

Abstract
Aims
Preterm birth remains a significant risk for later disability. The selective inhibition of the prostaglandin F2α receptor has significant advantages for a tocolytic. The prodrug OBE022 and its metabolite OBE002 are novel prostaglandin F2α receptor antagonists under development for treating preterm labour.
Methods
We performed a prospective, first in human, Phase I, dose escalation, placebo‐controlled, randomized trial at a clinical trial site in the UK. Placebo, single ascending doses of 10, 30, 100, 300, 1000 or 1300 mg, and multiple ascending doses over 7 days of 100, 300 or 1000 mg day–1; were administered to postmenopausal female volunteers. Food interaction was additionally evaluated.
Results
Subjects tolerated OBE022 well at all single and multiple doses. No clinically relevant changes in safety parameters were shown and there were no serious adverse events. Observations showed that prodrug OBE022 was readily absorbed and rapidly converted into its equally active stable metabolite OBE002. The plasma level of OBE002 rose with increasing doses, reaching exposure levels that were anticipated to be clinically relevant within 1 h following administration. There was no clinically significant food interaction, with peak exposures reduced to 80% and area under the curve staying bioequivalent. The mean half‐life of OBE002 ranged between 8 and 11 h following administration of a single dose and 22–29 h after multiple doses.
Conclusions
Administration of OBE022 was safe and had favourable pharmacokinetic characteristics and no clinically relevant interaction with food. Our results allow further investigation of OBE022 in preterm labour patients.
Keywords: OBE022, pharmacokinetics, prostaglandin F2α receptor antagonist, safety, tocolytic
What is Already Known about this Subject
Preterm birth remains the major cause of perinatal mortality and morbidity.
OBE022 is a novel, oral prostaglandin F2α receptor antagonist under development for treating preterm labour. It acts by specifically antagonizing myometrial prostaglandin F receptors.
In human myometrial strips, OBE022 reduces the strength and duration of contractions induced by either prostaglandin F2α or oxytocin.
OBE022 is being developed to reduce uterine contractions without the risk of serious fetal complications observed with nonspecific prostaglandin inhibitors.
What this Study Adds
OBE022 was well tolerated in this first‐in‐human trial in healthy women at single (10–1300 mg) and multiple doses (100–1000 mg day–1) allowing subsequent evaluation of OBE022 in preterm labour patients.
Exposures to the prodrug OBE022 and its active stable metabolite OBE002 increased with dose and half‐life was compatible with once daily dosing.
Introduction
Preterm birth remains the major cause of perinatal mortality and morbidity. Global figures indicate that 5.9 million children aged <5 years died in 2015; 2.7 million of these deaths occurred during the neonatal period, with preterm birth complications being the leading cause 1. Neonates who survive have a high risk of immaturity of multiple organ systems and neurodevelopmental disorders 2. Overall, prematurity, particularly birth before 28 weeks, places a heavy burden on healthcare and society 3, 4.
Term and preterm labour are similar processes, sharing common physiological endpoints characterized by uterine contractions, cervical dilation and activation of the fetal membranes, the differences being the gestational age at which they occur and the mechanisms through which they are initiated 5. Prostaglandins appear to play a key role in the initiation of parturition. In uterine tissues, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1883 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1884 (PGF2α) have been shown to exert cervical changes and elicit uterine contractility, two key events in the physiology of parturition 6, 7. Prostaglandin F2α acts via its receptor, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=344, to promote myometrial contractility. Activation of the FP receptor in the human myometrium by PGF2α results in the elevation of intracellular calcium concentration, which, in turn, leads to contraction of the uterine smooth cell muscle. The FP receptor is upregulated in uterine tissues towards term 8. Inhibition of prostaglandin formation with http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=269 inhibitors such as indomethacin appear to suppress premature labour; however, the associated safety concerns preclude use beyond short‐term exposure or use at all before 32 weeks 9. Antagonism of the PGF2α receptor reduces inflammation, decreases uterine contractions and prevents membrane ruptures and cervical changes, which are key features of preterm labour resulting in preterm birth.
OBE002 is a potent and selective, first‐in‐class, PGF2α receptor antagonist that is administered as the orally active, small molecule prodrug OBE022, and is being developed for the treatment of preterm labour and to prevent preterm delivery in pregnant women. Tocolytic activity has been confirmed previously with other PGF2α receptor antagonists in rats, mice and sheep 10, 11. This early phase trial investigated the safety, tolerability and pharmacokinetics (PK) of OBE022 in healthy postmenopausal female subjects following the administration of single and multiple ascending doses.
Methods
Trial population
The trial enrolled healthy postmenopausal female subjects aged 50–65 years, with a body mass index (BMI) of 18–32 kg m–2. Subjects were in good health based on physical examination, vital signs, laboratory parameters and 12‐lead electrocardiography (ECG).
Subjects with a history of clinically significant physical or psychiatric disease or with any abnormalities that may have interfered the interpretation of trial data were excluded. Subjects were also excluded if they had used prescription or over‐the‐counter medicines or any potent inhibitors or inducers of cytochrome P450 enzymes within 2 weeks of first trial drug administration. Subjects who had been using hormone replacement therapy were eligible for the trial after a sufficient wash‐out period, and concomitant medications for the treatment of common minor illnesses such as flu‐like symptoms were permitted within protocol‐defined limits. Caffeine or tobacco consumption was not permitted within 48 h and 3 months prior to the first study drug administration respectively.
All subjects provided written informed consent and the trial was approved by the Medicines and Healthcare products Regulatory Agency and a Research Ethics Committee (South Central‐Berkshire B, UK) and performed in accordance with the guidelines established by the Declaration of Helsinki and the International Conference on Harmonization Good Clinical Practice guidelines.
