

Abstract
Mitochondria are the source of cellular energy production and are present in different types of cells. However, their function is especially important for the heart due to the high demands in energy which is achieved through oxidative phosphorylation. Mitochondria form large networks which regulate metabolism and the optimal function is achieved through the balance between mitochondrial fusion and mitochondrial fission. Moreover, mitochondrial function is upon quality control via the process of mitophagy which removes the damaged organelles. Mitochondrial dysfunction is associated with the development of numerous cardiac diseases such as atherosclerosis, ischemia-reperfusion (I/R) injury, hypertension, diabetes, cardiac hypertrophy and heart failure (HF), due to the uncontrolled production of reactive oxygen species (ROS). Therefore, early control of mitochondrial dysfunction is a crucial step in the therapy of cardiac diseases. A number of anti-oxidant molecules and medications have been used but the results are inconsistent among the studies. Eventually, the aim of future research is to design molecules which selectively target mitochondrial dysfunction and restore the capacity of cellular anti-oxidant enzymes.
Keywords: Mitochondria, cardiovascular disease, oxidative stress, treatment
Introduction
Mitochondria are cellular organelles of maternal origin which are involved in energy production through the process of oxidative phosphorylation. Several organic substrates can be used in energy production, usually fatty acids and less commonly glucose (1). The origin of mitochondria is impressive since they are the consequence of bacterial endosymbiosis into the very early forms of eukaryotic cells (2). Moreover, mitochondria have their own DNA and a unique genetic code which differs from the nuclear DNA. However, the vast majority of mitochondrial proteins are produced from the translation of nuclear DNA (3).
Mitochondria are not isolated organelles but form complex networks which are under strict control by two distinct processes. The first one is mitochondrial fusion, which forms long filamentous mitochondria, and the second one is mitochondrial fission, which generates small spherical mitochondria. Both processes depend on the metabolic needs of the cell (4). Proper mitochondrial function is associated with a balance between the two previous processes. Additionally, another source of mitochondrial quality control is the selective degradation of the dysfunctional organelles through autophagy which is defined as mitophagy (5).
Mitochondria are the site of reactive oxygen species (ROS) generation during the enzymatic activity of electron transport chain (6). Uncontrolled production of ROS and impairment of mitochondrial dynamics result in mitochondrial dysfunction and ultrastructural changes of cellular lipids, proteins, enzymes and DNA which are the pathophysiologic background for the development of several cardiac diseases (7). Therefore, targeting of mitochondrial dysfunction is a crucial step in the treatment of a variety of cardiac diseases and several approaches have been tested in experimental and clinical studies with, however, controversial findings.
In this review article, we will discuss the role of mitochondria in the pathophysiology of atherosclerosis, ischemia-reperfusion (I/R) injury, hypertension, diabetes, cardiac hypertrophy and heart failure (HF) and discuss the potential therapeutic interventions against the development of cardiac diseases.
Cardiac mitochondria: description of origin, function, network and biogenesis
Mitochondria are highly present in cardiac cells due to the increased energy demands and are responsible for the daily production of approximately 6 kg of ATP through the process of oxidative phosphorylation (8) (Figure 1). Apart from energy production, mitochondria are involved in regulation of oxidative stress, cell survival and apoptotic death (9). It has been proved that in neonatal cardiac myocytes the main source of energy is obtained by glycolysis and glucose oxidation, and cardiac mitochondria exhibit great motility in the cytosol (10). On the other hand, in the adult heart the main source of energy is obtained by oxidation of fatty acids, and cardiac mitochondria exhibit reduced motility in the cytosol (11).
Figure 1.
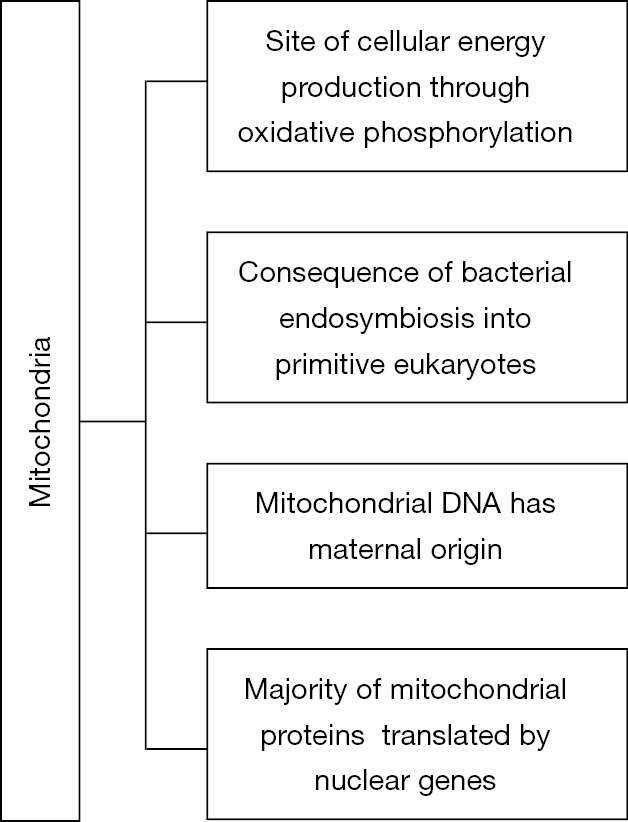
Important characteristics of mitochondria.
The origin of mitochondria and mitochondria-related organelles was one of the greatest mysteries addressed by the biologists of 20th century. Initiating from the landmark paper of Sagan (2), today it is widely accepted that mitochondria originated due to bacterial endosymbiosis of the primitive forms of eukaryotic cells. The phylogenetic classification of mitochondria has been particularly challenging due to the loss of DNA at the expense of cell nucleus and the evolutionary pressure they were put under as compartments of eukaryotic cells (12). However, scientific evidence now concludes that ancestors of mitochondria were closely related to either Rhodospirillales (13) or Rickettsiales (14) which belong to proteobacteria. An even greater debate takes place regarding the time-lapse of mitochondria endosymbiosis and the complexity of the initial host of mitochondria. The existing theories argue that endosymbiosis took place either early, in a simple host, or later in an already complex eukaryote, based on the extent of gene transfer between mitochondria and the host cell (15,16). The answer of this question is still elusive. Regarding the reasons that led to endosymbiosis, two models exist which are based on the role of mitochondria in current cells as a source of energy production and ROS detoxification which is referred as “hydrogen hypothesis” and “oxygen scavenger hypothesis” respectively.
Mitochondrial DNA is a double-stranded, circular deoxyribonucleic acid with an approximate length of 16.6 Kbp whose structure was deciphered in 1981 (17). During the evolutionary process significant fragments of mitochondrial DNA were transferred to nucleus where the vast majority of mitochondrial proteins are expressed. However, the remaining mitochondrial DNA still retains its utility and importance since it encodes 13 proteins which participate in the four complexes of oxidative phosphorylation, and a great number of proteins which are associated with mitochondrial function (18). It is critical to mention that human proteome presents distinct characteristics which are tissue specific and alterations are frequently correlated with certain diseases (19). Moreover, mitochondria encode their own tRNA and rRNA genes and genetic code is substantially different between mammalian mitochondria and nuclear sequences (20).
Mitochondria have a complex relationship with nucleus with both anterograde (from nucleus to mitochondria) and retrograde (from mitochondria to nucleus) signaling. Nucleus regulates mitochondrial dynamics as well as the expression of an important number of mitochondrial enzymes, mitochondrial DNA repair genes and mitochondrial proteins. Peroxisome proliferator-activated receptor (PPAR) gamma coactivator-1 (PGC-1α and PGC-1β) induces mitochondrial DNA replication and catalyzes mitochondrial biogenesis (21). Moreover, AMP kinase and Akt/mTOR signaling pathway regulate the processes of mitochondrial fission and degradation which will be discussed below, with AMP kinase as stimulator (22) and Akt/mTOR as inhibitor (23). Nevertheless, mitochondria regulate the expression of nuclear genes through changes in the cytoplasmic metabolites. NAD+/NADH ratio is directly dependent on mitochondrial activity. As a result, mitochondria control the NAD+-activated family of sirtuin (SIRT) proteins. Sirtuins act as histone deacetylases and epigenetically modify expression of nuclear genes (24). However, more studies are required to fully understand the complex pathways of cross-talking between mitochondria and nucleus.
Mitochondria are not isolated organelles but form large networks which regulate metabolism and involve two major processes; mitochondrial fusion, which forms long filamentous mitochondria, and mitochondrial fission, which generates small spherical mitochondria (25) (Figure 2). This order of organization has been studied extensively in heart muscle and its impairment is associated with a wide range of cardiovascular diseases (26). Several molecules are implicated in the regulation of these processes which aim at mitochondrial and cellular homeostasis; for example, an imbalance between fission and fusion results in the accumulation of non-functional organelles which produce excessive amounts of ROS (25).
Figure 2.

Mitochondrial biogenesis.
ROS induce membrane depolarization through the opening of anion channels which stimulate mitochondrial fission; also, mitochondrial fragmentation results in the release of cytochrome c in the cytoplasm which triggers cellular apoptosis and cell death (27). Mitochondrial fission is mediated through the cytosolic protein dynamin related protein 1 (DRP1) and its interaction with outer mitochondrial membrane receptors such as mitochondria fission factor (MIFF) and the proteins Fis1, Mid49 and Mid51. Specifically, polymers of DRP1 are formed around mitochondrial membranes which gradually shrink mitochondrial membranes till their complete separation (28).
Mitochondrial fusion is mainly mediated through the inner mitochondrial membrane protein optic atrophy 1 (OpA1) which preserves the membrane integrity and inhibits the formation of pores. It is part of the dynamin group of proteins which belong to the family of GTPases that are widely preserved across different species. After GTP hydrolysis, SNARE complexes are formed which pull the membranes together and result in the fusion of the organelles (29). Another set of proteins which co-ordinate fusion are mitofusins 1 and 2. Although the exact mechanism of action is still unspecified, ablation of mitofusins results in mitochondrial fragmentation and may be the consequence of many cardiovascular diseases (30). Nonetheless, mitofusins increase membrane permeability which augments mitochondrial susceptibility to injury by ROS. An important feature of these proteins is their dependence on calcium; specifically, they are activated by calcium entry into cells mediated by ryanodine related receptors and IP3. This is especially important in the heart muscle since it links mitochondrial network with myocardial contraction (31).
Mitochondrial fission and fusion are also associated with programmed cell death. In fact, the pro-apoptotic proteins Bak and Bax stimulate protease OMA1 which results in the cleavage of OpA1 and stimulates mitochondrial fission (32). Moreover, mitochondrial fusion and fission are under control by the nucleus. Specifically, mitochondrial proteins which belong to SIRT family trigger the expression of FOXO3A transcription factor which upregulates PTEN-induced kinase 1 (PINK-1) that promotes mitochondrial fission (33).
Last but not least, a unique finding in skeletal and heart muscles is the continuous inter-mitochondrial communication. It has been noted that “kissing junctions” exist between adjacent mitochondria which permit the exchange of proteins and ions (34). Moreover, nanotunnels are found between neighboring organelles which control mitochondrial response particularly during alterations in calcium dynamics (35).
Mitochondrial fission is closely related to the degradation of damaged mitochondria by autophagy in a process called mitophagy. The main protein implicated in mitophagy is the ubiquitin ligase protein parkin. In the case of mitochondrial damage, parkin translocates to mitochondrial membrane and induces the ubiquitination of mitochondrial proteins. As a result, the ubiquitinated mitochondrion gets in proximity to lysosomes where it gets engulfed and degraded (36). Mitophagy is one of the most effective strategies to remove damaged mitochondria and its impairment is frequently noted in the development of cardiovascular diseases. Mitophagy is a well-regulated process that is under the control of sympathetic nervous system, intracellular calcium dynamics and intracellular signaling pathways. Furthermore, heat shock protein 27 (hsp 27) increases lysosomal activity and protects cardiomyocytes from the damaged mitochondria (37). Lastly, Yan et al. revealed that mitophagy is associated with overexpression of calcineurin which is linked to the opening of mPTP channel and stimulation of parkin-mediated autophagy (38).
Cardiac mitochondria and regulation of oxidative stress
Mitochondria are the powerhouse of the cell. Utilization of oxygen as the final recipient of electrons at the electron transport chain complexes (ETC), mainly I and III, renders mitochondria important mediators of ROS production. Mitochondria-derived ROS influence both mitochondrial dynamics and cell adaptation to oxidative stress (Figure 3). Specifically, increased oxygen consumption raises the amount of reduced ubiquinone and cytochrome c and stimulates ROS formation in a procedure termed as reverse ETC. Moreover, ischemia ceases the conversion of fumarate to malate and increases the formation of succinate which is another donor of electrons (39). Complex I also creates ROS through utilization of NAD+ substrates and reduction of flavin mononucleotide (40). Recently, it has been indicated that complex II produces ROS through the opening of mPTP channel induced by cyclophilin D; more specifically, the influx of Ca2+ depolarizes mitochondrial membrane and forms reduced malate and fumarate which provoke cell dysfunction (41,42). Similarly, under ischemic conditions, ROS produced by complex III inhibit the opening of mPTP channel in comparison to those of complex I (43). Lastly, production of ROS induces changes in the structure of ETC complexes which accelerate a vicious cycle of ROS production (44).
Figure 3.

