

Summary
The proinflammatory cytokines interleukin (IL)‐17 and tumour necrosis factor (TNF)‐α are targets for treatment in many chronic inflammatory diseases. Here, we examined their role in liver inflammatory response compared to that of IL‐6. Human hepatoma cells (HepaRG, Huh7.5 and HepG2 cells) and primary human hepatocytes (PHH) were cultured with IL‐6, IL‐17 and/or TNF‐α. To determine the contribution of the IL‐6 pathway in the IL‐17/TNF‐α‐mediated effect, an anti‐IL‐6 receptor antibody was used. IL‐17 and TNF‐α increased in synergy IL‐6 secretion by HepaRG cells and PHH but not by Huh7.5 and HepG2 cells. This IL‐17/TNF‐α synergistic cooperation enhanced the levels of C‐reactive protein (CRP) and aspartate aminotransferase (ASAT) in HepaRG cell and PHH cultures through the induction of IL‐6. IL‐17/TNF‐α also up‐regulated IL‐8, monocyte chemoattractant protein (MCP)‐1 and chemokine (C‐C motif) ligand 20 (CCL20) chemokines in synergy through an IL‐6‐independent pathway. Interestingly, first exposure to IL‐17, but not to TNF‐α, was crucial for the initiation of the IL‐17/TNF‐α synergistic effect on IL‐6 and IL‐8 production. In HepaRG cells, IL‐17 enhanced IL‐6 mRNA stability resulting in increased IL‐6 protein levels. The IL‐17A/TNF‐α synergistic effect on IL‐6 and IL‐8 induction was mediated through the activation of extracellular signal‐regulated kinase (ERK)‐mitogen‐activated protein kinase, nuclear factor‐κB and/or protein kinase B (Akt)–phosphatidylinositol 3‐kinase signalling pathways. Therefore, the IL‐17/TNF‐α synergistic interaction mediates systemic inflammation and cell damage in hepatocytes mainly through IL‐6 for CRP and ASAT induction. Independently of IL‐6, the IL‐17A/TNF‐α combination may also induce immune cell recruitment by chemokine up‐regulation. IL‐17 and/or TNF‐α neutralization can be a promising therapeutic strategy to control both systemic inflammation and liver cell attraction.
Keywords: hepatocyte, inflammation, interleukin‐6, interleukin‐17, tumour necrosis factor‐α
Introduction
Interleukin (IL)‐6 is a systemic proinflammatory cytokine playing a pivotal role in the acute‐phase response to tissue injury, infection or inflammation 1, 2. This response is characterized by changes in the hepatic production of acute‐phase proteins such as increased C‐reactive protein (CRP) production. However, the acute‐phase protein response and the production of IL‐6 itself are also induced by a long list of cytokines, such as tumour necrosis factor‐α (TNF‐α), IL‐1β and IL‐17A, also known as IL‐17 3, 4.
Clinical inhibition of IL‐6 with an anti‐IL‐6 receptor (anti‐IL‐6R) antibody has shown beneficial effects on the joint manifestations of rheumatoid arthritis, with a massive decrease of CRP levels 5, 6, 7. However, IL‐6 also has anti‐inflammatory functions 8. Indeed, IL‐6‐deficient mice developed liver inflammation, steatosis and insulin resistance 9. Moreover, IL‐6 blockade has been associated with colon perforation 7, 10, transaminase elevation 11 and adverse lipid changes 12, 13, probably related to liver changes. Therefore, the IL‐6 effects on hepatocytes remain not well known and some of the liver changes in chronic inflammation may reflect the effects of other cytokines, independently or not of IL‐6.
Liver diseases are characterized by immune cell infiltrates following chemokine release 14. Neutrophils are key players in the initiation of the inflammatory response. Because IL‐17 is known to induce neutrophil‐attracting chemokines such as IL‐8, IL‐17 contributes to liver inflammation by inducing the local recruitment of neutrophils 15. As shown in various cells, including endothelial cells or skin and synovial fibroblasts, IL‐17 can cooperate with TNF‐α to induce in‐vitro IL‐8 and IL‐6 secretion in synergy 16, 17, 18, 19. These two cytokines are also involved in several liver disorders 15, 20, 21, 22. In the liver, IL‐17 was also able to activate hepatic stellate cells and CRP production by hepatocytes independently of IL‐6 3, 23.
The objective of this study was to clarify the effects of IL‐17 and TNF‐α on the induction of the inflammatory response in hepatocytes and to determine the contribution of IL‐6 in these effects. Because primary human hepatocytes (PHH) are from native liver, they are considered to be the gold standard approach to reflect the specific functionality and mediators of the human organ. Therefore, PHH and human hepatoma cell lines were used. IL‐17 and TNF‐α cooperated to increase CRP expression and aspartate aminotransferase (ASAT) level in hepatocyte cultures through the activation of the IL‐6 pathway. Independently of the IL‐6 pathway, IL‐17 and TNF‐α induced IL‐8, monocyte chemoattractant protein‐1 (MCP‐1) and chemokine (C‐C motif) ligand 20 (CCL20) expression and/or production synergistically. These differences may help understanding of the liver situation in chronic inflammation.
Materials and methods
Cell cultures
The human hepatoma Huh7.5, HepG2 and HepaRG cells were cultured as described previously 24, 25. Proliferative HepaRG cells were used after 15 days post‐plating and differentiated HepaRG cells were maintained in the same standard medium supplemented by dimethylsulphoxide (DMSO) 2% for 2 more weeks. PHH were isolated from surgical liver resections and cultured as reported 24. The samples were collected according to the local ethical committee and the Ministry of Research, which approved the study (reference number: AC‐2010‐1164).
Culture conditions
Hepatocytes were exposed to IL‐6 5 ng/ml (R&D Systems, Minneapolis, MN, USA) or IL‐17A 50 ng/ml (Dendritics, Lyon, France) and/or TNF‐α 1 ng/ml (R&D systems). To block the IL‐6, IL‐17 or TNF‐α pathways, tocilizumab (Roche, Welwyn, UK), anti‐IL‐17A (R&D Systems) and infliximab (MSD, Courbevoie, France) were used at 10 μg/ml. A monoclonal antibody against the BetV1 allergen (Dendritics) was used as a control antibody at the same concentration. Exposures to nuclear factor‐kappaB (NF‐κB) inhibitor pyrrolidine dithiocarbamate, phosphoinositide 3‐kinase (PI3K) inhibitor LY294002, protein kinase B (Akt) inhibitor A6730 (all from Sigma, St Louis, MO, USA) and mitogen‐activated protein kinase (MAPK) inhibitors SP6000125 [c‐Jun N‐terminal kinase (JNK) inhibitor], SB203580 (p38 inhibitor), U0125 [mitogen‐activated protein kinase kinase/extracellular signal‐regulated kinase (MEK/ERK) inhibitor] (all from Calbiochem, San Diego, CA, USA) at 1, 10, 20 and/or 100 μM were added 1 h prior to cytokine addition. Cells were treated for 12 and 24 h for mRNA expression, 24 h for cytokine production and 120 h for CRP and transaminase levels.
mRNA stability
HepaRG cells were treated with IL‐17 and/or TNF‐α for 12 h. Cells were then washed and incubated with 5 μg/ml actinomycin D (Orphan Europe, Puteaux, France) to inhibit further transcription. Total mRNA was extracted following 0, 1, 2 and 3 h incubation with actinomycin D. Results were presented as % mRNA remaining compared with the steady‐state level.