Trial design
This was a Phase 1, randomized, double‐blind, placebo‐controlled trial, which included the assessment of safety, tolerability and the PK of single (SAD) and multiple (MAD) ascending doses of OBE022 (prodrug), with and without food, in healthy postmenopausal women. The trial was conducted at Richmond Pharmacology Ltd. (London, UK). The trial parts discussed in this manuscript were performed between July and November 2016. The trial used an adaptive integrated design where various aspects of the trial could be adapted within protocol‐defined boundaries, and consecutive trial parts could be triggered once the relevant emerging data became available.
The starting dose of the SAD part and a PK plasma exposure limit for the trial were set in the protocol, based on nonclinical data. All other dosing regimens were adaptive and selected during the study, using emerging human data. The protocols contained rules stating the minimum safety, tolerability and PK data required to make decisions to escalate within the SAD and MAD parts and to progress from the SAD to the MAD part of the trial.
A decision to escalate doses in the SAD required a minimum of 48 h postdose safety and 24 h PK data from a minimum of three (of four) subjects who had received the prodrug from the cohort with the next lower exposure level. Similar rules applied to dose escalation in the MAD part.
To decide on the first MAD – when progressing from SAD to MAD – the safety review committee needed to review minimum data from a SAD level that was determined as relevant for this decision. The relevant dose level was defined as a dose that demonstrated acceptable safety/tolerability at a mean exposure that was not anticipated to be exceeded by the first dosing regimen in the combined MAD/food effect part, taking into account dose proportionality, potential accumulation and a potential food effect increasing exposures up to 3‐fold.
The PK exposure limits for maximum plasma concentration (Cmax) of 818 ng ml–1 and area under the curve (AUC) of 9329 ng h ml–1 were based on the no observed adverse event level of 180 mg kg–1 day–1 in the dog.
An SRC meeting occurred after each cohort. The SRC ensured that the minimum data requirements had been met and that the data had been reviewed prior to selecting the next dosing regimen(s). It also decided whether any other adaptive options were to be implemented.
The publication of this trial adheres to the Consolidated Standards of Reporting Trials (CONSORT) 2010 statement 12.
Determination of the starting dose and PK plasma exposure limit
The Investigational Medicinal Product's (IMP) maximum recommended human starting dose (MRSD) and the PK exposure limit for this first‐in‐human trial were set based on nonclinical data, applying the recommendations of the European Medicines Agency guidelines 13, the US Food and Drug Administration (FDA) algorithm 14 and using the IMP's anticipated therapeutic dose (ATD) range.
The MRSD of the prodrug and OBE002 were based on the no observed adverse‐event level of 180 mg kg–1 day–1 in the dog – the most sensitive and relevant species – and its human equivalent dose of 100 mg kg–1 day–1. A standard safety factor of 10 was then applied and the calculation also accounted for potential differences in bioavailability and saturation of absorption between dogs and humans by applying additional safety factors, leading to an overall safety factor of 60. In combination with an assumed standard weight of 60 kg for healthy women, the MRSD arrived at 100 mg.
The human ATD was estimated to be between 23 and 240 mg OBE022 (prodrug). The estimation accounted for differences in receptor affinity between rats and humans when applying the pharmacology model in rats to the calculation of effective doses in humans. It also accounted for differences between nonpregnant and pregnant status, estimated human clearance and assumed once daily oral dosing with a bioavailability of 20%.
It is recommended that starting doses in first‐in‐human trials should have minimal or no pharmacological activity. Therefore, based on the ATD range and supported by the MRSD, a starting dose of 10 mg was selected.
Part A: SADs
An alternate cohort design was used. Testing of six dose levels in two cohorts was planned, enrolling a total of 12 postmenopausal females. Each cohort was scheduled to participate in three ascending dose treatment periods separated by a washout, during which the alternate cohort received their doses. In each cohort subjects were randomly assigned to receive single, ascending doses of the IMP in two of the treatment periods and placebo in the remaining period. An adaptive design option was implemented introducing a fourth (separately randomized) period for Cohort 1 to gather more PK data on a dose level previously tested in Cohort 2.
Subjects were screened within 21 days before dosing. Subjects attended the clinical pharmacology unit (CPU) the day before dosing (Day –1). In each treatment period, dosing took place on Day 1 and subjects remained in the CPU until Day 3. Subjects attended outpatient visits on Days 4–7 and a follow‐up visit 14 ± 3 days after the last dose. During each period, four subjects in Cohorts 1 and 2 were randomized to receive a single dose of prodrug OBE022 (Cohort 1: 10, 100, 1000, 1300 mg; Cohort 2: 30, 300, 1300 mg), and two subjects received a single dose of matching placebo. All subjects fasted for at least 10 h predose and 4 h postdose. Venous blood samples were collected for PK analysis predose and 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 3.5, 4, 6, 7, 8, 12, 16, 24, 36, 48, 72, 96, 120 and 144 h postdose.
Part B: MADs
Three ascending dose levels of prodrug in three cohorts of eight subjects were planned (six randomized to receive active and two to matching placebo). An assessment of the effect of food on the PK properties of the prodrug and OBE002 was integrated. Six subjects in Cohorts 1 and 2 and five subjects in Cohort 3 received multiple doses of prodrug (100, 300 or 1000 mg respectively); two subjects in each cohort received matching placebo. Doses were administered once daily: in the fed condition on Day 1, and in the fasted condition from Day 3 to Day 9.
Subjects were screened within 21 days and attended the CPU on the day before the first dosing (Day –1) where they remained until Day 11. They returned for outpatient visits on Days 12–15 and attended a follow‐up visit 14 ± 3 days after their last dose.
