

Summary
Regulatory B (Breg) cells are characterized by various membrane markers and the secretion of different inhibitory cytokines. A new subset of Breg cells was identified as CD5hi Fas‐ligand (FasL)hi. Their main reported role is to suppress anti‐viral and anti‐tumour immune responses, and, hence they have been dubbed ‘killer’ B cells. In this study, we aim to assess the role of these cells in chronic hepatitis C virus (HCV) infection, and determine if they contribute to the increased viral load and persistence of HCV and its related autoimmunity. (i) FasL expression on CD5hi B cells is increased significantly in HCV‐infected patients compared to healthy individuals [28·06 ± 6·71 mean fluorescence intensity (MFI) ± standard error of the mean (s.e.m.), median = 27·9 versus 10·87 ± 3·97 MFI ± s.e.m., median = 10·3, respectively, P < 0·0001]. (ii) Killer B cells from HCV patients increased autologous CD4+ T cell apoptosis compared to the apoptosis in healthy individuals [39·17% ± 7·18% mean ± standard deviation (s.d.), median = 39·6 versus 25·92 ± 8·65%, mean ± s.d., median = 24·1%, P < 0·0001, respectively]. A similar increase was observed in CD8+ T cell apoptosis (54·67 ± 15·49% mean ± s.d., median = 57·3 versus 21·07% ± 7·4%, mean ± s.d., median = 20%, P = 0·0006, respectively). (iii) By neutralizing FasL with monoclonal anti‐FasL antibodies, we have shown that the induction of apoptosis by killer B cells is FasL‐dependent. (iv) Increased expression of FasL on CD5hi B cells is correlated positively with an increased viral load and the presence of anti‐nuclear antibodies and rheumatoid factor in HCV. This is the first study in which killer B cells have been suggested to play a pathogenic role in HCV. They seem to be involved in HCV's ability to escape efficient immune responses.
Keywords: autoimmunity, B cell, viral load
Introduction
Hepatitis C (HC) is the second most common infectious disease worldwide, with 130–150 million cases, and 500 000 deaths annually due to cirrhosis or liver cancer. The efficient host immune reaction against the various hepatitis C virus viral proteins determines viral persistence, the extent of infected hepatocytes and the severity of liver inflammation. It has been shown that the persistence of the hepatitis C virus (HCV) is associated with the increased presence of autoantibodies. Anti‐nuclear antibodies (ANA), rheumatoid factor (RF) and anti‐cardiolipin antibodies (aCL) are the most reported autoantibodies in HC patients with a prevalence of positivity in more than 20% among them 1. Once primary immunization has occurred, the repeated generation of apoptotic material (during persistent viral infection) might efficiently rechallenge the primed immune system. This capacity for immune‐derived autoamplification is possibly a critical principle underlying systemic autoimmune disorder 2. The first line of defence is always the efficient peripheral immune responses against HCV antigens, mainly cytotoxic T cell response and natural killer (NK) cell activity. Both T and NK cells were shown to eliminate HCV‐infected cells by over‐expressing Fas ligand (FasL) and/or producing granzyme B (GranB). In the early phase of infection, B cells produce neutralizing antibodies against different virus epitopes, mainly against envelope glycoproteins E1 and E2 3. Most of these antibodies fail to block the entry of the virus into host cells, and therefore the role of humoral anti‐viral responses remains limited. Chronic HCV infection and its persistence were reported to be the result of impaired NK cell function and insufficient interferon (IFN)‐γ production, in addition to increased secretion of interleukin (IL)‐10, leading to the failure of HCV cleaning. We reported previously that enhanced peripheral T cell apoptosis in chronic HCV infection was associated with an increased viral load, autoimmunity and liver disease severity 4, 5. Of the many possible mechanisms by which HCV infection increases the tendency of T cells to undergo apoptosis, worth mentioning are: (1) down‐regulation of major histocompatibility complex (MHC) class II and B7 molecules on HCV‐infected dendritic cells, which attenuates the delivery of co‐stimulation; (2) inhibition of the production of IFN‐γ and IL‐12, leading to an enhanced responsiveness to IL‐10 modulating effects and down‐regulation of T helper type 1 (Th1) responses; (3) induction of autoimmune responses due to cross‐reactions between HCV core proteins and cryptic epitopes on activated T cells; and (4) enhancement of T cell apoptosis by causing G1/S arrest and c‐myc up‐regulation 5. In order to facilitate an efficient process of anti‐viral cytotoxicity, a balance between regulatory T cell (Treg) function and efficient effector T cell function is required. In this respect, Treg expansion and their increased suppressive function (induced by HCV signalling) was found to be correlated with altered abilities of effector T cell responses and the persistence of HCV infection followed by its escape to the liver 6. In this regard, CD4+CD25hi cells from chronic HCV patients produce higher amounts of GranB, consequently suppressing autologous CD4+CD25low effector T cells and reducing CD4+ T cell responses against HCV 7. In parallel with Tregs, regulatory B cells (Bregs) are involved in suppressing autoimmunity and inflammation. These are characterized by different membrane markers and producing inhibitory cytokines, of which IL‐10 is the most dominant 8. Some studies describe Bregs as CD24hiCD38hiCD1dhi with an IL‐10‐dependent suppressive ability, and is altered in patients suffering from autoimmune diseases such as systemic lupus erythematosus 9. Other researchers have identified Bregs as CD25hiCD86hiCD1dhi, IL‐10 and transforming growth factor (TGF)‐β‐producing and showed an increase in Treg function through a cell‐to‐cell mechanism 10. In recent years, many studies have reported that CD5hi B cells are a main source of IL‐10 production, resulting in them being considered to be another member of the Breg cell family 11, 12. CD5hi Breg cells were also presented as expressing FasL, thereby showing that they play a role in regulating different immune responses by inducing effector T cell apoptosis 13. In one study, FasL expressing B lymphocytes were purified from the spleen of MRL/lpr mice, and were shown to be potent cytotoxic effectors against Fas‐positive targets. The level of FasL expression increased with the extent of the cell‐surface activation marker CD69, indicating that expression of FasL is up‐regulated in parallel with the activation state of B cells 14. Later, in a T cell‐receptor transgenic mouse model of collagen‐induced arthritis, altered T cell death and enhanced severity of arthritis correlated with reduced splenic CD5+FasL+ B cells. Appropriately dubbed ‘killer B’ cells, it was suggested that this subset of Bregs may provide a novel mechanism for inducing T cell death as a treatment for arthritis 15. With this in mind, CD5hiFasLhi B cells were reported to play a crucial role in the persistence of some pathogens and in their escape from efficient T cell immune responses. In a very early study, in a murine model of Schistosoma mansoni infection, a subset of splenic CD19+ B cells were shown to be FasL‐expressing and mediators of CD4+ T cell apoptosis, thus inhibiting anti‐pathogen immune responses 16. Several viral infections, including the human immunodeficiency virus and Epstein–Barr virus (EBV), have been reported to increase Fas (CD95) on effector CD4+ T cells and FasL expression on B cells, leading to increased T cell apoptosis and the evasion of viruses from cellular cytotoxic immune responses. The mechanisms by which viruses and parasites induce FasL expression are still not clear enough 17. Although it has been mentioned in the literature throughout the last decade, the issue of killer B cells and their contribution to viral persistence remains enigmatic 18. In this study we aim to analyse, for the first time, the status of this unique subset of Bregs in patients suffering from chronic HCV infection. We assume that increased numbers and expression of FasL on CD5hi B cells is followed by these cells' increased killing function, and are thus possibly associated with HCV persistence, increased viral load and disease severity.