Quantitative real time PCR
Total RNA was purified using an RNeasy® Plus Mini kit (Quiagen, Hilden, Germany). cDNA was synthesized using the iScript™ kit (Bio‐Rad, Hercules, CA, USA). Polymerase chain reaction (PCR) amplification was performed using the CFX96TM real‐time system instrument (Bio‐Rad) with the iTaqTM universal SYBR® green supermix (Bio‐Rad) and the Qiagen QuantiTect® primers. Expression of the genes of interest was normalized to the expression of the glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) housekeeping gene.
Enzyme‐linked immunosorbent assays (ELISA)
Supernatant IL‐6 and IL‐8 concentrations were quantified with human ELISA kits, according to the manufacturer's instructions (R&D Systems).
Western blotting
Proliferative HepaRG cells were exposed to high concentrations of TNF‐α alone (10 ng/ml) or a combination of IL‐17 (50 ng/ml) and TNF‐α (1 ng/ml) for 30 min. Cells were lysed using the HaltTM protease and phosphatase inhibitor cocktail kit (Thermo Scientific, Rockford, IL, USA) and the protein concentration was determined by the bicinchoninic acid (BCA) protein assay kit (Thermo Scientific). Equal amounts of protein (50 μg) were loaded and separated by 15% sodium dodecyl sulphate (SDS)‐polyacrylamide gel and transferred on nitrocellulose membranes (Bio‐Rad). Membranes were blocked for 1 h at room temperature with 5% non‐fat milk or 5% bovine serum albumin (BSA). Blots were incubated overnight at 4°C with specific antibodies against total and phosphorylated ERK1/2, Akt and NF‐κB inhibitor α (IκBα) (Cell Signaling Technology, Leiden, the Netherlands). Blots were washed three times and incubated with a secondary antibody for 1 h at room temperature. Protein bands were detected with an enhanced chemiluminescence (ECL) detection kit (Bio‐Rad). Blots were scanned and analysed using the Gel DocTM XR+ Gel system (Bio‐Rad) and Image Lag 5.2 software. Cyclophilin B quantification was used as loading controls. Results are shown as the ratio of the total and phosphorylated forms.
Laboratory automated analyser
The CRP level was quantified by the automated analyser BN ProSpec® (Siemems, Erlangen, Germany) and transaminases by the Architect analyser (Abbot Diagnostics, Chicago, IL, USA).
Statistical analysis
Calculations were performed with GraphPad Prism version 5.01 software. Data are the mean of at least three independent experiments ± standard error of the mean (s.e.m.). Statistical differences were analysed using the Wilcoxon paired t‐test. P‐values less than 0·05 were considered significant.
Results
IL‐17 and TNF‐α combination increases IL‐6 expression and production synergistically
IL‐17 and TNF‐α have been shown previously to induce a massive IL‐6 production by various cell types in synergy 16, 17. Here, the IL‐17/TNF‐α effect on IL‐6 was studied in hepatic cell lines and PHH. In HepaRG cells and PHH, the IL‐17/TNF‐α combination exposure increased the IL‐6 secretion synergistically compared to IL‐17 or TNF‐α alone. However, the IL‐6 production by Huh 7.5 and HepG2 cell lines was not affected by IL‐17/TNF‐α exposure (Fig. 1a). Based on these results, the HepaRG cell line was used for the following experiments. The IL‐17/TNF‐α synergistic effect was also observed on IL‐6 mRNA in proliferative HepaRG cells, which was up‐regulated by up to 44‐fold (Fig. 1b).
Figure 1.
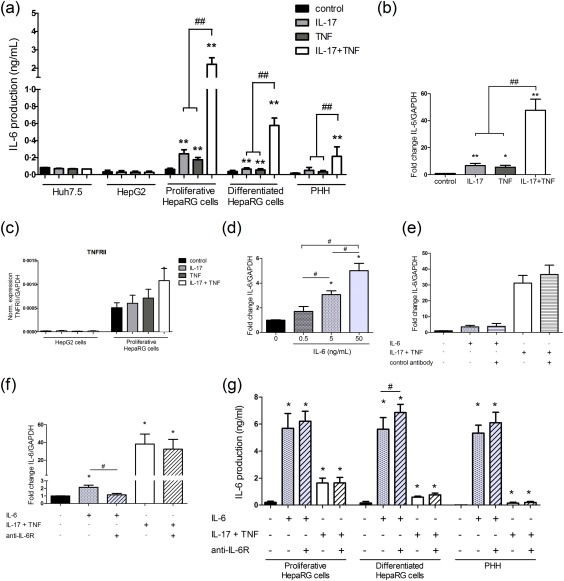
Interleukin (IL)‐17 and tumour necrosis factor (TNF)‐α combination increases IL‐6 expression and production synergistically. Hepatocytes were exposed to IL‐17 and/or TNF‐α or IL‐6 with/without the anti‐IL‐6R. (a,g) IL‐6 production by hepatocytes was quantified by enzyme‐linked immunosorbent assay (ELISA). (b,d,e,f) IL‐6 expression in proliferative human HepaRG cells at 12 h was expressed as fold changes compared to control. (c) TNF receptor II (TNF‐RII) expression at 12 h was normalized to that of glyceraldehyde 3‐phosphate dehydrogenase (GAPDH). The control antibody had no effect on IL‐6 mRNA and protein levels following IL‐6 or IL‐17/TNF‐α stimulation in HepaRG cells (e, data not shown). Data are the mean of three to 18 independent experiments ± standard error of the mean (s.e.m.); *P < 0·05 and **P < 0·01 versus control; #P < 0·05 and ##P < 0·01 versus other cytokine conditions.
Up‐regulation of IL‐17 and TNF‐α receptors may contribute to this IL‐17/TNF‐α synergistic effect. IL‐17 binds the heterodimer receptor complex, composed of the IL‐17 receptor subunit A (IL‐17RA) and the IL‐17RC, whereas TNF‐α acts through two independent receptors, the TNF receptor I (TNF‐RI) and the TNF‐RII. Only the TNF‐RII was up‐regulated in HepaRG cells following IL‐17/TNF‐α stimulation (Fig. 1c and data not shown). IL‐17R and TNF‐R expressions were also compared between the HepG2 and Huh7.5 cells, which did not produce IL‐6 after the IL‐17/TNF‐α stimulation, and the proliferative HepaRG cells, which released IL‐6 following the IL‐17/TNF‐α stimulation (Fig. 1a). The expression levels of TNF‐RI, IL‐17RA and IL‐17RC were similar between the HepG2 and the HepaRG cell lines (data not shown). However, the TNF‐RII mRNA levels were much lower in HepG2 (Fig. 1c) and Huh7.5 (data not shown) cell lines than in HepaRG cells. Therefore, TNF‐RII may have a crucial role in the IL‐6 production by IL‐17/TNF‐α.
To determine whether IL‐6 may regulate its own mRNA, proliferative HepaRG cells were treated with different IL‐6 concentrations and in the presence or not of an anti‐IL‐6R antibody. The specific effect of the anti‐IL‐6R antibody was verified with a control antibody which had no effect on the IL‐6 mRNA and protein levels induced by IL‐6 or IL‐17/TNF‐α (Fig. 1e and data not shown). Increasing IL‐6 concentrations up‐regulated IL‐6 expression dose‐dependently (Fig. 1d) and the IL‐6R blockade inhibited this effect in proliferative HepaRG cells (Fig 1f), indicating that IL‐6 regulated its own mRNA expression positively and directly. However, the anti‐IL‐6R did not reduce the IL‐6 up‐regulation‐induced IL‐17/TNF‐α significantly (Fig. 1f). The contribution of the IL‐6‐positive feedback loop in the IL‐6 induction by IL‐17/TNF‐α was therefore very weak compared to the IL‐17/TNF‐α direct effect on IL‐6. The IL‐6 supernatant levels in hepatocyte cultures stimulated with IL‐6 or IL‐17/TNF‐α were similar or slightly higher in the presence of the anti‐IL‐6R antibody (Fig. 1g). Because the anti‐IL‐6R blocks competitively the IL‐6 binding to its receptor, the IL‐6 free fraction level in supernatant increased in the presence of the anti‐IL‐6R antibody. This increase can be balanced by the IL‐6 positive‐feedback loop effect occurring in the absence of anti‐IL‐6R antibody.
