

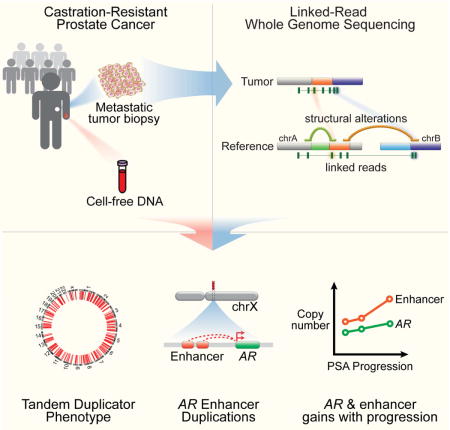
SUMMARY
Nearly all prostate cancer deaths are from metastatic castration-resistant prostate cancer (mCRPC) but there have been few whole genome sequencing (WGS) studies of this disease state. We performed linked-read WGS on 23 mCRPC biopsy specimens and analyzed cell-free DNA sequencing data from 86 patients with mCRPC. In addition to frequent rearrangements affecting known prostate cancer genes, we observed complex rearrangements of the AR locus in most cases. Unexpectedly, these include highly recurrent tandem duplications involving an upstream enhancer of AR in 70-87% of cases compared with <2% of primary prostate cancers. A subset of cases displayed AR or MYC enhancer duplication in the context of a genome-wide tandem duplicator phenotype associated with CDK12 inactivation. Our findings highlight the complex genomic structure of mCRPC, nominate alterations that may inform prostate cancer treatment, and suggest that additional recurrent events in the noncoding mCRPC genome remain to be discovered.
Keywords: 10X Genomics, long read sequencing, whole genome sequencing, structural variants, castration-resistant prostate cancer, enhancer, non-coding, androgen receptor, AR, CDK12, MYC, germline risk, tandem duplicator phenotype, cell-free DNA
In brief
Linked-read genome sequencing data from patients highlight that amplification of an enhancer upstream of the androgen receptor locus is a key feature for metastatic castration-resistant prostate cancer.
INTRODUCTION
Genomic studies have uncovered multiple recurrent genetic alterations that drive clinically localized prostate cancer, including mutations, copy number events, gene fusions, and more complex structural genomic rearrangements (Baca et al., 2013; Boysen et al., 2015; Cancer Genome Atlas Research Network, 2015; Fraser et al., 2017; Shenoy et al., 2017; Tomlins et al., 2005). There have been fewer genomic studies (particularly WGS studies) of metastatic castration-resistant prostate cancer (mCRPC), but emerging data suggest important distinctions between the mutational profiles of mCRPC and primary prostate cancer. Perhaps most notably, multiple studies have demonstrated that somatic mutations in AR pathway genes are pervasive in mCRPC but nearly absent in primary disease (Armenia et al., 2018; Cancer Genome Atlas Research Network, 2015; Grasso et al., 2012; Kumar et al., 2016; Robinson et al., 2015; Taplin et al., 1995; Visakorpi et al., 1995). Whole genome studies have also revealed functionally convergent rearrangements leading to AR copy gain in distinct metastases from the same patient with prostate cancer, indicating persistent selective pressure on AR signaling in mCRPC (Gundem et al., 2015). Still, our understanding of the full spectrum of genome-wide alterations in this disease state, including those that arise in the setting of treatment with the newest-generation androgen pathway inhibitors, is incomplete.
Recent WGS studies in diverse tumor types have begun to reveal recurrent alterations in regulatory regions of the genome, such as those that activate promoters or enhancers. For example, enhancers of oncogenes can be somatically activated by several mechanisms, including point mutations that induce transcription factor binding (Mansour et al., 2014), duplication of existing enhancers (Glodzik et al., 2017; Herranz et al., 2014; Shi et al., 2013; Zhang et al., 2016), or structural alterations that relocate a strong enhancer in proximity to a proto-oncogene (Hnisz et al., 2016; Northcott et al., 2014; Weischenfeldt et al., 2017).
Toward a more comprehensive understanding of somatic alterations in mCRPC with emphasis on alterations in the noncoding genome and/or those involving structural variants (SVs), we performed WGS employing a newly developed long-range, linked-read sequencing platform (10X Genomics, “10XG”) (Greer et al., 2017; Zheng et al., 2016). This approach produces barcoded short-read libraries from high-molecular weight DNA fragments, and is well-suited for the study of mCRPC for several reasons: 1) it allows for haplotype-resolved SV calling and improved SV detection, particularly of complex events; 2) barcode-aware alignment may offer superior mappability in certain regions of the genome, such as in repetitive regions harboring breakpoints for AR-related SVs (Nyquist et al., 2013); and 3) it requires as little as 1 ng of tumor DNA input, allowing for study of small metastatic biopsy samples.
In this study, we performed linked-read WGS on 23 biopsy specimens from individuals with mCRPC, including several obtained from heavily pre-treated patients after progression on next-generation androgen pathway inhibitors. We identified a number of novel and biologically important structural alterations in mCRPC, including highly recurrent duplications involving a newly-described long-range enhancer of AR expression (Takeda et al.), complex rearrangements driving AR gene and enhancer copy gain in mCRPC (both before and after treatment with next-generation AR pathway antagonists), and a genome-wide tandem duplicator phenotype (TDP) associated with CDK12 inactivation and recurrent duplications at the MYC and AR loci. Overall, these results highlight the diverse mechanisms by which structural alterations, particularly in the noncoding genome, act to sustain AR signaling in advanced prostate cancer.
RESULTS
Linked-read WGS of mCRPC
We performed 10XG linked-read WGS to an average depth of 31X on 23 metastatic biopsy specimens and matched germline controls. Cases were selected from a previously described cohort of mCRPC-affected individuals treated with either standard-of-care or on clinical trials (Armenia et al., 2018; Robinson et al., 2015). Eleven samples were taken from individuals prior to beginning treatment on a regimen including next-generation androgen synthesis inhibitor or AR pathway antagonist (enzalutamide, abiraterone, or apalutamide), while the remaining 12 samples were collected upon progression on one of these agents. Three pairs of samples were obtained from metastatic sites in the same individual prior to and after progression on treatment (Figure 1a and Table S1).
Figure 1. WGS of mCRPC tumors on the 10XG platform.
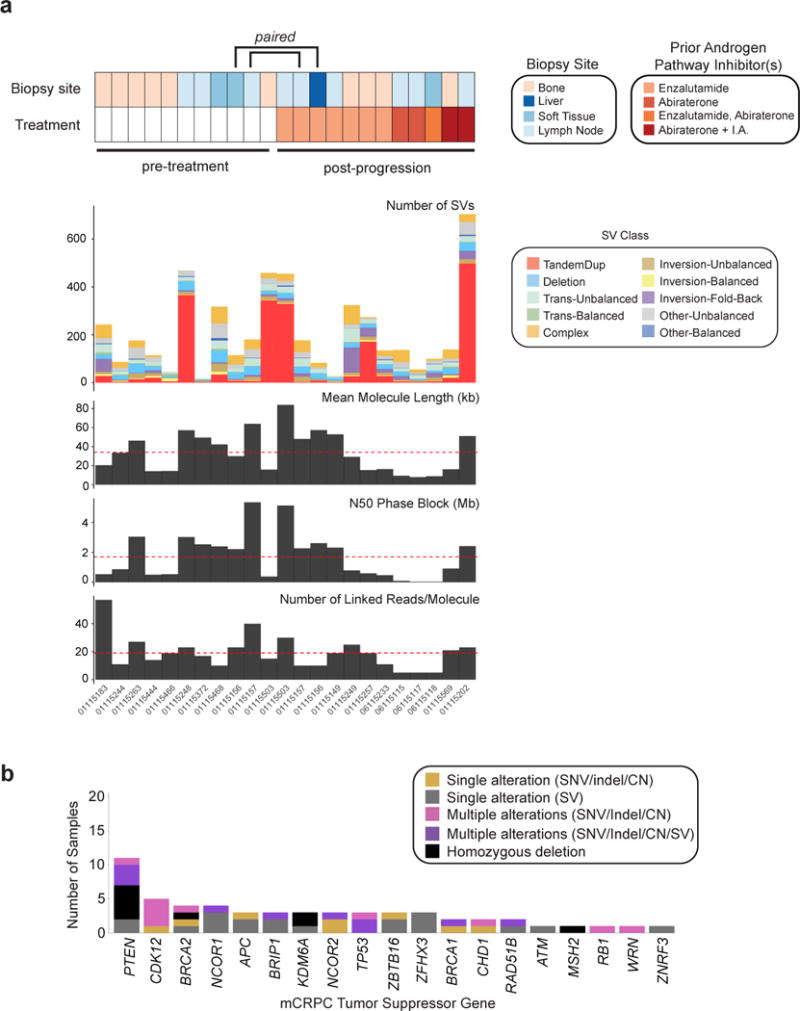
(a) Landscape of rearrangements and sequencing metrics across the 10XG WGS mCRPC cohort. Structural variant classification defined in STAR Methods. I.A., investigational agent.
(b) Number of samples containing one or multiple alterations in significantly inactivated mCRPC genes (Robinson et al., 2015). Alterations are classified as being due to SNVs, indels, or copy number loss or due to transecting SV.
See also Figures S1–S3 and Tables S1–S5.
By extracting high molecular weight DNA from frozen biopsy specimens, we achieved a mean molecule length of 34 kB with an average N50 phase block of 1.7 Mb (1.0 Mb in matched normal samples). Full sequencing metrics are provided in Table S2. We detected an average rate of single nucleotide variants (SNVs) of 3.6 SNVs/Mb per sample and indel rate of 1.8 indels/Mb per sample; leveraging 10XG linked-reads, 78% of these mutations could be phased to a haplotype (Table S3 and STAR Methods). Given the relatively small cohort size, we limited mutation (SNV and indel) analysis to genes previously reported to be significantly altered in mCRPC and observed alterations in multiple driver genes at frequencies roughly comparable to those reported using whole exome sequencing (WES) (Grasso et al., 2012; Pritchard et al., 2016; Robinson et al., 2015) (Figure S1 and Tables S3-S4).
SV Classes and Intragenic SVs in mCRPC Cohort
We identified SVs by incorporating support from read alignments, local assembly, and barcodes using three independent SV detection methods (STAR Methods). We observed an average of 230 SVs/sample. Structural alterations were classified into simple, complex, balanced, and unbalanced rearrangement types using breakpoint orientation and corroborating copy number information (Figures 1a, S2, Table S5, and STAR Methods). We found that SVs are a notable mechanism of inactivation of tumor suppressor genes (Figure 1b). For example, two cases without detectable SNVs, indels, or copy number alterations in PTEN were noted to have transecting rearrangements involving the gene body (paired samples from individual 01115503); in one additional sample (01115468), a transecting 24 kB deletion of PTEN occurred together with a splice site mutation and the two events were present on different haplotypes (Figures 1b and S1). In a fourth case (01115156), phasing afforded by linked-reads allowed for haplotype-specific resolution of rearrangement events ultimately resulting in homozygous deletion of PTEN (Figure S3). Overall, 19 of 23 cases had biallelic inactivation of tumor suppressor genes when inactivating rearrangements were considered (either via an apparently solitary transecting rearrangement or as part of a larger chromoplexy chain), as compared with 15 cases when inactivation was called on the basis of SNVs, indels, and gene-level copy number alone (Figures 1b and S1; Tables S3-S5). Thus, rearrangements represent an important mechanism of alteration in known mCRPC genes.
A Genome-wide Tandem Duplicator Phenotype Associated with CDK12 Inactivation
Five samples (22%) from 4 unique patients displayed a preponderance of tandem duplications compared with other SV classes (Figures 1a, 2a, S2). These tandem duplications occurred on both haplotypes within a chromosome (Figure 2b), suggesting a mechanistic distinction from other processes such as chromoplexy and chromothripsis. In addition to rearrangements discovered by 10XG WGS, copy number profiles displaying a high frequency of interstitial gains, consistent with dispersed tandem duplications, were observed in multiple samples of WES of tumor biopsies and of ultra low pass (ULP) WGS of cell-free DNA (Figure 2c).
Figure 2. A genome-wide TDP in mCRPC.
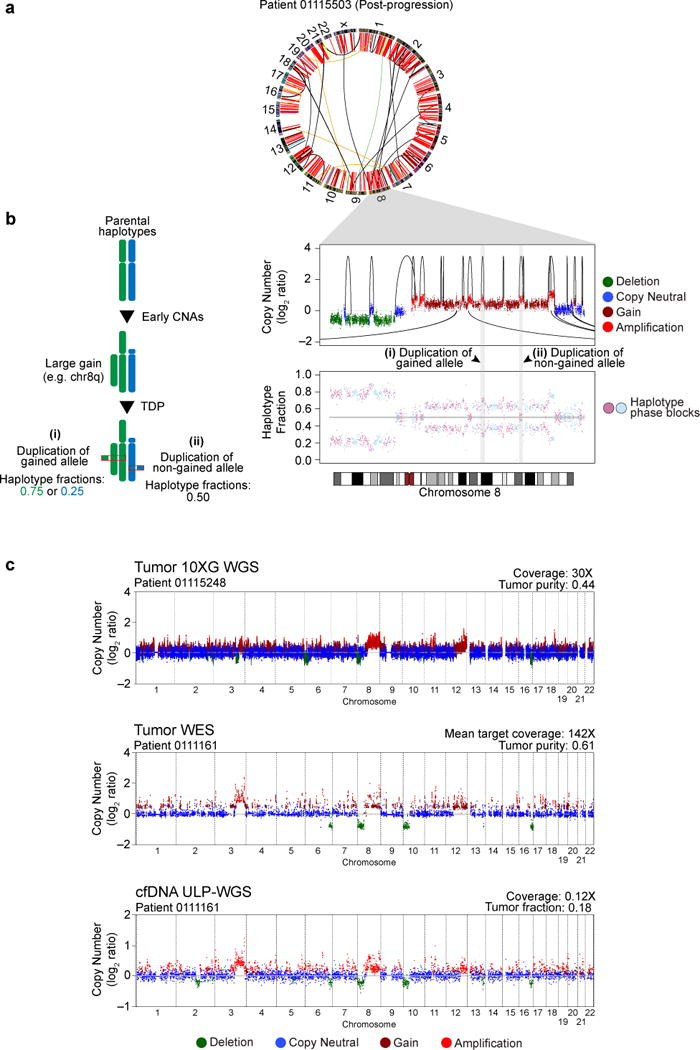
(a) CIRCOS plot for a representative TDP sample profiled by 10XG WGS. Red arcs, tandem duplications.
(b) Left: Duplications that occur on one haplotype in the setting of prior chromosome 8q gain lead to predicted haplotype fractions of either 0.75 or 0.25, depending on which allele is duplicated. Right: Chromosome 8 copy number profile (top) and haplotype fraction (bottom, alternating phase blocks colored). Intra-chromosomal tandem duplications shown by arcs on top of data points; inter-chromosomal events shown by arcs below the data points.
(c) Genome-wide copy number profiles (log2 ratio) for representative TDP samples profiled by 10XG WGS (top), WES (middle), or ULP-WGS of cfDNA (bottom).
See also Figures S1, S2, S4 and Table S6.
Four of the five TDP samples profiled by 10XG WGS had bi-allelic inactivation of CDK12, confirmed by phasing (Figure 3a). The 5th case had mono-allelic CDK12 inactivation (nonsense mutation) without evidence of a second inactivating event. Consistent with previous reports of increased rearrangements in tumors negative for ETS gene fusions (Baca et al., 2013; Wyatt et al., 2014), we observed mutual exclusivity between the TDP samples and samples with ETS fusions (p=0.0373, Fisher’s exact test) (Figure 3a).
Figure 3. The TDP in mCRPC is associated with biallelic and clonal CDK12 inactivation.
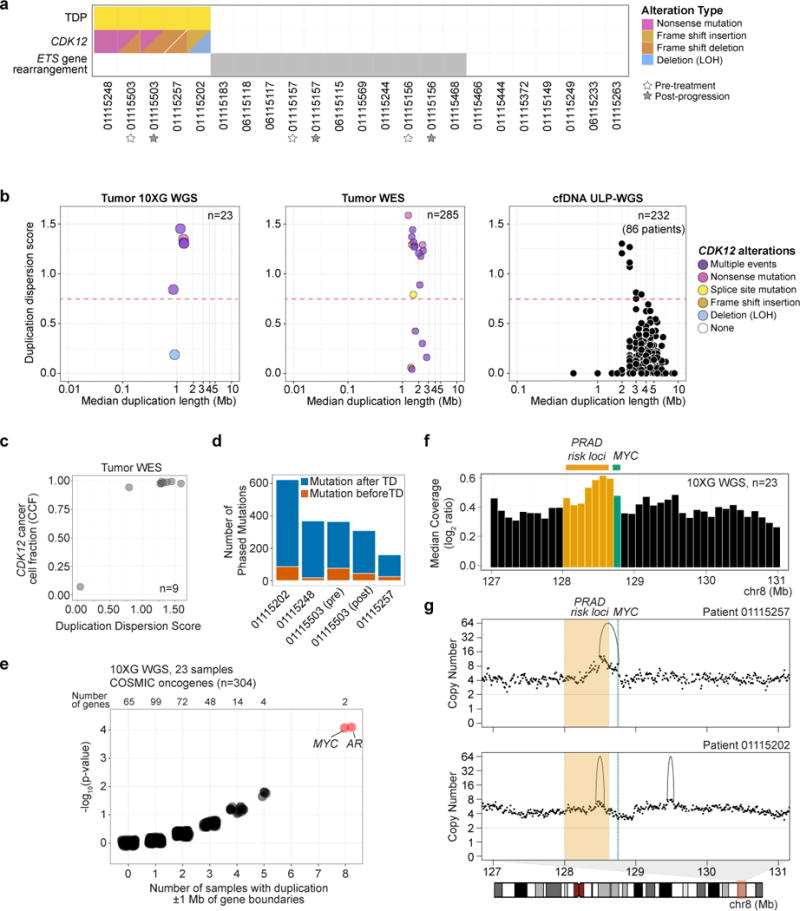
(a) TDP, CDK12 alteration, and ETS-rearrangement status in 10XG WGS mCRPC cohort.