The balance between oxidative stress and anti-oxidant actions.
Another important source of mitochondrial ROS is the NOX family of NADPH oxidase. Recently, Dalal et al. discovered that NOX4 localizes to mitochondria and provokes the opening of mPTP channel (45). Furthermore, the binding of angiotensin-II to vascular endothelium results in the expression of NOX2 which opens an ATP-dependent mitochondrial potassium channel (mitoKATP) that stimulates the function of reverse ETC (46).
In addition, oxidative stress is associated with altered expression of mitochondrial and nuclear proteins. For instance, it has been demonstrated that oxidative stress increases the activity of COUP-TFII transcription factor which induces the expression of nuclear-encoded mitochondrial enzymes, favoring mitochondrial fragmentation (47). Similarly, ROS downregulate the activity of ETC complexes and decrease oxygen consumption in patients with metabolic syndrome which results in left ventricular hypertrophy and HF (48). Lastly, ROS provoke structural changes in mitochondrial proteins such as an imbalance between mitochondrial tyrosine kinase Src and phosphatase SPH2, which decreases tyrosine phosphorylation at the active region of many mitochondrial enzymes (49).
ROS induce damage in mitochondrial DNA which is a key characteristic of several cardiac diseases. It has been demonstrated that guanine residues are prone to oxidation and formation of 7,8-dihydro-8-oxoguanine which result in mutations of mitochondrial DNA (7). Decreased expression of mitochondrial DNA repair enzymes has been reported, too. For example, the expression of DNA polymerase gamma, AP endonuclease, oxoguanine glycosylase (Ogg1) and uracil-DNA glycosylase was significantly reduced in an experimental model of sepsis in heart muscle (49). Moreover, in the case of hyperglycemia and diabetes excessive amounts of glucose induce a shift towards the pentose monophosphate shunt and hexosaminidase pathway. In the former, NADPH is produced and is utilized by mitochondrial NOX in order to generate ROS (50). In the latter, 8-Glc-N interacts directly with Ogg1 and forms 8GlcNOgg, a dysfunctional enzyme which hinders the repair of mitochondrial DNA (51).
Furthermore, excessive production of mitochondrial ROS is linked to stimulation of ageing process and apoptosis. Aged cardiomyocytes display high levels of cytosolic p53 which adheres to parkin and inhibits translocation to mitochondria; consequently, ubiquitination is abolished and clearance of defective mitochondria through autophagy is impaired. Dysfunctional mitochondria produce less amounts of ATP and permit the leakage of cytochrome c to cytoplasm that triggers the cascade of apoptosis (52). Aged cells display impaired mitochondrial biogenesis which in turn accelerates ROS production (53). Additionally, aged cardiac cells have reduced gene expression of molecules implicated in fatty acid oxidation and Krebs cycle (54). Lastly, it is important to mention that excessive intracellular ROS stimulate inflammatory pathways such as leukocyte chemotaxis which enhance the ageing process (55).
On the other hand, cells have developed mechanisms to either decrease ROS production or neutralize their effects. Selective downregulation of the activity of ETC complexes prevents bursts of ROS production; specifically, Src kinase has been found to phosphorylate critical Ser/Thr residues of complex I enzymes which decrease ROS production by ETC complexes (56). Moreover, B-oxybutyrate dehydrogenase reduces oxidation of guanine in failing hearts; also, it has histone deacetylase inhibitor properties which increases the expression of anti-oxidant enzymes in the nucleus (57).
Another critical step in the control of oxidative stress is the blockage of the opening of mPTP channel. It has been demonstrated that protein kinase D (PKD), which is regulated by phospholipase C, diacylglycerol and Rhoα, inhibits the translocation of cofilins to mitochondria that open mPTP under ischemic conditions (58). Hsp 90 normally binds to cyclophilin D in the cytosol and prevents its degradation. However, it has been proved that HAX-1 antagonizes the binding of cyclophilin D to hsp 90; therefore, cyclophilin D is ubiquitinated and mPTP opening is blocked (42).
It has been previously described that the clearance of the damaged mitochondria through autophagy ameliorates mitochondrial function (59,60). On the one hand, macro-autophagy involves ubiquitination and degradation of mitochondrial parkin protein. On the other hand, micro-autophagy involves the fusion of phospholipids between mitochondrial and lysosomal membranes under the effects of 3-glycerol aldehyde phosphate dehydrogenase (GAPDH). It is noteworthy that micro-autophagy is independent of the inhibitory effects of PI3K/Akt/mTOR pathway (61).
Finally, several cellular anti-oxidant enzymes protect against oxidative stress. Mitochondrial catalase (mCAT) in particular neutralizes the damage from hydrogen peroxide production. In an experimental model in mice it was found that overexpression of mCAT improved mitochondrial biogenesis and dysfunction (62). Superoxide dismutase (SOD-2) is another cellular free radical scavenger. Das et al. demonstrated that knock-out mice for SOD-2 exhibited early onset of mitochondrial dysfunction and decreased cell survival (63). Proper function of the enzymes of mitochondrial DNA repair such as OGG1, AP ligase and type III endonuclease may increase the expression of mitochondrial proteins and improve energy production (64). For example, Pillai et al. demonstrated that the previous effects are mediated by activation of SIRT1 (7). In general, cardiac cells control the rate of mitochondrial biogenesis through SIRT1-dependent pathways which protect against oxidative stress and inhibit the intrinsic pathway of apoptosis (65).
Cardiac mitochondria and endothelial function
Mitochondrial dysfunction affects endothelial cells since aged mitochondria produce large amounts of ROS and have decreased expression of antioxidant enzymes such as SOD-2 and thioredoxin reductase. Excessive ROS enhance the formation of peroxynitrite which impairs endothelial nitric oxide (NO) synthase and NO mediated dilatation (66,67). Therefore, mitochondrial ROS are linked to endothelial dysfunction and their targeting improves endothelial function (68). Moreover, stimulation of renin angiotensin system (RAS) induces hyperpolarization of inner mitochondrial membrane and cell death especially in cells with defective autophagy systems (69). Additionally, mitochondrial ROS inhibit smooth muscle cell relaxation of the perivascular adipose tissue and induce the formation of endothelial extracellular vesicles which contain the proteins parkin and MFR1 (70).
In conclusion, proper mitochondrial function belongs to the most important compensatory mechanisms against vascular aging. Therefore, interventions which target calcium entry in mitochondria would prevent smooth muscle contraction and ameliorate arterial dilatation (71). However, the impact of mitochondrial ROS on endothelial function has to be studied intensively since overexpression of catalase, which is an important anti-oxidant enzyme, induces endothelial dysfunction in mice models (72).
Mitochondrial function in coronary atherosclerosis
Atherosclerosis is a chronic inflammatory process and the most common substrate of coronary artery disease (CAD). CAD is the leading cause of death in the developed world and is characterized by acute or chronic ischemia due to insufficient myocardial oxygen supply (73).
Mitochondria have an important role in the pathophysiology of atherosclerosis (Figure 4). Mitochondrial dysfunction results in excessive production of ROS which oxidize cellular proteins, lipids and DNA (74). Mitochondrial DNA is especially prone to oxidative damage since it lacks histones and has a minor capacity for repair; furthermore, mitochondrial DNA mutations trigger the induction of a vicious cycle of ROS production as mentioned in a previous section (75). For example, studies in apo-E deficient mice which lacked the anti-oxidant enzyme SOD-2 have demonstrated that excessive production of ROS damaged mitochondrial DNA and accelerated the progression of atherosclerosis and proliferation of vascular smooth muscle cells (VSMCs) (76). Moreover, a study in humans who underwent intravascular ultrasound characterization of coronary artery plaques indicated that mitochondrial DNA damage of leukocytes is associated with the existence of vulnerable plaques but not with plaque burden (77).
Figure 4.

Mitochondria and pathophysiology of cardiac diseases.
Oxidized LDL molecules (ox-LDL) are absorbed by macrophages which display scavenger receptors on their surface and form foam cells which are rich in lipids and cell debris (78). Foam cells release a number of pro-inflammatory mediators such as adhesion molecules and circulating cytokines which attract inflammatory cells to the damaged vascular wall. These mediators stimulate the formation of neo-intima through hyperplasia, migration and proliferation of VSMCs (79).
Moreover, atherosclerosis is characterized by increased apoptosis of VSMCs and vascular wall remodeling. Studies in cultures of human aortic endothelial cells indicate that ox-LDL or glycated ox-LDL decrease the expression of cellular anti-apoptotic proteins and stimulate mitochondrial apoptotic pathways (80,81). Similarly, a study in human microvascular endothelial cells indicated that ox-LDL molecules increased the influx of cytosolic Ca2+ which in turn activated two mitochondrial pathways of apoptosis; the first one which involves the release of apoptosis inducing factor and the second one which is associated with the opening of mitochondrial mPTP channel (82).
To sum up, mitochondrial dysfunction is responsible not only for the initiation but also the progression of the atherosclerotic vascular disease and is, therefore, an important target for the treatment of CAD.
Mitochondrial function in ischemia/reperfusion injury
Ischemic heart disease is the leading cause of mortality and morbidity in the modern world and its most common clinical presentation is acute ischemia. Early and successful reperfusion is the key against acute myocardial ischemia and can be achieved either pharmaceutically or mechanically through surgical intervention or coronary artery stenting. However, reperfusion upon ischemia is associated with damage at the molecular level though complicated mechanisms and the phenomenon is described as “acute ischemia-reperfusion injury” (acute I/R) (78).
I/R injury consists one of the best experimental models to evaluate the effects of oxidative stress on cardiomyocytes (Figure 4). Prolonged ischemia results in the death of cardiac cells due to insufficient oxygen supply. However, it has been proved that cells around the area of the infarct are at risk of further, delayed damage upon reperfusion, when oxygen supply is replenished. It is really interesting that in both conditions ROS formation are the determinants of the final damage which is comprised by increased fibrosis, angiogenesis, and vascular remodeling (78).
During ischemia cardiomyocytes become hypoxic and deteriorate the function of mitochondrial respiratory chain enzymes; therefore, superoxide, hydrogen peroxide, peroxynitrite and hydroxyl radical are formed. Specifically, I/R injury impairs the function of mitochondrial complexes I and III and stimulates the generation of ROS by NADH (78). Decreased functional capacity of complex I function is linked to damaged mitochondrial cardiolipin which accelerates electron leakage and stimulates a vicious cycle of free radical generation (83).
Another important source of ROS in the re-perfused myocardium are the two isoforms of monoamine oxidases (MAO), MAO-A and MAO-B which are located on the outer mitochondrial membrane. It has been demonstrated that during post-ischemic reperfusion the enhanced activity of MAO-A is responsible for the precipitation of hydrogen peroxide and the progression towards left ventricle hypertrophy and cardiac remodeling. Also, increased influx of mitochondrial iron stimulates the formation of more potent and deleterious hydroxyl radical groups from hydrogen peroxide (84).
Additionally, Li et al. indicated that excessive production of ROS affects the balance among mitochondrial fission and fusion (85). Specifically, protein kinase C (PKC) phosphorylates critical substrates which are involved in the clearance of damaged mitochondria and inhibit the apoptosis of dysfunctional organelles (61). Also, myocardial I/R injury is associated with de-phosphorylation of Drp1 at serine 637 which in turn translocates to the outer mitochondrial membrane protein receptors Fis1, Mff or MIEFl and induces mitochondrial fission (86). ROS impair the function of mitofusins and OPA1 and decrease mitochondrial fusion (87). Moreover, reperfusion induces the opening of mPTP channel which stimulates the release of cytochrome c and apoptosis. In general, overproduction of ROS in I/R injury has been associated with the senescence and death of endothelial cells due to compromised telomere integrity which eventually favors cellular apoptosis (88).
Lastly, ROS damage mitochondrial DNA which is poor in mechanisms of repair and several studies have indicated that mitochondrial DNA of circulating leukocytes could be diagnostic of I/R injury (78). Interestingly, in a recent study in patients with acute coronary syndromes it was found that free circulating mitochondrial DNA in blood could predict cardiovascular mortality at 30 days (89).
In conclusion, I/R injury induces detrimental effects on cardiovascular system due to excessive ROS formation. It is important to mention that ROS production by two different phenomena might be beneficial for cardiovascular system. The first one is ischemic preconditioning and is defined as the production of sublethal amounts of ROS during short cycles of ischemia and reperfusion which results in cardio-protection. It was first identified in 1986 by Murry et al. who exposed anesthetized, open-chest dogs to four periods of 5-minute coronary artery occlusions followed by a 5-minute period of reperfusion before the onset of a 40-minute sustained occlusion of the coronary artery; interestingly, the infarct sizes were smaller in the intervention group in comparison to the control group (90). In general, the pathophysiological mechanism involves activation of PKC epsilon which stimulates mitochondrial KATP channels; then increased production of H2O2 exerts protective actions through blockage of mPTP channel (91). The second one is remote ischemic conditioning and was firstly described in 1993 by Przyklenk et al. who revealed that brief episodes of ischemia in one vascular bed protect remote and virgin myocardium against damage from sustained coronary artery occlusion (92). The underlying mechanism is the synthesis of factors in a remote organ which induce protective actions in remote organs through complex neuro-humoral interactions as previously described for ischemic preconditioning (91).
