

Abstract
Significant progress in the molecular pathology of melanocytic tumors have revealed that benign neoplasms, so-called nevi, are initiated by gain-of-function mutations in one of several primary oncogenes, such as BRAF in acquired melanocytic nevi, NRAS in congenital nevi or GNAQ/GNA11 in blue nevi, with consequent MAPK and PI3K/AKT/mTOR activation. Secondary genetic alterations overcome tumor suppressive mechanisms and allow the progression to intermediate lesions characterized by TERT-p mutation or to invasive melanomas displaying disruption of tumor suppressor genes. Currently, melanoma is molecularly regarded as four different diseases, namely BRAF, NRAS, NF1 and the “triple wild type” subtypes, which are associated with particular clinicopathological features. Melanocytic Spitzoid lesions include benign Spitz nevus, atypical Spitz tumor (AST) and Spitzoid melanoma. This is a challenging diagnostic group, particularly with regard to the distinction between AST and Spitzoid melanoma on clinical and histological grounds. Molecular analysis has identified the presence of HRAS mutation, BAP1 loss (often accompanying by BRAF mutations) or several kinase fusions in distinct categories of Spitz tumors. These aberrations account for the rapid growth characteristic of Spitz nevi. Subsequent growth is halted by various tumor suppressive mechanisms abrogation of which allow the development of AST, now better classified as low-grade melanocytic tumor. Although at present ancillary genetic techniques have not been very helpful in the prediction of biological behavior of AST, they have defined distinct tumor subsets differing with regard to biology and histology. Finally, we discuss how novel molecular markers may assist the differential diagnosis of melanoma, particularly from malignant peripheral nerve sheath tumor (MPNST). It is anticipated that the significant progress in the field of molecular pathology regarding the various types of melanocytic tumors, will eventually contribute to a more accurate histologic categorization, prediction of biologic behavior and personalized treatment.
Keywords: Spitz tumor, atypical Spitz tumor (AST), melanoma, molecular pathology
Introduction
During the past 20 years, there has been a rapid development of sophisticated molecular techniques increasing tremendously the possibilities of genetic testing in all kinds of tumor. This has led to an accelerated gain in our knowledge on the development of melanocytic tumors, which will hopefully assist diagnosis, prognosis and treatment decisions.
The classification of melanocytic tumors has recently undergone considerable re-evaluation integrating genomic data, with regard to recurrent driver mutations that activate specific oncogenes or inactivate tumor suppressors. Several molecular abnormalities are linked with particular clinical and histological features supporting the existence of biologically distinct types of melanocytic neoplasms. This concept applies to both melanomas and melanocytic nevi. The current molecularly oriented classification scheme may constitute the first step towards a refinement of histological criteria and tailored therapeutic approaches (1).
We herein highlight the major advances in the molecular pathology of melanocytic tumors focusing on their potential usefulness as diagnostic tools and tumor biomarkers. Emphasis will be put on Spitzoid tumors because this is the group in which most progress has been recently achieved.
Major pathways involved in melanomagenesis
Melanocytic neoplasia is driven by somatic mutations that activate oncogenes which may induce benign melanocytic nevi as well as malignant melanomas. The proliferation of melanocytes within benign nevi is under tight control mechanisms, including cell cycle checkpoints and telomere length among others. Such mutations are not sufficient to cause melanoma, unless subsequent genetic alterations disrupt the tumor suppressive mechanisms operating in melanocytic nevi. These secondary alterations largely determine the various categories of melanocytic neoplasms (1).
Two widely studied intracellular molecular pathways seem to be involved in the development of melanocytic nevi and melanoma. These include the MAP/ERK and the PI3K/AKT/mTOR pathways, activation of both leading to enhanced cell proliferation (Figure 1). Their importance lies in the fact that several components of these pathways can be targeted for therapeutic purposes, although their usefulness in cutaneous melanomas remains to be further elucidated.
Figure 1.

MAPK and PI3K/AKT/mTOR pathway interactions in melanocytic tumors. Extracellular growth factors trigger the dimerisation of receptor tyrosine kinases (ALK, NTRK1, RET, ROS1, MET or KIT), leading to autophosphorylation and activation of intracellular signaling cascades such as the MAPK/ERK or the PI3K/AKT/mTOR pathway. Among a variety of other functions, these signaling pathways increase cell proliferation and survival. BRAF serves as a mitotic signal transporter in the MAPK pathway causing activation and phosphorylation of MEK which in turn phosphorylates and activates ERK. Activation of PI3K/AKT/mTOR pathway is propagated through PI3K which catalyses the production of PIP3. The later binds to PDK1 and AKT allowing of both proteins to the cell membrane, followed by their activation. As a consequence of this co localization, AKT is fully activated. PI3K is antagonized by PTEN through dephosphorylation of PIP3, thereby preventing AKT activation. Upon activation, AKT in turn activates mTOR involved in the translation machinery through inactivation of 4E-BP1, the repressor of mRNA translation, and activation of S6K1, the promoter of mRNA translation. PI3K/AKT/mTOR can interact directly or indirectly with the MAPK pathway. NRAS plays an important role in both pathways. Mutations in GNAQ, GNA11, and BRAF lead to activation of the MAPK pathway only, while mutations in KIT and NRAS can activate both pathways. In addition, GNAQ, GNA11 as well as KIT mutations activate PKC which is a negative regulator of AKT. Different inhibitors can be applied in targeted therapy of advanced melanoma patients, and their points of action are indicated as strokes. Dashed arrows indicate indirect or probable interactions between molecules.
Under normal conditions, extracellular growth factors bind to receptor tyrosine kinases (RTKs) such as ALK, NTRK1, RET, ROS1 and MET. This interaction triggers the dimerization of receptor molecules, leading to their auto- and heterophosphorylation, thus activating both aforementioned pathways (2) (Figure 1). During the early stages of tumorigenesis many components of both pathways may be targeted by genetic and epigenetic aberrations. These aberrations predominantly affect genes controlling cell proliferation, survival and differentiation, ultimately resulting in activation of MAPK pathway. N-RAS plays an important role in both pathways. GNA11, GNAQ and BRAF mutations lead to activation of MAPK pathway only, while KIT and RAS mutations can activate both pathways. Mutations affecting BRAF (7q34), NRAS (1p13), (11q15), GNAQ (9p21), GNA11 (19p13) and KIT (4q12) are largely mutually exclusive. They are not on their own responsible for malignant transformation but persist throughout malignant progression (3).
With regard to Spitzoid neoplasms, such aberrations include HRAS mutations, most frequently Q61K/R in exon 3, also BRAF V600E, which, in these cases, is almost always co-detected with pre-existing BAP1 inactivating germline mutations, as well as larger genomic rearrangements involving the ALK, NTRK1, RET, ROS1 or MET RTKs, or even the BRAF serine-threonine kinase. Such mutations drive the growth of benign Spitz nevi, being considered to represent the initiative events in a genetic sequence leading to metastatic melanoma, according to current progressive models, which cast doubt upon previous beliefs that separated benign from malignant Spitzoid proliferation (4).
In Spitz nevi, activation of previously mentioned signaling pathways trigger proliferation, explaining the initial rapid growth of these lesions. Subsequently, various tumor suppressive mechanisms participating in the p53 and p16 cascades block further growth. DNA-damage response mechanisms, such as telomere shortening and reactive oxygen species (ROS), trigger the former cascade, whereas the latter cascade is epigenetically activated through de-repression of CDKN2A genetic locus in response to activation of oncogenic pathways. Abrogation of these tumor-suppressor mechanisms is thought to be involved in the pathogenesis of histological low-grade or atypical Spitz tumors (AST) (5). Further genomic aberrations result in disease progression towards high-grade malignant Spitzoid melanoma. Such mutations occur within PTEN and ARID2A genes, as well as the TERT promoter region (6).