(b) Duplication dispersion score (>0.75 defined as TDP) among mCRPC samples profiled by 10XG WGS (left), WES (middle), or ULP-WGS of cfDNA (right). CDK12 alteration status shown for WGS and WES datasets.
(c) Duplication dispersion score and CDK12 cancer cell fraction among CDK12 mutant (SNV) samples profiled by WES.
(d) Number of mutations determined to be acquired before or after duplication events in the five TDP samples from the 10XG WGS cohort.
(e) Tandem duplications within 1 Mb upstream and downstream of COSMIC oncogene boundaries in the 10XG WGS cohort. For each oncogene, the frequency (X-axis) and the p-value (binomial exact test; Y-axis) are shown with random jitter noise. Red points, Benjamini-Hochberg q-value < 0.05.
(f) Median of normalized molecule coverage near MYC. Green, MYC coding sequence. Yellow, region containing some of the prostate cancer 8q24 germline risk variants. Bin size, 100 kB.
(g) Purity-adjusted copy number profiles from representative TDP samples with duplications near MYC. Shaded region (chr8, 128.0-128.62Mb) contains tandem duplications in 10XG WGS cohort and overlaps with 8q24 prostate cancer germline risk variants.
See also Figures S1, S2, S4 and Table S6.
We next identified TDP in cohorts profiled on other sequencing platforms. We developed a metric to quantify tandem duplication dispersion across the genome (STAR Methods and Table S6). In the 10XG WGS cohort, all 5 samples displaying a large number of tandem duplications had a dispersion score > 0.75 (Figure 3b, left). These cases had a median of 150 tandem duplications per sample and a median tandem duplication span size of 1.3 Mb. In a dataset of 285 WES samples from mCRPC patients in the PCF/SU2C cohort (Armenia et al., 2018), we observed TDP with a dispersion score > 0.75 in 15 cases (5%), with a median duplication length of 2.53 Mb (Table S4, S6). TDP samples showed an enrichment for alterations in CDK12 (13/15 [87%]; p=8.73 × 10−17, Fisher’s exact test), and multiple CDK12 alterations (presumed biallelic inactivation) were seen in a majority of cases (Figure 3b, middle). CDK12 alterations trended towards clonality in samples displaying TDP (Figure 3c) and TDP samples themselves displayed multiple subclonal clusters (Table S6). Moreover, mutation phasing in all 5 TDP samples in the 10XG WGS cohort revealed that more mutations were acquired after tandem duplication events than before (p=0.008, Wilcoxon rank sum test; Figure 3d, Figure S4, and Table S6), suggesting that CDK12 inactivation and acquisition of TDP are early events followed by subclonal mutational heterogeneity.
To determine whether the TDP genomic signature could also be detected in cfDNA, we next interrogated a recently described collection of 624 ULP-WGS cfDNA samples taken from 137 unique individuals with mCRPC (Adalsteinsson et al., 2017). Of these, 232 samples from 86 patients had tumor fraction > 0.05; evidence of a TDP based on a dispersion score > 0.75 was seen in 9 samples from 4 patients (5%) (Figure 3b, right, and Table S4, S6). In 64 samples (18 patients) with both ULP-WGS of cfDNA and WES on metastatic biopsy, we observed good concordance in duplication dispersion score between the two modalities (Spearman’s ρ=0.51, p=2.1 × 10−5) (Figure S4).
We then investigated whether oncogenes can be amplified in the context of a genome-wide TDP. In the 10XG WGS cohort, we surveyed 304 oncogenes (COSMIC Cancer Gene Census) for the presence of non-transecting tandem duplications within 1MB of gene boundaries. We observed that several oncogenes were altered by tandem duplications involving the gene or neighboring sequence in multiple samples, with the most recurrently altered gene neighborhoods being near MYC and AR (8 samples each, q=0.014, binomial exact test with Benjamini-Hochberg correction, Figure 3e and Table S6). Duplications near or involving MYC were present in all 5 TDP cases (p=0.0017, Fisher’s exact test). Across the entire cohort, we observed a local peak in coverage approximately 500 kB upstream of the MYC gene, outside of the coding region, and overlapping with several previously reported prostate cancer risk alleles and elements shown to function as enhancers of MYC expression (Ahmadiyeh et al., 2010; Yeager et al., 2007; Zhang et al., 2016) (Figure 3f-g and Figure S4). These recurrent duplications in germline susceptibility loci and tissue-specific enhancers are reminiscent of findings in other cancer types (Glodzik et al., 2017; Menghi et al., 2017).
Rearrangements Reveal Persistent Selective Pressure on AR Signaling in mCRPC
We next sought to determine whether structural alterations may play a role in activating the AR axis in mCRPC, given the nearly universal importance of sustained AR signaling in this phase of the disease. Structural analysis revealed numerous and diverse somatic rearrangements surrounding AR; these usually resulted in amplification of the AR gene but occasionally breakpoints occurred within the AR itself (Figure 4 and Figure S5) (Henzler et al., 2016; Li et al., 2012). For example, in case 01115503 (Figure 4a), which displays TDP, we observed nested tandem duplications resulting in copy number gain of the AR gene and a higher-level copy number gain of a segment approximately 700 kB centromeric to the AR gene body. In sample 01115202, we observed high-level AR amplification flanked by two interchromosomal breakpoints, suggestive of AR containment within an extra-chromosomal element (Figure 4b). In sample 0115257, we observed multiple rearrangements affecting the AR locus, including fold-back inversions that resulted in laddered copy number segments, suggestive of AR copy gain driven by breakage-fusion-bridge cycles (Figure 4c). And in sample 06115115, we observed large rearrangements involving AR and crossing the centromere, raising the possibility of a ringlike structure encompassing the highly amplified gene body (Garsed et al., 2014) (Figure 4d). Rearrangements transecting negative regulators of AR, such as ZBTB16, NCOR1, and NCOR2, were also observed (Figures 4e-f and Figure S1). Several of these transection events involved tandem duplications and occured within the context of a genome-wide TDP (Figure 4a,b,c,e,f). Although singly transecting duplications may not always result in loss of function, such events have been reported to be enriched among tumor suppressor genes within the context of large-span TDPs (Menghi et al., 2017). Finally, in patient 01115157, we observed a chromoplexy chain resulting in an in-frame fusion between the N-terminus of NCOR1 on chromosome 17 and YARS on chromosome 1. This chained event is predicted to lead to disruption of the NCOR1 C-terminal domain, which has been implicated in repression of AR (Cheng et al., 2002) (Figure 4g and Table S5).
Figure 4. Diverse structural rearrangements of the AR axis.
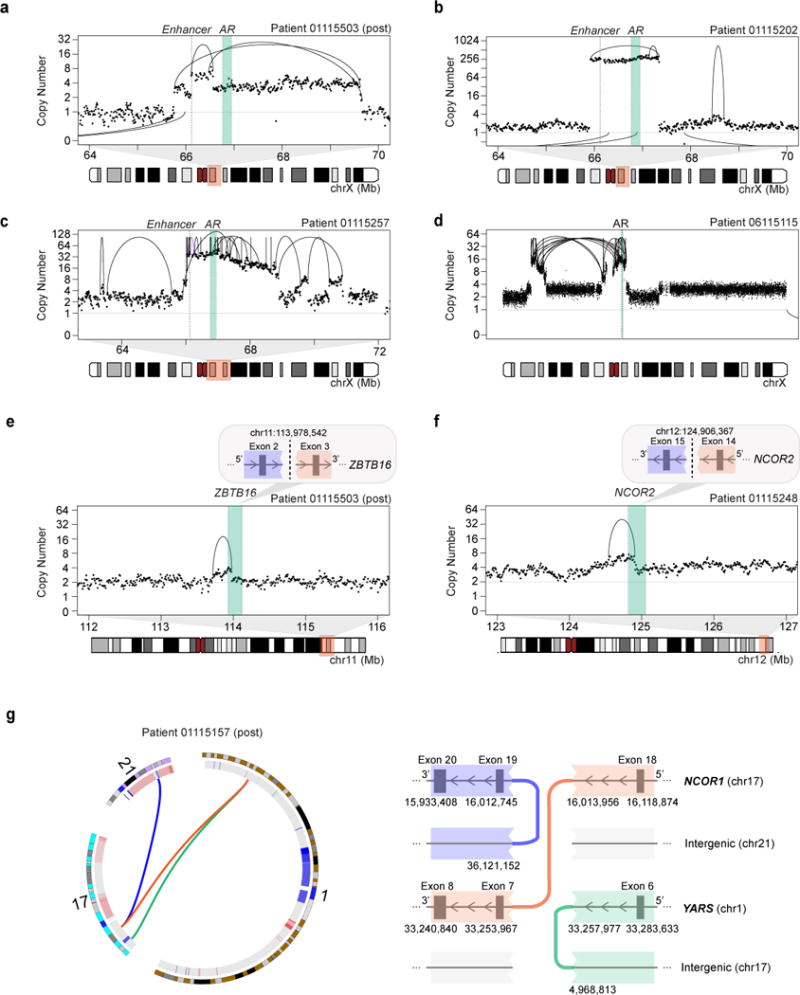
(a–d) Rearrangements involving the AR locus include: (a) simple and nested duplications, (b) high-level copy number gains, (c) amplification due to breakage-fusion-bridge cycles, and (d) trans-centromeric rearrangements. Copy number shown is purity-adjusted.
(e–g) Examples of rearrangements disrupting AR-related genes in mCRPC include duplications transecting (e) ZBTB16 and (f) NCOR2, and (g) a chained chromoplexy event resulting in disruption of the C-terminal domain of NCOR1 and production of an in-frame N-terminal NCOR1-YARS fusion transcript. Inter-chromosomal rearrangements are shown as arcs below the data points. Note: Samples 01115503, 01115202, 01115257, and 01115248 all display the TDP.
See also Figure S5 and Table S5.
Highly Recurrent Duplications of an Upstream Long-range Enhancer of the AR
Intriguingly, we noted that the peak region of copy number near the AR locus does not encompass the AR gene body (66.76-66.95 Mb) but is in fact located about 700 kB upstream (genomic bin, 66.10-66.20 Mb, Figure 5a), similar to findings described for MYC, above. This region overlaps with three DNase I hypersensitivity site (DHS) peaks in the androgen-dependent metastatic prostate cancer cell line, LNCaP, and has been shown to harbor an element that functions as a long-range enhancer of AR that is selectively activated in metastatic disease (Takeda et al.). In the 10XG WGS cohort, we noted copy number gain involving AR, most often arising via tandem duplications, in 16 (70%) samples. In all 16 cases, the upstream AR enhancer was also included in the gained segment. In addition, we observed highly selective copy number gain of the AR enhancer relative to the AR gene body in 4 (17%) additional cases, for a total of 20 cases (87%) with amplifications that include the AR enhancer (Figures 5b-d, S5; Table S7). Notably, we observed fewer gained copies of the AR enhancer in cases of selective enhancer gain as compared with cases of AR gene/enhancer co-amplification (median 2.9 vs. 8.0 copies, normalized to sample ploidy; p=0.011, two-tailed Wilcoxon rank sum test), raising the intriguing possibility that modest increases in enhancer copy number can drive AR expression comparable to higher-level gains of the AR gene (Figure 5d and Table S7). This is consistent with functional studies demonstrating that knock-in of a single additional copy of this enhancer can increase AR expression and confer castration resistance (Takeda et al.). By contrast, we observed a duplication involving the AR enhancer in only 1/54 (2%) of localized prostate adenocarcinoma specimens (Baca et al., 2013) (Figure 5f; Table S7). Thus, like other alterations involving AR, alterations in the AR enhancer are relatively specific to mCRPC.
Figure 5. Highly recurrent tandem duplications involving an enhancer of the AR in mCRPC.
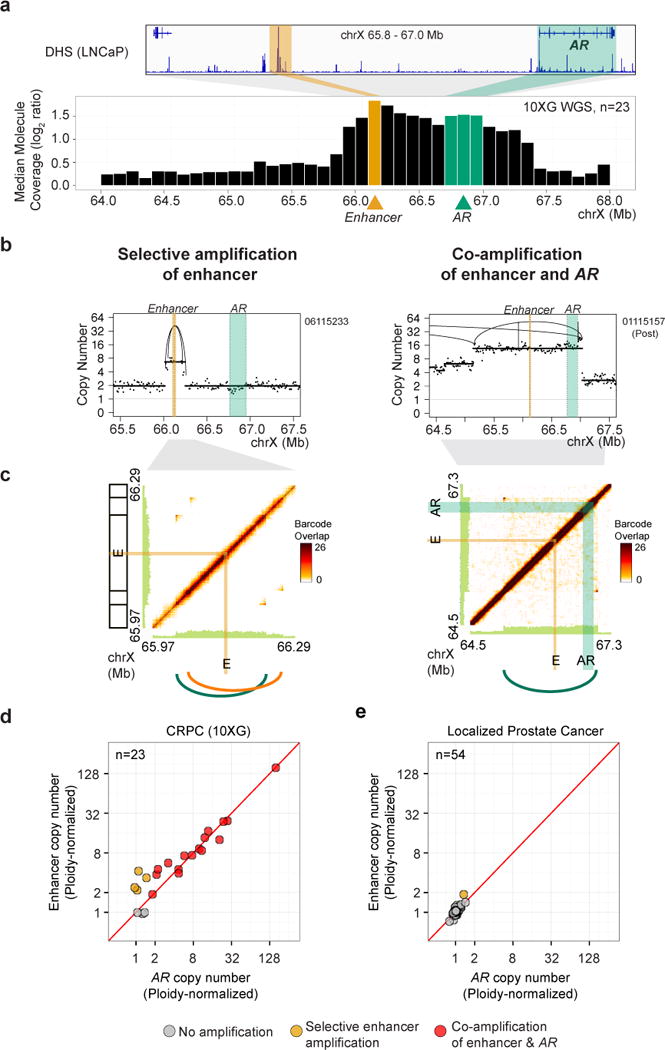
(a) Median of normalized molecule coverage near the AR gene and enhancer in the 10XG WGS mCRPC cohort; bins containing the enhancer overlaps with a DHS in LNCaP cells. Bin size, 100 kb.
(b) Purity-adjusted copy number profiles from representative samples displaying selective copy number gain involving the AR enhancer (left) and co-amplification of both the AR gene and enhancer (right). Intra-chromosomal rearrangements are shown by arcs.
(c) Barcode overlap plots for the samples shown in (b) demonstrating two tandem duplications spanning the AR enhancer (left) or a duplication involving both the AR gene and enhancer (right). Peaks in off-diagonal barcode overlap (dark orange) converge at rearrangement breakpoints.
(d) Purity-adjusted copy number (normalized to sample ploidy) at bins containing the AR enhancer (Y-axis) and AR gene body (X-axis) was used to identify samples containing gains of AR and/or AR enhancer in the 10XG WGS mCRPC cohort.
(e) Purity-adjusted copy number (normalized to sample ploidy) at bins containing the AR enhancer (Y-axis) and AR gene body (X-axis) in WGS samples from individuals with localized primary prostate cancer (Baca et al., 2013).
See also Figures S5–S7 and Table S4–S7.
We sought to validate our finding of highly recurrent duplications involving an AR enhancer in a larger cohort. By interrogating the ULP-WGS cfDNA mCRPC cohort described earlier, we were able to detect gains in the regions containing the AR enhancer and AR gene relative to surrounding regions despite low-coverage sequencing (0.1X) (Figures 6a-b). By sequencing 14 of these cfDNA samples to higher depth (median coverage 20.4X, range 15.9-61.1X), we observed excellent concordance of copy number between ULP-WGS and higher depth WGS at both the AR gene (Spearman’s ρ=1.00) and AR enhancer (Spearman’s ρ=0.96) loci (Figure 6b, 7, S6, and Tables S2, S5). Across ULP-WGS cfDNA samples from 86 patients, we observed selective enhancer amplification in 13 cases (15%) and co-amplification of the enhancer and the AR gene body in 47 cases (55%), for a total of 60 cases (70%) with amplifications involving the enhancer; additionally, selective amplification of the AR gene body was seen in 9 cases (10%) (Table S7).
Figure 6. Gains of the AR enhancer are detectable in ULP-WGS cfDNA and are associated with increased nucleosome spacing and higher AR expression.
