is colocalized with intron FISH spot. Signals outside nuclei (dashed white lines) are not shown for visual clarity (A, C). (D) For the 8 genes measured individually by DNA FISH (Phlpp1, Nck2, Irs1, Hdac4, Adora1, Parp1, Gli2 and Abl2 from left to right) shown in Figure 2G, the distance distributions from the CT surface are shown as a function of mean burst frequency for each gene measured. The mean and standard deviation are shown in the plot. In total, 480–1016 DNA FISH spots were analyzed in 406–563 cells per gene. (E) Chromosome paints of 4 chromosomes (green: chromosome 1, yellow: chromosome 2, red: chromosome 11 and purple: chromosome 19) in mESC nuclei (blue). The image is shown as a maximum projection of z stacks. Regions with pairs of chromosomes are intermingled are shown with white arrows. Scale bars (A, C, E), 5 μm. (F) Comparison of mean number of TAS contacts (all, intra-chromosomal and intra-TAD contacts) per cell as a function of searching radius, reflecting the spatial proximity of those contacts. Inter-chromosome contacts account for greater than 80% of the contacts for all search radius examined (n = 420 cells). (G) Concordance between the heatmaps of normalized contacts from intron seqFISH (upper right) and Hi-C (lower left) are shown for 624 genes (left) and 29 genes (right) as a zoomed-in view of the gray box in chromosome 7. Cyan boxes represent individual TADs assigned to each gene. Mean burst frequency of each gene is visualized above the contact heatmaps. (H) Histogram showing the fraction of intrachromosomal contacts relative to total number of contacts in single cells (n = 420 cells).
(A) Hierarchical clustering of G1/S phase cells in the 10,421 intron seqFISH experiment (E14 cells grown in serum/LIF and 2i, and NIH3T3 cells) with heatmap for the number of TAS for each gene normalized by z-score and clustering dendrogram of genes shown next to the heatmap. Different cells grown in different conditions are clustered together, and each condition has enrichment of a set of genes. Detail lists of the differentially expressed genes are in Table S2. (B, C) Differential expression of genes between two different conditions (B, E14 cells grown in serum/LIF vs. 2i; C, E14 cells grown in serum/LIF vs. NIH3T3 cells). p values were computed using single cell data set of each condition with Wilcoxon’s rank sum test. Top candidate genes for differential expression are labeled with gene names. (D) Comparison of changes in mean burst frequency and size between E14 cells grown in serum/LIF and NIH3T3 cells. Linear regression line with 95% confidence interval is overlaid. Changes in both mean burst frequency and size are generally correlated. (E) Representative images of cells at different cell cycle phases (G1/S, G2 and M) as determined by H3S10 phosphorylation immunofluorescence (green). The images are shown as a maximum projection of z stacks. Scale bars, 5 μm. (F) Cell cycle phases in mESCs (replicate 1, n = 314 cells; replicate 2, n = 382 cells), defined by H3S10 phosphorylation immunofluorescence. (G) Comparison between nuclear volume and total intron counts per cell decoded by the 10,421 gene intron seqFISH. Cells at different cell cycle phases are displayed with different colors. (H) Violin plots showing the distribution of nuclear volume (left), total number of introns per cell (middle) and Aurka counts per cell (right) across different cell cycle phases. We calculated nuclear volume by raising nuclear area, which were measured by DAPI signal, to the 3/2 power. Known G2/M phase marker gene, Aurka, is used as a positive control, showing the increase of expression levels at G2 and M phases compared to G1/S phases. (I) Coefficient of variation (CV) of the intron numbers per cell in each cell cycle phase in mESCs. Error bars represent s.e.m. between replicates.
(A) Distribution of the cytoplasmic polyA densities per cell detected by LNA dT probes. (B) Violin-plots of 5-EU click fluorescence intensity show heterogeneity amongst cells across different time points in the pulse chase experiment with E14 cells under serum/LIF. (C) Biological replicate of intron pulse chase experiments with E14 cells grown in serum/LIF condition. At each time point, 533–1081 cells were analyzed. (D) An alternative method for analyzing the pulse chase data. Cells are binned according to the 5-EU intensity and the intron density distribution is plotted for each bin. The mean and standard deviation for the intron (vertical bar) and 5-EU intensity (horizontal bar) are shown in each plot. Different plots represent different time points in the pulse chase experiments. Data from E14 cells grown in serum/LIF, in 2i and NIH3T3 cells are shown in blue, red and yellow, respectively. (E) The slopes of each plot (D) are extracted and shown as a function of time. Shaded regions represent standard error. (F) Confocal images with maximum intensity z projection of 5-EU signals detected by click linkage to an azide-dye, and 1,000 gene introns in single NIH3T3 cells shown for three time points. Both methods reflect the heterogeneity of nascent transcription states at different time points.