Patients and methods
Patients
Our study included 41 patients (15 females and 26 males, mean age = 53·4 years, range = 34–66 years) in whom chronic HCV infection was established by their having increased liver enzymes, positive serum anti‐HCV antibodies [assessed by enzyme‐linked immunosorbent assay (ELISA) II; Abbot Laboratories, North Chicago, IL, USA] and detectable HCV RNA (polymerase chain reaction). All patients were studied before any specific anti‐viral therapy was given. We excluded HCV patients in whom other immune‐mediated diseases were diagnosed and to whom any immune‐modulated treatments were given. HCV‐infected patients are followed at the Liver Disease Clinic, Bnai‐Zion Medical Center and at the Liver Disease Center of Carmel Medical Center, Haifa, Israel. All patients were assessed for the presence of liver enzymes, serum HCV antibodies, a panel of autoantibodies including ANA, anti‐smooth muscle antibodies (anti‐sm), RF and aCL. A viral load assessment was performed on all patients and was tracked periodically as part of the follow‐up. Forty‐five healthy age‐ and sex‐matched individuals served as a control group. Informed consent was obtained from all the participating individuals and the study was approved by the local ethics committee for clinical studies.
Methods
HCV quantification
HCV RNA concentration in serum was measured using an Amplicor Monitor Test Kit (Roche Diagnostic Systems, Basel, Switzerland). This test includes an RNA quantification standard of a known copy number that is co‐amplified with a target and is used to calculate the copy level of the sample by colorimetric assay following hybridization to a specific probe. All antibodies were detected using commercial ELISA kits as part of our routine evaluation of all HCV patients.
T and B cell isolation
Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll Paque density gradient. The CD4+ T cells or CD8+ T cells were positively isolated from PBMCs using anti‐human CD4 or CD8 microbeads, respectively [magnetic‐activated cell sorting (MACS) technology; Milteyni Biotec, Bergisch Gladbach, Germany], according to the manufacturer's instructions. Achieved purity was assessed by fluorescence activated cell sorter (FACS) analysis and was always found to be > 96%. The CD19+ B cells were positively isolated in order to characterize CD5low versus CD5hi B cells. However, B cells were also negatively isolated from PBMCs (to be used later for B cell sorting) using the EasySepTM human B cell enrichment kit (StemCell Technologies Inc., Vancouver, BC, Canada), according to the manufacturer's instructions. The desired unlabelled fraction was poured off into a final collection tube, thereby achieving a purity of > 96%.
Assessment of CD5hiFasLhi B cells by flow cytometry
Isolated CD19+ B cells (both naive and activated) were divided into three tubes, and stained as follows: (1) phycoerythrin/cyanin 5 (PE/Cy5) anti‐CD19+PE anti‐CD5+ Alexa Fluor 488 anti‐FasL; (2) fluorescein isothiocyanate (FITC) anti‐CD19+PE anti‐CD5, PE/Cy7 anti IL‐10; and (3) FITC anti‐CD19+PE/Cy5 anti‐CD5+PE anti‐perforin or +Alexa Fluor 647 anti‐granzyme B. The staining of intracellular markers was performed using the Fix and Perm Cell Permeabilization Kit™ (Invitrogen, Waltham, MA, USA), according to the manufacturer's instructions. The above markers were assessed on naive and activated purified B cells. Cells were cultured overnight with cytosine–phosphate–guanosine‐oligodeoxynucleotide (CpG‐ODN) and anti‐CD40L, as reported previously 10. Purified B cells were stained and gated on CD19+CD5hi and CD19+CD5low and then evaluated for the expression of FasL, GranB, perforin and IL‐10.
CD5hiFasLhi B cell sorting
Peripheral B cells were stained with anti‐human CD19 PE/Cy5, anti‐human CD5 PE and anti‐human CD3 peridinin chlorophyll (PerCP)‐Cy 5.5. Stained B cells were sorted by BD FACS ARIA III into two subpopulations: (1) cells defined as CD19+CD5hi but not CD3; and (2) cells defined as CD19+CD5low but not CD3. Following this sorting process, cells were washed twice in PBS and incubated overnight in a fresh sorting medium at 37°C. Sorted B cells (CD19+CD5low and CD19+CD5hi) were then stained with PE/Cy5 anti‐CD19+PE anti‐CD5+ Alexa Fluor 488 anti‐FasL, in order to check CD5hi purity and to double‐check FasL expression on each subpopulation. Both CD5hi purity and FasL expression were evaluated by flow cytometer (FC500 and CXP software; Beckman Coulter Life Sciences, Indianapolis, IN, USA).