Genetic alterations culminating in constitutive activation of MAPK or PI3K/AKT/mTOR pathway in cutaneous melanoma form the basis of the current molecular classification of cutaneous melanoma into four groups. Melanomas of the first group share BRAF mutations and account for almost one half of all cases (BRAF subtype). They tend to arise in younger patients with lox cumulative sun damage, commonly on trunk and lower extremities. The second group (25% of cases) consists of melanomas with RAS mutations (RAS subtype). NRAS mutations are seen in almost all of them, whereas KRAS and HRAS mutations are rarely identified. NRAS mutant melanomas are usually encountered in the head and neck region of older patients being associated with continuous or, less often, intermittent sun exposure. Ten percent of melanomas exhibit NF1 loss, constituting the third group (NF1 subtype), showing a strong association with severe sun exposure, older age and desmoplastic histology. Melanomas lacking any of the above mutations form a heterogeneous group commonly referred to as “triple wild-type subtype” which includes tumors with KIT mutations (mainly in acral and mucosal melanomas), GNAQ/GNA11 mutations (mainly in uveal melanoma) or rearrangements involving BRAF or RAF1. KIT mutant melanomas are not associated with sun exposure. This classification may form the basis for a tailored therapeutic approach (Figures 2,3) (1).
Figure 2.

Frequency of molecular alterations in the various types of melanocytic tumors.
Figure 3.

Identification of BRAF (V600E, V600K, V600R) and KIT (exon 17) mutations in Greek melanoma patients using Sanger sequencing, and Pyrosequencing.
Blue nevi and related melanocytic neoplasms share mutations in GNAQ and GNA11 genes. While GNAQ mutations are mostly found in cutaneous tumors, GNA11 and GNAQ mutations, each, accounts for almost 40% of uveal melanomas (7). In common nevi, BRAF and NRAS mutation are present in 60–87.5% and 20% of cases, respectively (8). In large congenital nevi, NRAS mutations are reported in up to 80% of the cases (Figure 2) (9). Interestingly BRAF V600E mutation is present at least in the majority of lesional cells in BRAF mutated nevi and is fully clonal being advanced as an initiating event in melanocytic neoplasia underpinning the formation of nevi (10). Table 1 summarizes the frequency of various molecular alterations in different types of melanocytic tumors.
Table 1. Frequency of various molecular alterations in different types of melanocytic tumors.
| Molecular alterations | Spitz nevi | AST | Spitzoid MM | Congenital nevi | Acquired nevi | Melanomas | Blue nevus like MM | Blue nevi |
|---|---|---|---|---|---|---|---|---|
| NRAS | 0–5% | 0–25% | 0–25% | Large/giant: 95%, small/medium: 70% | 6% | 25% primary cutaneous mm | 0–1% | |
| HRAS | 15–20% | 14% | 0% | <1% | ||||
| BRAF | 0–20% | 0–25% | 0–25% | Large/giant: 5%, small/medium: 30% | 78–81% | 80% primary mm, 68% metastatic mm | 25% | 0–12% |
| BAP1 | 15% | 4% uveal, 1.5% family history of cutaneous mm | 17% | |||||
| CDKN2a | Homozygous deletion of 9p21 more aggressive than heterozygous deletion | 5–72% (sporadic 1%, family history 9%, geographical areas 13–91%) | ||||||
| TERT-p | 22–71% | |||||||
| Kit fusions | 55% | 56% | 39% | Acral & mucosal | ||||
| ALK | 8% | 5% | 1% | |||||
| GNAQ | Uveal | 25% | 67–82% | |||||
| GNA11 | Uveal | 33% | 0–8% |
AST, atypical Spitz tumor; MM, malignant melanoma.
Development of melanoma from precursor lesions
The existence of intermediate lesion stages between obviously benign and clearly malignant melanocytic tumors has always been a controversial issue. The term “dysplastic nevus” was coined to describe enlarged acquired nevi, commonly found in patients with genetic predisposition to melanoma (11) and having certain atypical histopathological features, which however were later known to be commonly seen in acquired nevi and were unrelated to the size of the lesion (12,13). This term is used by clinicians and pathologists alike, although with varying connotations. In the clinical context, it refers to large acquired nevi with atypical dermoscopic features. In the pathological context, it implies irregular architectural and cytological features which fall short of melanoma. However, clinically atypical nevi may not be histologically dysplastic and vice-versa. The inconsistent clinical relevance of the histological term “dysplastic nevus” has been a nidus of controversy. Although the categorization of individuals presenting with multiple dysplastic nevi into a high-risk group for the development of melanoma has been extremely useful, the histopathological criteria that are assigned to dysplastic nevi are in debate by dermatopathologists (14).
Shain et al. (15) compared the mutational landscape of primary melanomas and their adjacent precursor lesions concluding that unequivocally benign lesions almost exclusively harbored the BRAF V600E mutation, as an apparent pathogenic alteration, as opposed to intermediate lesions that were enriched for additional mutations along with BRAF V600K or K601E and NRAS. In this study, intermediate lesions were histologically defined on the basis of probability of malignancy as an alternative to “dysplastic nevi”. These authors delineated two different modes of genetic evolutions of melanoma from precursor lesions. In particular, melanomas with BRAF V600E mutations tended to arise on preexisting benign lesions and were more common on intermittently sun-damaged skin of younger patients with distinct histopathological features (16). On the other hand, NRAS, BRAF V600K or K601E positive melanomas appeared to involve from intermediate lesions or melanomas in situ and occurred predominantly on chronically sun-damaged skin of older patients (17). It should be stressed, however, that most primary melanomas do not show an associated precursor nevus nor do they appear to develop for a benign precursor lesion (1).
In the study of Shain et al. (15) mutations of the TERT promoter region were amongst the earliest secondary genomic hits detected in 77/100 of intermediate lesions (alongside BRAF V600K or NRAS mutations) and melanomas in situ. Up-regulation of TERT gene, due to mutations within its promoter region, results in increased levels of the telomerase reverse transcriptase protein (TERT), thus assisting pathological melanocytes to avoid telomere shortening, eventually contributing to their immortality. Such mutations characterize 33% of primary melanomas and 85% of melanoma metastases, but no nevi cases whatsoever, suggesting their significant contribution to tumor progression. Although intermediate lesions are characterized by diverse point mutations associated with sun exposure, mutations acquired during the later stages of disease progression tend to present common patterns. Copy-number variations (CNVs) are absent in benign lesions and only occasionally occur in intermediate lesions or melanoma in situ cases, but prevail in invasive and metastatic melanomas. Chromosomal instability, caused by ultraviolet radiation, seems to function as an additional factor towards transition of tumor to the invasive stage. Loss of both CDKN2A copies, mutations in SWI/SNF chromatin remodeling genes and loss of PTEN or TP53 genes are restricted to invasive melanoma (15).
Classification of Spitzoid melanocytic tumors
In most melanocytic tumors, accurate pathological distinction between benign (nevus) and malignant (melanoma) is straightforward based on histological criteria. However, the clinical behavior of diagnostically challenging cases of Spitzoid melanocytic tumors with ambiguous morphological criteria is hard to be predicted with certainty, even by expert dermatopathologists. This is reflected in the use of the term “Spitz tumor of unknown malignant potential” (18).