The PK profile of the prodrug and OBE002 under fed condition was investigated on Day 1, when subjects ate a US FDA‐recommended high‐fat breakfast 15 starting 30 minutes before dosing. The PK profile of the prodrug and OBE002 under fasting conditions was investigated on Days 3 and 9, when subjects had been fasting for at least 10 h predose to 4 h postdose. On Days 4–8, breakfast was served 1 h after dosing. Blood samples were collected for PK analysis predose on Day 1 and Days 3–9. Samples were also collected at 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 3.5, 4, 6, 7, 8, 12, 16 and 24 h postdose on Day 1 (fed) and Days 3 and 9 (fasted), with additional samples taken at 48, 72, 96, 120 and 144 h post‐last dose (Day 9) on Days 10 to 15.
PK analysis
Blood samples for PK analysis were collected at each time point into NaF tubes and centrifuged for 10 minutes (≤1300 g, +5–10°C). The plasma supernatant was transferred to microtubes and snap‐frozen on dry ice. Samples were stored at –80°C until analysis.
Concentrations of prodrug and OBE002 in plasma were measured by SGS Cephac (Saint‐Benoît, France) using a validated liquid chromatography/tandem mass spectrometry method with a liquid–liquid extraction. The assay's lowest quantifiable concentration was 0.1 ng ml–1 for both OBE002 and prodrug and the method was validated on 0.1 ml of plasma samples for OBE002 and prodrug over the range 0.100 to 100 ng ml–1. Samples below the lower limit of quantification (LLOQ) prior to the first quantifiable concentration were set to zero. Samples with concentrations below LLOQ in the terminal phase (after the last quantifiable sample) were omitted from the analysis. Samples with concentrations above the limit of quantification were diluted and reanalysed. A dilution factor of 1/20 has been validated. Validation procedures were based on those outlined in the Guideline on Bioanalytical Method Validation (European Medicines Agency) 16 and Bioanalytical Method Validation (FDA) 17.
Chromatographic separation was performed through an Ascentis Express C8 (2.1 × 50 mm, 2.7 μm; Sigma–Aldrich, France) analytical column at 50°C at a flow rate of 0.6 ml min–1. The mobile phases were formic acid (0.1%) in water (A) and formic acid (0.1%) in acetonitrile (B) for which the elution gradient varied between 30% and 70%. Detection and quantitation were performed using a triple quadrupole mass spectrometer detector (ABSciex, Toronto, Canada). Quantitation was performed using the following transitions m/z 600.3 → 483.1 and m/z 501.3 → 349.1 for prodrug and OBE002, respectively.
The assay provided accurate and reproducible results within the defined limits of accuracy (standard deviation <10%) and precision (coefficient of variation <10%). Actual time points for blood sampling were used in the analysis. Noncompartmental analysis was used for estimation of PK parameters. PK modelling was also employed to assess potential time‐dependent PK.
The primary PK parameters for prodrug and OBE002 following single doses with prodrug in Part A of the trial were: peak plasma concentration (Cmax), time to Cmax (tmax), terminal elimination half‐time (t½), apparent total plasma clearance (CL/F) and volume of distribution (Vd/F). Area under the plasma concentration–time curve from administration to 24 h after dosing (AUC0–24h), to last sampling point (AUC0–t) and to infinite time (AUC0–inf) were calculated using the linear/log trapezoidal method, applying the linear trapezoidal rule up to Cmax and the log trapezoidal rule for the remainder of the curve.
The primary PK parameters for prodrug and OBE002 for each dose group were the same following multiple doses in Part B of the trial as these in Part A with the addition of minimal plasma concentration/pre‐dose values on multiple dosing days (Cmin). Plasma PK profiles of prodrug and OBE002 in the fasted state were evaluated in all cohorts; PK profiles of prodrug and OBE002 following single administration in the fed state vs. fasted state were also evaluated.
Safety assessments
Safety assessments included clinical laboratory parameters (clinical chemistry, coagulation, haematology and urinalysis), 12‐lead bedside ECG and telemetry, vital signs, physical examination and adverse event monitoring from screening until the final visit, coded in accordance with appropriate guidance 18, 19. Safety findings are summarized by treatment group.
Statistical analysis
Statistical analyses were performed using Statistical Analysis Software version 9.3 (SAS Institute Inc, USA). The safety set included all randomized subjects who received at least one dose of the trial drug and the PK set included those in the safety set who had sufficient blood samples taken for at least one of the PK parameters to be calculated. Data for subjects who withdrew before the last planned observation in a trial period were included up to the time of discontinuation. Safety data and PK parameters were summarized by treatment group using descriptive statistics.
Exposure ratios were calculated to evaluate parameters such as dose proportionality and accumulation. Dose accumulation ratio was calculated as AUC0–24h (Day 9)/AUC0–24h (Day 3) and steady state accumulation ratio was calculated as AUC0–24h (Day 9)/AUC0–inf (Day 3). To test dose proportionality, Cmax and all AUCs were normalized by dose. An estimate of the slope of the regression line of ‘1’ in a power model for repeated measurement corresponded to dose proportionality.
Food effect was analysed with a mixed effect model using a logarithm of the PK parameter as response variable and the logarithm of the dose and food status (fasted and fed) as fixed effects and subject as random effect. The model was applied to the following PK parameters: AUC0–t, AUC0–inf and Cmax. Based on the mixed effect model, the ratio between fasted and fed conditions and its two‐sided 90% confidence interval (CI) was estimated. Bioequivalence/absence of a food effect was concluded, if the linear scale 90% CI of this ratio was fully contained within the acceptance range for Cmax and AUCs (0.8, 1.25).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 20, and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 21, 22.