Induction of the IL‐6‐dependent CRP and ASAT level following IL‐17 and/or TNF‐α stimulation
IL‐6 was shown to control CRP production 2, 4. Here, CRP expression was up‐regulated significantly by IL‐6 (19‐fold), IL‐17 (6‐fold) and the synergistic IL‐17/TNF‐α combination (37‐fold) versus control in proliferative HepaRG cells. The IL‐6 pathway blockade reduced strongly the CRP mRNA level induced by IL‐17 and/or TNF‐α (Fig. 2a,b). Moreover, CRP production by PHH from different donors following IL‐17/TNF‐α stimulation correlated strongly with the PHH ability to produce IL‐6 (Fig. 2c,d). Therefore, CRP up‐regulation by IL‐17 and/or TNF‐α was mainly IL‐6‐dependent.
Figure 2.
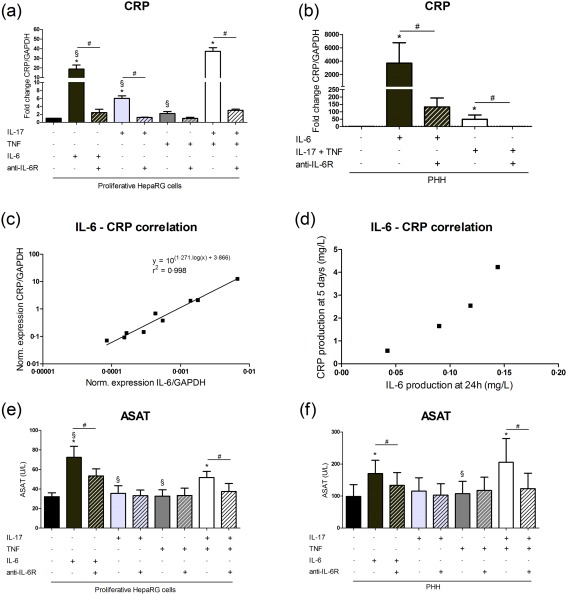
Induction of the interleukin (IL)‐6‐dependent C‐reactive protein (CRP) expression and aspartate aminotransferase (ASAT) activity level following IL‐17 and/or tumour necrosis factor (TNF)‐α stimulation. Hepatocytes were treated with IL‐17 and/or TNF‐α or IL‐6 with/without the anti‐IL‐6R. (a,b) CRP expression in the human HepaRG cell line and primary human hepatocytes (PHH) at 24 h was expressed as fold changes compared to control. The control antibody had no effect on CRP mRNA levels following IL‐6 or IL‐17/TNF‐α stimulation in HepaRG cells (data not shown). (c) Correlation between IL‐6 and CRP mRNA levels in PHH stimulated with IL‐17/TNF‐α for 24 h. mRNA levels were normalized to that of glyceraldehyde 3‐phosphate dehydrogenase (GAPDH). (d) Correlation between IL‐6 and CRP production in PHH cultures treated with IL‐17/TNF‐α. (e,f) ASAT supernatant levels were quantified at 120 h. Data are the mean of four to six independent experiments ± standard error of the mean (s.e.m.); *P < 0·05 versus control, §P < 0·05 versus IL‐17/TNF‐α condition, #P < 0·05 versus other cytokine conditions.
Liver inflammation may induce liver damage, as reflected by transaminase activity elevation in the clinic. In HepaRG cells and PHH cultures, ASAT levels increased after 5 days of IL‐6 and IL‐17/TNF‐α stimulation. These elevated ASAT levels returned to control level when an anti‐IL‐6R antibody was added (Fig. 2e,f). In these cultures, alanine aminotransferase (ALAT) level was lower than the detection limit (< 6 UI/l) (data not shown). The increase in ASAT level was therefore mainly IL‐6‐dependent.
IL‐17 and TNF‐α increase in synergy IL‐8 expression and production independently of the IL‐6 pathway
Cell recruitment is crucial for the inflammatory response. Liver biopsies in patients with active liver disease are characterized by the presence of inflammatory infiltrates 26. IL‐8 is associated with neutrophil recruitment involved in the acute‐phase inflammatory response, as in acute hepatitis 15. The IL‐17/TNF‐α co‐operation enhanced IL‐8 production by HepaRG cells and PHH versus IL‐17 alone, TNF‐α alone and control (Fig. 3a). IL‐8 mRNA expression in proliferative HepaRG cells was up‐regulated by IL‐17 alone (5‐fold) and TNF‐α alone (5‐fold), with a synergistic effect of both (24‐fold) (Fig. 3b). The IL‐17/TNF‐α synergistic interactions also increased IL‐8 mRNA levels by up to 14‐fold in PHH (Fig. 3b).
Figure 3.
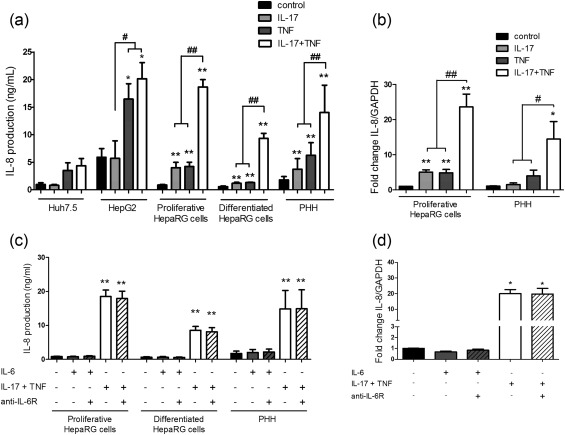
Interleukin (IL)‐17 and tumour necrosis factor (TNF)‐α combination increases IL‐8 expression and production synergistically independently of the IL‐6 pathway. Hepatocytes were exposed to IL‐17 and/or TNF‐α or IL‐6 with/without an anti‐IL‐6R antibody. (a,c). IL‐8 production was quantified by enzyme‐linked immunosorbent assay (ELISA). (b,d) IL‐8 expression at 12 h was expressed as fold change compared to control. The control antibody had no effect on the IL‐8 mRNA and protein levels following IL‐6 or IL‐17/TNF‐α stimulation in human HepaRG cells (data not shown). Data are the mean of three to 18 independent experiments ± standard error of the mean (s.e.m.); *P < 0·05 and **P < 0·01 versus control; #P < 0·05 and ##P < 0·01 versus other cytokine conditions.
However, the IL‐8 mRNA and protein level was unchanged after IL‐6 exposure. Moreover, the IL‐6 pathway inhibition had no effect on the induction of IL‐8 expression and production by IL‐17/TNF‐α (Fig. 3c,d). Therefore, IL‐8 induction by IL‐17 and TNF‐α was not mediated through IL‐6. The IL‐17 and TNF‐α combination may thus have a key role in the migration of neutrophils to the liver in the context of acute hepatitis.
IL‐17 and TNF‐α increase in synergy MCP‐1 and CCL20 expression mainly through an IL‐6‐independent pathway
MCP‐1 and CCL20 are two chemokines acting on mononuclear cells involved in the chronicity of the inflammatory response 27, 28, 29. MCP‐1 and CCL20 mRNA levels increased in the presence of IL‐17 or TNF‐α, with a clear synergistic effect of both cytokines (16‐ and 108‐fold, respectively, P < 0·01) versus control (Fig. 4a,b).