Spitzoid melanocytic tumors were first described in 1948 by Sophie Spitz as “juvenile melanoma”, because they mainly appeared in children and adolescents (19). Later on, it became clear that these tumors may occur anytime during lifetime, presenting a benign nature and were re-named “Spitz nevi”. Through sequential acquisition of genomic aberrations, a subset of these tumors becomes markedly atypical and eventually switches to Spitzoid melanoma, capable of producing distant metastasis.
An intermediate category, commonly referred to as AST exists between entirely benign Spitz nevi and Spitzoid melanomas, reflecting the continuous biological spectrum of Spitz tumors. In this context, debate still exists on whether Spitz nevus and Spitzoid melanoma represent the opposing ends of a single biological spectrum or two separate entities.
ASTs share morphological features of both Spitz nevi and malignant tumors, which fall short of their categorization as Spitzoid melanomas. Their spreading is limited to regional lymph nodes, with no clues for widespread metastasis to distant organs, supporting their classification as low-grade melanocytic tumors (20).
Nowadays, there remains a great diagnostic uncertainty in the field of Spitzoid melanocytic tumors, which in some cases may result in serious health consequences due to erroneous diagnosis. Therefore, interest focuses in identifying underlying genetic and epigenetic aberrations of Spitz tumors, which may serve as potential biomarkers and will eventually assist their histologic and biologic classification. Melanocytic Spitzoid tumors are currently classified according to their clinical and histological appearance, their biological behavior and their mitogenic driver aberrations. Recently published molecular analysis has revealed the existence of roughly three subgroups of Spitzoid melanocytic tumors based on distinct genetic alterations. The vast majority of tumors present with kinase fusions (ALK, ROS1, NTRK1, BRAF, RET, MET), rarely observed in other subtypes of melanocytic tumors. The remaining two groups comprise of patients with either HRAS, or BAP1 mutations. These are mainly characterized by a typical phenotype that can be easily recognized, or at least be suspected, upon histological grounds. In particular, HRAS mutations are associated with desmoplastic histological phenotype (desmoplastic Spitz nevus), whereas BAP1 mutations are associated with an epithelioid morphology (3) (Figure 2).
Risk assessment for AST
Recent studies suggest that identification of CNV in ASTs may play a significant role in risk assessment, tumor management (surgical and systemic therapy) and prognosis of the disease. Gerami et al. (21) have claimed that comparative genomic hybridization (CGH) or FISH methodologies enable the successful stratification of the risk of ASTs, through correlation of common chromosomal CNVs, particularly copy number gains of 1q, 6p, 7, 8q, 17q, 20q and copy number losses of 6q, 8p, 9p, 10q, with clinical behavior (21). This study has demonstrated that copy number gains in 6p25 or 11q13 and particularly homozygous deletion in 9p21, are associated with more aggressive behavior of ASTs.
ASTs with homozygous 9p21 deletion are more frequently identified amongst children (22). They display Spitzoid epithelioid cytomorphology and expansive nodular growth, deep dermal atypical mitoses, homozygous 9p21 deletions in the vast majority of lesional cells, complete loss of p16 protein expression as shown by immunohistochemistry and presence of BRAF mutations in a small subset of tumors. These frequently results in in-transit and often lymph node metastasis including non-sentinel lymph nodes (22). It has to be stressed, though, that in recent meta-analysis SLN positivity in AST was not an indication of biologic aggressiveness (23), nor is it considered a diagnostic procedure particularly in children (24).
AST with 6q23 deletion show less aggressive behavior, which frequently results in positive sentinel lymph node biopsy, but rarely progresses to distant metastasis (21). Other studies, however, have denied the prognostic usefulness of 9q21 status as a discriminating tool in ASTs (25,26) which implies that a panel with optimal sensitivity/specificity remains to be developed.
Notwithstanding, a diagnostic algorithm, based on immunohistochemical, cytological and molecular tests is proposed, in order to assess the malignant potential of ASTs tumors. In addition to histological evaluation, this algorithm includes a combination of immunohistochemistry assays (p16Ink4a, a dual-color Ki67/MART-1 and HMB45), a five-probe fluorescence in situ hybridization (FISH) protocol (6p25, 8q24, 11q13, CEN9 and 9p21) and an array-based CGH (array-CGH) panel. Although, the algorithm is still evolving, with the continuous incorporation of novel molecular targets implicated in ASTs development and evolution, it is considered as an advance in the management of ambiguous melanocytic tumors (27).
HRAS-mutated Spitz tumors
The majority of Spitz nevi have a normal karyotype, when analyzed by array-CGH. An isolated gain in chromosome 11p, where the HRAS genetic locus resides, is a recurrent finding in 20% of Spitz nevi (28). Importantly, no such gains reportedly exist in other nevi and are extremely rare in melanomas (29). Most tumors with 11p Copy Number Gains, present with activating HRAS mutations, display a characteristic histopathological appearance (Figure 4).
Figure 4.

HRAS mutated Spitz nevus. Desmoplastic Spitz tumours frequently show HRAS mutations and gains of the short arm of chromosome 11. (A) Quite symmetrical, intradermal tumour with low cellularity from the retro-auricular region of a 43-year-old man. (B) Desmoplasia with single cells and clusters of spindle-shaped and epithelioid melanocytes between collagen bundles. (C) Large, epithelioid melanocytes between collagen bundles with vesicular chromatin and prominent nucleoli (A,B,C: H&E). (D) The array-CGH profile indicates a gain of the short arm of chromosome 11, which harbours the HRAS gene. (E) The electropherogram shows a mutation affecting codon 61 of the HRAS gene (HRASQ61R). Obtained with permission from Wiesner et al. (3). CGH, comparative genomic hybridization.
Common mutations include the HRAS exon 3 amino acid substitution of position 61 glutamine towards lysine or arginine, whereas mutations within the exon 2 region are rare. HRAS mutations lead to a constitutively active protein triggering cell proliferation via activation of MAPK and PI3K/AKT/mTOR pathways (18).
Histologically, HRAS-mutated Spitz nevi are predominantly intradermal with a pronounced desmoplastic stroma reaction and relatively low cellularity. The melanocytes are larger and have pleomorphic nuclei. HRAS-mutated Spitz tumors have a common pattern of infiltrating growth at their base; they show little or no melanin pigment and are also characterized by extensive growth of single cells between collagen bundles. Finally, deep mitoses present in the lower half and/or at the base of the lesion are frequently seen, especially in the elderly (18).
Mutation analysis in Spitz tumors has demonstrated that 15% of Spitz nevi harbor an activating mutation of this oncogene. So far, no cases of Spitzoid melanomas harboring HRAS mutation have been reported, nor malignant progression of a Spitz tumor with an HRAS mutation has been documented. In contrast, NRAS, which also belongs to the family of RAS genes, is mutated in 25% of primary cutaneous melanomas. Mutations of NRAS have also been described in congenital melanocytic nevi, which, depending on their size, possess increased risk of progression towards melanoma. However, there is no published data supporting that the presence of NRAS mutation per se confers a higher probability of malignant progression in a particular case. Based on these molecular features, Spitz nevus should not be regarded as a precursor lesion of melanoma and the presence of HRAS mutations is a pointer of benignity and/or favorable clinical outcome.
Although Spitz nevus shares many features with melanoma, it is believed that the major difference that separates the two entities is the genomic stability status. It has been postulated that RAS genes are insufficient to provoke tumorigenesis on their own, but require the co-occurrence of other aberrations (for example BRAF mutations, or inactivation of p53 or p16) to promote malignant transformation. This, however, remains to be further studied and elucidated in the context of Spitzoid tumors. Furthermore, future studies are needed in order to clarify the status of the INK4A genomic locus (9p21) in Spitz nevi. INK4A encodes for the p16 and p14ARF proteins, which are important components of the G1 checkpoint and p53 dependent apoptotic pathway (30).