(A–B) Distributions of raw counts per cell (A) and z-scores (B) for 1,000 gene introns and 48 mRNAs involved in pluripotency, signaling pathway and other processes detected by non-barcoded seqFISH (n = 605 cells). (C) Comparison of Bulk RNA-seq (Antebi et al., 2017) and non-barcoded mRNA seqFISH with 48 genes. Pearson correlation coefficient R = 0.92. (D) Histogram of G2/M marker Aurka mRNA counts per cell. Cell cycle phases were determined by Aurka mRNA counts based on Figure S4 results. (E) Heat maps showing Pearson gene-to-gene correlation coefficients between 1,000 gene total introns and 47 mRNAs with G1/S and G2/M phase cells determined by Aurka mRNA counts per cell. Correlation coefficients were computed after FISH count normalization with Eef2 counts per cell. The gene cluster (Red boxes; 1,000 gene total introns, Hes1, Stat3 and Socs3) is observed even after separating the cell cycle phases showing the gene-to-gene clusters are robust. Color bars represent Pearson correlation coefficient values. (F) Heatmap showing hierarchical clustering of cells using z-scores of the 1,000 gene total introns and mRNAs. Cells were divided into five subpopulations (cell cluster 1–5) based on the clustering of cells. Cluster 1–5 consist of 42, 128, 66, 133 and 236 cells, respectively. (G) Kernel density estimation plots showing gene expression distributions. Cells were divided into five subpopulations (cell cluster 1–5) based on hierarchical clustering (F). (H) Heat maps showing Pearson gene-to-gene correlation coefficients between total introns from 10,421 genes with mRNAs in E14 cells grown in serum/LIF (top) and 2i (bottom) conditions. Correlation coefficients were computed after mRNA FISH count normalization with Eef2 counts per cell. (I) Comparison of the mean copy number of the mRNAs of mESCs in serum/LIF vs 2i. Differentially expressed genes are labeled. Hes1 is 2-fold upregulated in 2i compared serum cells, while Dll1 and Gadd45g are almost 10-fold repressed, suggesting that up-regulation of Hes1, a repressor, can be linked to the suppression of oscillations in 2i conditions. (J) Comparison of the mean burst frequencies for 10,421 genes in G1/S phase E14 serum/LIF cells with high total number of introns (y-axis) vs the cells with low total number of introns (x-axis). On average, all genes are upregulated in the high total intron cells compared to the the low intron total cells, suggesting that nascent transcriptome is modulated global. (K) Single cell Hes1 pulse chase experiment of the biological replicate 1 (Figure 6C) subdivided into two cell cycle phases still show 2 hour oscillations. Dashed black lines show traces from all cells. Cell cycle phases were determined by H3S10 phosphorylation immunofluorescence intensity. Shaded regions represent 95% confidence intervals.
Highlights.
Intron seqFISH allows in situ visualization of nascent transcription in single cells
Single cell nascent transcriptome can distinguish cell types and states
Transcriptionally active loci are positioned at the surface of chromosome territories
Nascent transcriptome oscillates with a 2-hour period asynchronously in many cells
Acknowledgments
We thank A. Raj for discussion on the intron experiments, W. Reik, B. Simons, S. Rulands, and H. Lee for discussion on the 2 hour oscillation, M. Elowitz, M. Guttman, M. Lawson for comments and suggestions on the manuscript, the Elowitz Lab and the Reik Lab for kindly providing cell lines, and the Gradinaru Lab for use of Imaris software. Y.T. is supported by a Graduate Fellowship from the Nakajima Foundation. This project is funded by NIH EB021240, HD075605, MH113508 and a Paul G. Allen Foundation Discovery Center grant.