Figure 4.
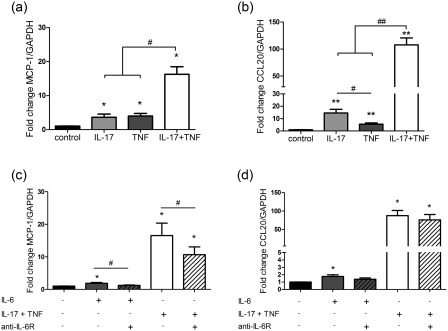
Interleukin (IL)‐17 and tumour necrosis factor (TNF)‐α combination increases monocyte chemoattractant protein (MCP)‐1 and chemokine (C‐C motif) ligand 20 (CCL20) chemokine expression synergistically, mainly through an IL‐6‐independent pathway. Proliferative human HepaRG cells were exposed to IL‐17 and/or TNF‐α or IL‐6 with/without an anti‐IL‐6R antibody. (a–d) MCP‐1 and CCL20 expression at 12 h in proliferative HepaRG cells was expressed as fold change compared to control. The control antibody had no effect on the MCP‐1 and CCL20 mRNA levels following IL‐6 or IL‐17/TNF‐α stimulation (data not shown). Data are the mean of seven to eight independent experiments ± standard error of the mean (s.e.m.); *P < 0·05 and **P < 0·01 versus control; #P < 0·05 and ##P < 0·01 versus other cytokine conditions.
To determine whether the IL‐17/TNF‐α effect on MCP‐1 and CCL20 expression was mediated through the IL‐6 pathway, IL‐6 and an anti‐IL‐6R antibody were used. By comparison with IL‐17/TNF‐α stimulation, IL‐6 had a very minimal effect on MCP‐1 and CCL20 mRNA levels. Similarly, the anti‐IL‐6R antibody did not abrogate the MCP‐1 and CCL20 expression induced by the IL‐17 and TNF‐α combination in proliferative HepaRG cells (Fig. 4c,d). Therefore, IL‐17 and TNF‐α can act independently of the IL‐6 signalling pathway on the induction of MCP‐1 and CCL20 expression. IL‐17/TNF‐α may thus have a key role in the migration of immune cells, including T helper type 17 (Th17) cells, to the liver in the context of chronic hepatitis.
IL‐17 initiates the IL‐17 and TNF‐α synergistic effect on IL‐6 and IL‐8 production
To understand more clearly the contribution of each cytokine on the IL‐17 and TNF‐α synergistic effect, antibodies blocking the IL‐17, TNF‐α and IL‐6 pathways were used. Anti‐IL‐17 or anti‐TNF‐α antibody exposure in conjunction of the IL‐17/TNF‐α combination reduced the synergistic effect on IL‐6 and IL‐8 production in HepaRG cells, whereas the IL‐6R inhibition had no significant effect on IL‐6 and IL‐8 supernatant levels (Fig. 5a,b).
Figure 5.
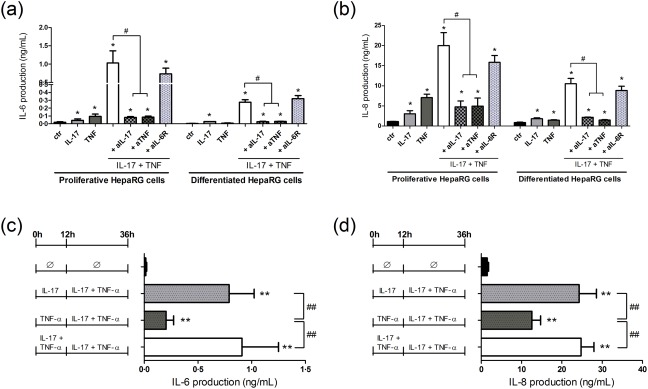
Interleukin (IL)‐17 initiates the IL‐17 and tumour necrosis factor (TNF)‐α synergistic effect on IL‐6 and IL‐8 production. (a,b) To evaluate the contribution of IL‐17, TNF‐α and IL‐6 pathways in the IL‐17/TNF‐α synergistic effect, human hepatoma cells (HepaRG) cells were treated with IL‐17 and/or TNF‐α with/without anti‐IL‐17A (aIL‐17), anti‐TNF‐α (aTNF) or anti‐IL‐6R (aIL‐6R) antibody. The control antibody had no effect on the IL‐6 and IL‐8 protein levels following IL‐17/TNF‐α stimulation (data not shown). (c,d) Proliferative HepaRG cells were pre‐exposed to IL‐17 and/or TNF‐α overnight (for 12 h) before IL‐17 and/or TNF‐α addition to have both cytokines in the culture medium (except for the control (Ø)). The IL‐6 and IL‐8 production was measured by enzyme‐linked immunosorbent assay (ELISA). Data are the mean of five to seven independent experiments ± standard error of the mean (s.e.m.); *P < 0·05 and **P < 0·01 versus control; #P < 0·05 and ##P < 0·01 versus other cytokine conditions.
We next investigated whether first exposure to IL‐17, TNF‐α or both initiated the IL‐17 and TNF‐α synergistic effects on IL‐6 and IL‐8 release. Proliferative HepaRG cells were pre‐exposed overnight to IL‐17 and/or TNF‐α and then IL‐17 and/or TNF‐α were added to both cytokines in the culture medium. Pre‐incubation first with IL‐17 followed by the addition of TNF‐α induced two‐fold higher IL‐6 and IL‐8 production at 24 h than pre‐incubation with TNF‐α, then with IL‐17 (Fig. 5c,d). First exposure to IL‐17, but not to TNF‐α, was thus crucial for initiation of the IL‐17/TNF‐α synergistic effect.
IL‐17 enhanced the stability of IL‐6 mRNA
Post‐transcriptional regulations could contribute to the IL‐17 and TNF‐α synergistic effect. To determine the IL‐17 and TNF‐α effect on mRNA stabilization, HepaRG cells were treated with IL‐17 and/or TNF‐α for 12 h and then transcription was inhibited. TNF‐α‐induced IL‐6 transcripts had a half‐life of 46 min, whereas IL‐17 and the IL‐17/TNF‐α combination increased the half‐life up to 124 and 82 min, respectively (Fig. 6). IL‐17 may thus increase IL‐6 mRNA stabilization in hepatocytes.
Figure 6.
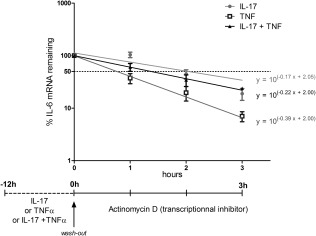
Interleukin (IL)‐17 enhances the stability of IL‐6 mRNA. Proliferative human HepaRG cells were incubated with IL‐17 (filled circles), tumour necrosis factor (TNF)‐α (empty squares) or the IL‐17/TNF‐α combination (filled triangles) for 12 h. Actinomycin D was added to inhibit further transcription. The IL‐6 and IL‐8 expression during the next 3 h were quantified. Results are presented as % of mRNA remaining over time compared with the steady‐state level (at 0 h). Data are the mean of four independent experiments ± standard error of the mean (s.e.m.).