Results
Patient disposition and baseline characteristics
Part A: Thirteen postmenopausal female subjects were enrolled in two cohorts, in which six SAD doses levels were tested over seven treatment periods. The mean age of the subjects was 56.92 years and their mean BMI was 26.50 kg m–2 (Table 1). Seven subjects were enrolled in Cohort 1, as one subject received 10 mg of the prodrug in Period 1 before being withdrawn from the trial because of a urinary tract infection (unrelated to treatment). A replacement subject was included for Periods 2–4. Consequently, six subjects participated in each of the four treatment periods, during which 10, 100, 1000 and 1300 mg of the prodrug or placebo were administered. Six subjects were enrolled in Cohort 2 and received 30, 300 and 1300 mg of the prodrug or placebo. All 13 subjects were included in the safety, ECG and PK analysis sets. (Figure 1A).
Table 1.
Demographic and baseline characteristics
| Parameter | SAD Part A | MAD Part B |
|---|---|---|
| (n = 13) | (n = 23) | |
| Age, years; mean (SD) | 56.9 (3.9) | 56.5 (4.0) |
| Race; n (%) | ||
| Asian | 2 (15.4) | 2 (8.7) |
| Black African | 2 (15.4) | 4 (17.4) |
| Caucasian | 6 (46.2) | 16 (69.6) |
| Other | 3 (23.1) | 1 (4.3) |
| Body Height, cm; mean (SD) | 163.5 (8.0) | 162.9 (4.8) |
| Body Weight, kg; mean (SD) | 70.8 (9.4) | 67.00 (9.3) |
| BMI, kg m –2 ; mean (SD) | 26.5 (3.1`) | 25.3 (3.4) |
SAD = single ascending dose; MAD = multiple ascending dose; BMI = body mass index; SD = standard deviation
Figure 1.
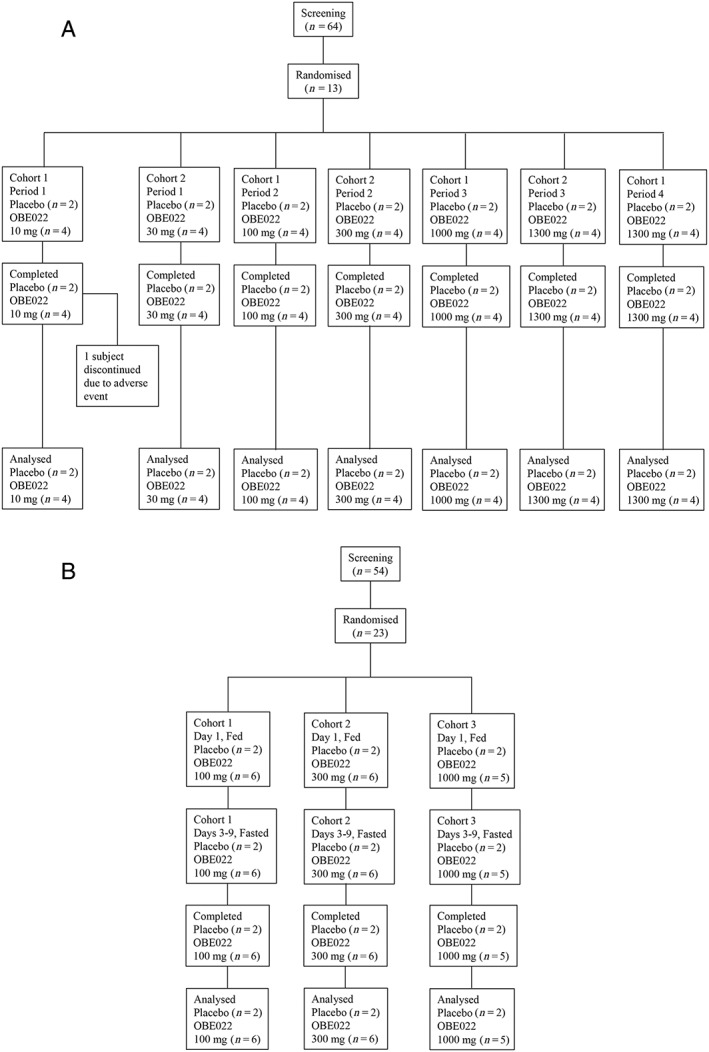
Patient CONSORT flow diagram. Part A – single ascending doses; Part B – multiple ascending doses
Part B: MADs were investigated in three cohorts totalling 23 subjects. Their mean age was 56.52 years and their mean BMI was 25.27 kg m–2 (Table 1). Prodrug doses of 100 (n = 6), 300 (n = 6) and 1000 mg (n = 5), were used for the full PK analysis of prodrug and OBE002 on Days 1, 3 and 9, while only Cmin was determined on Days 4 to 8 and following the final dose on Day 9 on all days up to Day 15. Two subjects in each cohort received matching placebo. All doses were given in the fed state on Day 1 and in the fasted state on Days 3 to 9. All 23 subjects were included in the safety and ECG analysis sets. All 17 subjects who received prodrug were included in the PK analysis set (Figure 1B).