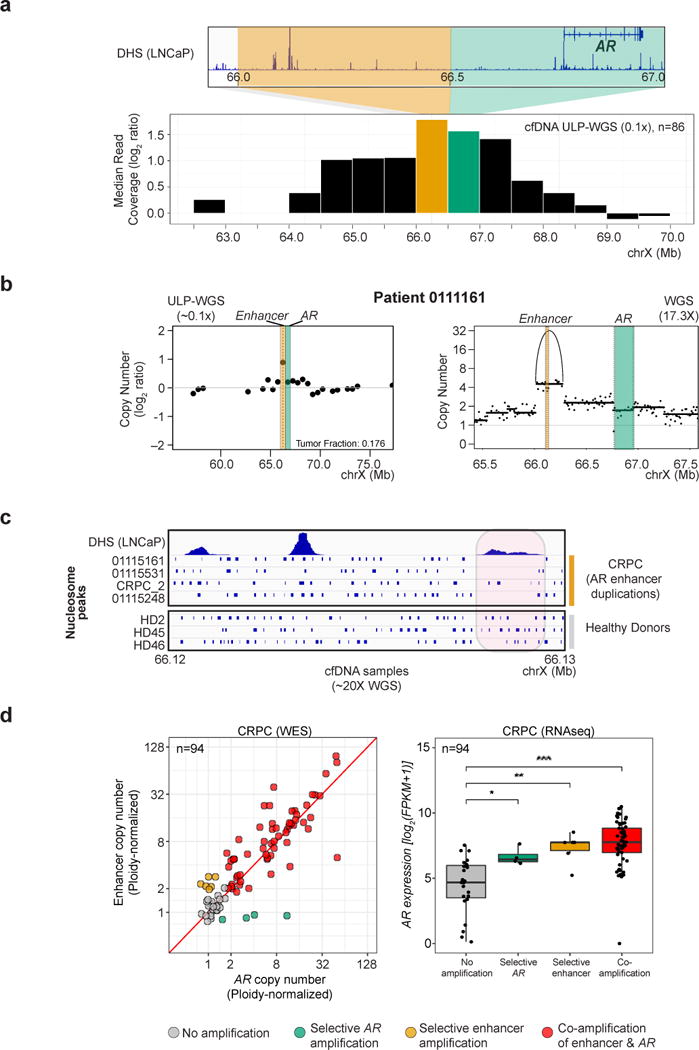
(a) Median of normalized read coverage near AR gene and enhancer in the ULP-WGS cfDNA mCRPC cohort (maximum tumor fraction per patient used); bin containing the enhancer overlaps with a DHS in LNCaP cells. Bin size, 500 kb.
(b) Copy number profile of a representative sample displaying selective gain involving the AR enhancer in cfDNA. For ULP-WGS data (~0.1X coverage, left), each point represents copy number (log2 ratio) within a 500 kb genomic bin; bins containing the AR gene and enhancer are shaded in green and orange, respectively. For deeper WGS data (17.3X coverage, right), the purity-adjusted copy number profile at 10 kb genomic bins is annotated with copy number segments (lines) as well as rearrangements (arcs).
(c) Nucleosome position (blue bars) inferred from cfDNA fragmentation pattern in the region of the AR enhancer in 4 patients with selective gain of the AR enhancer region (top) and 3 healthy donors (bottom), using deep WGS (~20X) of cfDNA.
(d) (left): Purity-adjusted copy number (normalized to sample ploidy) at bins containing the AR enhancer (Y-axis) and AR gene body (X-axis) in WES samples from individuals with mCRPC (Robinson et al., 2015). Only samples with available paired transcriptome data are shown. (right): AR expression in samples shown at left, as determined from paired transcriptome data. *, p < 0.05; **, p < 0.01; ***, p < 0.0001 by Wilcoxon rank sum test.
See also Figure S5–S7 and Table S4–S7.
Figure 7. Rearrangement pressure on the AR locus in the setting of androgen pathway blockade.
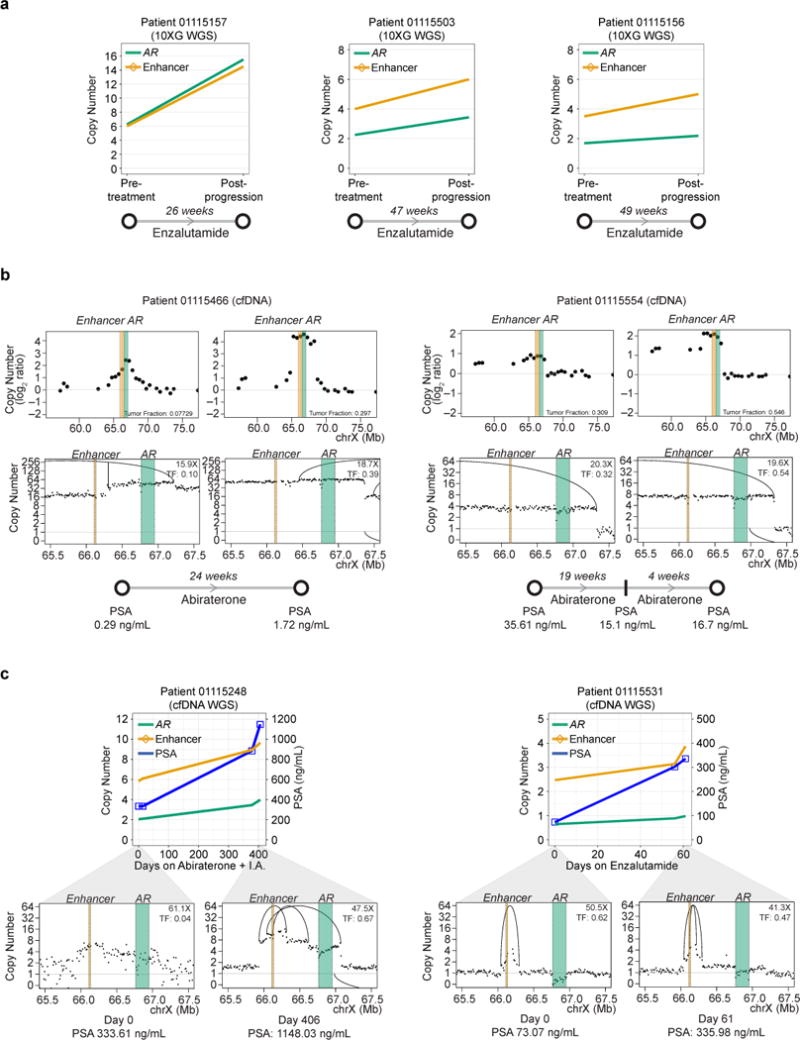
(a) Purity-adjusted copy number status at the AR gene and enhancer loci in three paired 10XG WGS tumor biopsy samples taken from patients prior to and after progression on enzalutamide.
(b) Copy number profiles at AR locus in cfDNA of two patients collected either early on treatment with abiraterone (left panels) or shortly after PSA progression (right panels). Top panels show ULP-WGS log2 ratio copy number profiles (~0.1X coverage). Lower panels show tumor-fraction-adjusted copy number for deep WGS of these samples (15-20X coverage).
(c) Top, Tumor-fraction-adjusted copy number in WGS of cfDNA at the AR gene and enhancer loci during treatment with abiraterone (Patient 01115248) or enzalutamide (Patient 01115531). Bottom, Tumor-fraction-adjusted copy number profiles at the first and last time points for each patient. Rearrangements are indicated by arcs. I.A, investigational agent; TF, tumor fraction.
See also Figure S5-S7 and Tables S4-S5.
As a complement to functional characterization of the AR enhancer region (Takeda et al.), we sought genomic evidence that this locus functions as an enhancer. We inferred nucleosome positions across the enhancer region in deep sequencing data of cfDNA samples in 4 individuals with duplications involving the AR enhancer and 3 healthy donors (STAR Methods). In individuals with AR enhancer gain, but not healthy donors, nucleosome spacing was increased in regions of the enhancer element that overlapped with DHS peaks in LNCaP cells (Figure 6c). We surveyed Hi-C interaction data on LNCaP cells from the ENCODE project and found that the AR enhancer and AR gene body lie within a putative topologically-associated domain (TAD) suggesting that duplication of the enhancer element, either selectively or in tandem with the AR gene body, allows for increased AR expression without disrupting topological boundaries (Figure S7).
Finally, we sought to determine whether alterations in the AR enhancer are associated with higher AR expression. As the majority of samples subjected to 10XG WGS did not have sufficient material remaining for transcriptome profiling, we turned to a recently reported mCRPC cohort of paired WES and transcriptome sequencing (Robinson et al., 2015). Although capture probe sets used for WES covered the AR gene and not the enhancer region, we were able to detect an appreciable number of off-target reads aligning to the AR enhancer locus in 205 samples (median of 732 reads across 50 kB bin containing the AR enhancer, Figure S6, STAR Methods). We compared the WES copy number with results from 10XG WGS for 9 samples that were profiled on both platforms, and observed excellent concordance between AR gene (on-target, Spearman’s ρ=0.88) and AR enhancer (off-target, Spearman’s ρ=0.85) copy number (Figure S6).
Overall, using this approach, we found 21 (10.3%) cases with selective AR enhancer amplification and 124 (60.5%) cases with co-amplification of AR gene and enhancer; thus, overall, 145 cases (71%) involved alterations of the AR enhancer. Selective gains of the AR gene were seen in 6 cases (3%). These frequencies of alteration were similar to those seen in ULP-WGS and 10X WGS cohorts (Figure S6, Table S7). Analysis of paired transcriptome sequencing data on 94 individuals revealed that AR expression was significantly increased in the cases with amplification of the AR enhancer, the AR gene, or both, as compared with cases without amplification at these loci (p=0.0012, p=0.021 and p=5.9×10−8, respectively by Wilcoxon rank sum test), and confirmed by multivariable analysis (Figure 6d, STAR Methods). This supports the notion that gains of the AR enhancer and AR gene both drive increased AR expression during the castration-resistant phase of prostate cancer.
Persistent Selective Pressure on AR and AR Enhancer During Androgen Pathway Inhibition
We next more closely interrogated several cases in which paired biopsy or cfDNA samples were available from the same individual at various time points during treatment with a next-generation AR pathway inhibitor, including at the time of progression. Strikingly, in all three paired metastatic biopsy samples and four paired cfDNA samples, we observed persistent selective pressure on the AR locus in the setting of potent androgen pathway blockade. For example, analysis of three paired 10XG WGS metastatic biopsy samples showed gains at both the AR gene and enhancer loci upon progression on enzalutamide in all cases (mean 1.77-fold increase in AR copy number and 1.78-fold increase in AR enhancer copy number adjusted for sample purity, Figure 7a).
In cases 01115466 and 01115554, we observed progressive increase in copy number (corrected for differences in purity between time points) at the AR enhancer locus (01115466) or AR enhancer and gene loci (01115554) with progression on abiraterone; these gains were detected in ULP-WGS of cfDNA and confirmed on deeper sequencing (Figure 7b). Finally, in two additional cases, we sequenced cfDNA samples from multiple time points during therapy with abiraterone or enzalutamide (patient 01115248, 4 time points; patient 01115531, 3 time points) to deep coverage (Figure 7c). In both cases, we observed copy number gains of both the AR gene body and AR enhancer tracking with PSA progression (Figure 7c). These copy number increases – which were seen despite correcting for differences in tumor fraction between time points – may be a result either of new rearrangements occurring under pressure of androgen pathway blockade, or of outgrowth of pre-existing clones under selective pressure. The variable tumor content of our samples and sensitivity of SV calling (particularly in cfDNA) do not allow us to reliably distinguish these possibilities.
Overall, enhancer gain was observed in all 12 post-progression 10XG WGS biopsy samples. Relative increases in enhancer copy number were noted during progression on treatment in all 3 paired 10XG WGS biopsy samples and in all 4 individuals from whom multiple timepoints of cfDNA were analyzed (Figures 7 and S1). Thus, gains of this regulatory region may be nearly universal when responses to next-generation androgen pathway inhibitors are lost. Interestingly, we also observed that all five 10XG WGS and all 15 WES cases displaying TDP harbored gains involving AR gene and/or enhancer (p=0.0135, Fisher’s exact test, Tables S6-S7). Although somatic alterations in AR region are pervasive even outside of TDP, the data suggest that TDP may be one possible mechanism leading to AR locus duplication events involving the AR enhancer. In sum, rearrangements leading to increased AR locus copy number or disruption of AR negative regulators are ubiquitous in mCRPC, both during the initial development of castration-resistance and in the setting of progression on the newest androgen pathway inhibitors.
DISCUSSION
Copy number gains near the AR locus have long been noted to be pervasive in mCRPC and the presumed target of these gains has been the AR gene (Visakorpi et al., 1995). Our study sheds light on the complexity of rearrangements resulting in such gains and provides insight into the mechanisms of AR activation in mCRPC. Moreover, we show here that amplification of the AR gene body most often co-occurs with duplication of a newly-characterized long-range enhancer of the AR (Takeda et al.). Both elements are responsible for maintaining increased AR expression and activated AR signaling in mCRPC. The frequent amplification of both elements in response to androgen pathway inhibitors has implications for our understanding of the dominant mechanisms of resistance to potent androgen pathway blockade.
Our findings add to a growing list of oncogenes activated by alterations in enhancer elements (Glodzik et al., 2017; Herranz et al., 2014; Mansour et al., 2014; Northcott et al., 2014; Shi et al., 2013; Weischenfeldt et al., 2017; Zhang et al., 2016). The AR enhancer appears to be predominantly altered by tandem duplications that occur alone or in combination with duplication of the gene body. As both elements appear to be located within the same TAD, this likely results in activation of AR expression without disruption of underlying topological domains, reminiscent of alterations in lineage-specific super-enhancers in other tumor types (Glodzik et al., 2017; Zhang et al., 2016) but distinct from enhancer-hijacking mechanisms (Hnisz et al., 2016; Northcott et al., 2014; Weischenfeldt et al., 2017).
Selective activation of the AR enhancer in mCRPC provides further evidence that a stepwise amplification of AR signaling is key in the transition to castration-resistance (Chen et al., 2004; Visakorpi et al., 1995). It will be of interest to characterize the factors that bind to and activate the AR enhancer, and to determine whether AR and/or its ligand-independent splice variants are capable of binding to the enhancer to increase AR expression under castrate conditions. Targeting enhancer-bound factors or chromatin readers (Asangani et al., 2014) may prove therapeutically effective in enhancer-duplicated cases. By contrast, enhancer duplication and rearrangements at the AR locus may be biomarkers of primary or acquired resistance to androgen-pathway inhibition, analogous to tumors expressing androgen-receptor splice variants (Antonarakis et al., 2014). Further interrogation of these and other alterations in the AR axis may allow for better stratification of patients likely to benefit from hormonal blockade versus cytotoxic chemotherapy.
The CDK12-associated TDP in mCRPC appears to represent a distinct structural class of prostate cancer, alongside ETS-rearranged and SPOP-mutant tumors (Baca et al., 2013; Barbieri et al., 2012; Boysen et al., 2015; Wyatt et al., 2014), and appears distinct from previously reported short-span BRCA1-associated TDPs (Glodzik et al., 2017; Menghi et al., 2016, 2017). Genome-wide TDPs have been variously described in multiple lineages and likely comprise a class of genomic configurations that differ in genetic background, duplication span-size, and mechanisms of driving oncogenesis; careful classification schemes will be important in distinguishing these phenotypes (Glodzik et al., 2017; McBride et al., 2012; Menghi et al., 2016, 2017; Ng et al., 2012; Wyatt et al., 2014).
While a similar CDK12-associated phenotype to that described here has been characterized in serous ovarian cancer (Popova et al., 2016), a recent pan-cancer analysis found TDP to be nearly absent in prostate cancer (Menghi et al., 2017). These studies have relied primarily on available genomic data from localized prostate cancer and the increased frequency of CDK12 alteration in mCRPC as compared with localized disease may explain this discrepancy (Armenia et al., 2018). Alterations in DNA repair pathway components (including CDK12) have been linked to sensitization to platinum agents and PARP inhibitors (Bajrami et al., 2014; Johnson et al., 2016; Mateo et al., 2015; Pomerantz et al., 2017; Pritchard et al., 2016; Riaz et al., 2017). Our ability to detect the TDP genomic instability signature through ULP-WGS of cfDNA may have implications for its use as a potential noninvasive biomarker in predicting response to these agents in mCRPC.
At this point, the correlative nature of our findings makes it difficult to conclude whether CDK12 loss directly contributes to oncogenesis via the tandem duplicator phenotype (i.e. by creating selective pressure for duplications that amplify oncogenes and/or transect tumor suppressor genes), or whether CDK12 loss drives prostate cancer through an independent mechanism. We favor the former explanation based on the compelling evidence for duplications of coding or regulatory regions of oncogenes (i.e. MYC, AR) and disruptive rearrangements involving tumor suppressor genes (i.e. NCOR1/2, ZBTB16, PTEN) in TDP samples. A global TDP may provide a means to coordinately activate or inactivate several genes, and various combinations of such events may therefore emerge under selective pressure. Our finding of recurrent duplications near MYC, in regions overlapping prostate cancer susceptibility loci, mirrors similar findings in breast (Glodzik et al., 2017) and ovarian cancers (Menghi et al., 2017). Regions containing germline risk variants may function as tissue-specific enhancers, and being in open chromatin, may be prone to double-strand breaks and repair by long-span tandem duplications.