Assessment of CD4+ and CD8+ T cell apoptosis
Purified CD4+ and CD8+ T cells were immune‐stained directly (one step) with propidium iodide (PI) and FITC‐labelled recombinant human annexin V (annexin V kit; MedSystems Diagnostics GmbH, Vienna, Austria). Flow cytometry was carried out with a FACS operating with CellQuest software (Becton Dickinson, Mountain View, CA, USA). The total population of viable cells was gated according to their typical forward‐ and right‐angle light‐scatter. The percentage of cells stained by annexin V alone or PI/annexin V was determined, taking into account only positive‐stained cells. The data were displayed on a dot‐plot where each was generated from at least 104 events.
The induction of T cell apoptosis following co‐culture with killer B cells
Autologous CD4+ and CD8+ T cells were activated overnight with anti‐CD3 and anti‐CD28 in a medium containing 5 units/ml of IL‐2. T cells were then cultured alone for the detection of spontaneous apoptosis, or co‐cultured with sorted CD5hiFasLhi or with CD5lowFasLlow (non‐killer B cells) at a 1 : 1 ratio in a 96‐well plate for 48 h at a final concentration of 105 cells/well. Cells undergoing early apoptosis were those stained with annexin V only, whereas those co‐stained with annexin/PI were considered cells in late apoptosis or necrosis.
The induction of apoptosis by killer B cells is FasL‐dependent
Aiming to establish that the induction of T cell apoptosis by killer B cells is FasL‐dependent, we added anti‐FasL neutralizing antibody [FasL monoclonal antibody (mAb) (3C82); Alexis Biochemicals, Nottingham, UK] or isotype control antibody (Alexis Biochemicals) to the co‐culture of the cells. When comparing the efficacy of three doses in neutralizing FasL (1, 2·5 and 5 μg ml−1), we found the dosage of 2·5 μg ml−1 to be the ideal choice.
Statistical analysis
The assessment of data normality was performed using the D'Agostino–Pearson test, and a comparison of the differences between two groups and assessment of the median values was performed using the Mann–Whitney non‐parametric test. A comparison among three groups was performed using Kruskal–Wallis one‐way analysis of variance (anova), followed by Dunn's post‐hoc test. In order to determine the difference between the different autoantibody levels (negative ≤ low ≤ high), anova and post‐hoc tests using Tukey's procedure were performed. A two‐tailed P‐value of 0·05 or less was considered to be statistically significant.
The correlation coefficient (r s) of the correlation between the percentage of killer B cells and viral load was determined using Spearman's correlation test. In order to determine the difference between the different autoantibody levels (negative ≤ low ≤ high), anova and post‐hoc tests using Tukey's procedure were performed. A two‐tailed P‐value of 0·05 or less was considered to be statistically significant.
Results
Hepatitis C patients
All patients were studied after HCV infection was confirmed by detecting HCV RNA genomes, and by recording increased levels of antibodies to the hepatitis C virus (anti‐HCV). All patients had elevated liver enzymes to at least one of the following: alanine aminotransferase (ALT), aspartate aminotransferase (AST) and gamma glutamyltransferase (GGT). In all patients, serology was performed for the detection of HCV‐related autoantibodies. Positive RF was found in 16 of 41 patients (39%), but only in three of 45 healthy controls (7%). ANA positivity was found in 17 of 41 patients (41%), but only in four of 45 healthy controls (9%). For detailed results, see Table 1.
Table 1.
Clinical and laboratory characteristics of hepatitis C virus (HCV)‐infected patients and healthy individuals
| HCV patients n = 41 | Healthy individuals n = 45 | |
|---|---|---|
| Age range | 34–66 years | 29–65 years |
| Sex F/M | 15/26 | 21/24 |
| ALT (U/l) | 123 ± 95 | 35 ± 12 |
| HCV RNA titre (×105 copies/ml) | Mean: 14 ± 11 (range: 5–40) | 0 |
| RF | 16 (39%) | 3 (7%) |
| ANA | 17 (41%) | 4 (9%) |
| Anti‐cardiolipin | 3 (7%) | 0 |
F/M = female/male; RF = rheumatoid factor; ANA = anti‐nuclear antibodies; ALT = alanine transaminase.
Characterization of killer B cells
In order to define the phenotypical markers of killer B cells, purified B cells from 12 healthy controls were positively selected from PBMCs using CD22 microbeads (purity > 98%) and assessed for 48 h as either non‐stimulated or stimulated cells, as described above. In normal controls, the amount of CD19+CD5low cells was comparable to that of CD19+CD5hi cells. CD5low and CD5hi B cells were compared in terms of their expression of FasL, GranB, perforin and IL‐10. These were found to be expressed significantly more highly on CD5hi B cells. Specifically, the expression of FasL on CD19+CD5hi cells was significantly higher than that on CD19+CD5low cells [12·18 ± 2·77 mean fluorescence intensity (MFI) ± standard error of the mean (s.e.m.), median = 12·3 versus 4·14 ± 2·0 MFI ± s.e.m., median = 3·2, P < 0·0001, respectively]. (Fig. 1). The expression of IL‐10 and the death molecules GranB and perforin by CD5low and CD5hi B cells were also significantly higher on CD5hi B cells (2·37 ± 1·04 MFI ± s.e.m., median = 2, versus 5·41 ± 1·82 MFI ± s.e.m., median = 5·9, P = 0·0003; 2·07 ± 0·80 MFI ± s.e.m., median = 1·9 versus 11·59 ± 8·94 MFI ± s.e.m., median = 8·6, P < 0·0001; 1·82 ± 1·02 MFI ± s.e.m., median = 1·8 versus 4·15 ± 2·25 MFI ± s.e.m., median = 3·8, P = 0·0069, respectively). This expression was increased significantly when B cells were stimulated. Again, the above‐mentioned markers, namely FasL, IL‐10, GranB and perforin, were increased further on both stimulated CD5low and CD5hi when compared to that on naive B cells. This expression was significantly higher on CD5hi compared to CD5low B cells; see detailed data in Table 2.
Figure 1.