Mitochondrial function in hypertension
Hypertension is one of the most important risk factors for the development of CAD (93). Mitochondrial dysfunction in hypertension results in impaired energy production and deficient calcium homeostasis (82,94); moreover, the decreased activity of anti-oxidant enzymes and oxidative modification of cellular compartments is associated with damage in the heart, the brain, the kidneys and the vessels (79,95) (Figure 4).
To begin with, SIRT 3 is a histone deacetylase which depends on NAD+ activity and displays crucial anti-oxidant properties. Interestingly, it has been found that the increased prevalence of hypertension in aged people is linked to impaired mitochondrial metabolism and reduction in the activity of SIRT3 protein. A study in humans indicated that the expression of SIRT3 is reduced by 40% at the age of 65 especially in sedentary adults in contrast to the higher levels which were found in trained subjects independently of age (96). Augmented levels of angiotensin-II produced by RAS system are also associated with the down-regulation of SIRT3 gene expression (97).
Well-functioning mitochondrial anti-oxidant systems such as SOD-2 prevent the damage induced by excessive ROS formation in hypertension due to ageing or high-salt diet (98). However, mutations in mitochondrial tRNA result in the development of hypertension, hypercholesterolemia and hypomagnesemia especially in subjects of 30 years of age indicating the detrimental effects of environmental factors and ageing on mitochondrial anti-oxidant capacity (99).
Activation of RAS system has an important role in the pathophysiology of hypertension. Interestingly, it has been demonstrated that angiotensin-II stimulates NADPH oxidase and acceleration of oxidative stress (100). Additionally, blood pressure levels regulate the production of ROS through the functions of mechano-sensitive receptors (101).
Furthermore, hypertension is associated with structural mitochondrial abnormalities which involve decreased mitochondrial mass, density and mitochondrial dwelling that result in impaired energy production and accelerated formation of ROS through instability of ETC complexes (102,103). Moreover, hypertension is linked to decreased functional capacity of complex-I system, and stimulation of of fibrosis and extracellular cell matrix expansion which further deteriorate myocardial contractility (104). Similarly, the raised expression of biomarkers of mitophagy promotes altered expression of calcium cycling proteins which result in interstitial fibrosis of left ventricle and diastolic dysfunction (105).
Hypertension also affects mitochondrial biogenesis and dynamics which affect energy production (106). For instance, studies in hypertensive rats have indicated decreased mRNA expression of the fusion proteins mitofusin-1 and -2, and optic atrophy-1 which implicated in mitochondrial fragmentation and stimulation of oxidative stress (107).
Moreover, oxidative stress provokes the expression of several pro-inflammatory molecules in several models of hypertension. Interleukin (IL)-1 and tumor necrosis factor (TNF)-α, for instance, which are important pro-inflammatory cytokines, decrease the function of mitochondrial aldehyde dehydrogenase-2 and reduce membrane potential and ATP production in adipocytes (108,109).
Finally, it has been demonstrated that hypertension is associated with the activation of apoptosis. Reduced expression of cardiolipin, which is an important phospholipid for the balanced function of mitochondria, induces the release of cytochrome c to cytosol and triggers the pathway of apoptosis (110,111). Similarly, in a study in hypertensive rats it was revealed that activation of RAS system decreased the functions of complex-III system, ATP synthase, creatine kinase and it enhanced the release of cytochrome c and caspase-3 from the dysfunctional organelles (112).
In conclusion, mitochondrial dysfunction is highly present in hypertension which indicates the importance of early therapeutic management based on the molecular level.
Mitochondrial function in obesity, metabolic syndrome and diabetes
Metabolic syndrome and diabetes mellitus (DM) belong to the most challenging medical problems of the 21st century. DM is a chronic disease which affects numerous people independently of age, race and sex and is characterized by hyperglycemia and altered lipid, protein and carbohydrate metabolism (113,114). The most common type of diabetes worldwide is type 2 DM which is attributed to deficient function of β-cell resulting in insulin resistance and deteriorated insulin secretion. Type 1 DM is attributed to autoimmune destruction of the β-cell and ends in total deficiency of insulin secretion (115). DM affects almost every tissue including vascular system, heart, retina, kidneys and peripheral nerves and several mechanisms have been implicated in the pathophysiology of the disease such as decreased physical activity, obesity, elevated free fatty acids, genetic factors, oxidative stress and mitochondrial dysfunction (116,117) (Figure 4).
Mitochondria are the central organelles for ATP production through oxidative phosphorylation (116). Recent studies have indicated that impaired mitochondrial function is linked to altered glucose and fatty acid metabolism, lower ATP production in muscle cells, impaired insulin secretion from β-cells and stimulation of ROS production (118). In general, the mechanisms of mitochondrial dysfunction involve the decrease in mitochondrial content in tissues such as muscles, liver and adipose tissue, the absence or dysfunction of mitochondrial proteins and reduced mitochondrial biogenesis (119).
Beta oxidation is the main source of energy for the heart. However, excess delivery of fatty acids in diabetes and metabolic syndrome results in decreased oxygen utilization by mitochondria and uncoupling of ETC systems. Moreover, NADH triggers ROS generation and impaired expression of thioredoxin and glutathione (120). Also, decreased mitochondrial density and reduced production of mitochondrial proteins due to mitochondrial DNA mutations or deletions is evident (116,119,121). Additionally, alterations in lipid extent and metabolism and the impairment of oxidative phosphorylation increase the accumulation of diacylglycerols and ceramides which block insulin secretion and favor the progression to metabolic syndrome and type 2 DM (119). Similarly, the impaired mitochondrial protein content activates stress related serine/threonine kinases which block glucose transport and favor the formation of fatty acids (122,123). Lastly, the excessive amounts of lipids target the downstreams of insulin receptor substrate (IRS 1-2) and Akt pathways resulting in insulin resistance (124).
Furthermore, cumulative DNA damage is linked to the loss of telomeres which shortens the lifespan of β-cells and provokes insufficient insulin production (116,125). However, this damage is usually found in a rare kind of diabetes initially described as maternally inherited diabetes and deafness syndrome induced by A3243G mutation of mitochondrial DNA (126).
The unopposed production of ROS elicits decreased NO synthesis in diabetic hearts which leads in structural alterations of cardiac proteins that negatively affect cardiac muscle relaxation and diastolic dysfunction in diabetic mice (127). Moreover, evidence from human studies indicates that patients with type 2 DM display dysfunctional myocardial contractility due to mitochondrial dysfunction. Interestingly, this finding does not happen at the early stages of this metabolic disorder which is described as metabolically “healthy” obesity status (128).
ROS alter mitochondrial dynamics and specifically increase mitochondrial fission and fragmentation at the expense of mitochondrial fusion (124). Little is known about the exact mechanisms but nuclear respiratory factors (NRF 1 and 2) are considered to play an important role (121). NRF1 and 2 regulate the expression of Tfam, a transcription factor of mitochondrial genome, and numerous other mitochondrial genes implicated in oxidative phosphorylation. Although NRFs and Tfam are necessary for mitochondrial biogenesis, Tfam knockout experimental studies have not yet linked Tfam and NRF disorders with the development of insulin resistance and metabolic syndrome despite the significant changes in mitochondria morphology and density (123). According to new studies, mitochondrial dysfunction and insulin resistance are linked to altered gene expression of PCG1 in muscle and liver tissues (129,130). Specifically, it has been demonstrated that downregulation of PGC1a leads to impaired mitochondrial biogenesis and the induction of insulin resistance (116).
In conclusion, mitochondrial dysfunction is closely related to the pathophysiology of DM; therefore, targeting mitochondrial function belongs to the most prominent therapeutic interventions against the spectrum of metabolic disorders.
Mitochondrial function in cardiac hypertrophy and HF
HF is the result of numerous cardiac diseases and has a rapidly increasing prevalence due to the effectiveness is primary and secondary prevention of cardiovascular diseases (131,132). HF is a clinical syndrome and involves structural and/or functional cardiac abnormalities which end in reduced cardiac output and/or elevated intra-cardiac pressures at rest or during stress (133,134). The initial response to increased cardiac workload is cardiac hypertrophy which is defined as the thickening of ventricular wall and reduction in ventricular volume (135). Physiological heart hypertrophy is a normal mechanism of adaptation which may regress through the time such as in the athletes’ heart; however, pathological hypertrophy is attributed to divergent cardiac diseases and is characterized by an initial phase of compensation and the acute or chronic progression to decompensation (134). Interestingly, mitochondrial dysfunction is an object of intense investigation in order to understand this complex clinical syndrome and discover novel molecular therapeutic targets (136) (Figure 4).
To begin with, PGC-1α is one of the most important regulators of mitochondrial biogenesis, as described above. PGC-1α co-activates NRF-1 and NRF-2 and increases the transcription of several mitochondrial DNA genes which participate in the respiratory chain (137). In normal cardiac hypertrophy, PGC-1α expression is enhanced and, as a result, mitochondrial DNA duplication is stimulated as well as fatty acid oxidation (137,138). Normal hypertrophy is also controlled by the pathways of PI3K and its downstream products Akt and GSK-3β which control cellular growth and preservation of heart function. In fact, there is evidence that the actions of Akt, despite being a component of PI3K pathway, differ from those of PI3K (138,139).
However, pathologically hypertrophied hearts display a metabolic shift to increased utilization of glucose than fatty acid oxidation, which is a metabolic pattern of the fetal age (140-142). Specifically, Heather et al. measured the expression of proteins implicated in glucose and fatty acid metabolism in samples obtained by cardiac biopsies in 18 patients with aortic stenosis. Their measurements showed a negative correlation between fatty acid translocase (FAT/CD36) and glucose uptake membrane transporters (GLUT) such as GLUT 1 and 4 and a further decrease in other proteins involving β-oxidation, Krebs cycle and oxidative phosphorylation (ATP synthase and complex I of ETC). In fact, the higher the left ventricular mass index, the more downregulated were FAT/CD36 and complex I and the more upregulated was GLUT 4 (140). Similarly, expression of PGC-1α is reduced in pathologically hypertrophied hearts and occurs at the early stages of the disease (141,143). Moreover, de las Fuentes et al. have also demonstrated a significant reduction in fatty acid metabolism in patients with hypertensive left ventricular hypertrophy which is linked to higher left ventricular mass and lower myocardial contractility (142). In general, the reverse of the energy production to the previously described pattern of fetal life is responsible for the deficits in energy production of pathologic cardiac hypertrophy since glycolysis produces less amount of ATP than β-oxidation of fatty acids (142,144,145).
Excessive ROS production and impaired function of the anti-oxidant systems are associated with the development of cardiac hypertrophy, remodeling and HF (146,147). In a model of PGC-1α knockout mice it was demonstrated that the expression of anti-oxidant enzymes such as mitochondrial Cu/Zn-SOD1, SOD-2 and peroxisomal catalase were notably decreased (148). Similarly, in a model of experimental myocardial infarction in rats it was observed a significant increase in the formation of hydroxyl radicals and reduced copies of mitochondrial DNA and transcripts of I, III and IV complexes (149). Lower activity or defects in mitochondrial complexes I, III, IV, V in dogs, rats and human frozen-thawed cardiac-muscle samples have been demonstrated too (149,150).
Another important feature of HF is the development of fibrosis and cardiac remodeling. Several mediators are implicated in this process such as angiotensin-II, norepinephrine, β-adrenergic agonists, TNF-α, endothelin-I as well as mechanical forces which activate PKC, MAPK, PI3K, JNK and nuclear factor kappa beta (NF-κB) (147). Additionally, stimulation of matrix metalloproteases (MMP) via the pathway of NF-κB is implicated in the degradation of extracellular cell matrix, cell proliferation and apoptosis which leads to remodeling and fibrosis (151,152). For example, in a model of pacing-induced supraventricular tachycardia in pigs it was demonstrated that MMP 1, 2 and 3 induce left ventricle dysfunction and dilation of left ventricle at 7 days (153).
Mitochondrial dysfunction in HF also induces impaired mitochondrial biogenesis. Studies in both human and rat models of HF have indicated small and fragmented mitochondria and lower levels of OPA1 implying the participation of mitochondrial fission in cardiac remodeling (154). Similarly, calcium overload stimulates mitochondrial fission and formation of fragmented mitochondria (155). Moreover, defective mitophagy in HF impairs myocardial function since damaged and non-functional mitochondria are important sources of ROS (41). For example, in an experimental model of parkin knockout Drosophila, it was indicated that blockage of mitophagy increased the number of dysfunctional mitochondria in heart tubes which resulted in the development of dilated cardiomyopathy (156).
Lastly, it is important to note that systolic and diastolic dysfunction of the failing myocardium is provoked by the detrimental effects of ROS on sarcomeric and excitation-contraction coupling proteins. For instance, oxidative modification of thiol groups of critical cysteine residues result in inhibition of L-type calcium channel and Ca2+ ATPase in the sarcoplasmic reticulum which reduces the rate of contractility. Other vulnerable sarcomeric proteins are myosin light chain-2, myosin light polypeptide-3, alpha-actin, troponin T, actin, desmin and tropomyosin (157,158).
In conclusion, normal cardiac hypertrophy is a totally different phenotype from pathologic cardiac hypertrophy based on the molecular level. Subsequently, further research on mitochondrial function in HF is crucial due to the central role of mitochondria in energy production.