IL‐17 and TNF‐α increase IL‐6 and IL‐8 production in synergy via the activation of ERK and/or PI3K/Akt signalling pathways and/or NF‐κB transcription factor
The effect of the TNF‐α/IL‐17 combination on the downstream signalling pathways was then investigated, focusing on the MAPKs (JNK, p38, ERK), NF‐κB and Akt/PI3K pathways 22, 30, 31. Results are shown as the ratio of the total and phosphorylated forms (Fig. 7a). In HepaRG cells, the IL‐17/TNF‐α association induced phosphorylation of IκBα (leading to NF‐κB release and activation) and ERK. The IL‐17/TNF‐α effect of Akt phosphorylation was weaker (Fig. 7a). Proliferative HepaRG cells were therefore treated with chemical inhibitors of these pathways. Quantification of transaminase secretion and cell viability were monitored to select the concentrations that did not induce cell death and other cytotoxic effects (data not shown).
Figure 7.
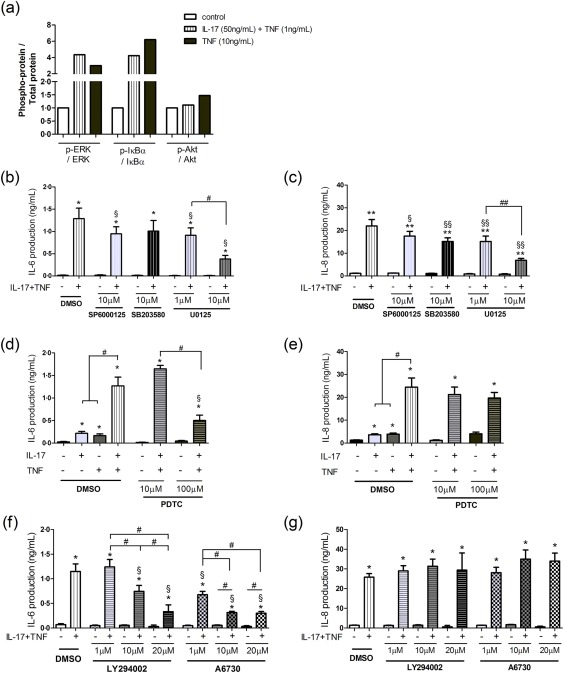
Interleukin (IL)‐17 and tumour necrosis factor (TNF)‐α combination increases IL‐6 and IL‐8 production synergistically through the activation of extracellular signal‐regulated kinase (ERK), mitogen‐activated protein kinase (MAPK) and/or phosphatidylinositide 3‐kinase/protein kinase B (PI3K/Akt) signalling pathways and nuclear factor kappa B (NF‐κB) transcription factor. (a) Proliferative human HepaRG cells were incubated with IL‐17 and/or TNF‐α for 30min. The total and phosphorylated forms of ERK, NF‐κB inhibitor α (IκBα) and Akt were quantified by Western blotting and their densitometry values were normalized to the cyclophilin content. Data are presented as the fold induction change of the ratio of phosphorylated to total protein in control samples. One representative experiment is shown. (b–g) Proliferative HepaRG cells were pre‐exposed to MAPKs [Janus kinase (JNK), p38, ERK], NF‐κB, Akt and PI3K pathway inhibitors for 1 h followed by the IL‐17 and/or TNF‐α addition. IL‐6 and IL‐8 production at 24 h was measured by enzyme‐linked immunosorbent assay (ELISA). SP6000125: JNK inhibitor, SB203580: p38 MAPK inhibitor, U0125: MEK/ERK inhibitor; pyrrolidine dithiocarbamate (PDTC): NF‐κB inhibitor; LY294002: PI3K inhibitor and A6730: Akt inhibitor. Data are the mean of at least four to eight independent experiments ± standard error of the mean (s.e.m.); *P < 0·05, **P < 0·01 versus control in dimethylsulphoxide (DMSO) condition, §P < 0·05 and §§P < 0·01 versus IL‐17/TNF‐α condition in DMSO medium; #P < 0·05 and ##P < 0·01 versus other inhibitor conditions.
The IL‐17/TNF‐α synergistic effect on IL‐6 and IL‐8 production was reduced slightly using JNK or p38 MAPK inhibitors. In contrast, ERK inhibition decreased strongly the production of IL‐6 (29 and 70% of inhibition for 1 and 10 μM, respectively) and IL‐8 (31 and 69% of inhibition for 1 and 10 μM, respectively) in a dose‐dependent manner induced by the IL‐17/TNF‐α combination (Fig. 7b,c). IL‐6 production induced by the IL‐17/TNF‐α combination was also inhibited in the presence of the NF‐κB inhibitor (60% of inhibition), the Akt inhibitor (more than 70% of inhibition for 10 and 20 μM) and the PI3K inhibitor (40 and 65% of inhibition for 1 and 10 μM, respectively), whereas IL‐8 production was not impacted significantly (Fig. 7d–g). Activation of ERK, NF‐κB and/or PI3K/Akt signalling pathways was therefore involved in the IL‐17/TNF‐α synergistic effect on IL‐6 and/or IL‐8 production.
Discussion
This study shows how the IL‐17/TNF‐α synergistic interactions mediate a hepatic inflammatory response mainly through IL‐6 for CRP and ASAT induction, and independently of IL‐6 for IL‐8, MCP‐1 and CCL20 chemokine up‐regulation.
IL‐6 is a systemic inflammatory mediator, which plays a key role in triggering the acute‐phase response to injury or inflammation. IL‐17 was shown previously to induce IL‐6 production by the human hepatoma Huh7, HepG2 and Hep3B cell lines 3, 32, 33. Here, IL‐17 and TNF‐α increased the production of IL‐6 in HepaRG cells and PHH in synergy. Because IL‐6 promotes the generation and differentiation of Th17 cells 28, the main IL‐17‐producing cells, the increase of IL‐6 production by the IL‐17/TNF‐α synergistic effect and IL‐6 autoinduction could exacerbate the inflammatory IL‐6/Th17/IL‐17 amplification. Interestingly, IL‐6 was also able to up‐regulate directly its own expression in vitro leading to a positive feedback loop of IL‐6. However, the contribution of this positive autoregulation of IL‐6 in the IL‐6 induction following the IL‐17/TNF‐α synergistic interaction was very weak at 12 h (Fig. 1g) and 24 h (data not shown) compared to the direct IL‐17/TNF‐α effect. Because an anti‐IL‐6R antibody blocks the IL‐6 binding to its receptor competitively, a higher IL‐6 protein level was expected in supernatants of hepatic cultures exposed to the anti‐IL‐6R antibody. For this reason, the IL‐6‐positive feedback loop after IL‐6 treatment was observed only on IL‐6 mRNA levels and not on IL‐6 protein levels in supernatants.
High CRP levels are associated with an increased risk of cardiovascular events 34. In‐vitro and in‐vivo studies have demonstrated the potent role of IL‐6 on CRP production 3, 7. Here, IL‐17 and/or TNF‐α enhanced CRP expression in HepaRG cells, showing a synergistic effect with the combination. The IL‐6 pathway blockade inhibited CRP induction strongly by IL‐17 and/or TNF‐α. In PHH treated with the IL‐17/TNF‐α combination, CRP levels correlated perfectly with those of IL‐6. Moreover, CRP mRNA level was lower at 12 than 24 h (data not shown), indicating that IL‐17 and TNF‐α act first on the induction of IL‐6 production, which leads in turn to CRP up‐regulation. Therefore, the induction of CRP appears mainly IL‐6‐dependent. However, one study has demonstrated that IL‐17 can stimulate CRP expression independently of IL‐6 in Hep3B cells 3, suggesting that a possible minor pathway independent of IL‐6 could exist.
In chronic inflammatory diseases, liver changes are common and may lead to transaminase elevation 35. The IL‐17/TNF‐α combination increased the ASAT activity level through the IL‐6 pathway. These results appear consistent with our in‐vivo studies, which showed that TNF‐α neutralization decreased serum levels of transaminase in mice and patients with autoimmune hepatitis 35, 36. Moreover, IL‐17 deficiency or IL‐17 neutralization reduced transaminase levels in various mouse models of liver injury 37, 38. Therefore, controlling IL‐17 and TNF‐α levels and functions may be protective and reduce liver damage.