In conclusion, HRAS mutation analysis seems to be useful in the differential diagnosis between Spitz nevus and Spitzoid melanoma and might be the first step towards a more reproducible classification of Spitz tumors combining histological and genetic data.
Spitzoid tumors with TERT promoter mutation
TERT promoter mutation is encountered in 22–71% of adult melanomas and in the majority of pediatric melanomas including Spitzoid melanomas (31,32). In a recent study of 56 Spitzoid neoplasms (6), all four patients developing haematogenous metastases were TERT promoter mutation positive whereas all remaining patients were TERT promoter mutation negative and fared well. Thus, TERT promoter mutation warrants validation as a potential biomarker of aggressive behavior in Spitzoid lesions.
BAP1-deficient Spitzoid tumors (BAPomas)
BAP1 protein functions as a de-ubiquitination enzyme located in the cell nucleus involved in chromatin remodeling. BAP1 loss sensitizes the cells to ionizing radiation and associates with the development of cutaneous melanocytic tumors, which share some common histopathologic characteristics with Spitz nevi (Figure 5) (33).
Figure 5.

BAPoma. (A) Polypoid, relatively symmetrical, dermal tumor consisting of melanocytes arranged in nests and sheets (H&E, ×20). (B,C) Large epithelioid melanocytes with a few scattered lymphocytes are surrounded by conventional small melanocytes. The larger cells are characterized by moderate amounts of amphophilic cytoplasm, well-defined cytoplasmic borders, and eccentric nuclei with prominent inclusions (H&E; B, ×100, C, ×200). (D) Large epithelioid melanocytes with a few scattered lymphocytes are surrounded by conventional small melanocytes. The large melanocytes lack labeling for BAP1 expression (short arrows), while the nuclei of small melanocytes (long arrows) and nonmelanocytic normal cells (keratinocytes and lymphocytes) are immunoreactive (BAP1 IHC, ×120).
Wiesner et al. (34) recently described a hereditary autosomal dominant tumor syndrome caused by inactivating germline mutations of the BAP1 gene (3p21). Several members of two unrelated families in whom this syndrome was first described developed multiple cutaneous epithelioid Spitzoid melanocytic neoplasms and were predisposed to increased risk of developing cutaneous and uveal melanoma and other tumors, such as mesothelioma and renal cell carcinoma and were found to harbor a BRAF V600E mutation (34,35). Later, Wiesner et al. (35) described sporadic AST with epithelioid morphology, BRAF mutation and biallelic BAP1 loss (36).
The epithelioid melanocytic tumors are predominantly located on sun-exposed skin and the number of lesions per affected individual ranges from a few up to more than 50. The characteristic cutaneous melanocytic tumors are skin-colored papules or nodules, 5–10 mm large, commonly intradermal, with frequent involvement of the dermal-epidermal junction. Histologically, they consist of plump epithelioid cells with amphophilic cytoplasm and well-demarcated cytoplasmic borders, enlarged round-oval nuclei with vesicular chromatin and variably prominent nucleoli, multinucleated giant cells and a small common nevus component at the periphery (35). Moreover, TILs are often prominent (37). Although many of the cytological characteristics of tumor cells are reminiscent of epithelioid cells of Spitz nevi, other features characteristically shown in Spitz nevi, such as clefting around junctional melanocytic nests, spindle-shaped melanocytes, epidermal hyperplasia, hypergranulosis and Kamino bodies, are clearly absent. Instead, some tumors show markedly atypical characteristics, such as nuclear pleomorphism, high cellularity and increased mitotic activity which hinders their histological designation as benign or malignant (38).
The majority of familiar epithelioid Spitz tumors show bi-allelic loss of BAP1. In the majority of the cases, losses are interstitial, involving a part of chromosome 3p, rather than a monosomy of chromosome 3 or loss of the entire short arm of chromosome 3p.
One BAP1 allele is inactivated through a BAP1 germline mutation, whereas the second one is somatically inactivated via three different mechanisms. These include either chromosomal deletion of the remaining wild-type BAP1 locus at 3p21, acquired uniparental disomy of the specific chromosome 3 where the mutated BAP1 gene is found, or an additional inactivating mutation of the second wild-type BAP1. The biallelic BAP1 gene inactivation is reliably identified by the loss of nuclear BAP1 protein expression in large epithelioid melanocytes using immunohistochemistry. In contrast, the population of small epithelioid melanocytes retains nuclear BAP1 expression. All non-lesional cells such as epithelial, stromal and inflammatory cells are also immunoreactive for BAP1. In contrast to classic Spitz nevi, the lesions showing a loss of BAP1, harbor the BRAF V600E mutation and are immunohistochemically positive for BRAF V600E protein in the vast majority of the cases, whereas a small subset are NRAS mutated (36). BRAF V600E mutation is identified in both Spitzoid and common nevus components. Thus, on the basis of discrete histologic and molecular features, these BAP1 deficient lesions are not true Spitz nevi.
Although the risk of malignant progression in BRAF V600E melanocytic tumors with BAP1 inactivation seems to be low, some tumors may progress to melanoma. An isolated loss of BAP1 does not necessarily provoke tumor progression but further caution is warranted about the biological behavior of the BAP1 deficient Spitzoid lesions with close follow-up of these patients (37). Consequently, in individuals with isolated lesion a conservative complete excision is indicated, whereas patients with multiple melanocytic tumors as previously described, should be screened for BAP1 and BRAF expression. If multiple tumors show loss of BAP1 expression, genetic counseling and testing for germline BAP1 mutations might be indicated. In case of positive testing, analytical screening for other type of cancers should follow with a close clinical follow-up of these patients being recommended with surgical intervention whenever there is a marked change of lesions (37).
The sporadic BAP1 inactivated tumors, share the same histological features with those seen in tumors with germline BAP1 mutations, albeit lacking the nevus component. In these sporadic BAP1 deficient lesions, epigenetic silencing of the BAP1 or other genes, rather than BAP1 deletions or mutations is responsible for BAP1 inactivation. In combined melanocytic tumors, all melanocytes harbor the BRAF V600E mutation, whereas BAP1 loss is restricted to the Spitzoid component (35).
Finally, if a lesion with morphologic features of BAP1 loss is identified, immunohistochemical staining for BAP1 is recommended. If there is a loss of nuclear expression, and the lesion has features which best suit to nevi with no atypical melanocytes, no large sheets of cells or increased mitoses, the diagnosis of either Spitz nevus or combined Spitz nevus with features of BAP1 loss, is rendered. If atypical features are present, molecular diagnosis with CGH is needed. The display of additional chromosomal gains or losses would be worrisome for melanoma, depending on the genetic aberrations that are involved (37).
Spitz tumors with kinase fusions
Latest studies report that Spitz tumors without mutations in either HRAS or BRAF, frequently display genomic translocations involving wild-type serine-threonine kinase BRAF or RTKs ALK, ROS1, NTRK1 and RET. The fusion kinases are formed by intra- or inter-chromosomal rearrangements, which result in various chimeric genes. The 3' portion of the intact kinase domain is linked to the 5' portion of a constitutively transcribed gene. The promoter of the 5' partner is responsible for the expression of the fusion transcript (39).
ALK fusions
ALK fusions are estimated in 8% of Spitz nevi, 5% of AST and 1% of Spitzoid melanomas (Figure 6). Since Kinase fusions are present across the entire spectrum of Spitzoid tumors, they are considered to represent initiating oncogenic events in Spitzoid tumorigenesis. As a diagnostic tool they are not useful in discriminating benign and malignant melanocytic tumors. They largely occur in a mutually exclusive pattern with HRAS mutations of BAP1 inactivation (39).