Single dose PK
The prodrug was rapidly absorbed and converted into the OBE002 metabolite. No quantifiable levels of prodrug were observed in the 10 mg dose group and only one subject in the 30 mg cohort showed any prodrug concentrations. Sufficient plasma concentrations permitted PK evaluations of prodrug in all subjects at ≥300 mg doses. Maximum prodrug concentrations tended to occur within 1 h at doses below 1300 mg and only slightly exceeded 1 h at the 1300 mg dose (Table 2). Quantifiable OBE002 levels were shown in all four subjects receiving prodrug from 0.25 h up to 24 h, starting at the lowest dose (10 mg). Variable plasma concentration results were observed after the first 1300 mg dose (Cohort 2 Period 3) and led its repetition (Cohort 1 Period 4). Most subjects showed bi‐exponential elimination of OBE002 and tmax was reached within 0.7 h at 10 mg of prodrug and 4 h at 1300 mg (Figure 2A). Values of Cmax for OBE002 exceeded that of the prodrug by 100‐ to 200‐fold and AUC values were up to 1000‐fold higher (Table 2). Both CL/F and Vd/F of prodrug were high, which was in line with the low AUC, the short t½ and the fact that neither the fraction of absorbed prodrug nor the total metabolic activity was known. AUC0–24h was often considerably lower than AUC0–inf for both prodrug and OBE002, which may have contributed to an over estimation of CL/F and Vd/F.
Table 2.
Pharmacokinetic parameters for the prodrug and OBE002 following a single dose (pharmacokinetic set)
| Parameter | Prodrug dose | ||||||
|---|---|---|---|---|---|---|---|
| 10 mg | 30 mg | 100 mg | 300 mg | 1000 mg | 1300 mg | 1300 mg | |
| Cohort 1 | Cohort 2 | Cohort 1 | Cohort 2 | Cohort 1 | Cohort 2 | Cohort 1 | |
| Period 1 | Period 1 | Period 2 | Period 2 | Period 3 | Period 3 | Period 4 | |
| (n = 4) | (n = 4) | (n = 4) | (n = 4) | (n = 4) | (n = 4) | (n = 4) | |
| Prodrug | |||||||
| C max (ng ml –1 ) | –a | –a | 0.31 (0.21) | 1.13 (0.17) | 3.66 (1.26) | 1.71 (1.4) | 4.88 (1.64) |
| t max (h) | –a | –a | 0.31 (0.13) | 0.44 (0.24) | 0.26 (0.02) | 1.28 (1.24) | 1.38 (1.36) |
| t ½ (h) | –a | –a | 0.59b | 3.17 (2.96)c | 1.03 (0.34) | 1.75 (1.35) | 1.45 (0.61) |
| AUC 0–24h (h × ng ml –1 ) | –a | 0.0b | 0.21 (0.08) | 0.81 (0.66) | 4.70 (2.53) | 2.87 (1.10) | 7.27 (3.02) |
| AUC 0–t (h × ng ml –1 ) | –a | 0.0b | 0.21 (0.08) | 0.81 (0.66) | 4.70 (2.53) | 2.87 (1.10) | 7.27 (3.02) |
| AUC 0–inf (h × ng ml –1 ) | –a | –a | 0.42b | 1.61 (0.23)c | 4.96 (2.61) | 3.23 (1.13) | 7.55 (3.04) |
| CL/F (l h –1 ) | –a | –a | 235 897b | 188 361 (25379)c | 327 148 (327669) | 457 537 (212406) | 207 070 (119419) |
| V d /F (l) | –a | –a | 199 208b | 875 617 (831784)c | 372 168 (184757) | 1 010 012 (811091) | 394 591 (165104) |
| OBE002 | |||||||
| C max (ng ml –1 ) | 7.5 (7.1) | 8.8 (5.9) | 35.5 (20.3) | 163.6 (123.1) | 519.8 (67.3) | 376.0 (320.1) | 848.0 (60.1) |
| t max (h) | 0.70 (0.13) | 1.06 (0.32) | 1.25 (0.61) | 2.94 (2.20) | 3.00 (0.57) | 4.00 (1.47) | 2.38 (1.11) |
| t ½ (h) | 37.7 (28.8) | 26.7 (7.1) | 19.4 (6.1) | 19.8 (7.08) | 17.4 (2.7) | 19.5 (4.7) | 16.5 (2.6) |
| AUC 0–24h (h × ng ml –1 ) | 20.0 (13.9) | 68.2 (35.4) | 222.3 (97.5) | 1083 (787.7) | 4181.9 (1082.8) | 2527.4 (2253.1) | 6740 (1864.5) |
| AUC 0–t (h × ng ml –1 ) | 27.4 (13.0) | 110.8 (44.8) | 415.9 (205.1) | 1670.4 (1135) | 6381.4 (1852.8) | 3930.6 (2877.4) | 9379.1 (2649) |
| AUC 0–inf (h × ng ml –1 ) | 34.2 (8.2) | 115.3 (43.8) | 422.5 (207.9) | 1687.9 (1141.7) | 6412.8 (1872.9) | 3964.2 (2893.3) | 9419.6 (2686.6) |
| CL/F (l h –1 ) | 305 (72) | 292 (112) | 318 (235) | 354 (400) | 170 (65) | 537 (454) | 147 (44) |
| V d /F (l) | 18 511 (16997) | 11 742 (6332) | 7718 (3386) | 9646 (10561) | 4220 (1592) | 14 613 (12018) | 3466 (992) |
No subject in this cohort available for pharmacokinetic parameter.
One subject in this cohort available for pharmacokinetic parameter.
Two subjects in this cohort available for pharmacokinetic parameter. All data are expressed as mean (standard deviation).
Figure 2.