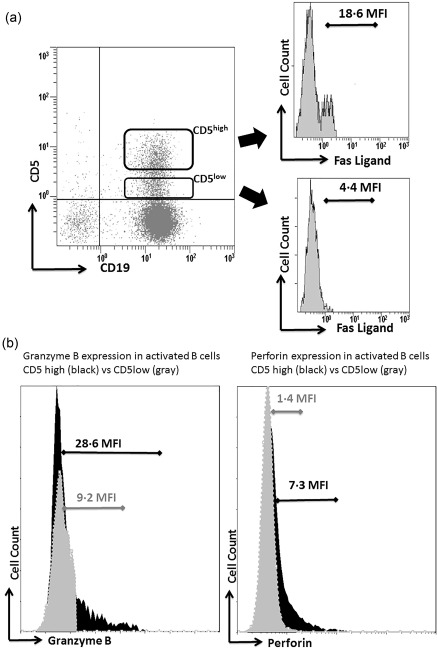
(a) Fas‐ligand (FasL) expression on CD5low and CD5hi B cells in normal individuals. A representative dot‐plot experiment presenting CD5low and CD5hi B cells in normal individuals. As can be seen, FasL expression is increased significantly on CD5hi B cells [18·6 mean fluorescence intensity (MFI)] when compared to that on CD5low (4·4 MFI) (on histogram). (b) Left panel: a representative fluorescence activated cell sorter (FACS) histogram showing increased expression of granzyme B in CD5high activated B cells (black) versus CD5low activated B cells (grey). Right panel: a representative FACS histogram showing increased expression of perforin in CD5high activated B cells (black) versus CD5low activated B cells (grey).
Table 2.
The expression of regulatory cytokines and death‐signal markers (MFI) in primary and activated CD5low vs. CD5hi B cells.
| Primary | Activated | |||||
|---|---|---|---|---|---|---|
| CD19+CD5low | CD19+CD5hi | P‐value | CD19+CD5low | CD19+CD5hi | P‐value | |
| FasL | 4·14 ± 2·0 | 12·18 ± 2·77 | P < 0·0001 | 11·58 ± 3·59 | 32·03 ± 8·65 | P < 0·0001 |
| Interleukin‐10 | 2·37 ± 1·04 | 5·41 ± 1·82 | P = 0·0003 | 18·36 ± 4·15 | 36·44 ± 5·45 | P < 0·0001 |
| Granzyme B | 2·07 ± 0·80 | 11·59 ± 8·94 | P < 0·0001 | 8·48 ± 3·77 | 27·68 ± 12·98 | P < 0·0001 |
| Perforin | 1·82 ± 1·02 | 4·15 ± 2·25 | P = 0·0069 | 1·33 ± 0·48 | 6·98 ± 2·34 | P < 0·0001 |
Killer B cells in healthy controls versus HCV patients
Aiming to establish the difference in the amount and level of Fas‐ligand expression of killer B cells in HCV patients compared to healthy controls, B cells were isolated from the peripheral blood of 41 patients suffering from chronic HCV infection and from 45 healthy controls and then sorted to CD5high/CD5low B cells (see Methods section). Using flow cytometry analysis, we could not detect any difference in FasL expression on the surface of CD19+CD5low B cells in HCV patients compared to healthy controls (6·91 ± 1·33 MFI ± s.e.m., median = 6·3 versus 4·62 ± 1·98 MFI ± s.e.m., median = 4·5 respectively, P = 0·053; data not shown). The expression of FasL on the surface of sorted CD19+CD5hi cells, however, was found to be significantly higher in HCV patients when compared to the healthy controls (28·06 ± 6·71 MFI ± s.e.m., median = 27·9 versus 10·87 ± 3·97 MFI ± s.e.m., median = 10·3, respectively, P < 0·0001); see Fig. 2a. Furthermore, killer B cells (CD19+CD5hiFasLhi), were found to be increased significantly in HCV patients when compared to the amount present in healthy controls [44 ± 15·95% mean ± standard deviation (s.d.), median = 41·7 versus 29·46 ± 12·58%, mean ± s.d., median = 26·4%, respectively, median = 41·7 versus 26·4, respectively, P = 0·0038]; see Fig. 2b.
Figure 2.
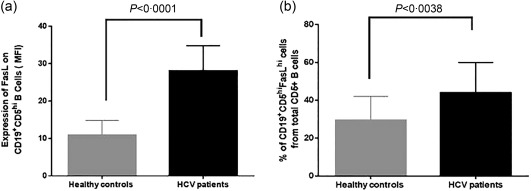
Characterization of killer B cells in hepatitis C (HC) patients versus healthy controls. Flow cytometry analysis of B cells from healthy controls (n = 41) and hepatitis C virus (HCV) patients (n = 45) showed that (a) Fas‐ligand (FasL) expression on CD19+CD5hi B is significantly higher in HCV patients compared to healthy controls [28·06 ± 6·71 versus 10·87 ± 3·97 mean fluorescence intensity (MFI) ± standard error of the mean (s.e.m.), respectively, P < 0·0001). (b) The percentage of killer B cells subpopulation is higher in HCV patients compared to healthy controls [44 ± 15·95 versus 29·46 ± 12·58%, mean ± standard deviation (s.d.), respectively, P = 0·0038].
The effect of killer B cells on autologous T cells
Killer B cells were assessed for their ability to induce increased CD4+ and CD8+ T cell apoptosis. To evaluate this, we co‐cultured sorted CD19+CD5hiFasLhi or CD19+CD5lowFasLlow cells with purified and activated CD4+ and CD8+ T cells, as described above in the Methods section. We analysed 17 experiments from both HCV patients and healthy controls and assessed the apoptosis of T cells using fluorescent antibodies to CD4/CD8+ T cells and annexin V and PI, as described above in the Methods section. In healthy controls, as can be seen in the representative Fig. 3a, early spontaneous apoptosis of activated CD4+ T cells was 1·5% and late apoptosis/necrosis was 7·4%. Induced early apoptosis in co‐cultured CD4+ T cells with CD19+CD5lowFasLlow B cells was 4·5%, while the late apoptosis/necrosis rate was 11·7%. When CD4+ T cells were co‐cultured with CD19+CD5hiFasLhi, however, the rate of early apoptosis rose to 17%, and the late apoptosis/necrosis rate was 20·7%. Figure 3b summarizes the apoptosis rate of co‐cultured CD4+ T cells with CD5low versus CD5hi in normal individuals. In HCV patients, Fig. 3c represents FACS results of the CD4+ T cell apoptosis rate following their co‐culture with CD5low versus CD5hi in HCV patients. Here as well, a significant increase of T cell apoptosis was noticed following the co‐culture of CD4+ T cells with CD19+CD5hiFasLhi.