Footnotes
Author Contributions
S.S, Y.T., W.Z., and L.C. designed the intron seqFISH experiments. W.Z., S.S., and E.L. performed the initial intron seqFISH experiments with the help of N.K., E.J.L., and M.A.. Y.T., and C.-H.L.E. optimized and Y.T. collected the intron seqFISH data. S.S., and J.Y. carried out the pulse chase experiments. C.C., and C.K. built the automated fluidics. Y.T., S.S., and L.C. carried out the analysis. S.S., Y.T., W.Z., and L.C. wrote the manuscript.
Declaration of Interest
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ang YS, Tsai SY, Lee DF, Monk J, Su J, Ratnakumar K, Ding J, Ge Y, Darr H, Chang B, et al. Wdr5 mediates self-renewal and reprogramming via the embryonic stem cell core transcriptional network. Cell. 2011;145:183–197. doi: 10.1016/j.cell.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antebi YE, Linton JM, Klumpe H, Bintu B, Gong M, Su C, McCardell R, Elowitz MB. Combinatorial Signal Perception in the BMP Pathway. Cell. 2017;170:1184–1196. e1124. doi: 10.1016/j.cell.2017.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beliveau BJ, Joyce EF, Apostolopoulos N, Yilmaz F, Fonseka CY, McCole RB, Chang Y, Li JB, Senaratne TN, Williams BR, et al. Versatile design and synthesis platform for visualizing genomes with Oligopaint FISH probes. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:21301–21306. doi: 10.1073/pnas.1213818110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolzer A, Kreth G, Solovei I, Koehler D, Saracoglu K, Fauth C, Muller S, Eils R, Cremer C, Speicher MR, et al. Three-Dimensional Maps of All Chromosomes in Human Male Fibroblast Nuclei and Prometaphase Rosettes. PLoS Biology. 2005;3:e157. doi: 10.1371/journal.pbio.0030157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle S, Rodesch MJ, Halvensleben HA, Jeddeloh JA, Bickmore WA. Fluorescence in situ hybridization with high-complexity repeat-free oligonucleotide probes generated by massively parallel synthesis. Chromosome Res. 2011;19:901–909. doi: 10.1007/s10577-011-9245-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L, Dalal CK, Elowitz MB. Frequency-modulated nuclear localization bursts coordinate gene regulation. Nature. 2008;455:485–490. doi: 10.1038/nature07292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmanis S, Sloan SA, Zhang Y, Enge M, Caneda C, Shuer LM, Hayden Gephart MG, Barres BA, Quake SR. A survey of human brain transcriptome diversity at the single cell level. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:7285–7290. doi: 10.1073/pnas.1507125112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dequeant ML, Glynn E, Gaudenz K, Wahl M, Chen J, Mushegian A, Pourquie O. A Complex Oscillating Network of Signaling Genes Underlies the Mouse Segmentation Clock. Science. 2006;314:1595. doi: 10.1126/science.1133141. [DOI] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolmetsch RE, Xu K, Lewis RS. Calcium oscillations increase the efficiency and specificity of gene expression. Nature. 1998;392:933. doi: 10.1038/31960. [DOI] [PubMed] [Google Scholar]
- Eng CHL, Shah S, Thomassie J, Cai L. Profiling the transcriptome with RNA SPOTs. Nature Methods. 2017;14:1153. doi: 10.1038/nmeth.4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Femino AM, Fay FS, Fogarty K, Singer RH. Visualization of Single RNA Transcripts in Situ. Science. 1998;280:585. doi: 10.1126/science.280.5363.585. [DOI] [PubMed] [Google Scholar]
- Hirata H, Yoshiura S, Ohtsuka T, Bessho Y, Harada T, Yoshikawa K, Kageyama R. Oscillatory Expression of the bHLH Factor Hes1 Regulated by a Negative Feedback Loop. Science. 2002;298:840. doi: 10.1126/science.1074560. [DOI] [PubMed] [Google Scholar]
- Hoffmann A, Levchenko A, Scott ML, Baltimore D. The IκB-NF-κB Signaling Module: Temporal Control and Selective Gene Activation. Science. 2002;298:1241. doi: 10.1126/science.1071914. [DOI] [PubMed] [Google Scholar]
- Hu X, Zhang L, Mao SQ, Li Z, Chen J, Zhang RR, Wu HP, Gao J, Guo F, Liu W, et al. Tet and TDG Mediate DNA Demethylation Essential for Mesenchymal-to-Epithelial Transition in Somatic Cell Reprogramming. Cell Stem Cell. 2014;14:512–522. doi: 10.1016/j.stem.2014.01.001. [DOI] [PubMed] [Google Scholar]