Mitochondrial dysfunction: therapeutic implications
It is undeniable that the breakthroughs in primary and secondary prevention of cardiovascular disease have improved the lives of million people worldwide. The aim of current research is to develop novel therapeutic molecules which target mitochondrial function and excessive ROS production implicated in the progression of atherosclerosis, I/R injury, DM, hypertension and HF (159). Several therapeutic strategies have been examined such as dietary changes, exercise and medications which target the mechanisms of oxidative stress, inflammation, cardiac hypertrophy, fibrosis and apoptosis with, however, controversial results (160-162) (Figure 5).
Figure 5.

Therapeutic interventions against mitochondrial dysfunction.
To begin with, certain dietary interventions have been tested in both animal and human models. For example, the administration of 2 gr L-carnitine daily had a survival benefit in patients with HF (163). Similarly, in patients with mild diastolic HF the consumption of 9 g L-carnitine per day for a period of 3 months resulted in improvement of diastolic dysfunction (164). Also, in a model of experimental hypertension the administration of L-carnitine improved cardiac remodeling through decreased ROS production (165).
Polyphenols such as flavolons, theaflavin and epicatechin are chemical compounds present in a variety of natural sources such as red wine, green tea, olive oil and dark chocolate (166). Polyphenols have important anti-oxidant actions against several chronic diseases including cardiovascular disease (167,168). For example, quercetin decreases the levels of superoxide and increases urinary excretion of nitrate, endothelial NO synthase activity and heme oxygenase-1 protein which has anti-oxidant actions (169). Moreover, polyphenols of olive oil and red wine reduce intracellular ROS levels (170) whereas epicatechin of green tea lowers the expression of pro-inflammatory molecules (171).
Vitamins C and E are popular anti-oxidant molecules and have beneficial actions against a large group of chronic diseases (172). Large clinical trials have exhibited disappointing results against mitochondrial dysfunction. Possibly, mitochondria absorb only a small percent of these anti-oxidants or there are unknown interactions with other therapeutic, regimens unspecified pro-oxidant actions or genetic variability response among the subjects which explain these inconsistent findings (173-177).
On the other hand, several studies have examined the role of co-enzyme Q10 (CoQ10) in animal and human trials. CoQ10 is present in the inner mitochondrial membrane and is important for the production of ATP; also, it possesses anti-thrombotic and anti-oxidant actions and improves hypertension and hyperglycemia (178). Administration of CoQ10 in hypertensive rats improved endothelial function and decreased cardiac hypertrophy (179). Though, CoQ10 does not increase mitochondrial DNA replication and does not hinder the degradation of mitochondrial cardiolipin (180-182). CoQ10 has been administrated in humans to alleviate muscle pain upon treatment of statins (183). Interestingly, it was found that supplementation of 100 mg daily CoQ10 for 30 days improved muscle pain in patients receiving statins (183). In short, CoQ10 is considered a safe option for the treatment of mitochondrial dysfunction in humans and can be administrated alone or along with other medications against hypertension and HF although its properties are not fully elucidated in ischemic heart disease (184).
Moreover, a current strategy is to administrate anti-oxidant molecules which are conjugated to lipophilic molecules in order to selectively target mitochondria (185,186). For example, the recently developed MitoQ10 improves endothelial NO bioavailability and blood pressure in a model of spontaneously hypertensive rats (179). Addition of MitoQ10 to treatment with losartan has revealed beneficial actions against target organ damage development in hypertension (187). Lastly, other synthetized molecules such as EUK-8 and EUK-134 mimic endogenous inorganic SOD activity and have displayed direct mitochondrial anti-oxidant actions against I/R injury (188).
Regular physical activity has beneficial actions on cardiovascular system (189). Aerobic exercise increases the production of NO, lowers the levels of superoxide and hydrogen peroxide and improves endogenous enzymatic anti-oxidant systems (190,191). Also, it reduces systolic and diastolic blood pressure in hypertensive subjects, whereas isometric exercise affects only systolic blood pressure (192,193). Nevertheless, the exact mechanisms of exercise on mitochondrial function are not yet elucidated.
Another therapeutic target is stimulation of NAD+ biosynthetic pathway which increases protein deacetylation through SIRT (194). Treatment with the NAD+ precursor NMN has indicated normalization of NAD+/NADH ratio and protection against diet- or age-induced diabetes (195). Moreover, in animal studies of hypertension it has been demonstrated that activation of SIRT-1-PGC1α signaling by resveratrol improves mitochondrial biogenesis (196). Similarly, in animal models of diabetes resveratrol hindered the progression to diabetic cardiomyopathy (197).
Excessive fatty acid oxidation is linked to the development of several metabolic disorders such as obesity and DM. Therefore, it seems reasonable that decreased uptake and utilization of fatty acids could be beneficial. For example, exercise, calorie restriction and weight loss such as that achieved through bariatric surgery improve insulin sensitivity and mitochondrial function (198,199). Moreover, inhibition of CD36 (which mediates lipid uptake through plasma membrane) decreases mitochondrial oxidative stress in animal models; however, targeting of CD36 has not been tested in humans due to the pleiotropic functions of this receptor in humans (200). Lastly, ligands of PPARα have been used in order to stimulate the impaired fatty acid metabolism in HF; however, their efficacy has to be evaluated (160).
Apart from the above agents, widely prescribed medications have demonstrated beneficial actions against mitochondrial dysfunction. For instance, carvedilol, which belongs to β-blockers, has indicated anti-oxidant and anti-apoptotic properties in patients with HF (201).
Targets of RAS system activation such as ACE inhibitors and angiotensin receptor-II blockers improve mitochondrial dysfunction; for instance, captopril increased mitochondrial biogenesis in an experimental study in dogs (202-204). Also, treatment with losartan and amlodipine reduced blood pressure in spontaneously hypertensive rats whereas only losartan restored mitochondrial dysfunction and kidney damage through preservation of glutathione and SOD activity (204).
Statins apart from inhibition of endogenous cholesterol synthesis display important pleiotropic effects (205-207). Specifically, they decrease oxidative stress in various tissues targeting mitochondrial function (208). Parihar et al. indicated that the administration of atorvastatin and simvastatin in rats reduced the activity of mitochondrial NO synthase, cytochrome c release and lipid peroxidation (209).
Thiazolideniones are oral antidiabetic drugs against type 2 DM that improve insulin resistance through activation of PPARγ, which is implicated in the transcription of genes involved in glucose and lipid metabolism (210). Studies in animals have indicated that rosiglitazone reduces lipid oxidation and hinders the development of atherosclerosis through upregulation of PPARγ (211).
Metformin, the first line therapy for patients with type 2 DM, has exhibited several beneficial actions on cardiovascular system (212). Recent studies point that metformin reduces mitochondrial ROS production, enhances the activity of anti-oxidant enzymes and decreases inflammation attributed to I/R injury (213).
Recent research has shed light to the role of micro-RNAs (miR) which are involved in transcription of cellular genes that either promote or prevent the development of disease (214-216). Interestingly, a study in rats indicated that overexpression of miR145 is linked to improved cardiac function and decreased infarct size post myocardial infarction; also, low levels of miR145 were confirmed in vitro in hypoxia-treated cardiomyocytes. Moreover, miR145 targets PDCD4 gene which is involved in the apoptotic pathway and it seems that mimics of this agent could be used in the treatment of myocardial infarction (217).
Edaravone is a novel free radical scavenger. Edaravone reduced pressure overload-induced left ventricular hypertrophy in mice through inhibition of Ask1 and its downstream kinases (218). Also, it diminished perivascular and intermuscular fibrosis and improved cardiac hypertrophy even when treatment was initiated after the onset of cardiac hypertrophy (219).
Elamipretide (SS-31) is a novel, water-soluble tetrapeptide which enhances mitochondrial energy production. Elamipretide binds selectively to cardiolipin and preserves the structure of mitochondrial cristae and the function of oxidative phosphorylation (220). Interestingly, in a study in dogs with advanced HF Elamipretide improved left ventricular function and enlargement, plasma natriuretic peptides and biomarkers of inflammation through decreased ROS formation (221). Moreover, in a randomized, placebo-controlled trial in humans with HF and reduced ejection fraction it ameliorated left ventricular end-diastolic and end-systolic volume (222). Lastly, in a study in rats it improved mitochondrial oxidative stress mediated by angiotensin-II through inhibition of p38 MAPK pathway and hindered cardiac remodeling and inflammation post myocardial infarction (223).
In conclusion, several medications have been tested for the therapy of mitochondrial dysfunction with controversial results so far. Novel therapeutic strategies involve the design of molecules which target specific pathways of mitochondrial function.
Conclusions
Mitochondria are cellular organelles which produce energy through oxidative phosphorylation and their function is crucial for the heart due to the high energy demands. Mitochondrial dynamics consist of a balance between mitochondrial fusion and mitochondrial fission which controls energy production. On the other hand, mitophagy is an important mechanism of removal of the dysfunctional organelles. Several cardiac diseases such as atherosclerosis, ischemia-reperfusion injury, hypertension, diabetes and heart failure are linked to improper mitochondrial function and excessive production of ROS which damage cellular lipids, proteins, enzymes and DNA. Mitochondrial dysfunction is also associated with apoptosis which accelerates cardiovascular damage. Numerous therapeutic interventions against mitochondrial dysfunction have been tested in both animal and human models and research in this field is constantly advancing.
Acknowledgements
None.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Balaban RS. Perspectives on: SGP symposium on mitochondrial physiology and medicine: metabolic homeostasis of the heart. J Gen Physiol 2012;139:407-14. 10.1085/jgp.201210783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sagan L. On the origin of mitosing cells. J Theor Biol 1967;14:255-74. 10.1016/0022-5193(67)90079-3 [DOI] [PubMed] [Google Scholar]
- 3.Ott M, Amunts A, Brown A. Organization and Regulation of Mitochondrial Protein Synthesis. Annu Rev Biochem 2016;85:77-101. 10.1146/annurev-biochem-060815-014334 [DOI] [PubMed] [Google Scholar]
- 4.Miettinen TP, Bjorklund M. Mitochondrial Function and Cell Size: An Allometric Relationship. Trends Cell Biol 2017;27:393-402. 10.1016/j.tcb.2017.02.006 [DOI] [PubMed] [Google Scholar]
- 5.Lemasters JJ. Variants of mitochondrial autophagy: Types 1 and 2 mitophagy and micromitophagy (Type 3). Redox Biol 2014;2:749-54. 10.1016/j.redox.2014.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galluzzi L, Kepp O, Trojel-Hansen C, et al. Mitochondrial control of cellular life, stress, and death. Circ Res 2012;111:1198-207. 10.1161/CIRCRESAHA.112.268946 [DOI] [PubMed] [Google Scholar]
- 7.Pillai VB, Bindu S, Sharp W, et al. Sirt3 protects mitochondrial DNA damage and blocks the development of doxorubicin-induced cardiomyopathy in mice. Am J Physiol Heart Circ Physiol 2016;310:H962-72. 10.1152/ajpheart.00832.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hall CJ, Sanderson LE, Crosier KE, et al. Mitochondrial metabolism, reactive oxygen species, and macrophage function-fishing for insights. J Mol Med (Berl) 2014;92:1119-28. 10.1007/s00109-014-1186-6 [DOI] [PubMed] [Google Scholar]