Although the linked-read 10XG platform has been previously applied to a limited number of clinical specimens (Greer et al., 2017; Spies et al., 2017; Zheng et al., 2016), this, to our knowledge, is the first use for comprehensive molecular characterization of a clinical cohort. Our initial results indicate that 10XG WGS can be readily applied to clinical specimens, including those with limited input DNA. We leveraged the use of phasing afforded by this platform in several instances, including to confirm biallelic mutations (i.e. of CDK12), to reconstruct haplotype-resolved complex SVs (i.e. of PTEN), and to confirm that the TDP simultaneously affects both haplotypes of a given sample. Overall, our data suggest that linked-read sequencing holds promise in improving read alignment, mutation/indel calling, and SV detection and resolution over current methods. Future studies will be required for more formal benchmarking of 10XG WGS against traditional short-read sequencing.
In summary, this study highlights the power of linked-read WGS to define the structural alterations driving castration-resistance and therapeutic resistance to androgen pathway blockade in mCRPC. A picture emerges of complex and diverse genetic alterations converging on a central need to sustain AR signaling in the face of highly potent androgen pathway blockade in mCRPC. Additional studies will be helpful in revealing the extent to which persistent addiction on AR versus an escape to androgen indifference contribute to resistance to androgen pathway inhibitors (Arora et al., 2013; Bluemn et al., 2017; Mu et al., 2017), and will help in framing strategies for developing the next generation of targeted therapeutics for mCRPC. More broadly, the novel alterations we have identified and significant differences from localized prostate cancer, even with a relatively small cohort, suggest a rich future for genomic discovery in the noncoding mCRPC genome.
STAR METHODS
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by Lead Contact Matthew Meyerson (matthew_meyerson@dfci.harvard.edu).
Experimental Model and Subject Details
Human Subjects
Cancer genome sequence data were generated through informed consent as part of previously published sequencing cohorts for cell-free DNA (Adalsteinsson et al., 2017) and metastatic tumor biopsies from individuals with mCRPC (Armenia et al., 2018; Robinson et al., 2015). Site-specific protocols under which individuals were accrued to each cohort are described in these publications (Adalsteinsson et al., 2017; Robinson et al., 2015). Affected individuals provided written informed consent to obtain fresh tumor biopsies and/or blood for genomic analysis of tumor and germline samples.
For metastatic tumor biopsies subjected to 10X Genomics WGS, samples came from individuals with mCRPC being considered for either standard of care enzalutamide or abiraterone acetate, or clinical trial investigating abiraterone acetate in combination with investigational agents (either ARN-509 or cabozantinib) (Armenia et al., 2018; Robinson et al., 2015). Sample size was 23 tumors, each with matched germline control (peripheral blood). 12 samples were from individuals with mCRPC taken prior to initiation of next-generation androgen pathway inhibitor; 11 samples were from individuals with mCRPC and were taken after progression on next-generation androgen pathway inhibitor. Of these samples, 6 (3 pairs) were pre- and post-progression samples taken from metastatic sites in the same individual. Age and gender characteristics of the cohort were as follows: 100% male, median age 70 (range 54–79 years old).
Method Details
Sequence Data and Sample Processing
HMW DNA preparation for 10X Genomics WGS
High molecular weight DNA was extracted from prostate tumor tissue samples using the MagAttract HMW DNA Kit (Qiagen), with the exception of the following four cases in which previously extracted DNA was used: 6115115, 6115117, 6115118, and 6115233. Starting with ~25 mg of tissue from a frozen core biopsy, samples were lysed overnight with proteinase K, and subsequently treated with RNase A to remove RNA. DNA was then bound to magnetic beads in the Magattract Suspension and washed with buffer and water before elution from the beads into buffer AE (10 mM Tris-Cl. 0.5 mM EDTA). The extracted genomic DNA was quantified using the Quant-it Picogreen assay kit (Thermo Fisher) on a Varioskan Flash Microplate Reader (Thermo Fisher).
For germline DNA samples, pre-extracted DNA was size selected using 750 ng of DNA on the PippinHT platform (Sage Science) according to the manufacturer’s instructions, using an 0.75% agarose cassette with a target range of 40 kb to 80 kb. After size selection, samples were quantified in triplicate using the Quant-it Picogreen assay kit (Thermo Fisher) on a Varioskan Flash Microplate Reader (Thermo Fisher) and normalized to a concentration of 0.5 ng/uL with TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0). Prior to 10X Genomics WGS library construction, genomic DNA fragment size distributions were determined with a Caliper Lab Chip GX (Perkin Elmer) to quantify DNA above 40 kb in length.
10X Genomics WGS library construction
DNA samples were normalized to a starting concentration of 0.5 ng/ul with TE (10 mM Tris-HCl, 1 mM EDTA, pH 8.0). 10X WGS Libraries were constructed using the 10X Chromium protocol (10X Genomics), starting with 1.2 ng of DNA for each sample. Resulting library fragment sizes were determined using the DNA 1000 Kit and 2100 BioAnalyzer (Agilent Technologies) and quantified using qPCR (KAPA Library Quantification Kit, Kapa Biosystems). The finished libraries were sequenced to ~30X coverage on an Illumina HiSeqX platform, using paired 151 bp reads with a single 8 bp index read. The resulting sequencing BCL files were processed by the Long Ranger Pipeline (10X Genomics) for alignment, variant discovery, and phasing.
WGS of cfDNA
Cell-free DNA (cfDNA) samples were collected and libraries prepared as previously described (Adalsteinsson et al., 2017). We selected 10 cfDNA samples for sequencing to higher depth (~20-48X) with 100/101 bp paired-end reads on an Illumina HiSeq2500/HiSeq4000 in high-output mode or Illumina Novaseq (200 cycles, 100 bp reads paired-end).
Quantification and Statistical Analysis
Sequence data processing
WES and cfDNA data
Data processing and read alignment of tumor and normal samples were performed using the Broad Institute Picard pipeline with the hg19 human genome build as described (Armenia et al., 2018; Robinson et al., 2015). For cfDNA ULP-WGS using Illumina, sequenced reads were analyzed by the Broad Picard pipeline with bwa 0.5.9, resulting in BAM files aligned to hg19 with calibrated quality scores.
10X Genomics WGS - Long Ranger pipeline
Samples were demultiplexed and paired end fastq files with matching barcode index files were generated with the Long Ranger (v2.1.2) mkfastq function. The Long Ranger pipeline (10X Genomics) was run on Google cloud using the Atmo GCE Instance Launcher (v0.2.1) for Long Ranger (v2.1.2) with the pre-built b37-GRCh37 reference available from 10XG and GATK (v3.5) as the default variant caller mode. The matched normal and tumor samples were run separately, as the pipeline currently does not support running in paired analysis mode. The Long Ranger pipeline performs alignment using the “Lariat” aligner, which bins read-pairs containing the same molecular barcode identifier into read clouds and performs the alignment of these read-pairs simultaneously with the prior knowledge that these read-pairs originate from a small number of larger DNA molecules.
Mutation and Indel Analysis
10X Genomics WGS - Somatic single nucleotide variants
Somatic SNV calling was performed using MuTect v1.1.6 (Cibulskis et al., 2013) with the Lariat aligned tumor and normal bam files as matched input from each sample. The variants were further annotated using Oncotator v1.9.1.0 (Ramos et al., 2015). The resulting wig files generated by MuTect were used to calculate the coverage of sites with sufficient power for SNV detection. The total number of SNVs were divided by this number of sites to calculate the mutation rate as mutations/Mb.
To eliminate systematic errors in SNV calling due to 10X Genomics technology and to avoid calling unannotated germline variants in previously inaccessible genomic regions, we applied a modified version of LoLoPicker (Carrot-Zhang and Majewski, 2017) adapted to 10XG data to further filter MuTect called variants. We used 50 in-house normal samples sequenced by the 10X technology as a panel of normals for LoLoPicker’s algorithm. Moreover, we implemented a new filter in LoLoPicker to exclude low-confidence variants from reads flagged to originate from separate haplotypes (HP tags), under the assumption that low-allelic fraction variants or poorly covered variants from both haplotypes are likely germline variants or false positives. In brief, for variants with less than 5 reads supporting the variant allele, we extract the variant reads with high phasing quality (PC >= 30) and count the number of reads originating from HP_1 and HP_2. If a variant site has low support (< 5 reads) for the altered (variant) allele and has high phasing quality reads from both haplotypes (HP_1 > 0 and HP_2 > 0), the variant site is excluded. Approximately 5% MuTect called variants were excluded by the haplotype filter. Finally, calls by Lolopicker were intersected with all variants passing filter called by Mutect to generate a combined SNV call set.
10X Genomics WGS - Indels
SvABA (Wala et al., 2018) (May 16, 2017 revision [commit 4a0606e]) was run on the Lariat/Long Ranger aligned BAM files with matched tumor and normal input. The resulting breakpoints were refiltered to require dbSNP sites to have a log odds ratio of > 6 and other sites a score of > 2.5 to be classified as somatic variants. The resulting somatic indels passing filter were then intersected with all somatic indels called by Strelka v1.0.15 (Saunders et al., 2012) with the following parameters different from default: sindelNoise = 0.000001, minTier1Mapq = 20 and extraStrelkaArguments = –ignore-conflicting-read-names. Indels were annotated with Oncotator (Ramos et al., 2015). For tumor and matched normal samples, the alternate and reference allele counts are given as the number of tier 1 reads supporting an indel and reference as counted by Strelka. The total number of Indels were divided by the number of base pairs with sufficient power, as estimated by MuTect to calculate the Indel rate as Indels/Mb.
10X Genomics WGS - Germline variants
Candidate germline variants were called in the matched normal samples using Long Ranger (for SNVs and indels) and SvABA (for indels). Variants in any of the 72 genes associated with mCRPC or DNA repair pathways were annotated with ANNOVAR (v2017Jun01)(Wang et al., 2010), including annotations of population allele frequencies from the Exome Aggregation Consortium (ExAC v0.3), status in dbSNP (version 147) and predictions of functional effects by MutationTaster, Polyphen2, SIFT and CADD v1.3. Synonymous SNVs, in-frame indels, as well as variants predicted to have non-deleterious functional effects or population allele frequencies greater than 10% were not reported. The remaining variants were cross-referenced with the ClinVar database (accessed Oct 6th, 2017), and only variants annotated as pathogenic or frameshift indels not annotated in ClinVar are reported.
WES Data - Mutation and Indel Analysis
SNV and indel calls were available from 311/325 whole exome sequenced metastatic prostate cancer samples from the SU2C as previously described (Adalsteinsson et al., 2017; Armenia et al., 2018; Robinson et al., 2015). To investigate the clonality of mutations in the WES tumors, we applied PyClone v0.13.0 (Roth et al., 2014) to analyze the SNVs. Copy number results generated from TITAN was used as input (see Copy Number Analysis - WES data), and the parameters used were -iterations 10000, -minDepth 50, -burnin 1000. Out of the 311 samples, 298 had sufficient number of SNVs, read depth, and available copy number results to produce PyClone results. For each sample, we summarized the number of clusters as output by PyClone, based on the cancer cell fraction (CCF) of the SNVs, but required each cluster to have a minimum of two SNVs to be counted. For CDK12 mutations, the mean CCF was calculated for the clusters containing the CDK12 SNVs. All samples reported passed purity and median absolute deviation thresholds and had at least one CDK12 non-silent SNV with read depth > 50.
Copy number analysis
10X Genomics WGS data
We modified the standard workflow of TITAN (Ha et al., 2014) to perform copy number analysis of 10X Genomics data. The code for this workflow can be found here: https://github.com/gavinha/TitanCNA/. A schematic showing use of 10X Genomics WGS to analyze haplotype-based copy number is shown at the following https://github.com/gavinha/TitanCNA/wiki/Haplotype-based-copy-number.
The workflow consists of the following steps:
Molecule coverage was used to represent the abundance of DNA at specific loci, instead of read-based coverage. This approach takes advantage of the high molecular weight molecules during the 10X sample processing. We used the bxtools tile (https://github.com/walaj/bxtools#tile) tool to extract the number of unique barcodes from reads aligned within each non-overlapping 10 kb window (bin) across the genome. A molecule was counted as overlapping a bin if there are >= 2 reads with the same barcode that are aligned within the bin. Molecule coverage was extracted for both tumor and matched normal samples.
The molecule coverage was corrected for GC-content and mappability biases, independently for tumor and normal using ichorCNA v0.1.0 (Adalsteinsson et al., 2017). The log2 copy ratio at each bin lt was computed as the corrected molecule coverage of the tumor (rTum) normalized by the matched normal molecule coverage (rNormal) at each bin, . For autosomes, only the log2 copy ratios were retained from the intermediate step of ichorCNA, while for chromosome X, ichorCNA segment boundaries were retained. Note that the copy number predictions by ichorCNA are not used. ichorCNA parameters used: –includeHOMD TRUE, –normal 0.5, –ploidy 3, –txnE 0.9999999 –txnStrength 10000000 –lambda 10000. Remaining parameters used are defaults as specified in https://github.com/gavinha/TitanCNA/blob/master/scripts/TenX_scripts/getMoleculeCoverage.R
Haplotype-based copy number analysis requires the phasing information from the Long Ranger output. Long Ranger provides phased heterozygous SNPs, along with haplotype information indicating the consecutive series of SNPs linked to the same phase block (denoted by PS tags in BAM file). From the Long Ranger output of the matched normal, heterozygous SNPs are selected based on minimum depth >= 10, minimum QUAL >= 100, variant allele frequency (VAF) between 0.25 and 0.75. See the script to extract these SNPs: https://github.com/gavinha/TitanCNA/blob/master/scripts/TenX_scripts/getPhasedHETSitesFro mLLRVCF.R from the Long Ranger VCF.
Tumor haplotype-based coverage was computed in the tumor sample using the phased SNPs and the haplotype phase block information from the normal sample. (i) For each normal heterozygous SNP identified in the previous step, the allele counts, a, were extracted from the tumor sample. Reads with mapping quality < 20 and base quality (at the SNP locus) < 10 were excluded. (ii) The genome was divided into 100 kb non-overlapping bins. If a bin overlapped multiple phase blocks, then the bin was split into smaller bins such that each bin overlaps only one haplotype phase block. The final set of bins should each only overlap one haplotype phase block. (iii) For each bin i, we computed the sum of the allele counts for all SNPs phased to haplotype 1 and haplotype 2 . The allele count sums for the major haplotype is the and minor haplotype is the for bin i, which we use to compute the haplotype fraction .
Extensions to TITAN (molecule coverage and haplotype-based CN) are included in TitanCNA R package v1.15.0. The log-transformed normalized molecule coverage lt for each 10 kb window t is modeled using a Gaussian distribution. The haplotype fraction hfi for bin i replaces the allelic fraction model and is assumed to follow a Gaussian distribution. TitanCNA still analyzes the observed data at the level of each SNP j, which are assigned the normalized molecule coverage of the overlapping window t and the haplotype fraction of the overlapping bin i. The resulting emission model is a bivariate Gaussian model for the joint distribution of the two data types, Dj|G = g ∼ N(μg, Σg), where Dj = (lj, hfj) for SNP j, μg is the joint mean using the 3-component mixture to represent tumor purity and subclonal proportions (Ha et al., 2014), and Σg is the covariance for allelic copy number state g. The mean is modeled using a Gaussian prior distribution and the covariance is modeled using the inverse-Wishart distribution. Inference of parameters is performed using the expectation-maximization algorithm, and Viterbi was used for segmentation following parameter estimation.
Solutions were generated for 1 to 3 number of clonal clusters and ploidy initializations for 2 to 4. Optimal solutions were first selected by determining the optimal ploidy initialization. This was done by finding the consistently larger log-likelihood between the different ploidy initializations when comparing the solutions with the same number of clonal clusters. Then, when the optimal ploidy initialization is determined, the solution with the optimal number of clonal cluster is selected using the minimum S_Dbw validity index (using both log ratio and allele ratio). The script for this analysis is found in https://github.com/gavinha/TitanCNA/blob/master/scripts/R_scripts/selectSolution.R. The TitanCNA arguments used for the 10X analysis: maxCN = 8, alphaK = 5000, txn_exp_len = 1e20, txn_z_strength = 1, minDepth=10, maxDepth=1000, –haplotypeBinSize 1e5 –phaseSummarizeFun sum –alleleModel Gaussian –alphaR 5000. Default values were used for remaining arguments. For more details of other arguments and the TitanCNA analysis, see https://github.com/gavinha/TitanCNA/blob/master/scripts/TenX_scripts/titanCNA_v1.15.0_TenX.R.
The copy number results from TITAN (autosomes) and ichorCNA (chromosome X) were combined. We specified TITAN to model up to 8 as the maximum number of copies, which may not reflect potentially higher number of copies. Therefore, we modified the copy number prediction for events with 8 copies by transforming the original log2 ratio value lt at bin t, while adjusting for tumor purity (α) and tumor ploidy (ϕ), as described in the TITAN model (Equation 1) into a corrected copy number ĉTum,
| (1) |
| (2) |
where cNorm = 2 for autosomes. All of chromosome X copy number results from ichorCNA were also corrected in order to use TITAN estimates of purity and ploidy. For chromosome X correction, cNorm = 1. Note that this correction only applies to total copy number, not allelic copy number.
Output SCNA state definitions: HET—heterozygous diploid, two copies; DLOH—deletion LOH, one copy; NLOH—copy neutral LOH, two copies; GAIN—copy number gain, three copies; ALOH—amplified LOH; three or more copies, ASCNA—allele-specific copy number amplification; four or more copies; BCNA—balanced copy number amplification; four or eight copies; UBCNA—unbalanced copy number amplification; five or more copies. The parameters for the optimal solutions are listed in Table S4.