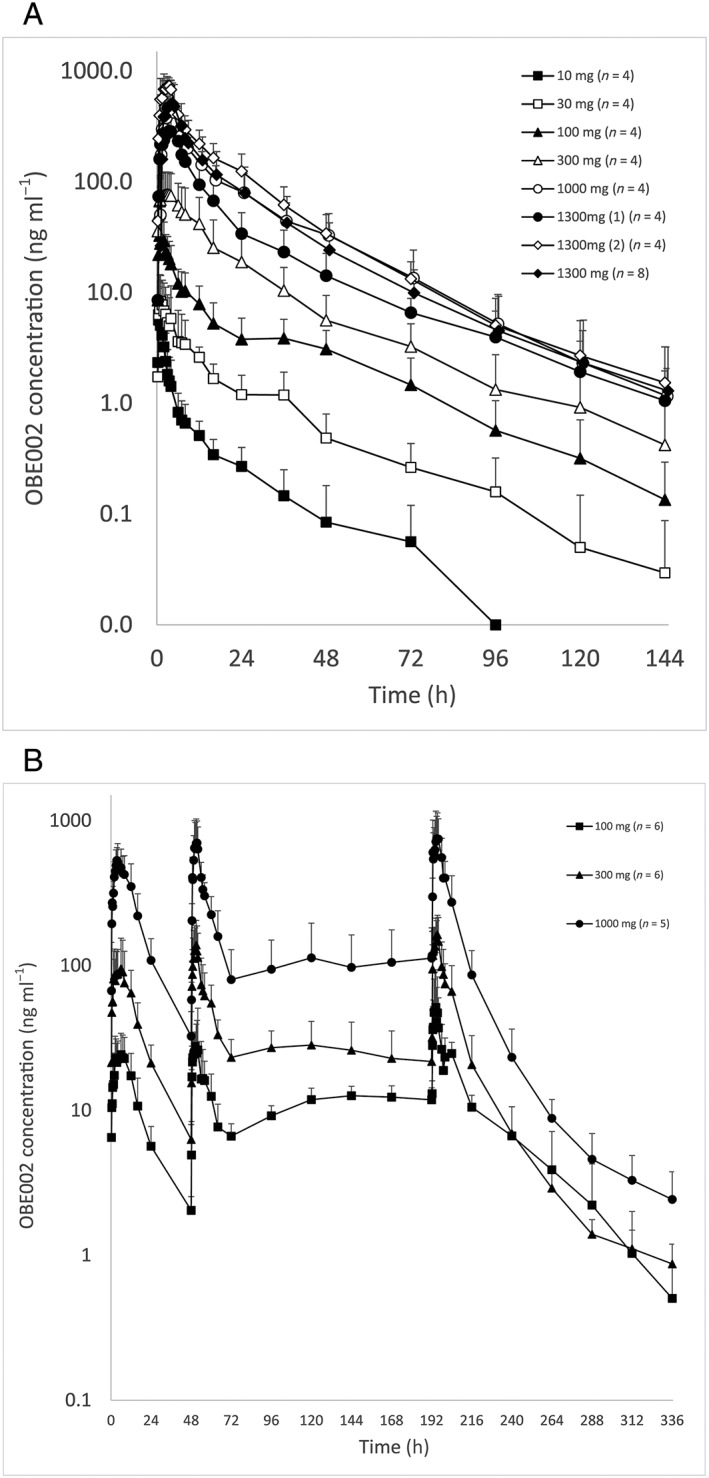
Mean (standard deviation) OBE002 plasma concentrations vs. nominal time – all cohorts and periods overlaid (log‐linear scale). Part A – single ascending doses; Part B – multiple ascending doses
Multiple dose PK
Plasma concentration profiles after multiple dosing are presented in Figure 2B. Values of prodrug Cmin were below LLOQ for all doses, which was in line with the short half‐life (1–4 h) and indicated that there was no accumulation. Neither Cmax nor AUC showed increased prodrug exposure between Day 3 and Day 9 (Table 3A). OBE002 showed a 2.5–3‐fold increase in half‐life from 8.27–10.66 h on Day 3 to 22.20–29.17 h on Day 9. Increases in Cmin from Day 3 to Day 9 became smaller with increasing doses: 6‐fold at 100 mg of prodrug, 3.5‐fold at 300 mg and 1000 mg of prodrug (Figure 2B). Similarly, increases in both Cmax and AUC for OBE002 from Day 3 to Day 9 were greatest at the 100 mg dose (Table 3B). Apparent total plasma clearance CL/F was variable and much higher for the prodrug in line with its rapid metabolism to OBE002. Both CL/F and Vd/F of the prodrug decreased substantially with time for the 300 and 1000 mg doses and in particular between Day 3 and Day 9 for OBE002.
Table 3.