- 9.Pellegrini L, Scorrano L. A cut short to death: Parl and Opa1 in the regulation of mitochondrial morphology and apoptosis. Cell Death Differ 2007;14:1275-84. 10.1038/sj.cdd.4402145 [DOI] [PubMed] [Google Scholar]
- 10.Lopaschuk GD, Russell JC. Myocardial function and energy substrate metabolism in the insulin-resistant JCR:LA corpulent rat. J Appl Physiol (1985) 1991;71:1302-8. 10.1152/jappl.1991.71.4.1302 [DOI] [PubMed] [Google Scholar]
- 11.Stanley WC. Rationale for a metabolic approach in diabetic coronary patients. Coron Artery Dis 2005;16 Suppl 1:S11-5. 10.1097/00019501-200511001-00003 [DOI] [PubMed] [Google Scholar]
- 12.Wang Z, Wu M. An integrated phylogenomic approach toward pinpointing the origin of mitochondria. Sci Rep 2015;5:7949. 10.1038/srep07949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Esser C, Ahmadinejad N, Wiegand C, et al. A genome phylogeny for mitochondria among alpha-proteobacteria and a predominantly eubacterial ancestry of yeast nuclear genes. Mol Biol Evol 2004;21:1643-60. 10.1093/molbev/msh160 [DOI] [PubMed] [Google Scholar]
- 14.Georgiades K, Madoui MA, Le P, et al. Phylogenomic analysis of Odyssella thessalonicensis fortifies the common origin of Rickettsiales, Pelagibacter ubique and Reclimonas americana mitochondrion. PLoS One 2011;6:e24857. 10.1371/journal.pone.0024857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pittis AA, Gabaldon T. Late acquisition of mitochondria by a host with chimaeric prokaryotic ancestry. Nature 2016;531:101-4. 10.1038/nature16941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martin WF, Roettger M, Ku C, et al. Late Mitochondrial Origin Is an Artifact. Genome Biol Evol 2017;9:373-9. 10.1093/gbe/evx027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anderson S, Bankier AT, Barrell BG, et al. Sequence and organization of the human mitochondrial genome. Nature 1981;290:457-65. 10.1038/290457a0 [DOI] [PubMed] [Google Scholar]
- 18.Guda P, Subramaniam S, Guda C. Mitoproteome: human heart mitochondrial protein sequence database. Methods Mol Biol 2007;357:375-83. [DOI] [PubMed] [Google Scholar]
- 19.Mercer TR, Neph S, Dinger ME, et al. The human mitochondrial transcriptome. Cell 2011;146:645-58. 10.1016/j.cell.2011.06.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Armengod ME, Meseguer S, Villarroya M, et al. Modification of the wobble uridine in bacterial and mitochondrial tRNAs reading NNA/NNG triplets of 2-codon boxes. RNA Biol 2014;11:1495-507. 10.4161/15476286.2014.992269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yao K, Zhang WW, Yao L, et al. Carvedilol promotes mitochondrial biogenesis by regulating the PGC-1/TFAM pathway in human umbilical vein endothelial cells (HUVECs). Biochem Biophys Res Commun 2016;470:961-6. 10.1016/j.bbrc.2016.01.089 [DOI] [PubMed] [Google Scholar]
- 22.Coronado M, Fajardo G, Nguyen K, et al. Physiological Mitochondrial Fragmentation Is a Normal Cardiac Adaptation to Increased Energy Demand. Circ Res 2018;122:282-95. 10.1161/CIRCRESAHA.117.310725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andres AM, Hernandez G, Lee P, et al. Mitophagy is required for acute cardioprotection by simvastatin. Antioxid Redox Signal 2014;21:1960-73. 10.1089/ars.2013.5416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kong D, Zhan Y, Liu Z, et al. SIRT1-mediated ERbeta suppression in the endothelium contributes to vascular aging. Aging Cell 2016. [Epub ahead of print]. 10.1111/acel.12515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parra V, Verdejo H, del Campo A, et al. The complex interplay between mitochondrial dynamics and cardiac metabolism. J Bioenerg Biomembr 2011;43:47-51. 10.1007/s10863-011-9332-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goh KY, Qu J, Hong H, et al. Impaired mitochondrial network excitability in failing guinea-pig cardiomyocytes. Cardiovasc Res 2016;109:79-89. 10.1093/cvr/cvv230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang HK, Wang YH, Sun L, et al. Aerobic interval training attenuates mitochondrial dysfunction in rats post-myocardial infarction: roles of mitochondrial network dynamics. Int J Mol Sci 2014;15:5304-22. 10.3390/ijms15045304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Long B, Wang K, Li N, et al. miR-761 regulates the mitochondrial network by targeting mitochondrial fission factor. Free Radic Biol Med 2013;65:371-9. 10.1016/j.freeradbiomed.2013.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koshiba T, Detmer SA, Kaiser JT, et al. Structural basis of mitochondrial tethering by mitofusin complexes. Science 2004;305:858-62. 10.1126/science.1099793 [DOI] [PubMed] [Google Scholar]
- 30.Papanicolaou KN, Ngoh GA, Dabkowski ER, et al. Cardiomyocyte deletion of mitofusin-1 leads to mitochondrial fragmentation and improves tolerance to ROS-induced mitochondrial dysfunction and cell death. Am J Physiol Heart Circ Physiol 2012;302:H167-79. 10.1152/ajpheart.00833.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eisner V, Cupo RR, Gao E, et al. Mitochondrial fusion dynamics is robust in the heart and depends on calcium oscillations and contractile activity. Proc Natl Acad Sci U S A 2017;114:E859-68. 10.1073/pnas.1617288114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang X, Jiang H, Shen Z, et al. Activation of mitochondrial protease OMA1 by Bax and Bak promotes cytochrome c release during apoptosis. Proc Natl Acad Sci U S A 2014;111:14782-7. 10.1073/pnas.1417253111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Das S, Mitrovsky G, Vasanthi HR, et al. Antiaging properties of a grape-derived antioxidant are regulated by mitochondrial balance of fusion and fission leading to mitophagy triggered by a signaling network of Sirt1-Sirt3-Foxo3-PINK1-PARKIN. Oxid Med Cell Longev 2014;2014:345105. 10.1155/2014/345105 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.Huang X, Sun L, Ji S, et al. Kissing and nanotunneling mediate intermitochondrial communication in the heart. Proc Natl Acad Sci U S A 2013;110:2846-51. 10.1073/pnas.1300741110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lavorato M, Iyer VR, Dewight W, et al. Increased mitochondrial nanotunneling activity, induced by calcium imbalance, affects intermitochondrial matrix exchanges. Proc Natl Acad Sci U S A 2017;114:E849-58. 10.1073/pnas.1617788113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang W, Ren H, Xu C, et al. Hypoxic mitophagy regulates mitochondrial quality and platelet activation and determines severity of I/R heart injury. Elife 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin S, Wang Y, Zhang X, et al. HSP27 Alleviates Cardiac Aging in Mice via a Mechanism Involving Antioxidation and Mitophagy Activation. Oxid Med Cell Longev 2016;2016:2586706. 10.1155/2016/2586706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yan L, Yang H, Li Y, et al. Regulator of calcineurin 1-1L protects cardiomyocytes against hypoxia-induced apoptosis via mitophagy. J Cardiovasc Pharmacol 2014;64:310-7. 10.1097/FJC.0000000000000121 [DOI] [PubMed] [Google Scholar]
- 39.Bazil JN, Beard DA, Vinnakota KC. Catalytic Coupling of Oxidative Phosphorylation, ATP Demand, and Reactive Oxygen Species Generation. Biophys J 2016;110:962-71. 10.1016/j.bpj.2015.09.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuka S, Tatarkova Z, Racay P, et al. Effect of aging on formation of reactive oxygen species by mitochondria of rat heart. Gen Physiol Biophys 2013;32:415-20. 10.4149/gpb_2013049 [DOI] [PubMed] [Google Scholar]
- 41.Korge P, John SA, Calmettes G, et al. Reactive oxygen species production induced by pore opening in cardiac mitochondria: The role of complex II. J Biol Chem 2017;292:9896-905. 10.1074/jbc.M116.768325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lam CK, Zhao W, Liu GS, et al. HAX-1 regulates cyclophilin-D levels and mitochondria permeability transition pore in the heart. Proc Natl Acad Sci U S A 2015;112:E6466-75. 10.1073/pnas.1508760112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Madungwe NB, Zilberstein NF, Feng Y, et al. Critical role of mitochondrial ROS is dependent on their site of production on the electron transport chain in ischemic heart. Am J Cardiovasc Dis 2016;6:93-108. [PMC free article] [PubMed] [Google Scholar]
- 44.Jang S, Javadov S. Association between ROS production, swelling and the respirasome integrity in cardiac mitochondria. Arch Biochem Biophys 2017;630:1-8. 10.1016/j.abb.2017.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dalal S, Zha Q, Singh M, et al. Osteopontin-stimulated apoptosis in cardiac myocytes involves oxidative stress and mitochondrial death pathway: role of a pro-apoptotic protein BIK. Mol Cell Biochem 2016;418:1-11. 10.1007/s11010-016-2725-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nazarewicz RR, Dikalova AE, Bikineyeva A, et al. Nox2 as a potential target of mitochondrial superoxide and its role in endothelial oxidative stress. Am J Physiol Heart Circ Physiol 2013;305:H1131-40. 10.1152/ajpheart.00063.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu SP, Kao CY, Wang L, et al. Increased COUP-TFII expression in adult hearts induces mitochondrial dysfunction resulting in heart failure. Nat Commun 2015;6:8245. 10.1038/ncomms9245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sverdlov AL, Elezaby A, Qin F, et al. Mitochondrial Reactive Oxygen Species Mediate Cardiac Structural, Functional, and Mitochondrial Consequences of Diet-Induced Metabolic Heart Disease. J Am Heart Assoc 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yao X, Carlson D, Sun Y, et al. Mitochondrial ROS Induces Cardiac Inflammation via a Pathway through mtDNA Damage in a Pneumonia-Related Sepsis Model. PLoS One 2015;10:e0139416. 10.1371/journal.pone.0139416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hicks S, Labinskyy N, Piteo B, et al. Type II diabetes increases mitochondrial DNA mutations in the left ventricle of the Goto-Kakizaki diabetic rat. Am J Physiol Heart Circ Physiol 2013;304:H903-15. 10.1152/ajpheart.00567.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cividini F, Scott BT, Dai A, et al. O-GlcNAcylation of 8-Oxoguanine DNA Glycosylase (Ogg1) Impairs Oxidative Mitochondrial DNA Lesion Repair in Diabetic Hearts. J Biol Chem 2016;291:26515-28. 10.1074/jbc.M116.754481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Din S, Konstandin MH, Johnson B, et al. Metabolic dysfunction consistent with premature aging results from deletion of Pim kinases. Circ Res 2014;115:376-87. 10.1161/CIRCRESAHA.115.304441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Song M, Franco A, Fleischer JA, et al. Abrogating Mitochondrial Dynamics in Mouse Hearts Accelerates Mitochondrial Senescence. Cell Metab 2017;26:872-83.e5. 10.1016/j.cmet.2017.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jian B, Yang S, Chen D, et al. Aging influences cardiac mitochondrial gene expression and cardiovascular function following hemorrhage injury. Mol Med 2011;17:542-9. 10.2119/molmed.2010.00195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Manskikh VN, Gancharova OS, Nikiforova AI, et al. Age-associated murine cardiac lesions are attenuated by the mitochondria-targeted antioxidant SkQ1. Histol Histopathol 2015;30:353-60. [DOI] [PubMed] [Google Scholar]
- 56.Ge H, Zhao M, Lee S, et al. Mitochondrial Src tyrosine kinase plays a role in the cardioprotective effect of ischemic preconditioning by modulating complex I activity and mitochondrial ROS generation. Free Radic Res 2015;49:1210-7. 10.3109/10715762.2015.1050013 [DOI] [PubMed] [Google Scholar]
- 57.Uchihashi M, Hoshino A, Okawa Y, et al. Cardiac-Specific Bdh1 Overexpression Ameliorates Oxidative Stress and Cardiac Remodeling in Pressure Overload-Induced Heart Failure. Circ Heart Fail 2017;10. pii: e004417. 10.1161/CIRCHEARTFAILURE.117.004417 [DOI] [PubMed] [Google Scholar]