- Jao CY, Salic A. Exploring RNA transcription and turnover in vivo by using click chemistry. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:15779–15784. doi: 10.1073/pnas.0808480105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers I, Kwak H, Lis JT. Genome-wide dynamics of Pol II elongation and its interplay with promoter proximal pausing, chromatin, and exons. eLife. 2014;3:e02407. doi: 10.7554/eLife.02407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, Peshkin L, Weitz DA, Kirschner MW. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell. 2015;161:1187–1201. doi: 10.1016/j.cell.2015.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Mizuno H, Imayoshi I, Furusawa C, Shirahige K, Kageyama R. The cyclic gene Hes1 contributes to diverse differentiation responses of embryonic stem cells. Genes & Development. 2009;23:1870–1875. doi: 10.1101/gad.1823109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodziejczyk AA, Kim JK, Tsang JC, Ilicic T, Henriksson J, Natarajan KN, Tuck AC, Gao X, Buhler M, Liu P, et al. Single Cell RNA-Sequencing of Pluripotent States Unlocks Modular Transcriptional Variation. Cell Stem Cell. 2015;17:471–485. doi: 10.1016/j.stem.2015.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Manno G, Soldatov R, Hochgerner H, Zeisel A, Petukhov V, Kastriti M, Lonnerberg P, Furlan A, Fan J, Liu Z, et al. RNA velocity in single cells. 2017 doi: 10.1038/s41586-018-0414-6. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Daugharthy ER, Scheiman J, Kalhor R, Ferrante TC, Yang JL, Terry R, Jeanty SSF, Li C, Amamoto R, et al. Highly multiplexed subcellular RNA sequencing in situ. Science. 2014;343:1360–1363. doi: 10.1126/science.1250212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letsou W, Cai L. Noncommutative Biology: Sequential Regulation of Complex Networks. PLoS Comput Biol. 2016;12:e1005089. doi: 10.1371/journal.pcbi.1005089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levesque MJ, Raj A. Single-chromosome transcriptional profiling reveals chromosomal gene expression regulation. Nat Methods. 2013;10:246–248. doi: 10.1038/nmeth.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. Comprehensive mapping of long range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lignell A, Kerosuo L, Streichan SJ, Cai L, Bronner ME. Identification of a neural crest stem cell niche by Spatial Genomic Analysis. Nature Communications. 2017;8:1830. doi: 10.1038/s41467-017-01561-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubeck E, Coskun AF, Zhiyentayev T, Ahmad M, Cai L. Single-cell in situ RNA profiling by sequential hybridization. Nat Methods. 2014;11:360–361. doi: 10.1038/nmeth.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161:1202–1214. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maharana S, Iyer KV, Jain N, Nagarajan M, Wang Y, Shivashankar GV. Chromosome intermingling—the physical basis of chromosome organization in differentiated cells. Nucleic Acids Research. 2016;44:5148–5160. doi: 10.1093/nar/gkw131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahy NL, Perry PE, Gilchrist S, Baldock RA, Bickmore WA. Spatial organization of active and inactive genes and noncoding DNA within chromosome territories. The Journal of Cell Biology. 2002;157:579–589. doi: 10.1083/jcb.200111071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahy NL, Perry PE, Bickmore WA. Gene density and transcription influence the localization of chromatin outside of chromosome territories detectable by FISH. The Journal of Cell Biology. 2002;159:753–763. doi: 10.1083/jcb.200207115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks H, Kalkan T, Menafra R, Denissov S, Jones K, Hofemeister H, Nichols J, Kranz A, Stewart AF, Smith A, et al. The transcriptional and epigenomic foundations of ground state pluripotency. Cell. 2012;149:590–604. doi: 10.1016/j.cell.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano T, Lubling Y, Stevens TJ, Schoenfelder S, Yaffe E, Dean W, Laue ED, Tanay A, Fraser P. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature. 