Pharmacokinetic parameters for the prodrug and OBE002 in plasma following repeat dosing
| A. Prodrug | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 100 mg Prodrug | 300 mg Prodrug | 1000 mg Prodrug | |||||||
| N = 6 | N = 6 | N = 5 | |||||||
| Parameter (unit) | Day 1 | Day 3 | Day 9 | Day 1 | Day 3 | Day 9 | Day 1 | Day 3 | Day 9 |
| C min (ng ml –1 ) | |||||||||
| n | 2 | 4 | 5 | 6 | 6 | 6 | 5 | 5 | 3 |
| Mean (SD) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) | 0.0 (0.0) |
| C max (ng ml –1 ) | |||||||||
| n | 2 | 4 | 5 | 6 | 6 | 6 | 5 | 5 | 5 |
| Mean (SD) | 0.90 (0.83) | 0.22 (0.11) | 0.19 (0.05) | 0.72 (0.40) | 0.92 (0.38) | 1.09 (0.77) | 5.52 (4.09) | 3.47 (1.84) | 2.64 (1.23) |
| t max (h) | |||||||||
| n | 2 | 4 | 5 | 6 | 6 | 6 | 5 | 5 | 5 |
| Mean (SD) | 0.73 (0.74) | 0.28 (0.17) | 0.48 (0.58) | 1.86 (2.67) | 1.02 (1.31) | 0.69 (0.90) | 0.89 (0.83) | 0.36 (0.36) | 1.00 (1.41) |
| t ½ (h) | |||||||||
| n | 1 | 0 | 0 | 5 | 4 | 6 | 5 | 5 | 4 |
| Mean (SD) | 3.50 | ‐ | ‐ | 4.31 (4.14) | 0.97 (0.57) | 2.03 (1.22) | 2.68 (1.33) | 1.57 (0.58) | 0.85 (0.19) |
| AUC 0–24h (h × ng ml –1 ) | |||||||||
| n | 2 | 4 | 5 | 6 | 6 | 6 | 5 | 5 | 5 |
| Mean (SD) | 0.55 (0.58) | 0.30 (0.42) | 0.14 (0.09) | 1.55 (0.24) | 1.56 (0.76) | 1.45 (0.88) | 10.33 (3.53) | 6.42 (2.30) | 5.69 (2.77) |
| AUC 0–t (h × ng ml –1 ) | |||||||||
| n | 2 | 4 | 5 | 6 | 6 | 6 | 5 | 5 | 5 |
| Mean (SD) | 0.55 (0.58) | 0.30 (0.42) | 0.14 (0.09) | 1.55 (0.24) | 1.56 (0.76) | 1.45 (0.88) | 10.33 (3.53) | 6.42 (2.30) | 5.69 (2.77) |
| AUC 0–inf (h × ng ml –1 ) | |||||||||
| n | 1 | 0 | 0 | 5 | 4 | 6 | 5 | 5 | 4 |
| Mean (SD) | 1.47 | ‐ | ‐ | 2.61 (1.46) | 1.60 (0.38) | 1.90 (0.81) | 11.12 (3.53) | 6.77 (2.45) | 6.93 (2.31) |
| CL/F (l h –1 ) | |||||||||
| N | 1 | 0 | 5 | 5 | 4 | 6 | 5 | 5 | 5 |
| Mean (SD) | 194 294 | ‐ | 960 979 (503245) | 552 793 (297556) | 2 220 810 (1057216) | 251 300 (97806) | 1 415 310 (529772) | 3 677 317 (1429059) | 225 942 (136693) |
| V d /F (l) | |||||||||
| n | 1 | 0 | 0 | 5 | 4 | 6 | 5 | 5 | 4 |
| Mean (SD) | 980 392 | ‐ | ‐ | 2 080 265 (370201) | 2 484 445 (276258) | 774 892 (490929) | 5 346 109 (2660228) | 7 382 860 (2028898) | 196 753 (36898) |
| B. OBE002 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 100 mg Prodrug | 300 mg Prodrug | 1000 mg Prodrug | |||||||
| N = 6 | N = 6 | N = 5 | |||||||
| Parameter (unit) | Day 1 | Day 3 | Day 9 | Day 1 | Day 3 | Day 9 | Day 1 | Day 3 | Day 9 |
| C min (ng ml –1 ) | |||||||||
| n | 6 | 6 | 6 | 6 | 6 | 6 | 5 | 5 | 5 |
| Mean (SD) | 0.0 (0.0) | 2.0 (0.6) | 11.9 (3.1) | 0.0 (0.0) | 6.3 (2.1) | 21.8 (15.8) | 0.0 (0.0) | 32.5 (17.8) | 112.5 (79.4) |
| C max (ng ml –1 ) | |||||||||
| n | 6 | 6 | 6 | 6 | 6 | 6 | 5 | 5 | 5 |
| Mean (SD) | 29.3 (13.4) | 36.1 (25.9) | 54.8 (49.7) | 120.2 (72.1) | 156.1 (77.4) | 180.1 (76.1) | 589.4 (188.4) | 742.4 (361.1) | 926.2 (426.9) |
| t max (h) | |||||||||
| n | 6 | 6 | 6 | 6 | 6 | 6 | 5 | 5 | 5 |
| Mean (SD) | 5.50 (3.71) | 3.42 (1.88) | 3.46 (4.26) | 4.59 (3.97) | 2.59 (0.87) | 3.10 (0.80) | 3.92 (1.29) | 3.31 (0.57) | 2.46 (1.36) |
| t ½ (h) | |||||||||
| n | 5 | 6 | 6 | 5 | 6 | 6 | 5 | 5 | 5 |
| Mean (SD) | 7.5 (1.6) | 8.3 (1.9) | 22.3 (4.0) | 8.4 (2.4) | 10.7 (6.1) | 29.2 (12.3) | 7.9 (2.3) | 9.1 (5.5) | 22.2 (12.0) |
| AUC 0–24h (h × ng ml –1 ) | |||||||||
| n | 6 | 6 | 6 | 6 | 6 | 6 | 5 | 5 | 5 |
| Mean (SD) | 344.6 (175.4) | 262.9 (166.6) | 530.6 (283.4) | 1250.9 (775.5) | 1155.7 (592.0) | 1540.4 (910.8) | 6980.6 (3108.0) | 5619.4 (2285.6) | 7298.3 (3843.0) |
| AUC 0–t (h × ng ml –1 ) | |||||||||
| n | 6 | 6 | 6 | 6 | 6 | 6 | 5 | 5 | 5 |
| Mean (SD) | 360.2 (161.0) | 262.9 (166.6) | 559.4 (259.4) | 1364.3 (754.9) | 1155.7 (592.0) | 1651.6 (893.9) | 7285.0 (2751.3) | 5619.4 (2285.6) | 7802.6 (3900.0) |
| AUC 0–inf (h × ng ml –1 ) | |||||||||
| n | 5 | 6 | 6 | 5 | 6 | 6 | 5 | 5 | 5 |
| Mean (SD) | 430.1 (190.0) | 354.4 (219.7) | 575.6 (256.5) | 1764.3 (849.2) | 1633.8 (578.6) | 1691.8 (896.6) | 8513.9 (3216.1) | 8228.9 (5261.0) | 7918.0 (3875.1) |
| CL/F (l h –1 ) | |||||||||
| N | 5 | 6 | 6 | 5 | 6 | 6 | 5 | 5 | 5 |
| Mean (SD) | 2121 (929) | 1322 (489) | 205 (75) | 1415 (909) | 768 (381) | 217 (80) | 1030 (586) | 797 (515) | 154 (66) |
| V d /F (l) | |||||||||
| N | 5 | 6 | 6 | 5 | 6 | 6 | 5 | 5 | 5 |
| Mean (SD) | 22 045 (7469) | 15 211 (5128) | 6832 (3308) | 16 098 (9275) | 9868 (3053) | 8870 (5048) | 10 736 (4151) | 7813 (3447) | 5268 (3917) |
N = number of subjects in cohort. n = number of subjects available for pharmacokinetic parameter. SD = standard deviation.