WES Data
We obtained WES samples from published studies (Armenia et al., 2018; Robinson et al., 2015), totaling 325 tumor samples with matched normals. The standard workflow of TITAN for WES was used. Read counts were computed at 50 kb bins overlapping the Illumina exome bait set intervals. Centromeres were filtered based on chromosome gap coordinates obtained from UCSC for hg19, including bins that are 100 kb flanking up- and downstream of the gap. The read coverage in each bin across the genome was corrected for GC content and mappability biases independently for tumor and germline samples using ichorCNA v0.1.0. The loess curve fitting for GC-correction was performed on autosomes but chromosome X was also corrected using this fit. Heterozygous SNPs were identified from the matched germline normal sample using Samtools mpileup. Only SNPs overlapping HapMap3.37 were retained. The reference and non-reference allele read counts at each heterozygous SNP were extracted from the tumor sample. SNPs were not analyzed in chromosome X. Copy number analysis was performed using TitanCNA R package v1.15.0. Solutions were selected using the same approach as for 10XG data. The TitanCNA arguments used: maxCN = 8, alphaK = 1000, txn_exp_len = 1e15, txn_z_strength = 1, minDepth=10, maxDepth=1000. Default values were used for remaining arguments. Out of the 325 PCF/SU2C cohort with whole exome sequencing, copy number was successfully analyzed for 315 cases, with 10 samples failing due to high data variance and/or mismatching tumor-normal pairing.
Copy number analysis in WES data using off-target reads
In WES, DNA fragments outside of target regions (off-target) can be non-specifically captured during hybridization, and are also sequenced. We observed a median of 12% of aligned reads from off-target regions across 205 samples (out of 315) after excluding samples with high variability (median absolute deviation >= 0.35) and low tumor purity (<0.3). The remaining 205 samples were generally consistent for data variability (Figure S6). Next, to normalize the data, the genome was divided into 50kb bins, and bins that overlapped with any bait interval set was considered “on-target”, otherwise they were “off-target”. Then, similar to the normalization steps described above for the TITAN analysis of WES data, the on-target and off-target bins were normalized separately using ichorCNA v0.1.0, as well as separately for tumor and normal samples. Autosomes were used in the loess curve fitting for GC-content bias correction and chromosome X was corrected using the resulting model fit. Next, the tumor bins were divided by the bins from the matched normal to generate log2 ratios.
WGS of primary prostate cancer samples
We obtained WGS data for 57 primary prostate tumor samples from Baca et al. (2013). We used the tumor-normal paired workflow of ichorCNA v0.1.0 to analyze total copy number. Read counts were computed at 10 kb bins across the genome. Centromeres were filtered the same as above. The read coverage in each bin across the genome was corrected for GC content and mappability biases independently for tumor and germline samples. Segmentation and copy number predictions were generated for autosomes and chromosome X. The ichorCNA arguments used: –includeHOMD TRUE, –maxCN=8, –normal 0.5, –ploidy 2, –txnE 0.9999 –txnStrength 10000 –lambda 1000. After excluding samples with less than 10% tumor purity, 54 samples were used for subsequent analyses.
Structural rearrangements
Analysis of 10X Genomics data using SvABA
We called SVs using SvABA (Wala et al., 2018) (May 16, 2017 revision [commit 4a0606e]) using the tumor-normal paired setting, which generates events distinguished as somatic and germline. SvABA uses discordant read-pairs (DR) and split-reads (SR), along with local reassembly to improve specificity for calling breakpoints. SV events that span a length >= 10 kb are considered except for fold-back inversions (see Classification of structural rearrangement classes).
From the set of unfiltered SV calls, we further used 10X linked-read information to rescue events that originally had insufficient evidence from discordant read-pairs and split-reads only. For all SV events from the unfiltered SV list (except for FILTER categories “DUPREADS” and “LOCALMATCH”), the barcode (BX tags in BAM file) counts near breakpoints are extracted.
For each SV event and each of the two breakpoints, the number of unique barcodes (BXC) with >= 1 proper read-pair fully aligned within 1 kb window upstream or downstream of the breakpoint depending on the orientation of the breakends in the event.
Overlapping barcodes (BXOL) are counted as being observed at both breakpoints for the SV event. The barcode overlap fraction at breakpoint 1 (BXOLF1=BXOL/BXC1) and breakpoint 2 (BXOLF2=BXOL/BXC2) and the minimum barcode fraction (BXOLF.min=min(BXOLF1, BXOLF2)) are also computed. For the rescue of SV events that did not initially pass SvABA filters, we assessed whether the barcode fraction is higher than expected as a function of SV length. For computing the expectation, we used SvABA passed events. In the following steps for the rescue, only SVs with BXOL >= 2, and length of SV larger than 1.5 times the mean molecule length in the sample and at least 10 kb for intra-chromosomal events were considered.
Compute the expected barcode overlap fraction based on the length of intra-chromosomal SVs. A 2-dimensional non-linear fit (using loess with span=0.3) is generated for BXOLF.min and length values from SvABA passed events. The maximum length considered is the 95th percentile of lengths of SvABA passed events. This provides the model fit, BXOLF.fit(L), as a function of lengthL.
For each intra-chromosomal event that did not pass SvABA filters, the binomial test is performed for each breakpoint. For example, the p-value of breakpoint 1 is . The SV event is rescued if the maximum p-value between the two breakpoints is less than 0.05.
Compute the expected barcode overlap fraction for inter-chromosomal SVs as the median BXOLF.min of interchromosomal SvABA passed events.
For each inter-chromosomal event that did not pass SvABA filters, the binomial test is performed as above for each breakpoint, except the median BXOLF.min was used as the binomial probability.
Next, SV events that did not pass SvABA filters were also rescued based on corroborating copy number boundaries. For each SV event, if one of the two breakpoints is within 50 kb of a copy number boundary and the BXOLF.min > 0.05, then it is considered as overlapping the copy number segment.
Analysis of 10X Genomics data using GROC-SVS
SVs were called with the tool GROC-SVS (Spies et al., 2017) (May 16, 2017 revisions [commit 1c3e407]) using the two sample (tumor-normal pair) setting and three sample (pre-treatment, post-treatment, normal) setting where applicable (for paired samples). SV events are kept if they are specific to the tumor sample(s) and have FILTER categories: “PASS”, “NOLONGFRAGS”, “NEARBYSNVS”, or “NEARBYSNVS;NOLONGFRAGS”. In addition, events are kept if both breakpoints have BXOL >= 2 and are on the same predicted haplotype while the other haplotype has BXOL <= 1, and p-value <= 1×10−10.
Analysis of 10X Genomics data using Long Ranger
SV events are called independently for tumor and normal samples using Long Ranger v2.1.2 (https://support.10xgenomics.com/genome-exome/software/pipelines/latest/using/wgs). The large SVs (*large_sv_calls.bedpe) and deletions (*dels.vcf.gz) were combined for tumor and normal, separately. Next, germline events were identified in the tumor sample based on overlapping with events from the normal. An overlap between an event in the tumor sample and an event in the normal sample is determined when the first breakpoints of both events are within 1 kb and the second breakpoints of both events are within 1 kb. Germline events are excluded from the tumor sample. Only events with FILTER category “PASS” are kept.
Final structural rearrangement call set for 10X Genomics samples
The final SV call-set consists of the union of SvABA, GROC-SVS, and Long Ranger SV predictions. Intersection of events between two or all tools was determined based on matching the first breakpoint of the events from the tools within 5 kb and also matching the second breakpoint from the tools within 5 kb. For intersecting events, only one event from a single tool is retained to avoid redundancy in the final call-set; the priority of the retention is first SVaBA, followed by GROC-SVS and Long Ranger. The final call-set as well as the original calls for each of the independent tools are also provided in Table S5.
Classification of structural rearrangement types
To determine the rearrangement class/type, we jointly analyzed the final SV call-set and copy number results. For every SV event, we determined the copy number flanking the two breakpoints by using the bin-level (10 kb) corrected copy number based on Equation 2. For the first breakpoint b1, the upstream copy number is assigned with the corrected copy number of the 10 kb bin to the left of the breakpoint; the downstream copy number is assigned the right bin. Similarly, for the second breakpoint b2, and are assigned the left and right bins of the breakpoint.
In addition, for each intra-chromosomal SV event, the mean corrected copy number cmean across bins within and between the breakpoints b1 and b2, and the number of segments b overlapping the region between b1 and b2 is determined. The orientation of a breakpoint is defined as “up” or “+” for the sequence to the left of the breakpoint and as “down” or “−” for the sequence to the right of the breakpoint.
Inter-chromosomal events were classified as translocations that are balanced if and , and unbalanced if or Intra-chromosomal events were classified as deletions if orientation of b1 is “up” and b2 is “down”, and or , and or , and s ≤ 5. In addition, events with orientation (“up”, “down”) having both breakpoints overlapping within 1 Mb of a copy number deletion or LOH segment boundaries are considered deletions. Remaining SVs with deletion orientation (“up”, “down”) and between lengths of 10 kb and 1 Mb are considered deletions.
Intra-chromosomal events were classified as tandem duplications if orientation of b1 is “down” and b2 is “up”, and or , and or , and s ≤ 5. In addition, events with tandem duplication orientation (“down”, “up”) having both breakpoints overlapping within 1 Mb of a copy number segment ≥ 2 copies or copy neutral LOH are considered tandem duplications. Remaining SVs with orientation (“down”, “up”) and between lengths of 10 kb and 1 Mb are considered tandem duplications, except Long Ranger events.
Intra-chromosomal events were classified as inversions if orientation of b1 is the same as b2 (i.e. “up”, “up” or “down”, “down”). Fold-back inversions include events shorter than 30 kb with or , and CN(b1)/ploidy > 2 or CN(b2)/ploidy > 2 (i.e. overlaps an amplified region). Then, remaining inversions shorter than 5 Mb are classified as balanced if and , or and , and unbalanced if and , or and . Remaining inversions larger than 5 Mb with and , or and are classified as “balanced rearrangements”.
All remaining intra-chromosomal events larger than 10 kb are considered “unbalanced rearrangements”.
Chromoplexy analysis
Chromoplexy events were predicted for 10XG data using ChainFinder (Baca et al., 2013). TITAN segments were used as input data. The corresponding corrected copy number from Equation 2 was log transformed log2 (ĉTum/ploidy); copy neutral (2 copies) or heterozygous (HET) segments were set to 0. The SV events from the final call-set that were larger than 10 kb were used as input. For both copy number and SV inputs, tandem duplications in 01115248, 01115257, 01115202, 01115503 (pre and post-treatment) samples were excluded because these events skew the background frequency estimation of SVs by ChainFinder. The ChainFinder arguments used are copy_number_type: seq, summarize_gene: true, mu_window: 1000000, gene_test_window: 25000, deletion_thres: −0.1, bp_window: 10000, significance_thres: 0.01, test_distance_thres: 1000000. Default values were used for the remaining arguments.
Analysis of primary prostate WGS data using SvABA
We called SVs within chromosome X using SvABA (Wala et al., 2018)(May 16, 2017 revision [commit 4a0606e]). The tumor-normal paired setting was used to generate somatic and germline events. SV events with FILTER category “PASS” and larger than 10 kb were used.
Annotation of variants and copy number
Gene overlap
Predicted copy number segments and the final SV call set were annotated using known protein coding genes from GenCode v19 (hg19). For copy number, each gene was assigned the corrected total copy number (see Equation 2) and LOH status (LOH=1, not LOH=0) of the segment that has the largest overlap with the gene; copy number segments shorter than 1 kb were excluded; and LOH segments shorter than 1 Mb were excluded. The copy number of the gene was then normalized to account for sample-specific ploidy and allow for consistent comparison between samples. The copy number was normalized to the median corrected copy number across all autosomal genes (i.e. becomes a copy ratio); chromosome X genes were normalized by half the median copy number because patients are male,
where ĉgene is the corrected total copy number of the segment that overlaps the gene. Copy number alterations and LOH were defined as gain: cgene ≥ 2 and cgene < 2.5, amplification: cgene ≥ 2.5, homozygous deletion: cgene = 0, deletion (LOH): cgene < 1 and cgene > 0 and LOH status = 1, copy neutral LOH: cgene = 1 and LOH status = 1.
For structural rearrangements, each SV was assigned the genes that at least one of the two breakpoints may be transecting. In addition, for intra-chromosomal rearrangements, each SV was also assigned the set of genes that are fully contained within the breakpoints.
Cancer genes related to mCRPC
For single-gene alterations; SNVs, indels, copy number alterations, or structural rearrangements transecting (by one or both breakpoints) the promoter or gene-body, we focused on alterations in genes known to be important in mCRPC and DNA-repair (Grasso et al., 2012; Pritchard et al., 2016; Robinson et al., 2015); a total of 72 genes (40 genes annotated with a tumor suppressor role, 18 with a known oncogene role and 3 with a potential dual role according to COSMIC v83 and the cancer gene census). We further restricted the list of genes to those with a known tumor suppressor role when investigating the number of samples containing one or multiple alterations due to SNVs, indels and copy number loss, or transecting rearrangements of the gene promoter or gene-body.
Transecting SVs affecting genomic features
The final SV call set was annotated with the overlap of genomic features including genes, promoters, transcription start sites, and 1Mb upstream and downstream regions. The promoter region was defined as being 5 kb upstream (positive strand) or downstream (negative strand) of transcription start site for each known protein coding gene from GenCode v19 (hg19). Genomic features with at least one breakpoint from any SV event is considered as transecting the genomic region. For 1Mb upstream or downstream of a gene, we were interested in identifying oncogenes which may have duplications of nearby non-coding regulatory regions such as enhancers. For this analysis, we focused only on breakpoints from tandem duplication events and 304 oncogenes from the COSMIC Cancer Gene Census (https://cancer.sanger.ac.uk/census) (Fig 3e). The frequency for each oncogene was computed for the 10XG WGS cohort of 23 samples. The enrichment (p-value) for duplications nearby oncogenes was determined using the binomial exact test, with the expectation computed as the mean frequency across all 304 oncogenes.
AR gene and the AR enhancer region
For the 10XG data, the copy number of the AR gene cAR (chrX:66,764,465-66,950,461) and the AR enhancer region cEnh (chrX:66,115,000-66,130,000) was computed as the median corrected total copy number (see Equation 2) across the 10 kb bins overlapping these two regions. Similarly, for the WES off-target data, the AR gene and the AR enhancer region was computed as the median corrected total copy number from the overlapping 50kb bins. The copy number is further normalized by half the predicted tumor ploidy ϕ of the sample, cAR = ĉAR/(ϕ/2) and cEnh = ĉEnh/(ϕ/2).
Amplification status was determined for each sample by comparing the fold-change FC = log2(ĉEnh/ĉAR). A sample is assigned a status of “Selective AR” if FC ≤ −log2(1.5)(i.e. AR gene copy number is 1.5 times higher than AR enhancer) and cEnh < 1.75; “Selective Enhancer” if FC ≥ log2(1.5) and cAR < 1.75; “Coamplification” if cEnh > 1.75 and cAR > 1.75; remaining samples were assigned “No amplification”.
Comparison of AR gene and enhancer with AR expression from RNAseq
We obtained RNAseq expression values (fragment per kilobase per million reads, FPKM) from cBioPortal (https://github.com/cBioPortal/datahub/blob/master/public/prad_su2c_2015.tar.gz, accessed February 8, 2018). There were 94 patients with RNAseq data, overlapping the WES cohort of 205 samples with evaluable off-target analysis of AR enhancer. The expression values used in the analysis was computed as log2(FPKM + 1). To determine if AR expression was significantly different between the amplification status, we applied a Wilcoxon rank sum test (Figure 6). Next, to determine the effect of AR enhancer and AR gene copy number as independent variables on AR expression, we performed a multivariable regression analysis. The predictor variables (covariates) were purity (p=0.78), ploidy (p=0.56), percent-off-target reads (p=0.90), median absolute deviation of genome-wide corrected off-target log ratios (p=0.43), AR enhancer copy number (off-target), AR gene copy number (on-target), and the interaction between AR gene and AR enhancer. The coefficients for AR enhancer and AR gene, independently, were positive (0.20 and 0.11) and the resulting p-values were significant (p=4.3×10−8 and p=2.1×10−4, respectively); the interaction covariate was also statistically significant (p=1.97×10−14).
ETS transcription factor rearrangements
The final SV call-set combining all three tools were annotated with ENSEMBL (hg19, release 74, February 2014) gene names. Samples harboring unbalanced or balanced translocations or other SV types with a length > 1 Mb involving one of the ETS transcription factor family genes were called as ETS rearranged. In addition, fusion transcripts were called using STAR-fusion for 10XG samples that had available RNA-seq data from the PCF/SU2C (10/23 overlapping samples). Samples that were found to express fusion transcripts with an ETS family gene partner were also annotated as ETS rearranged.
Cell-free DNA
Analysis of copy number and structural rearrangements in deep WGS data
Copy number alterations were analyzed using ichorCNA v0.1.0 under the tumor-only setting. Read counts were computed at 10 kb bins across the genome. Centromeres were filtered the same as above. The read coverage in each bin across the genome was corrected for GC content and mappability biases. Segmentation and copy number predictions were generated for autosomes and chromosome X. The ichorCNA arguments used were –includeHOMD TRUE, –maxCN 15, –normal 0.5, –ploidy c(2,3), –txnE 0.99999 –txnStrength 100000 –lambda 100. Default values were used for the remaining arguments. SVs were predicted using SvABA (May 16, 2017 revision [commit 4a0606e]) for chromosome X. Only SV events with FILTER category “PASS” and larger than 10 kb were used.