2013;502:59–64. doi: 10.1038/nature12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, Peskin CS, Tranchina D, Vargas DY, Tyagi S. Stochastic mRNA synthesis in mammalian cells. PLoS Biol. 2006;4:e309. doi: 10.1371/journal.pbio.0040309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouillard JM, Zuker M, Gulari E. OligoArray 2.0: design of oligonucleotide probes for DNA microarrays using a thermodynamic approach. Nucleic Acids Research. 2003;31:3057–3062. doi: 10.1093/nar/gkg426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S, Lubeck E, Schwarzkopf M, He TF, Greenbaum A, Sohn CH, Lignell A, Choi HM, Gradinaru V, Pierce NA, et al. Single-molecule RNA detection at depth by hybridization chain reaction and tissue hydrogel embedding and clearing. Development. 2016;143:2862–2867. doi: 10.1242/dev.138560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S, Lubeck E, Zhou W, Cai L. In Situ Transcription Profiling of Single Cells Reveals Spatial Organization of Cells in the Mouse Hippocampus. Neuron. 2016;92:342–357. doi: 10.1016/j.neuron.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankaran H, Ippolito DL, Chrisler WB, Resat H, Bollinger N, Opresko LK, Wiley HS. Rapid and sustained nuclear–cytoplasmic ERK oscillations induced by epidermal growth factor. Molecular Systems Biology. 2009;5:332–332. doi: 10.1038/msb.2009.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharova LV, Sharov AA, Nedorezov T, Piao Y, Shaik N, Ko MS. Database for mRNA half-life of 19 977 genes obtained by DNA microarray analysis of pluripotent and differentiating mouse embryonic stem cells. DNA Res. 2009;16:45–58. doi: 10.1093/dnares/dsn030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimojo H, Ohtsuka T, Kageyama R. Oscillations in Notch Signaling Regulate Maintenance of Neural Progenitors. Neuron. 2008;58:52–64. doi: 10.1016/j.neuron.2008.02.014. [DOI] [PubMed] [Google Scholar]
- Shishova KV, Zharskaya O, Zatsepina O. The Fate of the Nucleolus during Mitosis: Comparative Analysis of Localization of Some Forms of Pre-rRNA by Fluorescent in Situ Hybridization in NIH/3T3 Mouse Fibroblasts. Acta Naturae. 2011;3:100–106. [PMC free article] [PubMed] [Google Scholar]
- Singer ZS, Yong J, Tischler J, Hackett JA, Altinok A, Surani MA, Cai L, Elowitz MB. Dynamic heterogeneity and DNA methylation in embryonic stem cells. Mol Cell. 2014;55:319–331. doi: 10.1016/j.molcel.2014.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens TJ, Lando D, Basu S, Atkinson LP, Cao Y, Lee SF, Leeb M, Wohlfahrt KJ, Boucher W, O’Shaughnessy-Kirwan A, et al. 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature. 2017;544:59–64. doi: 10.1038/nature21429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei Y, Shah S, Harvey S, Qi LS, Cai L. Multiplexed Dynamic Imaging of Genomic Loci by Combined CRISPR Imaging and DNA Sequential FISH. Biophys J. 2017;112:1773–1776. doi: 10.1016/j.bpj.2017.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yissachar N, Sharar Fischler T, Cohen Ariel A, Reich-Zeliger S, Russ D, Shifrut E, Porat Z, Friedman N. Dynamic Response Diversity of NFAT Isoforms in Individual Living Cells. Molecular Cell. 2013;49:322–330. doi: 10.1016/j.molcel.2012.11.003. [DOI] [PubMed] [Google Scholar]
- Yoshiura S, Ohtsuka T, Takenaka Y, Nagahara H, Yoshikawa K, Kageyama R. Ultradian oscillations of Stat, Smad, and Hes1 expression in response to serum. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:11292–11297. doi: 10.1073/pnas.0701837104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Yan R, Zhang Q, Li J, Kang X, Wang H, Huan L, Zhang L, Li F, Yang S, et al. Hes1, a Notch signaling downstream target, regulates adult hippocampal neurogenesis following traumatic brain injury. Brain Research. 2014;1583:65–78. doi: 10.1016/j.brainres.2014.07.037. [DOI] [PubMed] [Google Scholar]
- Zeisel A, Munoz-Manchado AB, Codeluppi S, Lonnerberg P, La Manno G, Jureus A, Marques S, Munguba H, He L, Betsholtz C, et al. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science. 2015;347:1138. doi: 10.1126/science.aaa1934. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Reconstruction of all introns detected in individual mESCs grown in serum/LIF. Introns on individual chromosomes are shown. Introns are colored with respect to the chromosome to which it belongs. The color legend for each chromosome is shown in Figure 2A.