Figure 3.
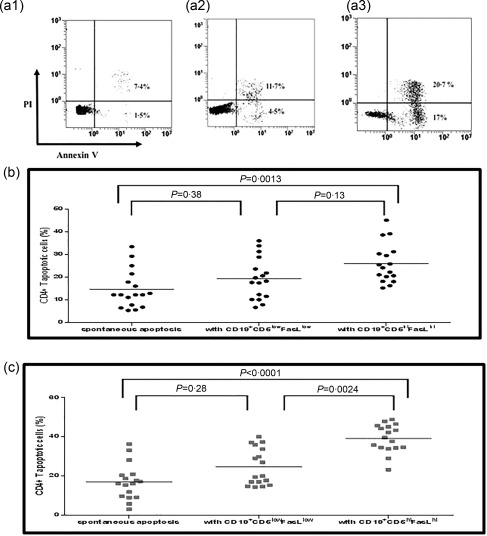
The effect of killer B cells on autologous CD4+ T cell apoptosis in healthy controls and hepatitis C virus (HCV) patients. (a) Representative fluorescence activated cell sorter (FACS) results of CD4+ T cells apoptosis: I. Spontaneous apoptosis II. Apoptosis induced by CD19+CD5low B cells. III. Apoptosis induced by CD19+CD5hiFas‐ligand (FasL)hi B cells. (b) Summary of 17 experiments, results of healthy controls [analysis of variance (anova) significance = 0·0019]: the spontaneous apoptosis of CD4+ T cells was 14·53 ± 8·36% mean ± standard deviation (s.d.), median = 12·2%. When CD4+ T cells were co‐cultured with CD19+CD5low the rate of apoptosis increased to 19·24 ± 9·13% mean ± s.d., median = 18·2%, P = 0·38. However, when CD4+ T cells were co‐cultured with CD19+ CD5hiFasLhi, the rate of apoptosis rose to 25·92 ± 8·65% mean ± s.d., median = 24.1%, P = 0·0013. (c) Summary of 17 experiments results of HCV patients (anova significance < 0·0001): the spontaneous apoptosis of CD4+ T cells was 16·9 ± 9·1% mean ± s.d., median = 16·1. When CD4+ T cells were co‐cultured with CD19+CD5low the rate of apoptosis increased to 24·66 ± 9·44% mean ± s.d., median = 19·9, P = 0·28. However, when CD4+ T cells were co‐cultured with CD19+CD5hiFasLhi, the rate of apoptosis rose to 39·17 ± 7·18% mean ± s.d., median = 39·6, P < 0·0001.
As can be seen in Fig. 4, CD19+CD5hiFasLhi B cells from HCV patients caused significantly more CD4+ T cell apoptosis compared to healthy controls (39·17 ± 7·18%, mean ± s.d., median = 39·6 versus 25·92 ± 8·65%, mean ± s.d., median = 24·1%, P < 0·0001, respectively). Spontaneous CD4+ T cell apoptosis and the rate of apoptosis when T cells were cultured with CD5lowFasLlow B cells were comparable in HCV patients and healthy individuals.
Figure 4.
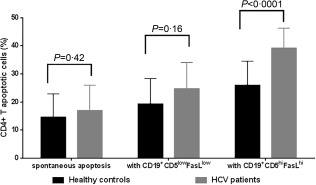
The effect of killer B cells on autologous CD4+ T cells, comparison between HCV patients and healthy controls. The results demonstrated that killer B cells induce more apoptosis of autologous CD4+ T cells compared to non‐killer B cells (CD19+CD5low) in both hepatitis C virus (HCV) patients and healthy controls. However, the rate of CD4+ T cell apoptosis induced by killer B cells was significantly greater in HCV patients than in healthy controls [39·17 ± 7·18% mean ± standard deviation (s.d.), median = 39·6 versus 25·92 ± 8·65% mean ± s.d., median = 24·1, P < 0·0001, respectively].
The same situation was found when we analysed the apoptotic rate in autologous CD8+ T cells: killer B cells induced more apoptosis of autologous CD8+ T cells in HCV patients compared to healthy controls. To determine this, we analysed seven experiments. In healthy controls, spontaneous apoptosis of CD8+ T was 8·6 ± 4·52%, mean ± s.d., median = 7·3%, but when cultured with CD19+CD5lowFasLlow B cells, the degree of apoptosis increased to 12·94 ± 5·82%, mean ± s.d., median = 11%, P = 0·68, and when CD8+ T cells were co‐cultured with CD19+CD5hiFasLhi the apoptosis rate rose to 21·07 ± 7·4%, mean ± s.d., median = 20%, P = 0·0067 (anova significance = 0·0087). In HCV patients, spontaneous apoptosis of CD8+ T cells was 16·91 ± 7·48%, mean ± s.d., median = 17·9%, but when co‐cultured with CD19+CD5lowFasLlow B cells, the apoptosis rate increased to 30·64 ± 14·93%, mean ± s.d., median = 28·7%, P = 0·43. When these cells were co‐cultured with CD19+CD5hiFasLhi, apoptosis rose to 54·67 ± 15·49%, mean ± s.d., median = 57·3%, P = 0·0017 (anova significance = 0·0005). Here as well, CD19+CD5hiFasLhi B cells induced more CD8+ T cell apoptosis in HCV patients compared to healthy controls (54·67 ± 15·49%, mean ± s.d., median = 57·3 versus 21·07 ± 7·4%, mean ± s.d., median = 20%, P = 0·0006, respectively).