Figure 6.
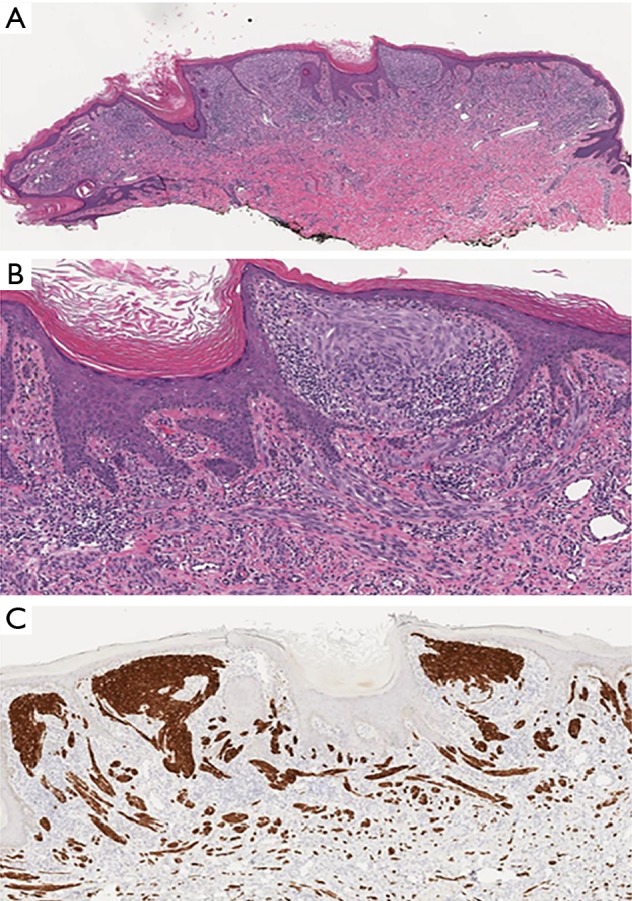
Spitz nevus with ALK-fusion. (A) Relatively symmetrical, exophytic, predominantly intradermal melanocytic tumour with focal epidermal hyperplasia (H&E, ×20). (B) Plexiform growth pattern and intersecting fascicles of fairly bland fusiform melanocytes (H&E, ×75). (C) Strong cytoplasmic ALK expression by fusiform melanocytes (ALK IHC, ×40).
Patients with fusion-positive Spitz tumors are of a younger age than those with non-translocated tumors. These tumors are usually dome-shaped or exophytic and occur mainly on the extremities. The majority of them are amelanotic, otherwise they frequently are heavily pigmented. Clinically, the lesions may be confused with irritated melanocytic nevi, virus-induced lesions (verruca, molluscum contagiosum) or vascular lesions (angioma, pyogenic granuloma) (39).
While BRAF mutations predominantly activate the MAPK pathway, activation of the PI3K/AKT/mTOR pathway, which is a key regulator of cell size, could explain why melanocytes are larger in Spitz tumors with kinase fusions, compared to common nevi with BRAF mutations. Deep knowledge of the activation pattern of these oncogenic signaling pathways is important for the development of ALK fusion inhibitors, such as crizotinib, which may prove useful in malignant lesions (39).
Most ALK fused tumors are compound and present some distinctive histological features. They demonstrate a plexiform growth pattern with large nests of fusiform to polygonal melanocytes arranged in elongated and radially vertically oriented nests. The majority shows a pronounced infiltrative growth pattern with deep dermal invasion. Dermal mitoses are not uncommon. The constituent melanocytes display amphophilic cytoplasm with a fibrillar quality, enlarged nuclei and prominent nucleoli, being devoid of marked pleomorphism. Spitz tumors with ALK fusions are easily identified by immunohistochemistry. ALK translocations can also be confirmed with FISH, reverse transcription polymerase chain reaction (RT-PCR) or next generation sequencing (NGS) (39).
Most prominent partners which may act as recurrent N-terminal fusion partners are TPM3 and DCTN1. ALK fusion positive Spitz tumors, analyzed by FISH, are negative for CNVs of 6p, 6q, 9p or 11q. Although CNVs are uncommon, recent data from a relative array-CGH analysis has identified a few chromosomal changes, such us loss of the chromosome 2 genomic region, where ALK gene is located, which is a common finding in most types of tumors, as well as loss of chromosome 1p, an aberration initially reported in a high percentage of Spitz nevi, although also present in AST and melanoma (39).
ROS1 fusions
ROS1 fusions are the second most frequent fusions seen in Spitz tumors (about 10% of cases). Spitz tumors are demonstrated as dome-shaped, well-circumscribed melanocytic compound lesions. Because there are no distinctive cytological and histological characteristics associated with ROS1 fusions, their identification is challenging. They mainly consist of large spindled and epithelioid melanocytes with vesicular nuclei and variable atypia. An irregular epidermal hyperplasia is frequently shown along with some Kamino bodies. Immunohistochemistry using an anti-ROS1 antibody can be helpful for the detection of the expression of the chimeric ROS1-protein. Although positive ROS1 immunohistochemistry is highly specific, it lacks sensitivity. Moreover ROS1-aberrations can be detected by FISH, RT-PCR or NGS. ROS1 fusions are correlated with both main signaling pathways, MAPK/ERK and PI3K/AKT/MTOR, offering different options for new inhibitors development (40).
NTRK1 fusions
The NTRK1 fusions occur in up to 10% of Spitz tumors. Spitz tumors with positive NTRK1 fusions show classical Spitzoid features, but no distinctive histological characteristics. Intersecting fascicles of fusiform melanocytes in the dermis, as seen in ALK positive tumors, is uncommon. These tumors show strong staining for NTRK1 by immunohistochemistry. Despite the high specificity and sensitivity, some melanocytic lesions, without NTRK1 fusion, might show weak background staining due to low endogenous expression. LMNA-NTRK1 fusion is the most frequent and associates with both signal pathways (40).
NTRK3 fusions
NTRK3 belongs to Trk family of neurotrophin receptors which as a transmembrane tyrosine kinase receptor mediates response to neurotrophins. During nervous system development Trk receptors signalling triggers cell fate, proliferation and axon and dendrite patterning. They play an important role in the development of the synaptic strength and plasticity of the adult nervous system. Melanocytes share a common neuroectodermal origin with the nervous system and Trk receptors are expressed in them too, triggering cell proliferation and migration.
The transition of the kinase gene results in duplication of the DNA fragment that encodes for the 3' portion of the kinase gene and/or loss of the fragment that carries the 5' portion. Specifically, during activating of NTRK3 fusions CNVs arise within the NTRK3 locus on chromosome 15q, with a relative gain of the NTRK3 kinase domain. The most frequent fusions, that have been described, are ETV6-NTRK3, MYO5A-NTRK3 and MYH9-NTRK3, which arise early during the oncogenesis of melanocytes in a mutually exclusive pattern with other MAPK activating melanoma oncogenes. The fusion proteins trigger a constitutive signal through the MAPK, AKT and PLCγ1 pathways and many inhibitors clinically tested as promising drug targets, similar to NTRK1/2, ALK and ROS1 fusions.
NTRK3 fusion positive Spitz tumors are mainly seen in young patients (<18 years) with indistinctive Spitzoid characteristics including dermal involvement by large nests of epithelioid to spindled melanocytes with increased amounts of cytoplasm as compared to small melanocytes of conventional nevi (41).