Hepatic infiltration of neutrophils is an early response to systemic inflammation crucial to initiate liver injury 39, 40. The link between IL‐17, neutrophil recruitment and hepatic necrosis was demonstrated in several mouse models 37, 41. IL‐8 has a key role in the neutrophil mobilization and activation 15. IL‐17 may stimulate IL‐8 production through IL‐6 up‐regulation in Huh7 cells 33. In our study, IL‐8 production was increased by the IL‐17/TNF‐α synergistic interaction independently of the IL‐6 pathway, as the exposure to an anti‐IL‐6R antibody had no effect (Fig. 3c,d). IL‐6 could nevertheless act in vivo on the IL‐8 level by promoting the Th17–IL‐17 cells axis leading to an increase of IL‐17 production and, in turn, of IL‐8.
Chronic hepatitis is characterized by the liver infiltration by various immune cells attracted by chemokines, such as CCL20 and MCP‐1. Here, IL‐17 and TNF‐α were able to up‐regulate CCL20 and MCP‐1 expression synergistically through an IL‐6‐independent pathway. In HepG2 culture and an autoimmune hepatitis mouse model, TNF‐α induced CCL20 expression and this up‐regulation was associated with the progression of fatal inflammation 27, 36. MCP‐1 was described as a central co‐ordinator of hepatocyte‐mediated inflammation 42, 43. MCP‐1 expression was up‐regulated in primary hepatocytes by activation of the IL‐6‐mediated signalling cascade 44. In turn, MCP‐1 induced IL‐6 production in mouse hepatocytes, suggesting a possible positive feedback loop between IL‐6 and MCP‐1 42. Taken together, CCL20 and MCP‐1 induction by IL‐17/TNF‐α can increase the accumulation of a Th17‐driven response and lead to a chronic inflammatory state.
Although the IL‐17/TNF‐α synergistic effect has been well described in several cell types 16, 17, 18, 19, 45, 46, the mechanism of this effect on IL‐6 and IL‐8 production has not yet been studied in hepatocytes. Various mechanisms may act at several levels: at a receptor level, at a post‐transcriptional level and at a promoter level. In synoviocytes, the TNF receptor II (TNF‐RII) contributed to the IL‐17 and TNF‐α synergistic effect on CCL20 production, and IL‐17 treatment alone up‐regulated its expression 47. Here, in HepaRG cells, stimulation with the IL‐17/TNF‐α combination but not with IL‐17 alone increased TNF‐RII expression. Regulation of the TNF‐RII expression may be different between synoviocytes and HepaRG cells. However, the TNF‐RII mRNA level was much lower in HepG2 and Huh7.5 cells than in HepaRG cells (data not shown). This could explain the lack of response of the HepG2 and Huh7.5 cells to the IL‐17/TNF‐α synergistic effect on IL‐6 and IL‐8 production.
Post‐transcriptional regulation is important to control cellular transcript abundance and, in turn, the levels of the secreted proteins. Several studies have reported that the IL‐17 and TNF‐α co‐operation modulates mRNA stability 45, 46, 48, 49, 50. In HepaRG cells, IL‐17 could enhance IL‐6 mRNA stability (Fig. 6). As the 3'‐untranslated region of the IL‐6 mRNA contains adenylate and uridylate (AU)‐rich elements, IL‐17 may promote the binding of stabilizing AU‐binding proteins over that of destabilizing AU‐binding proteins, prolonging IL‐6 mRNA half‐life 51. Activation of the ERK MAPK pathway may contribute to this IL‐6 mRNA stabilization 52.
To investigate the potential signalling pathways involved in IL‐17 and TNF‐α synergistic stimulation on hepatic IL‐6 and IL‐8 production, several chemical pathway inhibitors were used. MAPK, PI3K and its downstream mediator Akt were involved in the IL‐6 signalling, and also in the IL‐17‐induced production of IL‐6, IL‐8 and MCP‐1 in several cell types, including human hepatocellular carcinoma cell lines 32, 33, 53, 54, 55. In HepaRG cells, activation of the ERK MAPK pathway in the IL‐17–TNF‐α interaction appears crucial, as the ERK pathway inhibition reduced both IL‐6 and IL‐8 production. The PI3K/Akt signalling pathway contributes to the synergistic effect of IL‐17/TNF‐α on the induction of IL‐6 but not of IL‐8. Because the MAPK and PI3K/Akt pathways can be activated by IL‐6, their contribution on the IL‐6 production can be related to the positive autoregulation of IL‐6 (Fig. 1c). However, the anti‐IL‐6R antibody failed to reduce IL‐6 expression induced by IL‐17/TNF‐α (Fig. 1f) in HepaRG cells. Therefore, the IL‐17/TNF‐α combination could activate the ERK MAPK and PI3K/Akt pathways directly to induce IL‐6 and/or IL‐8 production.
Transcription factor‐binding sites for NF‐κB and C/EBP (CCAAT/enhancer‐binding protein) in the IL‐6 promoter were both involved in the IL‐17 and TNF‐α synergistic effect on IL‐6 in an osteoblastic cell line 56. Part of the IL‐17/TNF‐α synergistic effect on IL‐6 may occur at the gene transcription level through the up‐regulation of C/EBPδ by IL‐17/TNF‐α and the increase of C/EBPδ recruitment to the promoter by TNF‐α 31, 56, 57. In primary murine hepatocytes, NF‐κB contributed to the activation of many IL‐17 target genes related to inflammation 58. Here, in HepaRG cells, the NF‐κB pathway appears to be involved in IL‐6 release induced by the IL‐17/TNF‐α combination (Fig. 7d). Both IL‐17 and TNF‐α signalling pathways promote NF‐κB activation. However, the IL‐17/TNF‐α combination may induce a further increase of NF‐κB activation through IκBζ, which acts as a NF‐κB co‐activator 57, 59. In primary murine hepatocytes, IL‐17 and TNF‐α cooperate to enhance the IκBζ mRNA expression in synergy 58.
In conclusion, targeting IL‐17 and/or TNF‐α could be a promising therapeutic strategy to control systemic inflammation, as seen with IL‐6 inhibition, but also the local cell recruitment and associated liver cell injury. These results are summarized in Fig. 8. Furthermore, control of the CRP level could be critical to reduce the cardiovascular risks that represent a major cause of death in patients with chronic inflammatory diseases.
Figure 8.
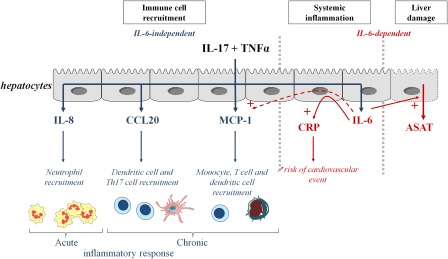
Summary of the effects of interleukin (IL)‐17 and tumour necrosis factor (TNF)‐α on hepatocytes. IL‐17 and TNF‐α have synergistic interactions on cytokine production by hepatocytes, with IL‐17 acting first to increase the effect of TNF. IL‐17 and TNF‐α induce C‐reactive protein (CRP) through the induction of IL‐6. IL‐17 and TNF‐α induce chemokine production independently of IL‐6.
Disclosure
None.
Author contributions
A. B. conducted the experiments and writing the manuscript; N. T., A. C. and B. B. developed the assays and experiments; and P. M. was responsible for the concept and writing the manuscript.