Analysis of cfDNA ULP-WGS using ichorCNA
Copy number alterations were analyzed using ichorCNA v0.1.0 as described in Adalsteinsson et al., (2017). Read counts were computed at 500 kb bins across the genome. Centromeres were filtered based on chromosome gap coordinates obtained from UCSC for hg19. The read coverage in each bin across the genome was corrected for GC content and mappability biases. The same panel of normals consisting of 27 healthy donors, provided by Adalsteinsson et al. (2017), was re-generated for 500 kb bins and used to further normalize the data. Segmentation and copy number predictions were generated for autosomes and chromosome X. The ichorCNA arguments used were –includeHOMD FALSE, –maxCN 6, –normal c(0.5,0.75.0.85), –ploidy c(2,3), –txnE 0.999 –txnStrength 1000, –chrTrain c(1:18). Default values were used for the remaining arguments.
A total of 624 samples from 137 patients were analyzed. Of these, 232 samples from 86 patients had an estimated tumor fraction ≥ 0.05 (5%) and median absolute deviation of the pairwise, adjacent copy ratio (not log transformed) differences for all bins being ≤ 0.15. These 232 samples were used for subsequent analyses.
AR enhancer and AR gene amplification in ULP-WGS
The AR enhancer region and AR gene body are fully contained within consecutive but separate 500 kb bins of chrX:66,000,001-66,500,000 and chrX:66,500,001-67,000,000, respectively. The corrected copy number (see Equation 2), rounded to the nearest integer or set to 0.01 if zero copies, of the AR enhancer bin and the AR gene bin. The AR enhancer and AR gene amplification status criteria were the same as previously described above.
Nucleosome positioning from WGS of cfDNA
We analyzed the occupancy and position of nucleosomes by assessing the coverage of WGS of cfDNA from mCRPC patients and healthy donors. We applied the Windowed Protection Score (WPS) approach described (Snyder et al., 2016). First, depth of coverage at every base pair position in chromosome X is collected for longer fragments greater than 100 bp in length and the WPS is computed based on comparing the number of fragments spanning 120 bp window minus the number of fragments within the window. Next, nucleosome peaks are called using the Savitzky-Golay filter as described in Snyder et al. (2015), using the same parameter settings including smoothing window size of 21; polynomial order 2; maximum single peak length of 150; regions < 50 bp or > 450 bp were discarded. The running median window size used was 3 kb, which helped to account for lower sequencing coverage of the WGS libraries.
Tandem Duplicator Phenotype
Tandem duplications were predicted from the copy number results genome-wide for each sample. For 10XG data, the tandem duplications were taken from the intersection of TITAN copy number segments and tandem duplication breakpoints from the final SV call set. For whole exome sequencing, duplication events must meet these criteria: segment is shorter than 10 Mb; have flanking segments with lower copy number; and the difference in copy number between the left and right flanking segment is ≤ 1. Out of the 325 whole exome sequencing samples, 285 samples that were successfully analyzed by TITAN and having tumor purity > 0.2 and MAD < 0.25 were used to identify duplications. For ULP-WGS data, duplication events must meet these criteria: segment is short than 10 Mb; tumor purity > 0.05; MAD < 0.15; have flanking segments with lower copy number and lower log ratio compared to MAD * tumor fraction; and the left and right flanking segments should have have equal copy number and log ratio difference less than 0.5 * MAD. MAD was used as a measure of data variability and was computed as the the median absolute deviation of the log ratio differences between all pairwise adjacent segments within a sample.
We devised a metric to predict whether a sample exhibits the tandem duplicator phenotype, characterized by numerous large tandem duplications across the genome. This metric was adapted from the Nearest Neighbor Index (NNI), which distinguishes the pattern of observed objects (e.g. tandem duplications) within a defined area as being clustered or dispersed/random. First, the inter-duplication distance (interDupDist) for each duplication to its neighbor duplication downstream were computed (in base pairs) per chromosome C; the distance to the chromosome start and end were used for the first and last duplications in each chromosome, respectively. The Nearest-Neighbor Index is computed for each chromosome as
where Nc is the number of duplication events and Lc is the length of chromosome c. The final duplication dispersion score is the average across all M chromosomes, . Samples with no duplication events will have .
Phasing of variants
Extracting phase of called variants
Although the Long Ranger pipeline outputs phase of variants, germline and somatic variants are not distinguished in the tumor sample. Additionally, not all SNVs as identified by our SNV calling pipeline described above are found in the Long Ranger annotated variants. Therefore, from the the combined SNV call set, Pysam pileup v0.8.4 was used to extract a count of reads supporting the alternative allele at each variant position together with the haplotype information of the molecule that generated the read (HP tag; if available) and the phase set containing the read (PS tag). This information was also extracted from each of the heterozygous SNPs to be used in the TITAN copy number pipeline (see Copy Number Analysis section). A mutation was determined to be phased if it had one or more alternative reads assigned to a haplotype and a PS tag. For indels, the phase information was extracted for the sites called by SvABA/Strelka that overlapped with the Long Ranger phased variants.
Phasing of variants within duplicated regions
To determine variants within duplicated regions, phased somatic SNVs, Indels and germline SNPs were identified in regions with copy number gains matching tandem duplications (as determined by structural variant analysis). The tandem duplication mutation rate was determined as the total number of mutations overlapping tandem duplication segments divided by their total length, whereas the non-duplicated mutation rate was determined as the total number of mutations overlapping the non-duplicated segments divided by their total length. A mutation was determined to be located on the duplicated haplotype if the mutation had a haplotype fraction > 0.5. Mutations phased to the duplicated haplotype with alternate allele fraction >= 0.5 were called as arising before the duplication event, whereas mutations phased to the duplicated haplotype with alternate allele fraction < 0.5 were called as arising after the duplication event.
Data visualization
For visualization of results, we applied custom R (build 3.3) code, R-packages ggplot2 (v2.2.1) and GenVisR (v1.4.1) (Skidmore et al., 2016). Specifically, for visualizing single-gene alterations; SNVs, indels, copy number alterations or structural rearrangements transecting the promoter or gene-body, we applied the waterfall function of GenVisR. For visualizing the 10XG cohort metrics as well as the summaries of SV classes, we used custom R code adapted from the GenVisR package. For visualizing the genome-wide landscape of SVs, including classes of SVs or chains of chromoplexy, we applied the perl Circos package v0.69-6 (Krzywinski et al., 2009), with copy number from TITAN (autosomes) and ichorCNA (chromosome X) and the final combined SV call set or the results from the chromoplexy analysis as input. Horizontal copy number profile plots were generated by TITAN/ichorCNA R packages.
Data and Software Availability
10X Genomics WGS data will be available via dbGAP controlled access (https://www.ncbi.nlm.nih.gov/gap) under study accession phs001577.v1.p1. The deep WGS of cfDNA data will be available via dbGAP controlled access under study accession phs001417.v1.p1. Modifications to TITAN to allow for copy number analysis of 10X data is available as open source R packages from the following URL: https://github.com/gavinha/TitanCNA/.
Supplementary Material
Figure S1. Somatic and germline alterations in 10XG WGS mCRPC cohort
CRPC patients (columns) and genes (rows) previously reported to be significantly altered in mCRPC (Grasso et al., 2012; Pritchard et al., 2016; Robinson et al., 2015) with at least one detectable alteration across our cohort. Cell colors indicate alteration type, which includes SNVs, indels, copy number alterations, and structural variants (SVs). SVs have been subcategorized into those with at least one breakpoint transecting gene body or gene promoter, and those in which a break occurs within the context of a larger chromoplexy chain. The top rows indicate samples with alterations in the AR enhancer and AR, samples displaying the CDK12-associated tandem duplicator phenotype (TDP), samples with ETS gene family rearrangements, and samples with widespread chromoplexy (here defined as having two or more chains, each harboring five or more rearrangements). The lower grid includes clinical annotation on the samples sequenced, including biopsy site and treatment status. The upper histogram indicates mutation rate per sample (mutations/Mb) while the histogram on the left indicates alteration frequency across the cohort.
Figure S2. Rearrangement profiles for mCRPC samples analyzed by 10XG WGS.
Rearrangements in each sample are visualized by CIRCOS plot. Line colors indicate rearrangement class. Color shading in the inner ring indicates copy number status.
Figure S3. Haplotype-based linked-read information is used to resolve a complex event resulting in PTEN inactivation in sample 01115156.
PTEN deletion on chr10:haplotype 1 appears to have occurred via a simple deletion event. An inter-chromosomal event results in loss of PTEN on chr10:haplotype 2 (summarized in schematic on left). Right panels (from top to bottom): chromosome-wide copy number, rearrangements, and haplotype fraction of chromosome 10; copy number profiles around breakpoint sites at chromosomes 1 and 10; and views of haplotype-assigned linked-reads around breakpoints on chromosome 1 (right).
See also Figure 1 and Figure S1.
Figure S4. CDK12-associated TDP is associated with tandem duplications near the MYC locus
(a) Comparison of duplication dispersion score between cfDNA and tumor in 64 samples from 18 mCRPC patients who had both metastatic biopsies (10XG WGS or WES) and cfDNA (ULP-WGS) samples profiled. Point color, CDK12 alteration status as determined by 10XG WGS or WES of tumors. Note, samples collected for WES and cfDNA may have been collected at different time points. The points are sized based on the cfDNA tumor fractions.
(b) Schematic for expected haplotype fractions of phased SNVs if mutations occur before after (left) or before (right) tandem duplication events.
(c) Purity-adjusted copy number profiles for additional samples around the MYC locus. Yellow shaded region containing tandem duplications harbors some of the known 8q24 prostate cancer germline risk variants (shaded region: chr8, 128.0-128.62Mb). MYC gene is colored in green.
Figure S5. Copy number and rearrangement profiles across the region containing AR and AR enhancer in 10X WGS of mCRPC metastases.
Tumor purity-adjusted copy number profile (10 kB bins) and rearrangements (arcs) are shown for each sample subjected to 10XG WGS in the region indicated. Purple arcs represent events rescued by manual inspection. Intra-chromosomal rearrangements are shown as arcs above data points; inter-chromosomal rearrangements are shown as arcs below data points.
Figure S6. Characterization of AR and AR enhancer profiles in ULP-WGS cfDNA, WGS-DNA, WES, and 10XG WGS datasets.
(a) Selected cfDNA samples with alterations in the vicinity of AR were sequenced using ULP-WGS (~0.1X coverage, top panels) or deeper WGS (bottom panels, coverage for each sample indicated at bottom). Duplication rearrangement breakpoints were identified in deeper coverage samples and are indicated by arcs.
(b) Correlation between AR (left) or AR enhancer (right) tumor purity-adjusted copy number as determined by deep WGS and ULP-WGS (0.1X) of cfDNA from 14 cases.
(c) Copy number (purity-adjusted and normalized to sample ploidy) at bins containing the AR enhancer (Y-axis) and AR gene body (X-axis) in 86 ULP-WGS cfDNA specimens (highest tumor fraction per patient; minimum tumor fraction > 0.05). Yellow points indicate samples with selective enhancer amplification; red points indicate samples with co-amplification of AR enhancer and AR gene body (see STAR Methods for classification criteria).
(d) Comparison of the proportion of all reads per sample that are off-target (left) and median absolute deviation per sample (right) for each of the amplification classes (n=205 WES samples).
(e) Correlation between AR (left) or AR enhancer (right) copy number as determined by either 10XG WGS or WES on 9 cases that were profiled by both platforms. Open circles indicate cases in which the same DNA aliquot was used for both WES and 10XG WGS. Filled circles indicate cases in which distinct biopsy cores from the same anatomic site were used for 10X WGS and WES and may thus differ in terms of sample purity and heterogeneity.
(f) Proportion of samples within each amplification class in each data type (numbers shown above each plot). 95% confidence intervals (Clopper-Pearson method) of each proportion is shown. Cohort sizes: 10XG WGS (n=23), WES (n=205), cfDNA (n=86).
Figure S7. AR enhancer and gene body are located within the same topologically-associated domain (TAD).
(a) Chromatin interaction in the region around the AR locus, as measured by Hi-C in LNCaP cells (ENCODE). TADs are indicated by shaded bars (beige and blue) and visualized at: http://promoter.bx.psu.edu/hi-c/view.php. Approximate position of the AR enhancer is indicated by a dotted line and coincides with a region of DNase hypersensitivity in LNCaP cells.
(b) Model for enhanced AR expression resulting from tandem duplications of an AR enhancer in mCRPC.
Table S1. Annotation information for 10X Genomics WGS mCRPC cohort.
See also Figure 1, Figure S1
Table S2. Sequencing metrics for the 10X Genomics WGS cohort
(A) 10X Genomics WGS metrics for mCRPC cohort (Long Ranger Pipeline). (B) Sequencing metrics for 14 mCRPC patient and 3 healthy donor cfDNA samples subjected to deep WGS (Picard).
See also Figure 1
Table S3. Mutation callset and phasing metrics for 10X Genomics WGS mCRPC cohort
(A) Mutation annotation file containing final mutation callset (STAR Methods). Phasing information from Long Ranger pipeline on the tumor sample only are provided. HP_1, HP_2, number of unfiltered altered (variant) reads phased to haplotype 1 or 2; PS, phase set identifier representing the haplotype phase block. (B) Metrics indicate the percentage of mutations and indels with phasing information (for tumor sample) from Long Ranger analysis.
See also Figures 1–3, Figures S1, S4
Table S4. Copy number results for the analyzed cohorts (10XG WGS, WES, ULP-WGS cfDNA, and WGS-cfDNA)
TITAN outputs for 10X Genomics WGS (A) and WES (B) mCRPC cohorts. ichorCNA outputs for ULP-WGS (C) and WGS (D) of cfDNA samples. Copy number segments from 10X WGS (E), WES (F), ULP-WGS cfDNA (G), and WGS cfDNA (H).
Selected column header descriptions: “MAD”, median absolute deviation of all pairwise log ratio differences between adjacent data points. “Cellular prevalence” prevalence of subclones as a proportion of the tumor cells; “logR_Copy_Number”, corrected log ratio based on purity and ploidy of the individual sample (see Equation 2 in STAR Methods); “Corrected_Copy Number”; same as Copy_Number except when Copy_Number >= 8, then it is the “logR_Copy_Number” rounded to the nearest integer. ichorCNA “subclonal.status” is indicated by 1 if event is subclonal. For WGS and WES data, segments for autosomes are from TITAN and segments for chromosome X are from ichorCNA.
See also Figures 1–7, Figure S1-S6
Table S5. Structural variant calls, chromoplexy status, ETS-fusion status, and genes/promoters affected by rearrangements.
(A) Number of structural variant calls in 10X Genomics data detected by SvABA, GROC-SVS and Long Ranger (LR) as well as overlap between tools. (B) Bedpe formatted final SV call set for 10X Genomics data produced by union of SvABA, GROC-SVS, and LR SV calls. (C) ChainFinder output for clusters of SVs involved in chromoplexy events in 10X Genomics data. (D) ETS rearrangement calls in 10XG WGS cohort. (E) Sample frequency for 304 oncogenes from the COSMIC Cancer Gene Census containing at least one breakpoint from tandem duplications 1Mb upstream or downstream. Binomial exact test was used for computing statistical significance; Benjamini-Hochberg (FDR) multiple test correction was applied (q-value). (F) SvABA SV calls for 14 cfDNA deep WGS samples. Only events passing default SvABA filters are shown.
Selected column header descriptions: “SV.combined”, SV identifier in final union call-set per sample. “alt_1” and “alt_2” refer to the VCF definition of orientation. “Orient_1” and “Orient_2” refer to orientations of sequence at breakpoints 1 and 2. FWD, sequence to right of breakpoint; REV, sequence to left of breakpoint. “FILTER”, string generated by the software. “Support”: SVABA, SvABA passed filter events; BXOL/BX, barcode rescued events; CN, overlapping copy number boundaries; “UNIQUE_HAPLOTYPE”, GROC-SVS evidence of unique haplotype read support for both breakpoints. “overlap.SVABA/overlap.GROC/overlap.LR”, “SV.id” of overlapping calls from specified tool. “Type”, basic SV classification based only on orientation. “CN_overlap_type”, detailed classification of SV event based on copy number information at breakpoints. “tss/promoter/Gene”, features broken by one or both breakpoints. “tss.within/promoter.within/Gene.within”, features contained within both breakpoints. “DR”, discordant read count. “SR”, split-read count. “AD”, total unique reads supporting event. Copy number are assigned to the first (“_1”) and second (“_2”) breakpoints and the left (“_prev”) and right (“_next”) flanking regions of the each breakpoint.
See also Figures 1–7, Figure S1-S6
Table S6. Duplication dispersion scores for 10X Genomics WGS, WES, and ULP-cfDNA cohorts and phased mutations within tandem duplications
Duplication dispersion scores for (A) 23 10XG WGS, (B) 315 WES, (C) 624 ULP-WGS cfDNA samples. (D), status of phased mutations (i.e. occurring before or after duplication event) within tandem duplications in TDP samples
Selected column header descriptions: “Num.Gains”, number of copy number gain segments that have corroborating breakpoints from the same tandem duplication event at both segment boundaries. “NNI”, duplication dispersion score as computed by nearest neighbor index (STAR Methods). “Median. InterDupDist”, median of the inter-duplication distance. “MAD”, median absolute deviation across all pairwise differences of adjacent data points within a sample. “CDK12”, single or multiple detected mutations or indels in CDK12; multiple events are assumed to be biallelic. “MUT.DUP.Phased”, phasing status of the mutation within tandem duplication. “MUT.DUP.order”, indicates whether the mutation occurred before or after the duplication. “Number.PyClone.clusters”, Number of distinct clonal clusters with a minimum of two SNVs, as called by PyClone. “Total.SNV.sample”, Number of SNVs included in the PyClone analysis.