The induction of T cell apoptosis by killer B cells is prevented by the anti‐ FasL neutralizing antibody
Aiming to prove the concept of apoptosis being dependent upon FasL expression on killer B cells, we used anti‐FasL neutralizing monoclonal antibody to block biological activity of FasL. As described above, we co‐cultured T cells with killer B cells, but this time added anti‐FasL neutralizing monoclonal mAbs or isotype control mAbs, as described in the Methods section. In healthy controls, the analysis of the results obtained from 10 experiments demonstrated that the spontaneous apoptosis of CD4+ T was comparable in the presence of the isotype control or in the presence of the anti‐FasL neutralizing antibody, P = 0·78. A similar degree of apoptosis was also recorded when CD4+ T cells were co‐cultured with CD19+CD5lowFasLlow B cells, either in the presence of the isotype control or anti‐FasL neutralizing antibody; P = 0·72. However, when CD4+ T cells were co‐cultured with CD19+CD5hiFasLhi, the apoptosis rate rose to 33·7 ± 9·76%, mean ± s.d., median = 32·9% in the presence of the isotype control, and decreased significantly to 21·17 ± 5·72%, mean ± s.d., median = 21·25% in the presence of anti‐FasL neutralizing antibody; P = 0·0039; see Fig. 5a. In HCV patients, the analysis of results obtained from seven experiments demonstrated that the same situation as was found when CD4+ T cells were cultured with the isotype control or with neutralizing monoclonal antibody in healthy controls. When CD4+ T cells were co‐cultured with CD19+CD5hiFasLhi, however, the apoptosis rate rose to 44·63 ± 10·13%, mean ± s.d., median = 45·9% in the presence of the isotype control, and decreased significantly to 22·33 ± 11·83%, mean ± s.d., median = 20·05% in the presence of the anti‐FasL neutralizing antibody; P = 0·0041; see Fig. 5b.
Figure 5.
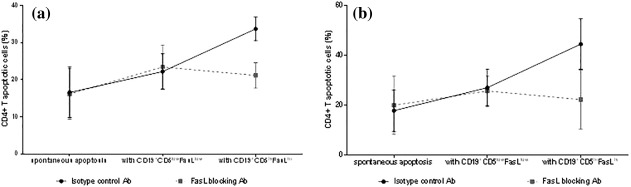
Killer B cells induce apoptosis of CD4+ T cells in a Fas‐ligand (FasL)‐mediated mechanism. The results showed that in the presence of isotype control antibody, killer B cells but not non‐killer B cells induce increase CD4+ T cells apoptosis. However, when anti‐FasL blocking antibody was added, the level of CD4+ T cell apoptosis induced by killer B cells was reduced significantly, with no significant change in the rate of apoptosis induced by non‐killer ones. These results are valid for both (a) healthy controls (n = 10) and (b) HCV patients (n = 7).
An increase in the number of killer B cells is associated with an increased viral load and autoimmunity
Aiming to prove the concept that an increased viral load and the presence of autoimmunity in HCV patients are associated strongly with increased killer B cells, the percentage of CD19+CD5hiFasLhi B cells was assessed in correlation with the viral load and with different autoantibodies in HCV patients. We were able to demonstrate that the viral load in HCV patients is correlated strongly and positively with the increase of killer B cells, r s = 0·55, P < 0·01; see Fig. 6a. One‐way anova was used to show how the presence of RF and ANA effects the percentage of killer B cells (overall anova significance: P = 0.0264 and P = 0·0424, respectively). Post‐hoc tests using Tukey's procedure show that the order of the levels are negative ≤ low ≤ high and that the difference between high and negative is significant (P = 0·0198, P = 0·0357, respectively). See Fig. 6b,c.
Figure 6.
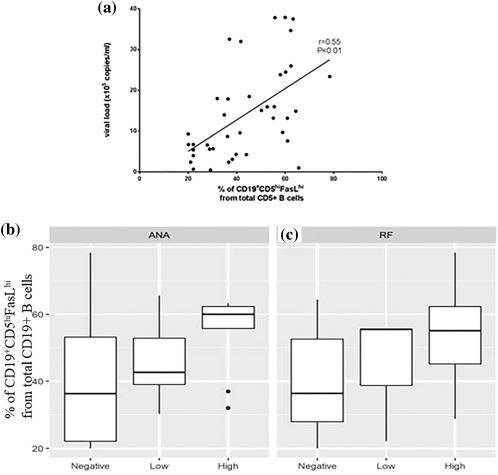
Killer B cells – association with increased viral load and autoantibodies. (a) Expansion of killer B cells is correlated highly with viral load (r s = 0·55, P < 0·01). Strong correlation was found between killer B cels percentage and viral load. Box‐plot analysis between the percentage of killer B cells and the level of (b) anti‐nuclear antibodies and (c) rheumatoid factor.
Discussion
Historically, most studies have focused upon understanding how HCV infection is cleared by efficient immune responses; namely, natural killer cells and cytotoxic T cells. These responses, however, sometimes fail, and when they do HCV infection becomes chronic, thereby damaging the liver by inducing hepatocyte apoptosis. Seeking to understand that HCV chronicity and its escape from efficient immune responses is frequent, researchers have looked for the mechanisms by which this evasion occurs. The exhaustion of HCV‐specific T cells and NK cells is usually balanced by an increase in immune regulatory function; namely, an increased level of anti‐inflammatory Tregs. Many researchers have linked the failure of HCV clearance and progression to chronic infection with significantly higher IL‐10 production and a relative absence of IFN‐γ and IL‐2 production. In contrast, deficient CD4+CD25+ cells increased HCV‐specific CD4+ and CD8+ T cell proliferation. The failure of CD4+CD25+ Treg suppressive function was shown to be TGF‐β‐dependent in a cell‐to‐cell manner. These data support the idea that high Treg cell frequency is correlated positively with HCV RNA titre 19. Both CD4+ and CD8+ effector T cells and CD4+CD25hi Tregs are HCV antigen‐specific. Subjects who cleared the virus had efficient HCV‐specific CD4+ T cell responses dominated by IFN‐γ‐producing cells and down‐regulation of IL‐10‐producing cells 20. In chronic HCV infection, IFN‐α‐based therapy gradually enhances CD4+ T cell responses, whereas IL‐10 and TGF‐β serum levels are decreased 21. In one of our early studies, we demonstrated that increased spontaneous CD4+ T cell apoptosis in peripheral blood may be an important mechanism by which HCV escapes efficient immune responses. In the last few years, the role of B cells in fighting HCV infection has been gaining more attention. Being