Acknowledgements
The authors thank Marie‐Nathalie Sarda and Annie Varennes for their technical support. This work has been supported by the OPeRa IHU program and the Institut Universitaire de France. P.M. is a senior member of the Institut Universitaire de France. A.B. is supported by the Ministry of Education and Research.
References
- 1. Zhou Z, Xu MJ, Gao B. Hepatocytes: a key cell type for innate immunity. Cell Mol Immunol 2016; 13:301–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yoshizaki K. Pathogenic role of IL‐6 combined with TNF‐alpha or IL‐1 in the induction of acute phase proteins SAA and CRP in chronic inflammatory diseases. Adv Exp Med Biol 2011; 691:141–50. [DOI] [PubMed] [Google Scholar]
- 3. Patel DN, King CA, Bailey SR et al Interleukin‐17 stimulates C‐reactive protein expression in hepatocytes and smooth muscle cells via p38 MAPK and ERK1/2‐dependent NF‐kappaB and C/EBPbeta activation. J Biol Chem 2007; 282:27229–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kramer F, Torzewski J, Kamenz J et al Interleukin‐1beta stimulates acute phase response and C‐reactive protein synthesis by inducing an NFkappaB‐ and C/EBPbeta‐dependent autocrine interleukin‐6 loop. Mol Immunol 2008; 45:2678–89. [DOI] [PubMed] [Google Scholar]
- 5. Choy E, Ganeshalingam K, Semb AG et al Cardiovascular risk in rheumatoid arthritis: recent advances in the understanding of the pivotal role of inflammation, risk predictors and the impact of treatment. Rheumatology (Oxf) 2014; 53:2143–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nishimoto N, Miyasaka N, Yamamoto K et al Long‐term safety and efficacy of tocilizumab, an anti‐IL‐6 receptor monoclonal antibody, in monotherapy, in patients with rheumatoid arthritis (the STREAM study): evidence of safety and efficacy in a 5‐year extension study. Ann Rheum Dis 2009; 68:1580–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Calabrese LH, Rose‐John S. IL‐6 biology: implications for clinical targeting in rheumatic disease. Nat Rev Rheumatol 2014; 10:720–7. [DOI] [PubMed] [Google Scholar]
- 8. Arango Duque G, Descoteaux A. Macrophage cytokines: involvement in immunity and infectious diseases. Front Immunol 2014; 5:491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Matthews VB, Allen TL, Risis S et al Interleukin‐6‐deficient mice develop hepatic inflammation and systemic insulin resistance. Diabetologia 2010; 53:2431–41. [DOI] [PubMed] [Google Scholar]
- 10. Taniguchi K, Wu LW, Grivennikov SI et al A gp130‐Src‐YAP module links inflammation to epithelial regeneration. Nature 2015; 519:57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Genovese MC, Kremer JM, van Vollenhoven RF et al Transaminase levels and hepatic events during tocilizumab treatment: pooled analysis of long‐term clinical trial safety data in rheumatoid arthritis. Arthritis Rheumatol 2017; 69:1751–61. [DOI] [PubMed] [Google Scholar]
- 12. Strang AC, Bisoendial RJ, Kootte RS et al Pro‐atherogenic lipid changes and decreased hepatic LDL receptor expression by tocilizumab in rheumatoid arthritis. Atherosclerosis 2013; 229:174–81. [DOI] [PubMed] [Google Scholar]
- 13. McInnes IB, Sieper J, Braun J et al Efficacy and safety of secukinumab, a fully human anti‐interleukin‐17A monoclonal antibody, in patients with moderate‐to‐severe psoriatic arthritis: a 24‐week, randomised, double‐blind, placebo‐controlled, phase II proof‐of‐concept trial. Ann Rheum Dis 2014; 73:349–56. [DOI] [PubMed] [Google Scholar]
- 14. Zimmermann HW, Tacke F. Modification of chemokine pathways and immune cell infiltration as a novel therapeutic approach in liver inflammation and fibrosis. Inflamm Allergy Drug Targets 2011; 10:509–36. [DOI] [PubMed] [Google Scholar]
- 15. Lemmers A, Moreno C, Gustot T et al The interleukin‐17 pathway is involved in human alcoholic liver disease. Hepatology 2009; 49:646–57. [DOI] [PubMed] [Google Scholar]
- 16. Hot A, Lenief V, Miossec P. Combination of IL‐17 and TNFalpha induces a pro‐inflammatory, pro‐coagulant and pro‐thrombotic phenotype in human endothelial cells. Ann Rheum Dis 2012; 71:768–76. [DOI] [PubMed] [Google Scholar]
- 17. Katz Y, Nadiv O, Beer Y. Interleukin‐17 enhances tumor necrosis factor alpha‐induced synthesis of interleukins 1,6, and 8 in skin and synovial fibroblasts: a possible role as a ‘fine‐tuning cytokine’ in inflammation processes. Arthritis Rheum 2001; 44:2176–84. [DOI] [PubMed] [Google Scholar]
- 18. Osta B, Roux JP, Lavocat F et al Differential effects of IL‐17A and TNF‐alpha on osteoblastic differentiation of isolated synoviocytes and on bone explants from arthritis patients. Front Immunol 2015; 6:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chiricozzi A, Guttman‐Yassky E, Suarez‐Farinas M et al Integrative responses to IL‐17 and TNF‐alpha in human keratinocytes account for key inflammatory pathogenic circuits in psoriasis. J Invest Dermatol 2011; 131:677–87. [DOI] [PubMed] [Google Scholar]
- 20. Hammerich L, Heymann F, Tacke F. Role of IL‐17 and Th17 cells in liver diseases. Clin Dev Immunol 2011; 2011:345803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang JY, Zhang Z, Lin F et al Interleukin‐17‐producing CD4(+) T cells increase with severity of liver damage in patients with chronic hepatitis B. Hepatology 2010; 51:81–91. [DOI] [PubMed] [Google Scholar]
- 22. Schwabe RF, Brenner DA. Mechanisms of Liver Injury. I. TNF‐alpha‐induced liver injury: role of IKK, JNK, and ROS pathways. Am J Physiol Gastrointest Liver Physiol 2006; 290:G583–9. [DOI] [PubMed] [Google Scholar]
- 23. Meng F, Wang K, Aoyama T et al Interleukin‐17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology 2012; 143:765–76.e1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grigorov B, Reungoat E, Gentil Dit Maurin A et al Hepatitis C virus infection propagates through interactions between Syndecan‐1 and CD81 and impacts the hepatocyte glycocalyx. Cell Microbiol 2017; 19. doi: 10.1111/cmi.12711. [DOI] [PubMed] [Google Scholar]
- 25. Lucifora J, Durantel D, Testoni B et al Control of hepatitis B virus replication by innate response of HepaRG cells. Hepatology 2010; 51:63–72. [DOI] [PubMed] [Google Scholar]
- 26. Quintin E, Scoazec JY, Marotte H et al Rare incidence of methotrexate‐specific lesions in liver biopsy of patients with arthritis and elevated liver enzymes. Arthritis Res Ther 2010; 12:R143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Miao H, Zhang Y, Lu Z et al FOXO1 increases CCL20 to promote NF‐kappaB‐dependent lymphocyte chemotaxis. Mol Endocrinol 2012; 26:423–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Miossec P, Korn T, Kuchroo VK. Interleukin‐17 and type 17 helper T cells. N Engl J Med 2009; 361:888–98. [DOI] [PubMed] [Google Scholar]
- 29. Melgarejo E, Medina MA, Sanchez‐Jimenez F et al Monocyte chemoattractant protein‐1: a key mediator in inflammatory processes. Int J Biochem Cell Biol 2009; 41:998–1001. [DOI] [PubMed] [Google Scholar]