- 58.Xiang SY, Ouyang K, Yung BS, et al. PLCepsilon, PKD1, and SSH1L transduce RhoA signaling to protect mitochondria from oxidative stress in the heart. Sci Signal 2013;6:ra108. 10.1126/scisignal.2004405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dutta D, Xu J, Kim JS, et al. Upregulated autophagy protects cardiomyocytes from oxidative stress-induced toxicity. Autophagy 2013;9:328-44. 10.4161/auto.22971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu P, Zhang J, Yu S, et al. Protective Effect of Sevoflurane Postconditioning against Cardiac Ischemia/Reperfusion Injury via Ameliorating Mitochondrial Impairment, Oxidative Stress and Rescuing Autophagic Clearance. PLoS One 2015;10:e0134666. 10.1371/journal.pone.0134666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yogalingam G, Hwang S, Ferreira JC, et al. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) phosphorylation by protein kinase Cdelta (PKCdelta) inhibits mitochondria elimination by lysosomal-like structures following ischemia and reoxygenation-induced injury. J Biol Chem 2013;288:18947-60. 10.1074/jbc.M113.466870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dai DF, Chen T, Wanagat J, et al. Age-dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria. Aging Cell 2010;9:536-44. 10.1111/j.1474-9726.2010.00581.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Das KC, Muniyappa H. Age-dependent mitochondrial energy dynamics in the mice heart: role of superoxide dismutase-2. Exp Gerontol 2013;48:947-59. 10.1016/j.exger.2013.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bradley JM, Li Z, Organ CL, et al. A novel mtDNA repair fusion protein attenuates maladaptive remodeling and preserves cardiac function in heart failure. Am J Physiol Heart Circ Physiol 2018;314:H311-21. 10.1152/ajpheart.00515.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li YG, Zhu W, Tao JP, et al. Resveratrol protects cardiomyocytes from oxidative stress through SIRT1 and mitochondrial biogenesis signaling pathways. Biochem Biophys Res Commun 2013;438:270-6. 10.1016/j.bbrc.2013.07.042 [DOI] [PubMed] [Google Scholar]
- 66.Costa RM, Filgueira FP, Tostes RC, et al. H2O2 generated from mitochondrial electron transport chain in thoracic perivascular adipose tissue is crucial for modulation of vascular smooth muscle contraction. Vascul Pharmacol 2016;84:28-37. 10.1016/j.vph.2016.05.008 [DOI] [PubMed] [Google Scholar]
- 67.Behringer EJ, Segal SS. Impact of Aging on Calcium Signaling and Membrane Potential in Endothelium of Resistance Arteries: A Role for Mitochondria. J Gerontol A Biol Sci Med Sci 2017;72:1627-37. 10.1093/gerona/glx079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen F, Chen B, Xiao FQ, et al. Autophagy protects against senescence and apoptosis via the RAS-mitochondria in high-glucose-induced endothelial cells. Cell Physiol Biochem 2014;33:1058-74. 10.1159/000358676 [DOI] [PubMed] [Google Scholar]
- 69.Chen Y, Lin JR, Gao PJ. Mitochondrial division inhibitor Mdivi-1 ameliorates angiotensin II-induced endothelial dysfunction. Sheng Li Xue Bao 2016;68:669-76. [PubMed] [Google Scholar]
- 70.Freed JK, Durand MJ, Hoffmann BR, et al. Mitochondria-regulated formation of endothelium-derived extracellular vesicles shifts the mediator of flow-induced vasodilation. Am J Physiol Heart Circ Physiol 2017;312:H1096-104. 10.1152/ajpheart.00680.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Morlino G, Barreiro O, Baixauli F, et al. Miro-1 links mitochondria and microtubule Dynein motors to control lymphocyte migration and polarity. Mol Cell Biol 2014;34:1412-26. 10.1128/MCB.01177-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hodara R, Weiss D, Joseph G, et al. Overexpression of catalase in myeloid cells causes impaired postischemic neovascularization. Arterioscler Thromb Vasc Biol 2011;31:2203-9. 10.1161/ATVBAHA.111.233247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kalampogias A, Siasos G, Oikonomou E, et al. Basic Mechanisms in Atherosclerosis: The Role of Calcium. Med Chem 2016;12:103-13. 10.2174/1573406411666150928111446 [DOI] [PubMed] [Google Scholar]
- 74.Birben E, Sahiner UM, Sackesen C, et al. Oxidative stress and antioxidant defense. World Allergy Organ J 2012;5:9-19. 10.1097/WOX.0b013e3182439613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ballinger SW, Patterson C, Yan CN, et al. Hydrogen peroxide- and peroxynitrite-induced mitochondrial DNA damage and dysfunction in vascular endothelial and smooth muscle cells. Circ Res 2000;86:960-6. 10.1161/01.RES.86.9.960 [DOI] [PubMed] [Google Scholar]
- 76.Ballinger SW, Patterson C, Knight-Lozano CA, et al. Mitochondrial integrity and function in atherogenesis. Circulation 2002;106:544-9. 10.1161/01.CIR.0000023921.93743.89 [DOI] [PubMed] [Google Scholar]
- 77.Yu E, Calvert PA, Mercer JR, et al. Mitochondrial DNA damage can promote atherosclerosis independently of reactive oxygen species through effects on smooth muscle cells and monocytes and correlates with higher-risk plaques in humans. Circulation 2013;128:702-12. 10.1161/CIRCULATIONAHA.113.002271 [DOI] [PubMed] [Google Scholar]
- 78.Panth N, Paudel KR, Parajuli K. Reactive Oxygen Species: A Key Hallmark of Cardiovascular Disease. Adv Med 2016;2016:9152732. 10.1155/2016/9152732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ahn SY, Choi YS, Koo HJ, et al. Mitochondrial dysfunction enhances the migration of vascular smooth muscles cells via suppression of Akt phosphorylation. Biochim Biophys Acta 2010;1800:275-81. 10.1016/j.bbagen.2009.09.005 [DOI] [PubMed] [Google Scholar]
- 80.Chen J, Mehta JL, Haider N, et al. Role of caspases in Ox-LDL-induced apoptotic cascade in human coronary artery endothelial cells. Circ Res 2004;94:370-6. 10.1161/01.RES.0000113782.07824.BE [DOI] [PubMed] [Google Scholar]
- 81.Vindis C, Elbaz M, Escargueil-Blanc I, et al. Two distinct calcium-dependent mitochondrial pathways are involved in oxidized LDL-induced apoptosis. Arterioscler Thromb Vasc Biol 2005;25:639-45. 10.1161/01.ATV.0000154359.60886.33 [DOI] [PubMed] [Google Scholar]
- 82.Postnov Iu V. The role of mitochondrial calcium overload and energy deficiency in pathogenesis of arterial hypertension. Arkh Patol 2001;63:3-10. [PubMed] [Google Scholar]
- 83.Paradies G, Petrosillo G, Pistolese M, et al. Decrease in mitochondrial complex I activity in ischemic/reperfused rat heart: involvement of reactive oxygen species and cardiolipin. Circ Res 2004;94:53-9. 10.1161/01.RES.0000109416.56608.64 [DOI] [PubMed] [Google Scholar]
- 84.Chang HC, Wu R, Shang M, et al. Reduction in mitochondrial iron alleviates cardiac damage during injury. EMBO Mol Med 2016;8:247-67. 10.15252/emmm.201505748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li Q, Su D, O'Rourke B, et al. Mitochondria-derived ROS bursts disturb Ca(2)(+) cycling and induce abnormal automaticity in guinea pig cardiomyocytes: a theoretical study. Am J Physiol Heart Circ Physiol 2015;308:H623-36. 10.1152/ajpheart.00493.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tsushima K, Bugger H, Wende AR, et al. Mitochondrial Reactive Oxygen Species in Lipotoxic Hearts Induce Post-Translational Modifications of AKAP121, DRP1, and OPA1 That Promote Mitochondrial Fission. Circ Res 2018;122:58-73. 10.1161/CIRCRESAHA.117.311307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shen T, Zheng M, Cao C, et al. Mitofusin-2 is a major determinant of oxidative stress-mediated heart muscle cell apoptosis. J Biol Chem 2007;282:23354-61. 10.1074/jbc.M702657200 [DOI] [PubMed] [Google Scholar]
- 88.Kurz DJ, Decary S, Hong Y, et al. Chronic oxidative stress compromises telomere integrity and accelerates the onset of senescence in human endothelial cells. J Cell Sci 2004;117:2417-26. 10.1242/jcs.01097 [DOI] [PubMed] [Google Scholar]
- 89.Sudakov NP, Apartsin KA, Lepekhova SA, et al. The level of free circulating mitochondrial DNA in blood as predictor of death in case of acute coronary syndrome. Eur J Med Res 2017;22:1. 10.1186/s40001-016-0241-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 1986;74:1124-36. 10.1161/01.CIR.74.5.1124 [DOI] [PubMed] [Google Scholar]
- 91.Muntean DM, Sturza A, Danila MD, et al. The Role of Mitochondrial Reactive Oxygen Species in Cardiovascular Injury and Protective Strategies. Oxid Med Cell Longev 2016;2016:8254942. 10.1155/2016/8254942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Przyklenk K, Bauer B, Ovize M, et al. Regional ischemic 'preconditioning' protects remote virgin myocardium from subsequent sustained coronary occlusion. Circulation 1993;87:893-9. 10.1161/01.CIR.87.3.893 [DOI] [PubMed] [Google Scholar]
- 93.Androulakis E, Tousoulis D, Papageorgiou N, et al. The role of matrix metalloproteinases in essential hypertension. Curr Top Med Chem 2012;12:1149-58. 10.2174/1568026611208011149 [DOI] [PubMed] [Google Scholar]
- 94.Stefanadi E, Tousoulis D, Androulakis ES, et al. Inflammatory markers in essential hypertension: potential clinical implications. Curr Vasc Pharmacol 2010;8:509-16. 10.2174/157016110791330870 [DOI] [PubMed] [Google Scholar]
- 95.Androulakis E, Tousoulis D, Papageorgiou N, et al. Inflammation in hypertension: current therapeutic approaches. Curr Pharm Des 2011;17:4121-31. 10.2174/138161211798764753 [DOI] [PubMed] [Google Scholar]
- 96.Lanza IR, Short DK, Short KR, et al. Endurance exercise as a countermeasure for aging. Diabetes 2008;57:2933-42. 10.2337/db08-0349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Capettini LS, Montecucco F, Mach F, et al. Role of renin-angiotensin system in inflammation, immunity and aging. Curr Pharm Des 2012;18:963-70. 10.2174/138161212799436593 [DOI] [PubMed] [Google Scholar]
- 98.Rodriguez-Iturbe B, Sepassi L, Quiroz Y, et al. Association of mitochondrial SOD deficiency with salt-sensitive hypertension and accelerated renal senescence. J Appl Physiol (1985) 2007;102:255-60. 10.1152/japplphysiol.00513.2006 [DOI] [PubMed] [Google Scholar]
- 99.Marx J. Medicine Metabolic defects tied to mitochondrial gene. Science 2004;306:592-3. 10.1126/science.306.5696.592b [DOI] [PubMed] [Google Scholar]
- 100.Dikalov SI, Ungvari Z. Role of mitochondrial oxidative stress in hypertension. Am J Physiol Heart Circ Physiol 2013;305:H1417-27. 10.1152/ajpheart.00089.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ichimura H, Parthasarathi K, Quadri S, et al. Mechano-oxidative coupling by mitochondria induces proinflammatory responses in lung venular capillaries. J Clin Invest 2003;111:691-9. 10.1172/JCI17271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Touyz RM. Reactive oxygen species, vascular oxidative stress, and redox signaling in hypertension: what is the clinical significance? Hypertension 2004;44:248-52. 10.1161/01.HYP.0000138070.47616.9d [DOI] [PubMed] [Google Scholar]
- 103.Cogliati S, Frezza C, Soriano ME, et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 2013;155:160-71. 10.1016/j.cell.2013.08.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ward ML, Pope AJ, Loiselle DS, et al. Reduced contraction strength with increased intracellular [Ca2+] in left ventricular trabeculae from failing rat hearts. J Physiol 2003;546:537-50. 10.1113/jphysiol.2002.029132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chaanine AH, Gordon RE, Kohlbrenner E, et al. Potential role of BNIP3 in cardiac remodeling, myocardial stiffness, and endoplasmic reticulum: mitochondrial calcium homeostasis in diastolic and systolic heart failure. Circ Heart Fail 2013;6:572-83. 10.1161/CIRCHEARTFAILURE.112.000200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dengjel J, Abeliovich H. Musical chairs during mitophagy. Autophagy 2014;10:706-7. 10.4161/auto.28150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rimbaud S, Ruiz M, Piquereau J, et al. Resveratrol improves survival, hemodynamics and energetics in a rat model of hypertension leading to heart failure. PLoS One 2011;6:e26391. 10.1371/journal.pone.0026391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang HJ, Kang PF, Wu WJ, et al. Changes in cardiac mitochondrial aldehyde dehydrogenase 2 activity in relation to oxidative stress and inflammatory injury in diabetic rats. Mol Med Rep 2013;8:686-90. 10.3892/mmr.2013.1524 [DOI] [PubMed] [Google Scholar]
- 109.Chen XH, Zhao YP, Xue M, et al. TNF-alpha induces mitochondrial dysfunction in 3T3-L1 adipocytes. Mol Cell Endocrinol 2010;328:63-9. 10.1016/j.mce.2010.07.005 [DOI] [PubMed] [Google Scholar]
- 110.Frey TG, Mannella CA. The internal structure of mitochondria. Trends Biochem Sci 2000;25:319-24. 10.1016/S0968-0004(00)01609-1 [DOI] [PubMed] [Google Scholar]
- 111.Osman C, Voelker DR, Langer T. Making heads or tails of phospholipids in mitochondria. J Cell Biol 2011;192:7-16. 10.1083/jcb.201006159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Walther T, Tschope C, Sterner-Kock A, et al. Accelerated mitochondrial adenosine diphosphate/adenosine triphosphate transport improves hypertension-induced heart disease. Circulation 2007;115:333-44. 10.1161/CIRCULATIONAHA.106.643296 [DOI] [PubMed] [Google Scholar]
- 113.Rani AJ, Mythili SV. Study on total antioxidant status in relation to oxidative stress in type 2 diabetes mellitus. J Clin Diagn Res 2014;8:108-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tousoulis D, Papageorgiou N, Androulakis E, et al. Diabetes mellitus-associated vascular impairment: novel circulating biomarkers and therapeutic approaches. J Am Coll Cardiol 2013;62:667-76. 10.1016/j.jacc.2013.03.089 [DOI] [PubMed] [Google Scholar]