the source of neutralizing anti‐HCV antibodies, they are believed to prevent envelope glycoproteins E1 and E2 from entering host cells and modifying the course of acute HCV infection. Chronic HCV infection, by itself, is not sufficient to activate mature memory B cells. The presence of RF in the serum of these patients with chronic HCV infection, however, was found to be associated with a more pronounced state of over‐representation of these memory B cells, and that by persisting in the blood, the latter probably play a role in the attempt to clean HCV from the host 22. So far, little is known about the role of Breg cells in the persistence of HCV. However, in chronic HBV, CD19+CD24hiCD38hi‐producing IL‐10 Breg cells were reported to increase when compared to healthy controls and were able to suppress the response of HBV‐specific CD8+ T cells 23. Another subset of Breg cells, CD19+CD5+CD1dhiIL10hi, was also found to be increased in both HCV and HBV patients, and to be correlated with HCV RNA in monocytes of patients' sera, as well as with poor virus elimination 24. Recently, studies have found that CD5hi Breg cells are important in producing IL‐10 when specifically activated 25. Similar to the above‐mentioned subsets of Breg cells, these were also reported to play a role in immune‐mediated disorders. Of great interest are CD5hi B cells highly expressing FasL, and are therefore called killer B cells. Their role in the spread of malignancy and the persistence of viral infections such as EBV and HIV has been emphasized in the literature 17, 26. Many studies have discussed the issue of HCV entry into immune cells, alluding to its importance for its spread and maintenance of infection. CD81, CLDN1 and other receptors were assessed for their ability to attach HCV. Their over‐expression on NK or T cells (although not fully proved) were suggested to be entry receptors for HCV. All this may explain why both FasR and FasL are over‐expressed on the T cells of chronic HCV patients, leading to their increased apoptosis and the escape of HCV from clearance. It is important to mention that increased T cell apoptosis is a result of antigen‐specific mediated killing, and therefore is not associated with any global T cell immune suppression. Our main aim in this study was to demonstrate the expansion of killer B cells in HCV‐infected patients and how they induce effector T cell apoptosis in a FasL‐dependent mechanism. It should be mentioned, however, that the well‐reported increase of FasR expression in virally infected T cells is an independent factor contributing to the increased effector T cell apoptosis. In addition, we demonstrate the important fact that increased FasL expression on CD5hi B cells and the expansion of this subpopulation of B cells in HCV patients is correlated positively with an increased viral load and with related HCV autoimmunity. Unfortunately, we were unable to show that this increase is correlated positively with HCV disease severity. This limitation in our study was due to the fact that the severity of HCV was evaluated using different assays in the two centres where HCV patients are followed. Future studies should evaluate this issue. In this respect, it is of importance to assess the level of killer B cell expansion vis‐à‐vis HCV severity and its possible down‐regulation following successful treatments and HCV elimination. The over‐expression of death molecules such as programmed cell death 1 (PD‐1), perforin, GranB and FasL on immune cells, mainly those infected with HCV, suggests that targeting them may become a successful therapeutic strategy in the battle against HCV. In this regard, HCV‐specific T cell function was assessed. Anti‐viral T cell responses were restored following their incubation with anti‐PD‐1 antibody. This suggests that PD‐1/programmed cell death ligand 1 (PD‐L1) blockade should be considered a beneficial therapeutic option in long‐lasting chronic HCV infection 27. Anti‐PD‐1 antibody was also given to chimpanzees with persistent HCV infection. Here, the control of HCV replication was associated with restoration of intra‐hepatic CD4+ and CD8+ T cell immunity against multiple HCV proteins 28. The beneficial effect of anti‐Fas ligand in animal models of chronic hepatitis was reported more than a decade ago. In an animal model of chronic hepatitis, the development of hepatocellular carcinogenesis, hepatocyte apoptosis, proliferation and liver inflammation were all prevented by the neutralization of FasL 29. Anti‐perforin neutralizing antibody was assessed for its ability in reducing myocardial damage in BALB/c mice (used as a model for viral myocarditis). The injection of anti‐perforin antibody into these mice reduced myocardial viral titres, the extent of cardiomyocyte apoptosis and down‐regulation of messenger ribonucleic acid and caspase‐3 expression 30. Our finding of FasL being over‐expressed on killer B cells strengthens the theory regarding their important role in the persistence of HCV and, hence, the therapeutic direction of combining anti‐FasL antibodies in the arsenal of anti‐viral therapy. Future studies should focus upon evaluating killer B cells following anti‐viral therapy and to determine whether this could restore the over‐activity of this B cell subset. It is possible that by adding anti‐FasL and anti‐PD‐1 antibodies to other anti‐viral therapies we could achieve better and, perhaps, longer‐lasting clearance of HCV. In conclusion, killer B cells should be taken into consideration when HCV persistence is studied, and their role in HCV chronicity and disease severity should be defined more clearly.
Disclosure
The authors declare that they have no conflicts of interest.