RET fusions
RET rearrangements are seen in less than 5% of Spitz tumors (main fusion partners KIF5B and GOLGA5). As RET positive Spitz tumors are rare and no known antibody exists for their reliable recognition, there is little information regarding their characteristics. Similar to other fusions, they activate both MAPK/ERK and P13/AKT/mTOR pathways as well as PLCγ-1 pathways. RET inhibitors suppress oncogenic activity (40).
MET fusions
MET rearrangements are quite rare and activate both main signaling pathways when fusioned. Kinase inhibitors such as cabozantinib block oncogenic MET signaling (42).
BRAF fusions and amplification
BRAF fusions are demonstrated in about 5% of Spitz tumors and they show no specific morphologic characteristics. As BRAF is constitutively expressed in melanocytic tumors, immunohistochemistry for BRAF fusions is not specific and kinase fusions should be detected with other methods such as FISH or NGS. BRAF amplifications of its wild type have also been described in a small number of cases (40).
Blue nevi and related neoplasms
Proliferation of melanocytes residing in the cutaneous dermis gives rise to benign blue nevi. They are distinguished in various subtypes such as epithelioid, cellular, sclerotic and plaque-type. However, blue nevi of the Jadassohn-Tièche type are most commonly detected.
An additional intermediate category of blue nevi representing tumors of uncertain risk also exists. The latter are characterized as ‘atypical cellular blue nevi’ presenting with atypical clinicopathologic features. Benign blue nevi should not be confused with blue nevus-like melanomas (Figure 7), which is a distinct category of malignant tumors that requires special treatment. Although, their histological discrimination can be rather challenging, the mutational profile of blue nevi is quite different from the respective molecular lesions identified in epidermal-derived nevi and melanomas. The latter are commonly characterized by BRAF and NRAS mutations, whereas blue nevi harbor mutations within GNAQ, GNA11 and less frequently the CYSLTR2 and PLCB4 genes, which are also found to be mutated in uveal melanomas (43). Uveal melanomas are characterized by additional mutations, assisting further classification of them into a poor prognosis BAP1 mutated category and a favorable prognosis SF3B1 R625 or EIF1AX mutated category. Mutations within the same gene set are identified in blue nevus-like melanomas and in atypical cellular blue nevi. Mutational screening of these genes could accompany histopathological evaluation in challenging cases. Furthermore, array-CGH analysis of blue nevus mimicking malignancies revealed deletions of chromosomes 1p, 3p, 4q, 6q, 8p, 16q and 17q and gains of chromosomes 6p, 8q and 21q (44).
Figure 7.

Blue nevus-like melanoma. (A) A dermal melanocytic tumor consisting of spindle cells (H&E, ×20). (B,C) Mitotic activity and prominent nucleoli are evident (B, H&E, ×100; C, H&E, ×200).
Molecular markers in the distinction between melanoma and its mimics
Malignant melanoma is often part of a broad differential diagnosis including a wide spectrum of epithelioid or spindle cell neoplasms. On such an occasion immunohistochemistry is imperative in order to avoid misdiagnosis. That is the reason why newer markers are being tested with regard to their potential usefulness in the differential diagnosis of melanoma and its mimics.
Quite often melanoma demonstrates an aberrant immunophenotype, although typically confined to a small number of neoplastic cells. It is also worthy of note that S100 protein-negative melanomas express intermediate filament proteins in a higher percentage in comparison to the rest of melanomas. Melanomas with an aberrant immunophenotype can be easily mistaken for carcinomas, rhabdomyosarcomas and neuroendocrine tumors. As a result, every pathologist assessing a poorly differentiated cutaneous tumor or a metastatic lesion should include in the immunohistochemistry panel all available melanocytic markers (45).
A quite challenging diagnostic problem is the distinction between melanoma and malignant peripheral nerve sheath tumor (MPNST). This difficulty has prompted the emergence of novel immunohistochemical and molecular markers with high specificity for MPNSTs. The use of comprehensive genomic approaches led to the identification of loss-of-function somatic alterations of PRC2 (polycomb repressive complex 2) core components, EED and SUZ12, in the majority of all types of MPNST (sporadic, NF-1 associated and radiotherapy-associated) (46). MPNSTs with PRC2 loss showed complete loss of H3K27me3 and aberrant transcriptional activation of multiple PRC2-repressed homeobox master regulators and their regulated development pathways. Apart from the above, it was also observed that somatic alterations of CDKN2A and NF1 often co-exist with PRC2 alterations. Based on these findings, the usefulness of loss of H3K27me3 expression as a potential sensitive immunohistochemical marker for MPNST was tested in a series of pathologically and genetically well-characterised MPNSTs, confirming the loss of H3K27me expression in the majority of sporadic (95%) and radiotherapy-related (91%) MPNSTs, as well as in a high percentage (60%) of NF1-related MPNSTs (47).
Although loss of H3K27me3 expression proved to be a highly sensitive marker for MPNST, especially for sporadic and radiation-induced subtypes, it failed to arise as a useful marker when trying to distinguish MPNST from melanoma. In a study of 387 cases including a cohort of pathologically and genetically well-characterised MPNSTs and a cohort of primary or metastatic melanomas, complete loss of expression of H3K27me3 was observed in 72% of MPNSTs and 37% of melanomas. These results indicate that complete loss of H3K27me3 is not an MPNST-specific feature and, therefore, cannot be used as a specific marker in the differential diagnosis between MPNSTs and melanomas (48).
It is possible that loss of H3K27me3 expression may also play a role in the distinction between nodular melanomas of childhood associated with congenital melanocytic nevi and proliferative nodules. In a study evaluating 5 cases of pediatric nodular melanomas and 20 cases of proliferative nodules loss of H3K27me3 expression was observed in 4 melanoma cases. In contrast H3K27me3 was retained or only mildly reduced in benign proliferative nodules. Thus, reduced H3K27me3 expression may be useful as a potential diagnostic marker in the differential diagnosis of pediatric nodular melanomas and proliferative nodules, favoring the former. However, a larger number of cases should be examined in order to establish its use as a diagnostic marker (49).
Targeted therapy
Apart from the advances in the field of diagnosis, great progress has also been made in the therapeutic approach of melanoma, especially melanoma of advanced stage (50). Currently, two orally bioavailable selective BRAF inhibitors (vemurafenib and dabrafenib) have been approved for the treatment of unresectable, metastatic BRAF V600-mutated melanomas leading to marked tumor regression.
Moreover, selective binding of the small molecule inhibitors to the active enzyme conformation results in a poor inhibition of wild type BRAF kinase and also causes a paradoxical MAPK cascade activation in BRAF wild-type keratinocytes that has important clinical implications, as a subset of treated patients develop squamous cell carcinomas (SCCs) and/or keratoacanthomas, as well as a variety of cutaneous toxic effects such as Grover disease (51).
These secondary cutaneous malignancies are successfully eliminated with local resection. Recently, a combination therapy of BRAF inhibitors with MEK inhibitors improved recurrence rate (RR) or progression free survival (PFS) in comparison with monotherapy as well as safety profile with regard to cutaneous adverse effects, due to reduced ERK MAPK activation. Nevertheless, melanoma patients treated with these regimens require ongoing accurate dermatologic monitoring to timely address any relevant complications (52)
Although these targeted agents greatly improved survival of BRAF V600E/K mutant melanoma patients over BRAF wild type ones, reinforcing the emerging role of predictive markers for molecular stratification of patients with regard to efficient therapy, the duration of responses is limited due to early onset of drug resistance. MAPK pathway reactivation in the presence of a BRAF inhibitor may occur due to several different alterations including BRAF amplification/overexpression, NRAS/MEK1/2 mutations and expression of various RTKs (53).