- 115.Pourvali K, Abbasi M, Mottaghi A. Role of Superoxide Dismutase 2 Gene Ala16Val Polymorphism and Total Antioxidant Capacity in Diabetes and its Complications. Avicenna J Med Biotechnol 2016;8:48-56. [PMC free article] [PubMed] [Google Scholar]
- 116.Jia G, Aroor AR, Sowers JR. Estrogen and mitochondria function in cardiorenal metabolic syndrome. Prog Mol Biol Transl Sci 2014;127:229-49. 10.1016/B978-0-12-394625-6.00009-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Antoniades C, Tousoulis D, Marinou K, et al. Effects of insulin dependence on inflammatory process, thrombotic mechanisms and endothelial function, in patients with type 2 diabetes mellitus and coronary atherosclerosis. Clin Cardiol 2007;30:295-300. 10.1002/clc.20101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jelenik T, Roden M. Mitochondrial plasticity in obesity and diabetes mellitus. Antioxid Redox Signal 2013;19:258-68. 10.1089/ars.2012.4910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Montgomery MK, Turner N. Mitochondrial dysfunction and insulin resistance: an update. Endocr Connect 2015;4:R1-15. 10.1530/EC-14-0092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cortassa S, Sollott SJ, Aon MA. Mitochondrial respiration and ROS emission during beta-oxidation in the heart: An experimental-computational study. PLoS Comput Biol 2017;13:e1005588. 10.1371/journal.pcbi.1005588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Patti ME, Corvera S. The role of mitochondria in the pathogenesis of type 2 diabetes. Endocr Rev 2010;31:364-95. 10.1210/er.2009-0027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rains JL, Jain SK. Oxidative stress, insulin signaling, and diabetes. Free Radic Biol Med 2011;50:567-75. 10.1016/j.freeradbiomed.2010.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wredenberg A, Freyer C, Sandstrom ME, et al. Respiratory chain dysfunction in skeletal muscle does not cause insulin resistance. Biochem Biophys Res Commun 2006;350:202-7. 10.1016/j.bbrc.2006.09.029 [DOI] [PubMed] [Google Scholar]
- 124.Watanabe T, Saotome M, Nobuhara M, et al. Roles of mitochondrial fragmentation and reactive oxygen species in mitochondrial dysfunction and myocardial insulin resistance. Exp Cell Res 2014;323:314-25. 10.1016/j.yexcr.2014.02.027 [DOI] [PubMed] [Google Scholar]
- 125.Passos JF, Saretzki G, von Zglinicki T. DNA damage in telomeres and mitochondria during cellular senescence: is there a connection? Nucleic Acids Res 2007;35:7505-13. 10.1093/nar/gkm893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhou MC, Min R, Ji JJ, et al. Analysis of association among clinical features and shorter leukocyte telomere length in mitochondrial diabetes with m.3243A>G mitochondrial DNA mutation. BMC Med Genet 2015;16:92. 10.1186/s12881-015-0238-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Jeong EM, Chung J, Liu H, et al. Role of Mitochondrial Oxidative Stress in Glucose Tolerance, Insulin Resistance, and Cardiac Diastolic Dysfunction. J Am Heart Assoc 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kubli DA, Gustafsson AB. Unbreak my heart: targeting mitochondrial autophagy in diabetic cardiomyopathy. Antioxid Redox Signal 2015;22:1527-44. 10.1089/ars.2015.6322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Aroor AR, Mandavia C, Ren J, et al. Mitochondria and Oxidative Stress in the Cardiorenal Metabolic Syndrome. Cardiorenal Med 2012;2:87-109. 10.1159/000335675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Scarpulla RC. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim Biophys Acta 2011;1813:1269-78. 10.1016/j.bbamcr.2010.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tousoulis D, Oikonomou E, Siasos G, et al. Predictive value of biomarkers in patients with heart failure. Curr Med Chem 2012;19:2534-47. 10.2174/092986712800492968 [DOI] [PubMed] [Google Scholar]
- 132.Tousoulis D, Kampoli AM, Siasos G, et al. Circulating biomarkers for the diagnosis and prognosis of heart failure. Curr Med Chem 2009;16:3828-40. 10.2174/092986709789178000 [DOI] [PubMed] [Google Scholar]
- 133.Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013;62:e147-239. 10.1016/j.jacc.2013.05.019 [DOI] [PubMed] [Google Scholar]
- 134.Weeks KL, McMullen JR. The athlete's heart vs. the failing heart: can signaling explain the two distinct outcomes? Physiology (Bethesda) 2011;26:97-105. 10.1152/physiol.00043.2010 [DOI] [PubMed] [Google Scholar]
- 135.Hunter JJ, Chien KR. Signaling pathways for cardiac hypertrophy and failure. N Engl J Med 1999;341:1276-83. 10.1056/NEJM199910213411706 [DOI] [PubMed] [Google Scholar]
- 136.Scheubel RJ, Tostlebe M, Simm A, et al. Dysfunction of mitochondrial respiratory chain complex I in human failing myocardium is not due to disturbed mitochondrial gene expression. J Am Coll Cardiol 2002;40:2174-81. 10.1016/S0735-1097(02)02600-1 [DOI] [PubMed] [Google Scholar]
- 137.LeBleu VS, O'Connell JT, Gonzalez Herrera KN, et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol 2014;16:992-1003, 1-15. [DOI] [PMC free article] [PubMed]
- 138.O'Neill BT, Kim J, Wende AR, et al. A conserved role for phosphatidylinositol 3-kinase but not Akt signaling in mitochondrial adaptations that accompany physiological cardiac hypertrophy. Cell Metab 2007;6:294-306. 10.1016/j.cmet.2007.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.McMullen JR, Shioi T, Zhang L, et al. Phosphoinositide 3-kinase(p110alpha) plays a critical role for the induction of physiological, but not pathological, cardiac hypertrophy. Proc Natl Acad Sci U S A 2003;100:12355-60. 10.1073/pnas.1934654100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Heather LC, Howell NJ, Emmanuel Y, et al. Changes in cardiac substrate transporters and metabolic proteins mirror the metabolic shift in patients with aortic stenosis. PLoS One 2011;6:e26326. 10.1371/journal.pone.0026326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lehman JJ, Kelly DP. Gene regulatory mechanisms governing energy metabolism during cardiac hypertrophic growth. Heart Fail Rev 2002;7:175-85. 10.1023/A:1015332726303 [DOI] [PubMed] [Google Scholar]
- 142.de las Fuentes L, Soto PF, Cupps BP, et al. Hypertensive left ventricular hypertrophy is associated with abnormal myocardial fatty acid metabolism and myocardial efficiency. J Nucl Cardiol 2006;13:369-77. 10.1016/j.nuclcard.2006.01.021 [DOI] [PubMed] [Google Scholar]
- 143.Finck BN, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) regulatory cascade in cardiac physiology and disease. Circulation 2007;115:2540-8. 10.1161/CIRCULATIONAHA.107.670588 [DOI] [PubMed] [Google Scholar]
- 144.Luptak I, Balschi JA, Xing Y, et al. Decreased contractile and metabolic reserve in peroxisome proliferator-activated receptor-alpha-null hearts can be rescued by increasing glucose transport and utilization. Circulation 2005;112:2339-46. 10.1161/CIRCULATIONAHA.105.534594 [DOI] [PubMed] [Google Scholar]
- 145.Garnier A, Fortin D, Delomenie C, et al. Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac and skeletal muscles. J Physiol 2003;551:491-501. 10.1113/jphysiol.2003.045104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Dai DF, Johnson SC, Villarin JJ, et al. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ Res 2011;108:837-46. 10.1161/CIRCRESAHA.110.232306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sabri A, Hughie HH, Lucchesi PA. Regulation of hypertrophic and apoptotic signaling pathways by reactive oxygen species in cardiac myocytes. Antioxid Redox Signal 2003;5:731-40. 10.1089/152308603770380034 [DOI] [PubMed] [Google Scholar]
- 148.St-Pierre J, Drori S, Uldry M, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006;127:397-408. 10.1016/j.cell.2006.09.024 [DOI] [PubMed] [Google Scholar]
- 149.Ide T, Tsutsui H, Hayashidani S, et al. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res 2001;88:529-35. 10.1161/01.RES.88.5.529 [DOI] [PubMed] [Google Scholar]
- 150.Marin-Garcia J, Goldenthal MJ, Moe GW. Abnormal cardiac and skeletal muscle mitochondrial function in pacing-induced cardiac failure. Cardiovasc Res 2001;52:103-10. 10.1016/S0008-6363(01)00368-6 [DOI] [PubMed] [Google Scholar]
- 151.Tousoulis D, Kampoli AM, Papageorgiou N, et al. Matrix metallopropteinases in heart failure. Curr Top Med Chem 2012;12:1181-91. 10.2174/1568026611208011181 [DOI] [PubMed] [Google Scholar]
- 152.Coker ML, Thomas CV, Clair MJ, et al. Myocardial matrix metalloproteinase activity and abundance with congestive heart failure. Am J Physiol 1998;274:H1516-23. [DOI] [PubMed] [Google Scholar]
- 153.Spinale FG, Coker ML, Thomas CV, et al. Time-dependent changes in matrix metalloproteinase activity and expression during the progression of congestive heart failure: relation to ventricular and myocyte function. Circ Res 1998;82:482-95. 10.1161/01.RES.82.4.482 [DOI] [PubMed] [Google Scholar]
- 154.Chen L, Gong Q, Stice JP, et al. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc Res 2009;84:91-9. 10.1093/cvr/cvp181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hom J, Yu T, Yoon Y, et al. Regulation of mitochondrial fission by intracellular Ca2+ in rat ventricular myocytes. Biochim Biophys Acta 2010;1797:913-21. 10.1016/j.bbabio.2010.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Bhandari P, Song M, Chen Y, et al. Mitochondrial contagion induced by Parkin deficiency in Drosophila hearts and its containment by suppressing mitofusin. Circ Res 2014;114:257-65. 10.1161/CIRCRESAHA.114.302734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Zima AV, Blatter LA. Redox regulation of cardiac calcium channels and transporters. Cardiovasc Res 2006;71:310-21. 10.1016/j.cardiores.2006.02.019 [DOI] [PubMed] [Google Scholar]
- 158.Bayeva M, Ardehali H. Mitochondrial dysfunction and oxidative damage to sarcomeric proteins. Curr Hypertens Rep 2010;12:426-32. 10.1007/s11906-010-0149-8 [DOI] [PubMed] [Google Scholar]
- 159.Bozaykut P, Karademir B, Yazgan B, et al. Effects of vitamin E on peroxisome proliferator-activated receptor gamma and nuclear factor-erythroid 2-related factor 2 in hypercholesterolemia-induced atherosclerosis. Free Radic Biol Med 2014;70:174-81. 10.1016/j.freeradbiomed.2014.02.017 [DOI] [PubMed] [Google Scholar]
- 160.Bayeva M, Gheorghiade M, Ardehali H. Mitochondria as a therapeutic target in heart failure. J Am Coll Cardiol 2013;61:599-610. 10.1016/j.jacc.2012.08.1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Papageorgiou N, Tousoulis D, Katsargyris A, et al. Antioxidant treatment and endothelial dysfunction: is it time for flavonoids? Recent Pat Cardiovasc Drug Discov 2013;8:81-92. 10.2174/15748901113089990018 [DOI] [PubMed] [Google Scholar]
- 162.Papageorgiou N, Tousoulis D, Androulakis E, et al. Lifestyle factors and endothelial function. Curr Vasc Pharmacol 2012;10:94-106. 10.2174/157016112798829788 [DOI] [PubMed] [Google Scholar]
- 163.Rizos I. Three-year survival of patients with heart failure caused by dilated cardiomyopathy and L-carnitine administration. Am Heart J 2000;139:S120-3. 10.1067/mhj.2000.103917 [DOI] [PubMed] [Google Scholar]
- 164.Serati AR, Motamedi MR, Emami S, et al. L-carnitine treatment in patients with mild diastolic heart failure is associated with improvement in diastolic function and symptoms. Cardiology 2010;116:178-82. 10.1159/000318810 [DOI] [PubMed] [Google Scholar]
- 165.O'Brien D, Chunduri P, Iyer A, et al. L-carnitine attenuates cardiac remodelling rather than vascular remodelling in deoxycorticosterone acetate-salt hypertensive rats. Basic Clin Pharmacol Toxicol 2010;106:296-301. 10.1111/j.1742-7843.2009.00480.x [DOI] [PubMed] [Google Scholar]
- 166.Kokkou E, Siasos G, Georgiopoulos G, et al. The impact of dietary flavonoid supplementation on smoking-induced inflammatory process and fibrinolytic impairment. Atherosclerosis 2016;251:266-72. 10.1016/j.atherosclerosis.2016.06.054 [DOI] [PubMed] [Google Scholar]
- 167.Siasos G, Tousoulis D, Kokkou E, et al. Favorable effects of concord grape juice on endothelial function and arterial stiffness in healthy smokers. Am J Hypertens 2014;27:38-45. 10.1093/ajh/hpt176 [DOI] [PubMed] [Google Scholar]
- 168.Siasos G, Tousoulis D, Tsigkou V, et al. Flavonoids in atherosclerosis: an overview of their mechanisms of action. Curr Med Chem 2013;20:2641-60. 10.2174/0929867311320210003 [DOI] [PubMed] [Google Scholar]
- 169.Loke WM, Proudfoot JM, Hodgson JM, et al. Specific dietary polyphenols attenuate atherosclerosis in apolipoprotein E-knockout mice by alleviating inflammation and endothelial dysfunction. Arterioscler Thromb Vasc Biol 2010;30:749-57. 10.1161/ATVBAHA.109.199687 [DOI] [PubMed] [Google Scholar]