References
- 1. Ferri S, Muratori L, Lenzi M et al HCV and autoimmunity. Curr Pharm Des 2008; 14:1678–85. [DOI] [PubMed] [Google Scholar]
- 2. Kessel A, Toubi E. Chronic HCV‐related autoimmunity: a consequence of viral persistence and lymphotropism. Curr Med Chem 2007; 14:547–54 [DOI] [PubMed] [Google Scholar]
- 3. Sabo MC, Luca VC, Prentoe J et al Neutralizing monoclonal antibodies against hepatitis C virus E2 protein bind discontinuous epitopes and inhibit infection at a postattachment step. J Virol 2011; 85:7005–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Toubi E, Kessel A, Goldstein L et al Enhanced peripheral T‐cell apoptosis in chronic hepatitis C virus infection: association with liver disease severity. J Hepatol 2001; 35:774–80. [DOI] [PubMed] [Google Scholar]
- 5. Sarih M, Bouchrit N, Benslimane A. Different cytokine profiles of peripheral blood mononuclear cells from patients with persistent and self‐limited hepatitis C virus infection. Immunol Lett 2000; 74:117–20. [DOI] [PubMed] [Google Scholar]
- 6. Sugimoto K, Ikeda F, Stadanlick J, Nunes FA, Alter HJ, Chang KM. Suppression of HCV‐specific T cells without differential hierarchy demonstrated ex vivo in persistent HCV infection. Hepatology 2003; 38:1437–48. [DOI] [PubMed] [Google Scholar]
- 7. Perrella A, D'Antonio A, Sbreglia C et al CD4+/CD25+T cells suppress autologous CD4+/CD25‐lymphocytes and secrete granzyme B during chronic hepatitis C. Hepatology; 2006; 44:308A. [DOI] [PubMed] [Google Scholar]
- 8. Zha B, Wang L, Liu X et al Decrease in proportion of CD19+CD24hiCD27+ B cells and impairment of their suppressive function in Graves' disease. PLOS ONE 2012; 7:e49835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Blair PA, Nore LY, Flores‐Borja F et al CD19+CD24hiCD38hi B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic lupus erythematosus patients. Immunity 2010; 32:129–40. [DOI] [PubMed] [Google Scholar]
- 10. Kessel A, Haj T, Peri R et al Human CD19+CD25high B regulatory cells suppress proliferation of CD4+ T cells and enhance Foxp3 and CTLA‐4 expression in T‐regulatory cells. Autoimmun Rev 2012; 11:670–7. [DOI] [PubMed] [Google Scholar]
- 11. Lee JH, Noh J, Noh G, Choi WS, Lee SS. IL‐10 is predominantly produced by CD19 (low) CD5(+) regulatory B cell subpopulation: characterization of CD19 (high) and CD19(low) subpopulations of CD5(+) B cells. Yonsei Med J 2011; 52:851–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Miyagaki T, Fujimoto M, Sato S. Regulatory B cells in human inflammatory and autoimmune diseases: from mouse models to clinical research. Int Immunol 2015; 27:495–504. [DOI] [PubMed] [Google Scholar]
- 13. Yanaba K, Bouaziz JD, Haas KM, Poe JC, Fujimoto M, Tedder TF. A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell‐dependent inflammatory responses. Immunity 2008; 28:639–50. [DOI] [PubMed] [Google Scholar]
- 14. Bonardelle D, Benihoud K, Kiger N, Bobe P. B lymphocytes mediate Fas‐dependent cytotoxicity in MRL/LPR mice. J Leukoc Biol 2005; 78:1052–9. [DOI] [PubMed] [Google Scholar]
- 15. Lundy SK, Fox DA. Reduced Fas ligand‐expressing splenic CD5+ B lymphocytes in severe collagen‐induced arthritis. Arthritis Res Ther 2009; 11:R128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lundy SK, Boros DL. Fas ligand‐expressing B‐1a lymphocytes mediate CD4+‐T‐cell apoptosis during schistosomal infection: induction by interleukin 4 (IL‐4) and IL‐10. Infect Immun 2002; 70:812–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tanner JE, Alfieri C. Epstein‐Barr virus induces Fas (CD95) in T cells and Fas ligand in B cells leading to T‐cell apoptosis. Blood 1999; 94:3439–47. [PubMed] [Google Scholar]
- 18. Lundy SK. Killer B lymphocytes: the evidence and the potential. Inflamm Res 2009; 58:345–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Flynn JK, Dore GJ, Hellard M et al Early IL‐10 predominant responses are associated with progression to chronic hepatitis C virus infection in injecting drug users. J Viral Hepat 2011; 18:549–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Boettler T, Spangenberg HC, Neumann‐Haefelin C et al T cells with a CD4+CD25+ regulatory phenotype suppress in vitro proliferation of virus‐specific CD8+ T cells during chronic hepatitis C virus infection. J Virol 2005; 79:7860–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. AP, Chalupa A, Davidova O, Beran S et al Effect of antiviral treatment of chronic hepatitis C on the frequency of regulatory T cells, T‐cell activation, and serum levels of TGF‐beta. APMIS 2016; 124:711–8. [DOI] [PubMed] [Google Scholar]
- 22. Reyes‐Avilés E, Kostadinova L, Rusterholtz A, Cruz‐Lebrón A, Falck‐Ytter Y, Anthony DD. Presence of rheumatoid factor during chronic HCV infection is associated with expansion of mature activated memory B cells that are hypo‐responsive to B‐cell receptor stimulation and persist during the early stage of IFN free therapy. PLoS One 2015; 10:e0144629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Das A, Ellis G, Pallant C et al IL‐10‐producing regulatory B cells in the pathogenesis of chronic hepatitis B virus infection. J Immunol 2012; 189:3925–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang L, Qiu J, Yu L, Hu X, Zhao P, Jiang Y. Increased numbers of CD5+CD19+CD1dhighIL‐10+ Bregs, CD4+Foxp3+ Tregs, CD4+CXCR5+Foxp3+ follicular regulatory T (TFR) cells in CHB or CHC patients. J Transl Med 2014; 12:251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Duddy ME, Alter A, Bar‐Or A. Distinct profiles of human B cell effector cytokines: a role in immune regulation? J Immunol 2004; 172:3422–7. [DOI] [PubMed] [Google Scholar]
- 26. Samuelsson A, Sönnerborg A, Heuts N, Cöster J, Chiodi F. Progressive B cell apoptosis and expression of Fas ligand during human immunodeficiency virus type 1 infection. AIDS Res Hum Retroviruses 1997; 13:1031–8. [DOI] [PubMed] [Google Scholar]
- 27. Urbani S, Amadei B, Tola D et al Restoration of HCV‐specific T cell functions by PD‐1/PD‐L1 blockade in HCV infection: effect of viremia levels and antiviral treatment. J Hepatol 2008; 48:548–58. [DOI] [PubMed] [Google Scholar]
- 28. Fuller MJ, Callendret B, Zhu B et al Immunotherapy of chronic hepatitis C virus infection with antibodies against programmed cell death‐1 (PD‐1). Proc Natl Acad Sci USA 2013; 110:15001–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nakamoto Y, Kaneko S, Fan H et al Prevention of hepatocellular carcinoma development associated with chronic hepatitis by anti‐Fas ligand antibody therapy. J Exp Med 2002; 196:1105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chun‐yan G, Bo H, Hong C, Hong‐lei J, Xiu‐zhen H. Anti‐perforin neutralizing antibody reduces myocardial injury in viral myocarditis. Cardiol Young 2009; 19:601–7. [DOI] [PubMed] [Google Scholar]