10,421 gene intron seqFISH and mRNA seqFISH data for 4 conditions (E14 cells grown in serum/LIF with replicate 1, 2, grown in 2i and NIH3T3 cells). Rows are cells and columns are genes. Values correspond to the number of TAS or mRNAs detected for each of the gene in the given cell.
Lists of differentially expressed genes enriched in one of the conditions of 10,421 gene intron seqFISH (E14 cells grown in serum/LIF, 2i and NIH3T3 cells).
Lists of gene pairs (introns and mRNAs), showing statistically significant Pearson correlation coefficient (p value < 0.01) in E14 cells grown in serum/LIF across two biological replicates.
(A) Detail primary probe design schematics for intron seqFISH experiments. Each gene is targeted by 25 primary probes with 35-nt gene specific sequence complementary to the intron region, four 15-nt barcode sites (a, b, c, d), 20-nt PCR primer binding sites and nucleotide spacers. Each barcode site (a, b, c, d) corresponds to one of the five barcoding rounds (I, II, III, IV and V). The 5 rounds of barcodes are distributed over 25 primary probes for each gene, such that each probe contains 4 barcode sites. (B) Schematic illustration of hybridization, stripping and re-hybridization of readout probes per one gene over 5 rounds of barcoding rounds. In each barcoding round, barcode sites (a, b, c, d) of the barcoding round (I, II, III, IV or V), are read out by a readout probe conjugated with one of the fluorophores (Alexa 647, Cy3B or Alexa 488). After imaging, readout probes are stripped off by 55% formamide solution, while primary probes remain bound to intron sequences due to longer probe length and higher DNA-RNA affinity. (C) Representative image of one of the channels (hyb1 channel 1; left) and its repeat after 20 rounds of hybridizations (hyb21 channel 1; middle) using the same readout probes as hyb1 channel 1. Merged image (right) shows many colocalized spots (white) between those two images (green and magenta), showing the robustness of the intron seqFISH protocol over 20 rounds of hybridizations without significant decrease of the signals. (D) Comparison of 10,421 gene intron seqFISH (n = 314 cells) and RNA-seq FPKM values with Pearson correlation coefficient of 0.40. (E) Comparison of 34 gene intron smFISH (n = 446–480 cells) and RNA-seq FPKM values with Pearson correlation coefficient of 0.63. Following 34 genes were used for this validation (Akt1s1, Fam120c, Pou5f1, Igf1r, Ap1s2, Lmx1a, Dlg2, Dock11, Scamp1, Wnt11, Mbtps2, Dnmt3b, Pdha1, Acsl4, Pgk1, Echdc3, Chm, Mras, Esrrb, Prrg1, Ric3, Sall4, Zfp42, Sox6, Src, Fgf1, Dusp8, Il6st, Dennd4c, 4933407K13Rik, Tet1, Zfp516, Eef2). Note that Dlg2 intron spots were not detected in our mESC population measured. (F) Fano factors as a function of mean burst frequency plotted for each gene in the 10,421 gene intron seqFISH using G1/S phase E14 cells grown in serum/LIF (n = 257 cells). Most genes have Fano factors close to unity. RNA-seq data from Antebi et al., (2017).
(A) Representative images showing intron spots from the Alexa 647 channel in the first hybridization of the 10,421 gene intron seqFISH (green), lncRNAs by lncRNA seqFISH (magenta) and nuclear stain by DAPI (blue) in mESCs. Images are a single confocal section. Introns are not necessarily colocalized with lncRNAs investigated here. (B) Representative images showing intron spots, polyA FISH, SC35 immunofluorescence, and nuclear stain by DAPI. Scale bars (A, B), 5 μm. (C) Distributions of localization correlation scores (Pearson correlation coefficient) in single cells (n = 437 nuclei). Solid lines display density plots and dashed lines indicate median correlation scores from our data. Note that Rex1 (mRNA FISH) & SC35 correlation score represents baseline correlation.