- 170.Scoditti E, Calabriso N, Massaro M, et al. Mediterranean diet polyphenols reduce inflammatory angiogenesis through MMP-9 and COX-2 inhibition in human vascular endothelial cells: a potentially protective mechanism in atherosclerotic vascular disease and cancer. Arch Biochem Biophys 2012;527:81-9. 10.1016/j.abb.2012.05.003 [DOI] [PubMed] [Google Scholar]
- 171.Natsume M, Baba S. Suppressive effects of cacao polyphenols on the development of atherosclerosis in apolipoprotein E-deficient mice. Subcell Biochem 2014;77:189-98. 10.1007/978-94-007-7920-4_16 [DOI] [PubMed] [Google Scholar]
- 172.Tousoulis D, Antoniades C, Vasiliadou C, et al. Effects of atorvastatin and vitamin C on forearm hyperaemic blood flow, asymmentrical dimethylarginine levels and the inflammatory process in patients with type 2 diabetes mellitus. Heart 2007;93:244-6. 10.1136/hrt.2006.093112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Bjelakovic G, Nikolova D, Gluud LL, et al. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Cochrane Database Syst Rev 2008;(2):CD007176. [DOI] [PubMed] [Google Scholar]
- 174.Cocheme HM, Murphy MP. Can antioxidants be effective therapeutics? Curr Opin Investig Drugs 2010;11:426-31. [PubMed] [Google Scholar]
- 175.Podmore ID, Griffiths HR, Herbert KE, et al. Vitamin C exhibits pro-oxidant properties. Nature 1998;392:559. 10.1038/33308 [DOI] [PubMed] [Google Scholar]
- 176.Michels AJ, Frei B. Myths, artifacts, and fatal flaws: identifying limitations and opportunities in vitamin C research. Nutrients 2013;5:5161-92. 10.3390/nu5125161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Michels AJ, Hagen TM, Frei B. Human genetic variation influences vitamin C homeostasis by altering vitamin C transport and antioxidant enzyme function. Annu Rev Nutr 2013;33:45-70. 10.1146/annurev-nutr-071812-161246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Garrido-Maraver J, Cordero MD, Oropesa-Avila M, et al. Clinical applications of coenzyme Q10. Front Biosci (Landmark Ed) 2014;19:619-33. 10.2741/4231 [DOI] [PubMed] [Google Scholar]
- 179.Graham D, Huynh NN, Hamilton CA, et al. Mitochondria-targeted antioxidant MitoQ10 improves endothelial function and attenuates cardiac hypertrophy. Hypertension 2009;54:322-8. 10.1161/HYPERTENSIONAHA.109.130351 [DOI] [PubMed] [Google Scholar]
- 180.Paradies G, Petrosillo G, Paradies V, et al. Role of cardiolipin peroxidation and Ca2+ in mitochondrial dysfunction and disease. Cell Calcium 2009;45:643-50. 10.1016/j.ceca.2009.03.012 [DOI] [PubMed] [Google Scholar]
- 181.Davies SM, Poljak A, Duncan MW, et al. Measurements of protein carbonyls, ortho- and meta-tyrosine and oxidative phosphorylation complex activity in mitochondria from young and old rats. Free Radic Biol Med 2001;31:181-90. 10.1016/S0891-5849(01)00576-7 [DOI] [PubMed] [Google Scholar]
- 182.Santos JH, Meyer JN, Mandavilli BS, et al. Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol Biol 2006;314:183-99. 10.1385/1-59259-973-7:183 [DOI] [PubMed] [Google Scholar]
- 183.Littarru GP, Langsjoen P. Coenzyme Q10 and statins: biochemical and clinical implications. Mitochondrion 2007;7 Suppl:S168-74. 10.1016/j.mito.2007.03.002 [DOI] [PubMed] [Google Scholar]
- 184.Pepe S, Marasco SF, Haas SJ, et al. Coenzyme Q10 in cardiovascular disease. Mitochondrion 2007;7 Suppl:S154-67. 10.1016/j.mito.2007.02.005 [DOI] [PubMed] [Google Scholar]
- 185.Smith RA, Porteous CM, Coulter CV, et al. Selective targeting of an antioxidant to mitochondria. Eur J Biochem 1999;263:709-16. 10.1046/j.1432-1327.1999.00543.x [DOI] [PubMed] [Google Scholar]
- 186.Adlam VJ, Harrison JC, Porteous CM, et al. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J 2005;19:1088-95. 10.1096/fj.05-3718com [DOI] [PubMed] [Google Scholar]
- 187.McLachlan J, Beattie E, Murphy MP, et al. Combined therapeutic benefit of mitochondria-targeted antioxidant, MitoQ10, and angiotensin receptor blocker, losartan, on cardiovascular function. J Hypertens 2014;32:555-64. 10.1097/HJH.0000000000000054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Pucheu S, Boucher F, Sulpice T, et al. EUK-8 a synthetic catalytic scavenger of reactive oxygen species protects isolated iron-overloaded rat heart from functional and structural damage induced by ischemia/reperfusion. Cardiovasc Drugs Ther 1996;10:331-9. 10.1007/BF02627957 [DOI] [PubMed] [Google Scholar]
- 189.Siasos G, Athanasiou D, Terzis G, et al. The Acute Impact of Different Types of Aerobic Exercise on Arterial Wave Reflections and Inflammation. Cardiology 2016;135:81-6. 10.1159/000445993 [DOI] [PubMed] [Google Scholar]
- 190.Skrypnik D, Bogdanski P, Madry E, et al. Effect of physical exercise on endothelial function, indicators of inflammation and oxidative stress. Pol Merkur Lekarski 2014;36:117-21. [PubMed] [Google Scholar]
- 191.Roberts CK, Won D, Pruthi S, et al. Effect of a diet and exercise intervention on oxidative stress, inflammation and monocyte adhesion in diabetic men. Diabetes Res Clin Pract 2006;73:249-59. 10.1016/j.diabres.2006.02.013 [DOI] [PubMed] [Google Scholar]
- 192.Korsager Larsen M, Matchkov VV. Hypertension and physical exercise: The role of oxidative stress. Medicina (Kaunas) 2016;52:19-27. 10.1016/j.medici.2016.01.005 [DOI] [PubMed] [Google Scholar]
- 193.Siasos G, Athanasiou D, Terzis G, et al. Acute effects of different types of aerobic exercise on endothelial function and arterial stiffness. Eur J Prev Cardiol 2016;23:1565-72. 10.1177/2047487316647185 [DOI] [PubMed] [Google Scholar]
- 194.Wang G, Han T, Nijhawan D, et al. P7C3 neuroprotective chemicals function by activating the rate-limiting enzyme in NAD salvage. Cell 2014;158:1324-34. 10.1016/j.cell.2014.07.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Yoshino J, Mills KF, Yoon MJ, et al. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab 2011;14:528-36. 10.1016/j.cmet.2011.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Chan V, Fenning A, Iyer A, et al. Resveratrol improves cardiovascular function in DOCA-salt hypertensive rats. Curr Pharm Biotechnol 2011;12:429-36. 10.2174/138920111794480552 [DOI] [PubMed] [Google Scholar]
- 197.Thirunavukkarasu M, Penumathsa SV, Koneru S, et al. Resveratrol alleviates cardiac dysfunction in streptozotocin-induced diabetes: Role of nitric oxide, thioredoxin, and heme oxygenase. Free Radic Biol Med 2007;43:720-9. 10.1016/j.freeradbiomed.2007.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Huang X, Vaag A, Hansson M, et al. Down-regulation of insulin receptor substrates (IRS)-1 and IRS-2 and Src homologous and collagen-like protein Shc gene expression by insulin in skeletal muscle is not associated with insulin resistance or type 2 diabetes. J Clin Endocrinol Metab 2002;87:255-9. 10.1210/jcem.87.1.8144 [DOI] [PubMed] [Google Scholar]
- 199.Lin CH, Kurup S, Herrero P, et al. Myocardial oxygen consumption change predicts left ventricular relaxation improvement in obese humans after weight loss. Obesity (Silver Spring) 2011;19:1804-12. 10.1038/oby.2011.186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Bessi VL, Labbe SM, Huynh DN, et al. EP 80317, a selective CD36 ligand, shows cardioprotective effects against post-ischaemic myocardial damage in mice. Cardiovasc Res 2012;96:99-108. 10.1093/cvr/cvs225 [DOI] [PubMed] [Google Scholar]
- 201.Cheng J, Kamiya K, Kodama I. Carvedilol: molecular and cellular basis for its multifaceted therapeutic potential. Cardiovasc Drug Rev 2001;19:152-71. 10.1111/j.1527-3466.2001.tb00061.x [DOI] [PubMed] [Google Scholar]
- 202.Sanbe A, Tanonaka K, Kobayasi R, et al. Effects of long-term therapy with ACE inhibitors, captopril, enalapril and trandolapril, on myocardial energy metabolism in rats with heart failure following myocardial infarction. J Mol Cell Cardiol 1995;27:2209-22. 10.1016/S0022-2828(95)91551-6 [DOI] [PubMed] [Google Scholar]
- 203.Bjelakovic G, Nikolova D, Gluud LL, et al. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Sao Paulo Med J 2015;133:164-5. 10.1590/1516-3180.20151332T1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.de Cavanagh EM, Toblli JE, Ferder L, et al. Renal mitochondrial dysfunction in spontaneously hypertensive rats is attenuated by losartan but not by amlodipine. Am J Physiol Regul Integr Comp Physiol 2006;290:R1616-25. 10.1152/ajpregu.00615.2005 [DOI] [PubMed] [Google Scholar]
- 205.Davignon J, Jacob RF, Mason RP. The antioxidant effects of statins. Coron Artery Dis 2004;15:251-8. 10.1097/01.mca.0000131573.31966.34 [DOI] [PubMed] [Google Scholar]
- 206.Vogiatzi G, Oikonomou E, Siasos G, et al. Statins and inflammation in cardiovascular disease. Curr Pharm Des 2017. [Epub ahead of print]. 10.2174/1381612823666171009141201 [DOI] [PubMed] [Google Scholar]
- 207.Tousoulis D, Oikonomou E, Siasos G, et al. Dose-dependent effects of short term atorvastatin treatment on arterial wall properties and on indices of left ventricular remodeling in ischemic heart failure. Atherosclerosis 2013;227:367-72. 10.1016/j.atherosclerosis.2013.01.015 [DOI] [PubMed] [Google Scholar]
- 208.Costa S, Reina-Couto M, Albino-Teixeira A, et al. Statins and oxidative stress in chronic heart failure. Rev Port Cardiol 2016;35:41-57. 10.1016/j.repc.2015.09.006 [DOI] [PubMed] [Google Scholar]
- 209.Parihar A, Parihar MS, Zenebe WJ, et al. Statins lower calcium-induced oxidative stress in isolated mitochondria. Hum Exp Toxicol 2012;31:355-63. 10.1177/0960327111429141 [DOI] [PubMed] [Google Scholar]
- 210.Hauner H. The mode of action of thiazolidinediones. Diabetes Metab Res Rev 2002;18 Suppl 2:S10-5. 10.1002/dmrr.249 [DOI] [PubMed] [Google Scholar]
- 211.Hernanz R, Martin A, Perez-Giron JV, et al. Pioglitazone treatment increases COX-2-derived prostacyclin production and reduces oxidative stress in hypertensive rats: role in vascular function. Br J Pharmacol 2012;166:1303-19. 10.1111/j.1476-5381.2012.01825.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Tousoulis D, Koniari K, Antoniades C, et al. Combined effects of atorvastatin and metformin on glucose-induced variations of inflammatory process in patients with diabetes mellitus. Int J Cardiol 2011;149:46-9. 10.1016/j.ijcard.2009.11.038 [DOI] [PubMed] [Google Scholar]
- 213.Cahova M, Palenickova E, Dankova H, et al. Metformin prevents ischemia reperfusion-induced oxidative stress in the fatty liver by attenuation of reactive oxygen species formation. Am J Physiol Gastrointest Liver Physiol 2015;309:G100-11. 10.1152/ajpgi.00329.2014 [DOI] [PubMed] [Google Scholar]
- 214.Siasos G, Tousoulis D, Tourikis P, et al. MicroRNAs in cardiovascular therapeutics. Curr Top Med Chem 2013;13:1605-18. 10.2174/15680266113139990109 [DOI] [PubMed] [Google Scholar]
- 215.Latsios G, Tousoulis D, Androulakis E, et al. MicroRNAs in the diagnosis and treatment of unstable angina. Curr Top Med Chem 2013;13:1596-604. 10.2174/15680266113139990108 [DOI] [PubMed] [Google Scholar]
- 216.Siasos G, Kollia C, Tsigkou V, et al. MicroRNAs: Novel diagnostic and prognostic biomarkers in atherosclerosis. Curr Top Med Chem 2013;13:1503-17. 10.2174/15680266113139990099 [DOI] [PubMed] [Google Scholar]
- 217.Xu H, Cao H, Zhu G, et al. Overexpression of microRNA-145 protects against rat myocardial infarction through targeting PDCD4. Am J Transl Res 2017;9:5003-11. [PMC free article] [PubMed] [Google Scholar]
- 218.Date MO, Morita T, Yamashita N, et al. The antioxidant N-2-mercaptopropionyl glycine attenuates left ventricular hypertrophy in in vivo murine pressure-overload model. J Am Coll Cardiol 2002;39:907-12. 10.1016/S0735-1097(01)01826-5 [DOI] [PubMed] [Google Scholar]
- 219.Tsujimoto I, Hikoso S, Yamaguchi O, et al. The antioxidant edaravone attenuates pressure overload-induced left ventricular hypertrophy. Hypertension 2005;45:921-6. 10.1161/01.HYP.0000163461.71943.e9 [DOI] [PubMed] [Google Scholar]
- 220.Szeto HH. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol 2014;171:2029-50. 10.1111/bph.12461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Sabbah HN, Gupta RC, Kohli S, et al. Chronic Therapy With Elamipretide (MTP-131), a Novel Mitochondria-Targeting Peptide, Improves Left Ventricular and Mitochondrial Function in Dogs With Advanced Heart Failure. Circ Heart Fail 2016;9:e002206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Daubert MA, Yow E, Dunn G, et al. Novel Mitochondria-Targeting Peptide in Heart Failure Treatment: A Randomized, Placebo-Controlled Trial of Elamipretide. Circ Heart Fail 2017;10. [DOI] [PubMed] [Google Scholar]
- 223.See F, Thomas W, Way K, et al. p38 mitogen-activated protein kinase inhibition improves cardiac function and attenuates left ventricular remodeling following myocardial infarction in the rat. J Am Coll Cardiol 2004;44:1679-89. 10.1016/j.jacc.2004.07.038 [DOI] [PubMed] [Google Scholar]