- 30. Beringer A, Noack M, Miossec P. IL‐17 in Chronic inflammation: from discovery to targeting. Trends Mol Med 2016; 22:230–41. [DOI] [PubMed] [Google Scholar]
- 31. Gaffen SL, Jain R, Garg AV et al The IL‐23‐IL‐17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol 2014; 14:585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhao L, Tang Y, You Z et al Interleukin‐17 contributes to the pathogenesis of autoimmune hepatitis through inducing hepatic interleukin‐6 expression. PLOS ONE 2011; 6:e18909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gu FM, Li QL, Gao Q et al IL‐17 induces AKT‐dependent IL‐6/JAK2/STAT3 activation and tumor progression in hepatocellular carcinoma. Mol Cancer 2011; 10:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kaptoge S, Di Angelantonio E, Pennells L et al C‐reactive protein, fibrinogen, and cardiovascular disease prediction. N Engl J Med 2012; 367:1310–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Toulemonde G, Scoazec JY, Miossec P. Treatment with etanercept of autoimmune hepatitis associated with rheumatoid arthritis: an open label proof of concept study. Ann Rheum Dis 2012; 71:1423–4. [DOI] [PubMed] [Google Scholar]
- 36. Iwamoto S, Kido M, Aoki N et al TNF‐alpha is essential in the induction of fatal autoimmune hepatitis in mice through upregulation of hepatic CCL20 expression. Clin Immunol 2013; 146:15–25. [DOI] [PubMed] [Google Scholar]
- 37. Stout‐Delgado HW, Du W, Shirali AC et al Aging promotes neutrophil‐induced mortality by augmenting IL‐17 production during viral infection. Cell Host Microbe 2009; 6:446–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nagata T, McKinley L, Peschon JJ et al Requirement of IL‐17RA in Con A induced hepatitis and negative regulation of IL‐17 production in mouse T cells. J Immunol 2008; 181:7473–9. [DOI] [PubMed] [Google Scholar]
- 39. Bonder CS, Ajuebor MN, Zbytnuik LD et al Essential role for neutrophil recruitment to the liver in concanavalin A‐induced hepatitis. J Immunol 2004; 172:45–53. [DOI] [PubMed] [Google Scholar]
- 40. Gujral JS, Farhood A, Bajt ML et al Neutrophils aggravate acute liver injury during obstructive cholestasis in bile duct‐ligated mice. Hepatology 2003; 38:355–63. [DOI] [PubMed] [Google Scholar]
- 41. O'Brien KM, Allen KM, Rockwell CE et al IL‐17A synergistically enhances bile acid‐induced inflammation during obstructive cholestasis. Am J Pathol 2013; 183:1498–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ziraldo C, Vodovotz Y, Namas RA et al Central role for MCP‐1/CCL2 in injury‐induced inflammation revealed by in vitro, in silico, and clinical studies. PLOS ONE 2013; 8:e79804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mandrekar P, Ambade A, Lim A et al An essential role for monocyte chemoattractant protein‐1 in alcoholic liver injury: regulation of proinflammatory cytokines and hepatic steatosis in mice. Hepatology 2011; 54:2185–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sarma NJ, Tiriveedhi V, Crippin JS et al Hepatitis C virus‐induced changes in microRNA 107 (miRNA‐107) and miRNA‐449a modulate CCL2 by targeting the interleukin‐6 receptor complex in hepatitis. J Virol 2014; 88:3733–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hartupee J, Liu C, Novotny M et al IL‐17 enhances chemokine gene expression through mRNA stabilization. J Immunol 2007; 179:4135–41. [DOI] [PubMed] [Google Scholar]
- 46. Henness S, Johnson CK, Ge Q et al IL‐17A augments TNF‐alpha‐induced IL‐6 expression in airway smooth muscle by enhancing mRNA stability. J Allergy Clin Immunol 2004; 114:958–64. [DOI] [PubMed] [Google Scholar]
- 47. Zrioual S, Ecochard R, Tournadre A et al Genome‐wide comparison between IL‐17A‐ and IL‐17F‐induced effects in human rheumatoid arthritis synoviocytes. J Immunol 2009; 182:3112–20. [DOI] [PubMed] [Google Scholar]
- 48. Henness S, van Thoor E, Ge Q et al IL‐17A acts via p38 MAPK to increase stability of TNF‐alpha‐induced IL‐8 mRNA in human ASM. Am J Physiol Lung Cell Mol Physiol 2006; 290:L1283–90. [DOI] [PubMed] [Google Scholar]
- 49. Shimada M, Andoh A, Hata K et al IL‐6 secretion by human pancreatic periacinar myofibroblasts in response to inflammatory mediators. J Immunol 2002; 168:861–8. [DOI] [PubMed] [Google Scholar]
- 50. Witowski J, Pawlaczyk K, Breborowicz A et al IL‐17 stimulates intraperitoneal neutrophil infiltration through the release of GRO alpha chemokine from mesothelial cells. J Immunol 2000; 165:5814–21. [DOI] [PubMed] [Google Scholar]
- 51. Chowdhury S, Dijkhuis A, Steiert S et al IL‐17 attenuates degradation of ARE‐mRNAs by changing the cooperation between AU‐binding proteins and microRNA16. PLOS Genet 2013; 9:e1003747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Andoh A, Shimada M, Bamba S et al Extracellular signal‐regulated kinases 1 and 2 participate in interleukin‐17 plus tumor necrosis factor‐alpha‐induced stabilization of interleukin‐6 mRNA in human pancreatic myofibroblasts. Biochim Biophys Acta 2002; 1591:69–74. [DOI] [PubMed] [Google Scholar]
- 53. Chen Y, Kijlstra A, Yang P. IL‐17A stimulates the production of inflammatory mediators via Erk1/2, p38 MAPK, PI3K/Akt, and NF‐kappaB pathways in ARPE‐19 cells. Mol Vis 2011; 17:3072–7. [PMC free article] [PubMed] [Google Scholar]
- 54. Shahrara S, Pickens SR, Mandelin AM 2nd et al IL‐17‐mediated monocyte migration occurs partially through CC chemokine ligand 2/monocyte chemoattractant protein‐1 induction. J Immunol 2010; 184:4479–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Huang F, Kao CY, Wachi S et al Requirement for both JAK‐mediated PI3K signaling and ACT1/TRAF6/TAK1‐dependent NF‐kappaB activation by IL‐17A in enhancing cytokine expression in human airway epithelial cells. J Immunol 2007; 179:6504–13. [DOI] [PubMed] [Google Scholar]
- 56. Ruddy MJ, Wong GC, Liu XK et al Functional cooperation between interleukin‐17 and tumor necrosis factor‐alpha is mediated by CCAAT/enhancer‐binding protein family members. J Biol Chem 2004; 279:2559–67. [DOI] [PubMed] [Google Scholar]
- 57. Zimmermann M, Aguilera FB, Castellucci M et al Chromatin remodelling and autocrine TNFalpha are required for optimal interleukin‐6 expression in activated human neutrophils. Nat Commun 2015; 6:6061. [DOI] [PubMed] [Google Scholar]
- 58. Sparna T, Retey J, Schmich K et al Genome‐wide comparison between IL‐17 and combined TNF‐alpha/IL‐17 induced genes in primary murine hepatocytes. BMC Genomics 2010; 11:226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Karlsen JR, Borregaard N, Cowland JB. Induction of neutrophil gelatinase‐associated lipocalin expression by co‐stimulation with interleukin‐17 and tumor necrosis factor‐alpha is controlled by IkappaB‐zeta but neither by C/EBP‐beta nor C/EBP‐delta. J Biol Chem 2010; 285:14088–100. [DOI] [PMC free article] [PubMed] [Google Scholar]