The multiple mechanisms of acquired resistance to BRAF inhibitors seem quite complex suggesting the possibility of multidrug combinations, including PI3K/AKT, mTOR inhibitors as well as immunotherapy for efficient and durable therapeutic intervention. It is also worthy of note that acquired drug resistance could be reversed by intermittent therapy schedules due to cessation of drug dependency of drug resistant cells (54).
Finally, a small subset of patients, mainly those with acral and mucosal melanomas, who display KIT mutations (especially in exon 11) may benefit from targeted therapies with imatinib, sunitinib, sorafenib, nilotinib as reported in various studies and clinical trials (55).
Conclusions and future perspectives
Significant advances in tumor genomics have provided considerable insight in the complex biology of melanocytic tumors particularly with regard to Spitzoid tumors. Integration of clinical, histological and molecular information has given rise to the specific types of HRAS, BAP1 and kinase fusion types of Spitz tumors which differ in terms of biologic behavior. HRAS mutated or ALK kinase fused Spitz tumors can be expected to behave indolently while the presence of BRAF mutation may be clinically worrisome in the context of a melanocytic tumor composed of large epithelioid or spindle cells arranged in a plexiform pattern.
The separation of melanocytic neoplasms based on reproducible association between pathogenetically relevant genetic alterations and clinical phenotypes may result in an improved understanding of these neoplasms facilitating future research and clinical personalised management.
Acknowledgements
We are grateful to Assistant Professor Konstantinos D. Linos for his valuable advice and for providing us with microphotographs
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Bastian BC. The molecular pathology of melanoma: an integrated taxonomy of melanocytic neoplasia. Annu Rev Pathol 2014;9:239-71. 10.1146/annurev-pathol-012513-104658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bennett DC. Genetics of melanoma progression: the rise and fall of cell senescence. Pigment Cell Melanoma Res 2016;29:122-40. 10.1111/pcmr.12422 [DOI] [PubMed] [Google Scholar]
- 3.Wiesner T, Kutzner H, Cerroni L, et al. Genomic aberrations in spitzoid melanocytic tumours and their implications for diagnosis, prognosis and therapy. Pathology 2016;48:113-31. 10.1016/j.pathol.2015.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wiesner T, Kutzner H. Morphological and genetic aspects of Spitz tumors. Pathologe 2015;36:37-43. 10.1007/s00292-014-1984-1 [DOI] [PubMed] [Google Scholar]
- 5.Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol 2014;15:482-96. 10.1038/nrm3823 [DOI] [PubMed] [Google Scholar]
- 6.Lee S, Barnhill RL, Dummer R, et al. TERT Promoter Mutations Are Predictive of Aggressive Clinical Behavior in Patients with Spitzoid Melanocytic Neoplasms. Sci Rep 2015;5:11200. 10.1038/srep11200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Raamsdonk CD, Bezrookove V, Green G, et al. Frequent somatic mutations of GNAQ in uvealmelanoma and blue naevi. Nature 2009;457:599-602. 10.1038/nature07586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pollock PM, Harper UL, Hansen KS, et al. High frequency of BRAF mutations in nevi. Nat Genet 2003;33:19-20. 10.1038/ng1054 [DOI] [PubMed] [Google Scholar]
- 9.Bauer J, Curtin JA, Pinkel D, et al. Congenital melanocytic nevi frequently harbor NRAS mutations but no BRAF mutations. J Invest Dermatol 2007;127:179-82. 10.1038/sj.jid.5700490 [DOI] [PubMed] [Google Scholar]
- 10.Yeh I, von Deimling A, Bastian BC. Clonal BRAF mutations in melanocytic nevi and initiating role of BRAF in melanocytic neoplasia. J Natl Cancer Inst 2013;105:917-9. 10.1093/jnci/djt119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reimer RR, Clark WH, Jr, Greene MH, et al. Precursor lesions in familial melanoma. A new genetic preneoplastic syndrome. JAMA 1978;239:744-6. 10.1001/jama.1978.03280350068019 [DOI] [PubMed] [Google Scholar]
- 12.Piepkorn M, Meyer LJ, Goldgar D, et al. The dysplastic melanocytic nevus: a prevalent lesion that correlates poorly with clinical phenotype. J Am Acad Dermatol 1989;20:407-15. 10.1016/S0190-9622(89)70050-5 [DOI] [PubMed] [Google Scholar]
- 13.Ackerman AB. What naevus is dysplastic, a syndrome and the commonest precursor of malignant melanoma? A riddle and an answer. Histopathology 1988;13:241-56. 10.1111/j.1365-2559.1988.tb02036.x [DOI] [PubMed] [Google Scholar]
- 14.Tucker MA, Halpern A, Holly EA, et al. Clinically recognized dysplastic nevi. A central risk factor for cutaneous melanoma. JAMA 1997;277:1439-44. 10.1001/jama.1997.03540420035026 [DOI] [PubMed] [Google Scholar]
- 15.Shain AH, Yeh I, Kovalyshyn I, et al. The Genetic Evolution of Melanoma from Precursor Lesions. N Engl J Med 2015;373:1926-36. 10.1056/NEJMoa1502583 [DOI] [PubMed] [Google Scholar]
- 16.Viros A, Fridlyand J, Bauer J, et al. Improving melanoma classification by integrating genetic and morphologic features. PLoS Med 2008;5:e120. 10.1371/journal.pmed.0050120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol 2011;29:1239-46. 10.1200/JCO.2010.32.4327 [DOI] [PubMed] [Google Scholar]
- 18.van Engen-van Grunsven AC, van Dijk MC, Ruiter DJ, et al. HRAS-mutated Spitz tumors: A subtype of Spitz tumors with distinct features. Am J Surg Pathol 2010;34:1436-41. 10.1097/PAS.0b013e3181f0a749 [DOI] [PubMed] [Google Scholar]
- 19.Spitz S. Melanomas of childhood. Am J Pathol 1948;24:591-609. [PMC free article] [PubMed] [Google Scholar]
- 20.Barnhill RL, Argenyi ZB, From L, et al. Atypical Spitz nevi/tumors: lack of consensus for diagnosis, discrimination from melanoma, and prediction of outcome. Hum Pathol 1999;30:513-20. 10.1016/S0046-8177(99)90193-4 [DOI] [PubMed] [Google Scholar]
- 21.Gerami P, Scolyer RA, Xu X, et al. Risk assessment for atypical spitzoid melanocytic neoplasms using FISH to identify chromosomal copy number aberrations. Am J Surg Pathol 2013;37:676-84. 10.1097/PAS.0b013e3182753de6 [DOI] [PubMed] [Google Scholar]
- 22.Yazdan P, Cooper C, Sholl LM, et al. Comparative analysis of atypical spitz tumors with heterozygous versus homozygous 9p21 deletions for clinical outcomes, histomorphology, BRAF mutation, and p16 expression. Am J Surg Pathol 2014;38:638-45. 10.1097/PAS.0000000000000160 [DOI] [PubMed] [Google Scholar]
- 23.Lallas A, Kyrgidis A, Ferrara G, et al. Atypical Spitz tumours and sentinel lymph node biopsy: a systematic review. Lancet Oncol 2014;15:e178-83. 10.1016/S1470-2045(13)70608-9 [DOI] [PubMed] [Google Scholar]
- 24.Stefanaki C, Stefanaki K, Chardalias L, et al. Differential diagnosis of Spitzoid melanocytic neoplasms. J Eur Acad Dermatol Venereol 2016;30:1269-77. 10.1111/jdv.13665 [DOI] [PubMed] [Google Scholar]