See also Figures 2–3, Figure S2, S4
Table S7. Copy number calls at the AR gene and AR enhancer loci in mCRPC and primary prostate cancer cohorts.
(A) 10X Genomics WGS copy number status for AR gene region corresponding to chrX:66764465-66950461 and AR enhancer region corresponding to chrX:66115000-66130000. (B) ULP-WGS cfDNA copy number status at 500 kb bins containing AR gene (chrX:66500001-67000000) and AR enhancer (chrX:66000001-665000000). (C) deep WGS cfDNA copy number status for AR gene and enhancer using the same coordinates as for 10XG analysis. (D) WES calls using off-target reads for AR enhancer (chrX:66150000-66200000) calls and on-target reads for AR gene (chrX:66700000-66900000) copy number at overlapping 50 kb bins. (E) Localized disease (Baca et al., 2013) copy number status for AR gene and enhancer using the same coordinates as for 10XG analysis. (F) Summary of AR gene and enhancer amplification status for three mCRPC (10XG, WES, cfDNA) and one localized disease cohort. Confidence intervals of the proportions for each amplification status group size was computed using Clopper-Pearson method are shown for each amplification class and each cohort.
Selected column header descriptions: For all tables, “AR.CN” and “enhancer.CN” refer to purity-corrected copy number. “AR.ploidy.normalized” and “enhancer.ploidy.normalized” refer to copy number calls normalized to half the sample ploidy. “*cor.AR” and “*cor.Enhancer” are GC-corrected log2 ratio values. “Tum.AR.CN” and “Tum.Enhancer.CN” are purity-corrected copy number. “Tum.AR.ploidy.normalized” and “Tum.Enhancer.ploidy.normalized” are purity-corrected copy number normalized to half the sample ploidy. “MAD”, data variability computed as the median absolute deviation across all pairwise log ratio differences between adjacent data points. “Sample.ID.1”, identifier the original publication (Adalsteinsson et al., 2017). “Tum.offTarget.genome.reads”, “Tum.onTarget.genome.reads”, “Norm.offTarget.genome.reads”, and “Norm.onTarget.genome.reads” refer to median per-bin (50 kB) genome-wide read coverage for bins overlapping on- or off-target regions in the tumor or paired normal sample. “OffTarget.readPct” refers the proportion of all reads in the WES sample that aligned to off-target bins. “OffTarget.MAD” and “OnTarget.MAD” refer to the median absolute deviation across all pairwise differences between adjacent data points for off- and on-target bins, respectively. “Log2.expression” refers to the cBioPortal expression matrix values previously published (Robinson et al., 2015).
Highlights.
Linked-read genome sequencing of mCRPC resolves haplotypes and rearrangements
CDK12 inactivation is associated with a global tandem duplication phenotype
A majority of cases have duplications of an enhancer of the androgen receptor
Progression on androgen pathway inhibitors is associated with gains in AR and enhancer
Acknowledgments
We thank the many patients and their families for their generosity in contributing to this study. Supported by: Department of Defense (W81XWH-17-1-0358; S.R.V), Prostate Cancer Foundation YIA (S.R.V.), Canadian Institutes for Health Research (MFE-140389; G.H.), Norwegian Cancer Society (A.M.H. PR-2007-0166), National Institutes of Health (K08 CA188615 to E.M.V.; R01CA174777 to S.M.D.; P01CA163227 to P.S.N.; NIH R01 CA215489 to R.B.); PCF-V Foundation (E.M.V), Movember/PCF (S.M.D.), Gerstner Family Foundation (V.A.A.), Prostate SPORE (P50CA097186), Fund for Innovation in Cancer Informatics (R.B.), R35 CA197568 (M.M.), and American Cancer Society Research Professorship (M.M.). Stand Up To Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research (SU2C-AACR-DT0712). We would like to dedicate this manuscript to the late Martin Meyerson.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
G.H., S.S.F., V.A.A.: Patent application WO2017161175A1 (ichorCNA)
C-Z.Z: Co-founder, advisor, and share-holder, Pillar Biosciences
E.M.V: Consultant, Tango Therapeutics, Genome Medical, Invitae; research funding, BMS and Novartis
A.D.C.: Receives research funding from Bayer
G.G.: Receives research funding from Bayer and IBM
M.M.: Scientific advisory board chair and equity holder, OrigiMed; research funding, Bayer; inventor of a patent for EGFR mutation diagnosis in lung cancer, licensed to LabCorp.
Author Contributions
Conceptualization: S.R.V., G.H., M.M.
Methodology: S.R.V., G.H.
Software: G.H., J.A.W., J.C-Z.
Sample preparation: M.C., S.C.R.
Formal Analysis: G.H., A.M.H., S.R.V., J.A.W., C.W.W., N.J.H., S.S.F., J.R., P.P., T.K., S.A.W.
Data Curation: Z.Z., A.W., A.D.C., S.R.V., S.C.R., D.R., G.J.G., S.A.W.
Writing, Original Draft: S.R.V., G.H., A.M.H., M.M.
Writing, Review & Editing: S.R.V., G.H., A.M.H., S.M.D., P.S.N., A.D.C., V.A.A., E.M.V., R.B., M-E.T., M.M.
Resources: J.C.L., G.G., S.G., C-Z.Z., S.M.D., P.S.N, E.M.V, A.D.C., V.A.A., R.B., M-E.T, M.M., International PCF/SU2C Prostate Cancer Dream Team Funding Acquisition: M.M.
Discussions and Feedback: S.M.D., P.S.N., R.B. M-E.T.
Supervision: M.M.
References
- Adalsteinsson VA, Ha G, Freeman SS, Choudhury AD, Stover DG, Parsons HA, Gydush G, Reed SC, Rotem D, Rhoades J, et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat Commun. 2017;8:1324. doi: 10.1038/s41467-017-00965-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmadiyeh N, Pomerantz MM, Grisanzio C, Herman P, Jia L, Almendro V, He HH, Brown M, Liu XS, Davis M, et al. 8q24 prostate, breast, and colon cancer risk loci show tissue-specific long-range interaction with MYC. Proc Natl Acad Sci U S A. 2010;107:9742–9746. doi: 10.1073/pnas.0910668107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL, et al. AR-V7 and Resistance to Enzalutamide and Abiraterone in Prostate Cancer. N Engl J Med. 2014;371:1028–1038. doi: 10.1056/NEJMoa1315815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armenia J, Wankowicz SAM, Liu D, Gao J, Kundra R, Reznik E, Chatila WK, Chakravarty D, Han GC, Coleman I, et al. The long tail of oncogenic drivers in prostate cancer. Nat Genet. 2018 doi: 10.1038/s41588-018-0078-z. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora VK, Schenkein E, Murali R, Subudhi SK, Wongvipat J, Balbas MD, Shah N, Cai L, Efstathiou E, Logothetis C, et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell. 2013;155:1309–1322. doi: 10.1016/j.cell.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, Escara-Wilke J, Wilder-Romans K, Dhanireddy S, Engelke C, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature. 2014;510:278–282. doi: 10.1038/nature13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, Park K, Kitabayashi N, MacDonald TY, Ghandi M, et al. Punctuated evolution of prostate cancer genomes. Cell. 2013;153:666–677. doi: 10.1016/j.cell.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajrami I, Frankum JR, Konde A, Miller RE, Rehman FL, Brough R, Campbell J, Sims D, Rafiq R, Hooper S, et al. Genome-wide profiling of genetic synthetic lethality identifies CDK12 as a novel determinant of PARP1/2 inhibitor sensitivity. Cancer Res. 2014;74:287–297. doi: 10.1158/0008-5472.CAN-13-2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, White TA, Stojanov P, Van Allen E, Stransky N, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44:685–689. doi: 10.1038/ng.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluemn EG, Coleman IM, Lucas JM, Coleman RT, Hernandez-Lopez S, Tharakan R, Bianchi-Frias D, Dumpit RF, Kaipainen A, Corella AN, et al. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell. 2017;32:474–489.e6. doi: 10.1016/j.ccell.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boysen G, Barbieri CE, Prandi D, Blattner M, Chae SS, Dahija A, Nataraj S, Huang D, Marotz C, Xu L, et al. SPOP mutation leads to genomic instability in prostate cancer. Elife. 2015;4 doi: 10.7554/eLife.09207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. The Molecular Taxonomy of Primary Prostate Cancer. Cell. 2015;163:1011–1025. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrot-Zhang J, Majewski J. LoLoPicker: detecting low allelic-fraction variants from low-quality cancer samples. Oncotarget. 2017;8:37032–37040. doi: 10.18632/oncotarget.16144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- Cheng S, Brzostek S, Lee SR, Hollenberg AN, Balk SP. Inhibition of the dihydrotestosterone-activated androgen receptor by nuclear receptor corepressor. Mol Endocrinol. 2002;16:1492–1501. doi: 10.1210/mend.16.7.0870. [DOI] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser M, Sabelnykova VY, Yamaguchi TN, Heisler LE, Livingstone J, Huang V, Shiah YJ, Yousif F, Lin X, Masella AP, et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature. 2017;541:359–364. doi: 10.1038/nature20788. [DOI] [PubMed] [Google Scholar]
- Garsed DW, Marshall OJ, Corbin VDA, Hsu A, Di Stefano L, Schröder J, Li J, Feng ZP, Kim BW, Kowarsky M, et al. The architecture and evolution of cancer neochromosomes. Cancer Cell. 2014;26:653–667. doi: 10.1016/j.ccell.2014.09.010. [DOI] [PubMed] [Google Scholar]
- Glodzik D, Morganella S, Davies H, Simpson PT, Li Y, Zou X, Diez-Perez J, Staaf J, Alexandrov LB, Smid M, et al. A somatic-mutational process recurrently duplicates germline susceptibility loci and tissue-specific super-enhancers in breast cancers. Nat Genet. 2017;49:341–348. doi: 10.1038/ng.3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer SU, Nadauld LD, Lau BT, Chen J, Wood-Bouwens C, Ford JM, Kuo CJ, Ji HP. Linked read sequencing resolves complex genomic rearrangements in gastric cancer metastases. Genome Med. 2017;9:57. doi: 10.1186/s13073-017-0447-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JMC, Papaemmanuil E, Brewer DS, Kallio HML, Högnäs G, Annala M, et al. The evolutionary history of lethal metastatic prostate cancer. Nature. 2015;520:353–357. doi: 10.1038/nature14347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha G, Roth A, Khattra J, Ho J, Yap D, Prentice LM, Melnyk N, McPherson A, Bashashati A, Laks E, et al. TITAN: inference of copy number architectures in clonal cell populations from tumor whole-genome sequence data. Genome Res. 2014;24:1881–1893. doi: 10.1101/gr.180281.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henzler C, Li Y, Yang R, McBride T, Ho Y, Sprenger C, Liu G, Coleman I, Lakely B, Li R, et al. Truncation and constitutive activation of the androgen receptor by diverse genomic rearrangements in prostate cancer. Nat Commun. 2016;7:13668. doi: 10.1038/ncomms13668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herranz D, Ambesi-Impiombato A, Palomero T, Schnell SA, Belver L, Wendorff AA, Xu L, Castillo-Martin M, Llobet-Navás D, Cordon-Cardo C, et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat Med. 2014;20:1130–1137. doi: 10.1038/nm.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Weintraub AS, Day DS, Valton AL, Bak RO, Li CH, Goldmann J, Lajoie BR, Fan ZP, Sigova AA, et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science. 2016;351:1454–1458. doi: 10.1126/science.aad9024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SF, Cruz C, Greifenberg AK, Dust S, Stover DG, Chi D, Primack B, Cao S, Bernhardy AJ, Coulson R, et al. CDK12 Inhibition Reverses De Novo and Acquired PARP Inhibitor Resistance in BRCA Wild-Type and Mutated Models of Triple-Negative Breast Cancer. Cell Rep. 2016;17:2367–2381. doi: 10.1016/j.celrep.2016.10.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Coleman I, Morrissey C, Zhang X, True LD, Gulati R, Etzioni R, Bolouri H, Montgomery B, White T, et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat Med. 2016;22:369–378. doi: 10.1038/nm.4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Hwang TH, Oseth LA, Hauge A, Vessella RL, Schmechel SC, Hirsch B, Beckman KB, Silverstein KA, Dehm SM. AR intragenic deletions linked to androgen receptor splice variant expression and activity in models of prostate cancer progression. Oncogene. 2012;31:4759–4767. doi: 10.1038/onc.2011.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour MR, Abraham BJ, Anders L, Berezovskaya A, Gutierrez A, Durbin AD, Etchin J, Lawton L, Sallan SE, Silverman LB, et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science. 2014;346:1373–1377. doi: 10.1126/science.1259037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, Nava Rodrigues D, Robinson D, Omlin A, Tunariu N, et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N Engl J Med. 2015;373:1697–1708. doi: 10.1056/NEJMoa1506859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride DJ, Etemadmoghadam D, Cooke SL, Alsop K, George J, Butler A, Cho J, Galappaththige D, Greenman C, Howarth KD, et al. Tandem duplication of chromosomal segments is common in ovarian and breast cancer genomes. J Pathol. 2012;227:446–455. doi: 10.1002/path.4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menghi F, Inaki K, Woo X, Kumar PA, Grzeda KR, Malhotra A, Yadav V, Kim H, Marquez EJ, Ucar D, et al. The tandem duplicator phenotype as a distinct genomic configuration in cancer. Proceedings of the National Academy of Sciences. 2016;113:E2373–E2382. doi: 10.1073/pnas.1520010113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menghi F, Barthel FP, Yadav V, Tang M, Ji B, Tang Z, Carter GW, Ruan Y, Scully R, Verhaak RGW, et al. The Tandem Duplicator Phenotype is a prevalent genome-wide cancer configuration driven by distinct gene mutations. 2017 doi: 10.1016/j.ccell.2018.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen CC, Wongvipat J, Ku SY, Gao D, Cao Z, et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science. 2017;355:84–88. doi: 10.1126/science.aah4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng CKY, Cooke SL, Howe K, Newman S, Xian J, Temple J, Batty EM, Pole JCM, Langdon SP, Edwards PAW, et al. The role of tandem duplicator phenotype in tumour evolution in high-grade serous ovarian cancer. J Pathol. 2012;226:703–712. doi: 10.1002/path.3980. [DOI] [PubMed] [Google Scholar]
- Northcott PA, Lee C, Zichner T, Stütz AM, Erkek S, Kawauchi D, Shih DJH, Hovestadt V, Zapatka M, Sturm D, et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature. 2014;511:428–434. doi: 10.1038/nature13379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyquist MD, Li Y, Hwang TH, Manlove LS, Vessella RL, Silverstein KAT, Voytas DF, Dehm SM. TALEN-engineered AR gene rearrangements reveal endocrine uncoupling of androgen receptor in prostate cancer. Proc Natl Acad Sci U S A. 2013;110:17492–17497. doi: 10.1073/pnas.1308587110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz MM, Spisák S, Jia L, Cronin AM, Csabai I, Ledet E, Sartor AO, Rainville I, O’Connor EP, Herbert ZT, et al. The association between germline BRCA2 variants and sensitivity to platinum-based chemotherapy among men with metastatic prostate cancer. Cancer. 2017;123:3532–3539. doi: 10.1002/cncr.30808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popova T, Manié E, Boeva V, Battistella A, Goundiam O, Smith NK, Mueller CR, Raynal V, Mariani O, Sastre-Garau X, et al. Ovarian Cancers Harboring Inactivating Mutations in CDK12 Display a Distinct Genomic Instability Pattern Characterized by Large Tandem Duplications. Cancer Res. 2016;76:1882–1891. doi: 10.1158/0008-5472.CAN-15-2128. [DOI] [PubMed] [Google Scholar]
- Pritchard CC, Mateo J, Walsh MF, De Sarkar N, Abida W, Beltran H, Garofalo A, Gulati R, Carreira S, Eeles R, et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N Engl J Med. 2016;375:443–453. doi: 10.1056/NEJMoa1603144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos AH, Lichtenstein L, Gupta M, Lawrence MS, Pugh TJ, Saksena G, Meyerson M, Getz G. Oncotator: cancer variant annotation tool. Hum Mutat. 2015;36:E2423–E2429. doi: 10.1002/humu.22771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riaz N, Blecua P, Lim RS, Shen R, Higginson DS, Weinhold N, Norton L, Weigelt B, Powell SN, Reis-Filho JS. Pan-cancer analysis of bi-allelic alterations in homologous recombination DNA repair genes. Nat Commun. 2017;8:857. doi: 10.1038/s41467-017-00921-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell. 2015;161:1215–1228. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth A, Khattra J, Yap D, Wan A, Laks E, Biele J, Ha G, Aparicio S, Bouchard-Côté A, Shah SP. PyClone: statistical inference of clonal population structure in cancer. Nat Methods. 2014;11:396–398. doi: 10.1038/nmeth.2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders CT, Wong WSW, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28:1811–1817. doi: 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- Shenoy TR, Boysen G, Wang MY, Xu QZ, Guo W, Koh FM, Wang C, Zhang LZ, Wang Y, Gil V, et al. CHD1 loss sensitizes prostate cancer to DNA damaging therapy by promoting error-prone double-strand break repair. Ann Oncol. 2017;28:1495–1507. doi: 10.1093/annonc/mdx165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Whyte WA, Zepeda-Mendoza CJ, Milazzo JP, Shen C, Roe JS, Minder JL, Mercan F, Wang E, Eckersley-Maslin MA, et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev. 2013;27:2648–2662. doi: 10.1101/gad.232710.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skidmore ZL, Wagner AH, Lesurf R, Campbell KM, Kunisaki J, Griffith OL, Griffith M. GenVisR: Genomic Visualizations in R. Bioinformatics. 2016;32:3012–3014. doi: 10.1093/bioinformatics/btw325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder MW, Kircher M, Hill AJ, Daza RM, Shendure J. Cell-free DNA Comprises an In Vivo Nucleosome Footprint that Informs Its Tissues-Of-Origin. Cell. 2016;164:57–68. doi: 10.1016/j.cell.2015.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spies N, Weng Z, Bishara A, McDaniel J, Catoe D, Zook JM, Salit M, West RB, Batzoglou S, Sidow A. Genome-wide reconstruction of complex structural variants using read clouds. Nat Methods. 2017 doi: 10.1038/nmeth.4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda DY, Spisák S, Seo JH, Bell C, O’Connor E, Ribli D, Csabai I, Solymosi N, Szállási Z, Cejas P, et al. A somatically acquired enhancer of the androgen receptor is a noncoding driver in advanced prostate cancer. Submitted to Cell. doi: 10.1016/j.cell.2018.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taplin ME, Bubley GJ, Shuster TD, Frantz ME, Spooner AE, Ogata GK, Keer HN, Balk SP. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N Engl J Med. 1995;332:1393–1398. doi: 10.1056/NEJM199505253322101. [DOI] [PubMed] [Google Scholar]
- Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinänen R, Palmberg C, Palotie A, Tammela T, Isola J, Kallioniemi OP. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995;9:401–406. doi: 10.1038/ng0495-401. [DOI] [PubMed] [Google Scholar]
- Wala JA, Bandopadhayay P, Greenwald NF, O’Rourke R, Sharpe T, Stewart C, Schumacher S, Li Y, Weischenfeldt J, Yao X, et al. SvABA: genome-wide detection of structural variants and indels by local assembly. Genome Res. 2018;28:581–591. doi: 10.1101/gr.221028.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weischenfeldt J, Dubash T, Drainas AP, Mardin BR, Chen Y, Stütz AM, Waszak SM, Bosco G, Halvorsen AR, Raeder B, et al. Pan-cancer analysis of somatic copy-number alterations implicates IRS4 and IGF2 in enhancer hijacking. Nat Genet. 2017;49:65–74. doi: 10.1038/ng.3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt AW, Mo F, Wang K, McConeghy B, Brahmbhatt S, Jong L, Mitchell DM, Johnston RL, Haegert A, Li E, et al. Heterogeneity in the inter-tumor transcriptome of high risk prostate cancer. Genome Biol. 2014;15:426. doi: 10.1186/s13059-014-0426-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeager M, Orr N, Hayes RB, Jacobs KB, Kraft P, Wacholder S, Minichiello MJ, Fearnhead P, Yu K, Chatterjee N, et al. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet. 2007;39:645–649. doi: 10.1038/ng2022. [DOI] [PubMed] [Google Scholar]
- Zhang X, Choi PS, Francis JM, Imielinski M, Watanabe H, Cherniack AD, Meyerson M. Identification of focally amplified lineage-specific super-enhancers in human epithelial cancers. Nat Genet. 2016;48:176–182. doi: 10.1038/ng.3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng GXY, Lau BT, Schnall-Levin M, Jarosz M, Bell JM, Hindson CM, Kyriazopoulou-Panagiotopoulou S, Masquelier DA, Merrill L, Terry JM, et al. Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nat Biotechnol. 2016 doi: 10.1038/nbt.3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Somatic and germline alterations in 10XG WGS mCRPC cohort
CRPC patients (columns) and genes (rows) previously reported to be significantly altered in mCRPC (Grasso et al., 2012; Pritchard et al., 2016; Robinson et al., 2015) with at least one detectable alteration across our cohort. Cell colors indicate alteration type, which includes SNVs, indels, copy number alterations, and structural variants (SVs). SVs have been subcategorized into those with at least one breakpoint transecting gene body or gene promoter, and those in which a break occurs within the context of a larger chromoplexy chain. The top rows indicate samples with alterations in the AR enhancer and AR, samples displaying the CDK12-associated tandem duplicator phenotype (TDP), samples with ETS gene family rearrangements, and samples with widespread chromoplexy (here defined as having two or more chains, each harboring five or more rearrangements). The lower grid includes clinical annotation on the samples sequenced, including biopsy site and treatment status. The upper histogram indicates mutation rate per sample (mutations/Mb) while the histogram on the left indicates alteration frequency across the cohort.