(A) Representative confocal images of a single z-section showing intron FISH targeting genes from individual chromosomes, DNA FISH targeting corresponding coding regions and chromosome paints in mESC nuclei stained by DAPI. Intron FISH probes targeting 736 genes, and DNA FISH probes targeting 380 genes in chromosome 11 are used. White arrow represents introns looped away from their core CT boundaries. Panel on the right displays quantified fluorescence intensity of the intron FISH, DNA FISH and chromosome paint along the yellow line within a single z section. Solid black arrow shows overlapped pattern between intron and DNA FISH peaks while dashed black arrow shows chromosome paint peak. (B) Violin plots showing the distance distribution of TAS relative to their chromosomal territory (CT) edge in mESC nuclei (n = 2880, 4609, 7372 and 1296 spots from 312, 301, 330 and 217 cells respectively) for chromosome 1, 2, 11 and 19 (intron probes targeting 77, 86, 79, and 30 genes on their chromosomes, respectively). Much larger displacements are observed if the introns and chromosome territories are scrambled (i.e. calculating distance from chromosome 1 introns to chromosome 19 CT). ****, significant with Wilcoxon’s rank sum test (P < 0.0001). (C) Representative confocal images from a single z-section showing loci looped away from their core CTs, imaged by DNA FISH (cyan) and intron FISH (red) targeting Ppp3r1 along with chromosome paint (yellow) in mESC nuclei (blue). DNA FISH spot confirms that coding genomic regions are looped away from the core CTs and is colocalized with intron FISH spot. Signals outside nuclei (dashed white lines) are not shown for visual clarity (A, C). (D) For the 8 genes measured individually by DNA FISH (Phlpp1, Nck2, Irs1, Hdac4, Adora1, Parp1, Gli2 and Abl2 from left to right) shown in Figure 2G, the distance distributions from the CT surface are shown as a function of mean burst frequency for each gene measured. The mean and standard deviation are shown in the plot. In total, 480–1016 DNA FISH spots were analyzed in 406–563 cells per gene. (E) Chromosome paints of 4 chromosomes (green: chromosome 1, yellow: chromosome 2, red: chromosome 11 and purple: chromosome 19) in mESC nuclei (blue). The image is shown as a maximum projection of z stacks. Regions with pairs of chromosomes are intermingled are shown with white arrows. Scale bars (A, C, E), 5 μm. (F) Comparison of mean number of TAS contacts (all, intra-chromosomal and intra-TAD contacts) per cell as a function of searching radius, reflecting the spatial proximity of those contacts. Inter-chromosome contacts account for greater than 80% of the contacts for all search radius examined (n = 420 cells). (G) Concordance between the heatmaps of normalized contacts from intron seqFISH (upper right) and Hi-C (lower left) are shown for 624 genes (left) and 29 genes (right) as a zoomed-in view of the gray box in chromosome 7. Cyan boxes represent individual TADs assigned to each gene. Mean burst frequency of each gene is visualized above the contact heatmaps. (H) Histogram showing the fraction of intrachromosomal contacts relative to total number of contacts in single cells (n = 420 cells).
(A) Hierarchical clustering of G1/S phase cells in the 10,421 intron seqFISH experiment (E14 cells grown in serum/LIF and 2i, and NIH3T3 cells) with heatmap for the number of TAS for each gene normalized by z-score and clustering dendrogram of genes shown next to the heatmap. Different cells grown in different conditions are clustered together, and each condition has enrichment of a set of genes. Detail lists of the differentially expressed genes are in Table S2. (B, C) Differential expression of genes between two different conditions (B, E14 cells grown in serum/LIF vs. 2i; C, E14 cells grown in serum/LIF vs. NIH3T3 cells). p values were computed using single cell data set of each condition with Wilcoxon’s rank sum test. Top candidate genes for differential expression are labeled with gene names. (D) Comparison of changes in mean burst frequency and size between E14 cells grown in serum/LIF and NIH3T3 cells. Linear regression line with 95% confidence interval is overlaid. Changes in both mean burst frequency and size are generally correlated. (E) Representative images of cells at different cell cycle phases (G1/S, G2 and M) as determined by H3S10 phosphorylation immunofluorescence (green). The images are shown as a maximum projection of z stacks. Scale bars, 5 μm. (F) Cell cycle phases in mESCs (replicate 1, n = 314 cells; replicate 2, n = 382 cells), defined by H3S10 phosphorylation immunofluorescence. (G) Comparison between nuclear volume and total intron counts per cell decoded by the 10,421 gene intron seqFISH. Cells at different cell cycle phases are displayed with different colors. (H) Violin plots showing the distribution of nuclear volume (left), total number of introns per cell (middle) and Aurka counts per cell (right) across different cell cycle phases. We calculated nuclear volume by raising nuclear area, which were measured by DAPI signal, to the 3/2 power. Known G2/M phase marker gene, Aurka, is used as a positive control, showing the increase of expression levels at G2 and M phases compared to G1/S phases. (I) Coefficient of variation (CV) of the intron numbers per cell in each cell cycle phase in mESCs. Error bars represent s.e.m. between replicates.