- 25.Cesinaro AM, Schirosi L, Bettelli S, et al. Alterations of 9p21 analysed by FISH and MLPA distinguish atypical spitzoid melanocytic tumors from conventional Spitz’s nevi but do not predict their biologic behavior. Histopathology 2010;57:515-27. 10.1111/j.1365-2559.2010.03653.x [DOI] [PubMed] [Google Scholar]
- 26.Massi D, Tomasini C, Senetta R, et al. Atypical Spitz tumors in patients younger than 18 years. J Am Acad Dermatol 2015;72:37-46. 10.1016/j.jaad.2014.09.049 [DOI] [PubMed] [Google Scholar]
- 27.Cho-Vega JH. A diagnostic algorithm for atypical spitzoid tumors: guidelines for immunohistochemical and molecular assessment. Mod Pathol 2016;29:656-70. 10.1038/modpathol.2016.70 [DOI] [PubMed] [Google Scholar]
- 28.Bastian BC, Wesselmann U, Pinkel D, et al. Molecular cytogenetic analysis of Spitz nevi shows clear differences to melanoma. J Invest Dermatol 1999;113:1065-9. 10.1046/j.1523-1747.1999.00787.x [DOI] [PubMed] [Google Scholar]
- 29.Jiveskog S, Ragnarsson-Olding B, Platz A, et al. N-RAS mutations are common in melanomas from sun-exposed skin of humans but rare in mucosal membranes of unexposed skin. J Invest Dermatol 1998;111:757-61. 10.1046/j.1523-1747.1998.00376.x [DOI] [PubMed] [Google Scholar]
- 30.Bastian BC, LeBoit PE, Pinkel D. Mutations and copy number increase of HRAS in Spitz nevi with distinctive histopathological features. Am J Pathol 2000;157:967-72 10.1016/S0002-9440(10)64609-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Horn S, Figl A, Rachakonda PS, et al. TERT promoter mutations in familial and sporadic melanoma. Science 2013;339:959-61. 10.1126/science.1230062 [DOI] [PubMed] [Google Scholar]
- 32.Lu C, Zhang J, Nagahawatte P, et al. The genomic landscape of childhood and adolescent melanoma. J Invest Dermatol 2015;135:816-23. 10.1038/jid.2014.425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Machida YJ, Machida Y, Vashisht AA, et al. The deubiquitinating enzyme BAP1 regulates cell growth via interaction with HCF-1. J Biol Chem 2009;284:34179-88. 10.1074/jbc.M109.046755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wiesner T, Obenauf AC, Murali R, et al. Germline mutations in BAP1 predispose to melanocytic tumors. Nat Genet 2011;43:1018-21. 10.1038/ng.910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wiesner T, Murali R, Fried I, et al. A distinct subset of atypical Spitz tumors is characterized by BRAF mutation and loss of BAP1 expression. Am J Surg Pathol 2012;36:818-30. 10.1097/PAS.0b013e3182498be5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yeh I, Mully TW, Wiesner T, et al. Ambiguous melanocytic tumors with loss of 3p21. Am J Surg Pathol 2014;38:1088-95. 10.1097/PAS.0000000000000209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Busam KJ, Wanna M, Wiesner T. Multiple epithelioid Spitz nevi or tumors with loss of BAP1 expression: a clue to a hereditary tumor syndrome. JAMA Dermatol 2013;149:335-9. 10.1001/jamadermatol.2013.1529 [DOI] [PubMed] [Google Scholar]
- 38.Murali R, Wiesner T, Scolyer RA. Tumours associated with BAP1 mutations. Pathology 2013;45:116-26. 10.1097/PAT.0b013e32835d0efb [DOI] [PubMed] [Google Scholar]
- 39.Yeh I, de la Fouchardiere A, Pissaloux D, et al. Clinical, histopathologic, and genomic features of Spitz tumors with ALK fusions. Am J Surg Pathol 2015;39:581-91. 10.1097/PAS.0000000000000387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wiesner T, He J, Yelensky R, et al. Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat Commun 2014;5:3116. 10.1038/ncomms4116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yeh I, Tee MK, Botton T, et al. NTRK3 kinase fusions in Spitz tumours. J Pathol 2016;240:282-90. 10.1002/path.4775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yeh I, Botton T, Talevich E, et al. Activating MET kinase rearrangements in melanoma and Spitz tumours. Nat Commun 2015;6:7174. 10.1038/ncomms8174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Costa S, Byrne M, Pissaloux D, et al. Melanomas Associated With Blue Nevi or Mimicking Cellular Blue Nevi: Clinical, Pathologic, and Molecular Study of 11 Cases Displaying a High Frequency of GNA11 Mutations, BAP1 Expression Loss, and a Predilection for the Scalp. Am J Surg Pathol 2016;40:368-77. 10.1097/PAS.0000000000000568 [DOI] [PubMed] [Google Scholar]
- 44.Griewank KG, Müller H, Jackett LA, et al. SF3B1 and BAP1 mutations in blue nevus-like melanoma. Mod Pathol 2017;30:928-39. 10.1038/modpathol.2017.23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Romano RC, Carter JM, Folpe AL. Aberrant intermediate filament and synaptophysin expression is a frequent event in malignant melanoma: an immunohistochemical study of 73 cases. Mod Pathol 2015;28:1033-42. 10.1038/modpathol.2015.62 [DOI] [PubMed] [Google Scholar]
- 46.Lee W, Teckie S, Wiesner T, et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat Genet. 2014;46:1227-32 10.1038/ng.3095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prieto-Granada CN, Wiesner T, Messina JL, et al. Loss of H3K27me3 Expression Is a Highly Sensitive Marker for Sporadic and Radiation-induced MPNST. Am J Surg Pathol 2016;40:479-89. 10.1097/PAS.0000000000000564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Le Guellec S, Macagno N, Velasco V, et al. Loss of H3K27 trimethylation is not suitable for distinguishing malignant peripheral nerve sheath tumor from melanoma: a study of 387 cases including mimicking lesions. Mod Pathol 2017;30:1677-87. 10.1038/modpathol.2017.91 [DOI] [PubMed] [Google Scholar]
- 49.Busam KJ, Shah KN, Gerami P, et al. Reduced H3K27me3 Expression Is Common in Nodular Melanomas of Childhood Associated With Congenital Melanocytic Nevi But Not in Proliferative Nodules. Am J Surg Pathol 2017;41:396-404. 10.1097/PAS.0000000000000769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Luke JJ, Flaherty KT, Ribas A, et al. Targeted agents and immunotherapies:optimizing outcomes in melanoma. Nat Rev Clin Oncol 2017;14:463-82. 10.1038/nrclinonc.2017.43 [DOI] [PubMed] [Google Scholar]
- 51.Holderfield M, Deuker MM, McCormick F, et al. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer 2014;14:455-67. 10.1038/nrc3760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carlos G, Anforth R, Clements A, et al. Cutaneous Toxic Effects of BRAF Inhibitors Alone and in Combination With MEK Inhibitors for Metastatic Melanoma. JAMA Dermatol 2015;151:1103-9. 10.1001/jamadermatol.2015.1745 [DOI] [PubMed] [Google Scholar]
- 53.Fiskus W, Mitsiades N. B-Raf Inhibition in the Clinic: Present and Future. Annu Rev Med 2016;67:29-43. 10.1146/annurev-med-090514-030732 [DOI] [PubMed] [Google Scholar]
- 54.Das Thakur M, Salangsang F, Landman AS, et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 2013;494:251-5. 10.1038/nature11814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lovly CM, Pao W, Sosman J. 2015. KIT in Melanoma. My Cancer Genome. Available online: https://www.mycancergenome.org/content/disease/melanoma/kit/