Figure S2. Rearrangement profiles for mCRPC samples analyzed by 10XG WGS.
Rearrangements in each sample are visualized by CIRCOS plot. Line colors indicate rearrangement class. Color shading in the inner ring indicates copy number status.
Figure S3. Haplotype-based linked-read information is used to resolve a complex event resulting in PTEN inactivation in sample 01115156.
PTEN deletion on chr10:haplotype 1 appears to have occurred via a simple deletion event. An inter-chromosomal event results in loss of PTEN on chr10:haplotype 2 (summarized in schematic on left). Right panels (from top to bottom): chromosome-wide copy number, rearrangements, and haplotype fraction of chromosome 10; copy number profiles around breakpoint sites at chromosomes 1 and 10; and views of haplotype-assigned linked-reads around breakpoints on chromosome 1 (right).
See also Figure 1 and Figure S1.
Figure S4. CDK12-associated TDP is associated with tandem duplications near the MYC locus
(a) Comparison of duplication dispersion score between cfDNA and tumor in 64 samples from 18 mCRPC patients who had both metastatic biopsies (10XG WGS or WES) and cfDNA (ULP-WGS) samples profiled. Point color, CDK12 alteration status as determined by 10XG WGS or WES of tumors. Note, samples collected for WES and cfDNA may have been collected at different time points. The points are sized based on the cfDNA tumor fractions.
(b) Schematic for expected haplotype fractions of phased SNVs if mutations occur before after (left) or before (right) tandem duplication events.
(c) Purity-adjusted copy number profiles for additional samples around the MYC locus. Yellow shaded region containing tandem duplications harbors some of the known 8q24 prostate cancer germline risk variants (shaded region: chr8, 128.0-128.62Mb). MYC gene is colored in green.
Figure S5. Copy number and rearrangement profiles across the region containing AR and AR enhancer in 10X WGS of mCRPC metastases.
Tumor purity-adjusted copy number profile (10 kB bins) and rearrangements (arcs) are shown for each sample subjected to 10XG WGS in the region indicated. Purple arcs represent events rescued by manual inspection. Intra-chromosomal rearrangements are shown as arcs above data points; inter-chromosomal rearrangements are shown as arcs below data points.
Figure S6. Characterization of AR and AR enhancer profiles in ULP-WGS cfDNA, WGS-DNA, WES, and 10XG WGS datasets.
(a) Selected cfDNA samples with alterations in the vicinity of AR were sequenced using ULP-WGS (~0.1X coverage, top panels) or deeper WGS (bottom panels, coverage for each sample indicated at bottom). Duplication rearrangement breakpoints were identified in deeper coverage samples and are indicated by arcs.
(b) Correlation between AR (left) or AR enhancer (right) tumor purity-adjusted copy number as determined by deep WGS and ULP-WGS (0.1X) of cfDNA from 14 cases.
(c) Copy number (purity-adjusted and normalized to sample ploidy) at bins containing the AR enhancer (Y-axis) and AR gene body (X-axis) in 86 ULP-WGS cfDNA specimens (highest tumor fraction per patient; minimum tumor fraction > 0.05). Yellow points indicate samples with selective enhancer amplification; red points indicate samples with co-amplification of AR enhancer and AR gene body (see STAR Methods for classification criteria).
(d) Comparison of the proportion of all reads per sample that are off-target (left) and median absolute deviation per sample (right) for each of the amplification classes (n=205 WES samples).
(e) Correlation between AR (left) or AR enhancer (right) copy number as determined by either 10XG WGS or WES on 9 cases that were profiled by both platforms. Open circles indicate cases in which the same DNA aliquot was used for both WES and 10XG WGS. Filled circles indicate cases in which distinct biopsy cores from the same anatomic site were used for 10X WGS and WES and may thus differ in terms of sample purity and heterogeneity.
(f) Proportion of samples within each amplification class in each data type (numbers shown above each plot). 95% confidence intervals (Clopper-Pearson method) of each proportion is shown. Cohort sizes: 10XG WGS (n=23), WES (n=205), cfDNA (n=86).
Figure S7. AR enhancer and gene body are located within the same topologically-associated domain (TAD).
(a) Chromatin interaction in the region around the AR locus, as measured by Hi-C in LNCaP cells (ENCODE). TADs are indicated by shaded bars (beige and blue) and visualized at: http://promoter.bx.psu.edu/hi-c/view.php. Approximate position of the AR enhancer is indicated by a dotted line and coincides with a region of DNase hypersensitivity in LNCaP cells.
(b) Model for enhanced AR expression resulting from tandem duplications of an AR enhancer in mCRPC.
Table S1. Annotation information for 10X Genomics WGS mCRPC cohort.
See also Figure 1, Figure S1
Table S2. Sequencing metrics for the 10X Genomics WGS cohort
(A) 10X Genomics WGS metrics for mCRPC cohort (Long Ranger Pipeline). (B) Sequencing metrics for 14 mCRPC patient and 3 healthy donor cfDNA samples subjected to deep WGS (Picard).
See also Figure 1
Table S3. Mutation callset and phasing metrics for 10X Genomics WGS mCRPC cohort
(A) Mutation annotation file containing final mutation callset (STAR Methods). Phasing information from Long Ranger pipeline on the tumor sample only are provided. HP_1, HP_2, number of unfiltered altered (variant) reads phased to haplotype 1 or 2; PS, phase set identifier representing the haplotype phase block. (B) Metrics indicate the percentage of mutations and indels with phasing information (for tumor sample) from Long Ranger analysis.
See also Figures 1–3, Figures S1, S4
Table S4. Copy number results for the analyzed cohorts (10XG WGS, WES, ULP-WGS cfDNA, and WGS-cfDNA)
TITAN outputs for 10X Genomics WGS (A) and WES (B) mCRPC cohorts. ichorCNA outputs for ULP-WGS (C) and WGS (D) of cfDNA samples. Copy number segments from 10X WGS (E), WES (F), ULP-WGS cfDNA (G), and WGS cfDNA (H).
Selected column header descriptions: “MAD”, median absolute deviation of all pairwise log ratio differences between adjacent data points. “Cellular prevalence” prevalence of subclones as a proportion of the tumor cells; “logR_Copy_Number”, corrected log ratio based on purity and ploidy of the individual sample (see Equation 2 in STAR Methods); “Corrected_Copy Number”; same as Copy_Number except when Copy_Number >= 8, then it is the “logR_Copy_Number” rounded to the nearest integer. ichorCNA “subclonal.status” is indicated by 1 if event is subclonal. For WGS and WES data, segments for autosomes are from TITAN and segments for chromosome X are from ichorCNA.
See also Figures 1–7, Figure S1-S6
Table S5. Structural variant calls, chromoplexy status, ETS-fusion status, and genes/promoters affected by rearrangements.
(A) Number of structural variant calls in 10X Genomics data detected by SvABA, GROC-SVS and Long Ranger (LR) as well as overlap between tools. (B) Bedpe formatted final SV call set for 10X Genomics data produced by union of SvABA, GROC-SVS, and LR SV calls. (C) ChainFinder output for clusters of SVs involved in chromoplexy events in 10X Genomics data. (D) ETS rearrangement calls in 10XG WGS cohort. (E) Sample frequency for 304 oncogenes from the COSMIC Cancer Gene Census containing at least one breakpoint from tandem duplications 1Mb upstream or downstream. Binomial exact test was used for computing statistical significance; Benjamini-Hochberg (FDR) multiple test correction was applied (q-value). (F) SvABA SV calls for 14 cfDNA deep WGS samples. Only events passing default SvABA filters are shown.
Selected column header descriptions: “SV.combined”, SV identifier in final union call-set per sample. “alt_1” and “alt_2” refer to the VCF definition of orientation. “Orient_1” and “Orient_2” refer to orientations of sequence at breakpoints 1 and 2. FWD, sequence to right of breakpoint; REV, sequence to left of breakpoint. “FILTER”, string generated by the software. “Support”: SVABA, SvABA passed filter events; BXOL/BX, barcode rescued events; CN, overlapping copy number boundaries; “UNIQUE_HAPLOTYPE”, GROC-SVS evidence of unique haplotype read support for both breakpoints. “overlap.SVABA/overlap.GROC/overlap.LR”, “SV.id” of overlapping calls from specified tool. “Type”, basic SV classification based only on orientation. “CN_overlap_type”, detailed classification of SV event based on copy number information at breakpoints. “tss/promoter/Gene”, features broken by one or both breakpoints. “tss.within/promoter.within/Gene.within”, features contained within both breakpoints. “DR”, discordant read count. “SR”, split-read count. “AD”, total unique reads supporting event. Copy number are assigned to the first (“_1”) and second (“_2”) breakpoints and the left (“_prev”) and right (“_next”) flanking regions of the each breakpoint.
See also Figures 1–7, Figure S1-S6
Table S6. Duplication dispersion scores for 10X Genomics WGS, WES, and ULP-cfDNA cohorts and phased mutations within tandem duplications
Duplication dispersion scores for (A) 23 10XG WGS, (B) 315 WES, (C) 624 ULP-WGS cfDNA samples. (D), status of phased mutations (i.e. occurring before or after duplication event) within tandem duplications in TDP samples
Selected column header descriptions: “Num.Gains”, number of copy number gain segments that have corroborating breakpoints from the same tandem duplication event at both segment boundaries. “NNI”, duplication dispersion score as computed by nearest neighbor index (STAR Methods). “Median. InterDupDist”, median of the inter-duplication distance. “MAD”, median absolute deviation across all pairwise differences of adjacent data points within a sample. “CDK12”, single or multiple detected mutations or indels in CDK12; multiple events are assumed to be biallelic. “MUT.DUP.Phased”, phasing status of the mutation within tandem duplication. “MUT.DUP.order”, indicates whether the mutation occurred before or after the duplication. “Number.PyClone.clusters”, Number of distinct clonal clusters with a minimum of two SNVs, as called by PyClone. “Total.SNV.sample”, Number of SNVs included in the PyClone analysis.
See also Figures 2–3, Figure S2, S4
Table S7. Copy number calls at the AR gene and AR enhancer loci in mCRPC and primary prostate cancer cohorts.
(A) 10X Genomics WGS copy number status for AR gene region corresponding to chrX:66764465-66950461 and AR enhancer region corresponding to chrX:66115000-66130000. (B) ULP-WGS cfDNA copy number status at 500 kb bins containing AR gene (chrX:66500001-67000000) and AR enhancer (chrX:66000001-665000000). (C) deep WGS cfDNA copy number status for AR gene and enhancer using the same coordinates as for 10XG analysis. (D) WES calls using off-target reads for AR enhancer (chrX:66150000-66200000) calls and on-target reads for AR gene (chrX:66700000-66900000) copy number at overlapping 50 kb bins. (E) Localized disease (Baca et al., 2013) copy number status for AR gene and enhancer using the same coordinates as for 10XG analysis. (F) Summary of AR gene and enhancer amplification status for three mCRPC (10XG, WES, cfDNA) and one localized disease cohort. Confidence intervals of the proportions for each amplification status group size was computed using Clopper-Pearson method are shown for each amplification class and each cohort.
Selected column header descriptions: For all tables, “AR.CN” and “enhancer.CN” refer to purity-corrected copy number. “AR.ploidy.normalized” and “enhancer.ploidy.normalized” refer to copy number calls normalized to half the sample ploidy. “*cor.AR” and “*cor.Enhancer” are GC-corrected log2 ratio values. “Tum.AR.CN” and “Tum.Enhancer.CN” are purity-corrected copy number. “Tum.AR.ploidy.normalized” and “Tum.Enhancer.ploidy.normalized” are purity-corrected copy number normalized to half the sample ploidy. “MAD”, data variability computed as the median absolute deviation across all pairwise log ratio differences between adjacent data points. “Sample.ID.1”, identifier the original publication (Adalsteinsson et al., 2017). “Tum.offTarget.genome.reads”, “Tum.onTarget.genome.reads”, “Norm.offTarget.genome.reads”, and “Norm.onTarget.genome.reads” refer to median per-bin (50 kB) genome-wide read coverage for bins overlapping on- or off-target regions in the tumor or paired normal sample. “OffTarget.readPct” refers the proportion of all reads in the WES sample that aligned to off-target bins. “OffTarget.MAD” and “OnTarget.MAD” refer to the median absolute deviation across all pairwise differences between adjacent data points for off- and on-target bins, respectively. “Log2.expression” refers to the cBioPortal expression matrix values previously published (Robinson et al., 2015).