(A) Distribution of the cytoplasmic polyA densities per cell detected by LNA dT probes. (B) Violin-plots of 5-EU click fluorescence intensity show heterogeneity amongst cells across different time points in the pulse chase experiment with E14 cells under serum/LIF. (C) Biological replicate of intron pulse chase experiments with E14 cells grown in serum/LIF condition. At each time point, 533–1081 cells were analyzed. (D) An alternative method for analyzing the pulse chase data. Cells are binned according to the 5-EU intensity and the intron density distribution is plotted for each bin. The mean and standard deviation for the intron (vertical bar) and 5-EU intensity (horizontal bar) are shown in each plot. Different plots represent different time points in the pulse chase experiments. Data from E14 cells grown in serum/LIF, in 2i and NIH3T3 cells are shown in blue, red and yellow, respectively. (E) The slopes of each plot (D) are extracted and shown as a function of time. Shaded regions represent standard error. (F) Confocal images with maximum intensity z projection of 5-EU signals detected by click linkage to an azide-dye, and 1,000 gene introns in single NIH3T3 cells shown for three time points. Both methods reflect the heterogeneity of nascent transcription states at different time points.
(A–B) Distributions of raw counts per cell (A) and z-scores (B) for 1,000 gene introns and 48 mRNAs involved in pluripotency, signaling pathway and other processes detected by non-barcoded seqFISH (n = 605 cells). (C) Comparison of Bulk RNA-seq (Antebi et al., 2017) and non-barcoded mRNA seqFISH with 48 genes. Pearson correlation coefficient R = 0.92. (D) Histogram of G2/M marker Aurka mRNA counts per cell. Cell cycle phases were determined by Aurka mRNA counts based on Figure S4 results. (E) Heat maps showing Pearson gene-to-gene correlation coefficients between 1,000 gene total introns and 47 mRNAs with G1/S and G2/M phase cells determined by Aurka mRNA counts per cell. Correlation coefficients were computed after FISH count normalization with Eef2 counts per cell. The gene cluster (Red boxes; 1,000 gene total introns, Hes1, Stat3 and Socs3) is observed even after separating the cell cycle phases showing the gene-to-gene clusters are robust. Color bars represent Pearson correlation coefficient values. (F) Heatmap showing hierarchical clustering of cells using z-scores of the 1,000 gene total introns and mRNAs. Cells were divided into five subpopulations (cell cluster 1–5) based on the clustering of cells. Cluster 1–5 consist of 42, 128, 66, 133 and 236 cells, respectively. (G) Kernel density estimation plots showing gene expression distributions. Cells were divided into five subpopulations (cell cluster 1–5) based on hierarchical clustering (F). (H) Heat maps showing Pearson gene-to-gene correlation coefficients between total introns from 10,421 genes with mRNAs in E14 cells grown in serum/LIF (top) and 2i (bottom) conditions. Correlation coefficients were computed after mRNA FISH count normalization with Eef2 counts per cell. (I) Comparison of the mean copy number of the mRNAs of mESCs in serum/LIF vs 2i. Differentially expressed genes are labeled. Hes1 is 2-fold upregulated in 2i compared serum cells, while Dll1 and Gadd45g are almost 10-fold repressed, suggesting that up-regulation of Hes1, a repressor, can be linked to the suppression of oscillations in 2i conditions. (J) Comparison of the mean burst frequencies for 10,421 genes in G1/S phase E14 serum/LIF cells with high total number of introns (y-axis) vs the cells with low total number of introns (x-axis). On average, all genes are upregulated in the high total intron cells compared to the the low intron total cells, suggesting that nascent transcriptome is modulated global. (K) Single cell Hes1 pulse chase experiment of the biological replicate 1 (Figure 6C) subdivided into two cell cycle phases still show 2 hour oscillations. Dashed black lines show traces from all cells. Cell cycle phases were determined by H3S10 phosphorylation immunofluorescence intensity. Shaded regions represent 95% confidence intervals.