Dose accumulation ratio was 1.35 for 100 mg of prodrug, but exposure decreased at the 300 mg and 1000 mg doses with ratios of 0.92 and 0.89, respectively. In contrast, dose accumulation ratios of OBE002 were >1 at all dose levels with ratios of 2.14, 1.34 and 1.25 at the 100 mg, 300 mg and 1000 mg dose levels, respectively. Steady‐state accumulation ratios were <1, indicating a decrease in exposure for prodrug and OBE002 at the 300 mg and 1000 mg dose levels. Meanwhile, a ratio of 1.56 for OBE002 at the 100 mg dose indicated steady state accumulation.
Dose proportionality
In the SAD part of the trial, all concentration‐dependent PK parameters of prodrug showed marked variability. None of the 90% CIs of the estimates (slopes) were contained in the acceptance range (0.80; 1.25) for dose proportionality (Table 4). Even the AUC0–inf with a value of 0.953, which was closest to 1, showed a 90% CI of 0.684–1.221, exceeding the lower limit of the acceptance range. Thus, dose proportionality could not be demonstrated for the prodrug. In contrast, for OBE002, although dose‐normalized Cmax and AUC values also showed some variability across the dose range from 10 to 1300 mg, all estimates were close to 1 and none of the 90% CIs exceeded the upper or lower limit (0.80; 1.25). Thus, dose proportionality could be claimed for OBE002.
Table 4.
Evaluation of dose proportionality for prodrug and OBE002 in plasma (SAD and MAD) – power model analysis
| Prodrug | OBE002 | |||||
|---|---|---|---|---|---|---|
| Parameter | No. of subjects/observations | Estimates | 90% CI | No. of subjects/observations | Estimates | 90% CI |
| SAD | ||||||
| C max | 12/20 | 1.09 | 0.91–1.26 | 13/28 | 1.01 | 0.90–1.12 |
| AUC 0–24h | 12/20 | 1.40 | 1.23–1.57 | 13/28 | 1.11 | 1.05–1.17 |
| AUC 0–t | 12/20 | 1.40 | 1.23–1.57 | 13/28 | 1.11 | 1.05–1.17 |
| AUC 0–inf | 12/16 | 0.95 | 0.68–1.22 | 13/28 | 1.05 | 0.99–1.12 |
| MAD– Day 1 | ||||||
| C max | 17/13 | 1.04 | 0.46–1.61 | 17/17 | 1.32 | 1.09–1.56 |
| AUC 0–24h | 17/13 | 1.46 | 1.17–1.76 | 17/17 | 1.31 | 1.07–1.54 |
| AUC 0–t | 17/13 | 1.46 | 1.17–1.76 | 17/17 | 1.31 | 1.10–1.52 |
| AUC 0–inf | 17/11 | 1.03 | 0.71–1.34 | 17/15 | 1.30 | 1.09–1.51 |
| MAD– Day 9 | ||||||
| C max | 17/16 | 1.15 | 0.90–1.41 | 17/17 | 1.30 | 1.05–1.55 |
| AUC 0–24h | 17/16 | 1.62 | 1.32–1.92 | 17/17 | 1.13 | 0.89–1.37 |
SAD = single ascending dose; MAD = multiple ascending dose; CI = confidence interval
In the MAD part of the trial, dose proportionality could not be claimed for Day 1, either due to significant over‐proportional increases in AUC0–24h of the prodrug and in all evaluated PK parameters of OBE002, or to variability in Cmax of the prodrug. For similar reasons, dose proportionality could not be concluded for AUC0–24 and Cmax of the prodrug and OBE002 on Day 9.
Food effect
The evaluation of the food effect on the prodrug and OBE002 PK was performed across all three MAD dose levels and all 17 subjects. The relevant PK parameters of prodrug (as far as available) and OBE002 determined from plasma concentrations in the fed and in the fasted state are summarised in Table 5A. The geometric mean of the prodrug AUC0–inf was not available for the 100 mg dose group on Day 3 because this parameter could be determined in one subject only. The analysis of the effects of food on the PK properties of OBE002 and the prodrug is summarized in Table 5B.
Table 5.
Geometric mean pharmacokinetic parameters (A) and evaluation of food effect (B) for the prodrug and OBE002 in plasma
| A. Geometric mean pharmacokinetic parameters | |||||||
|---|---|---|---|---|---|---|---|
| Geometric means of Prodrug | Geometric means of OBE002 | ||||||
| Parameter (unit) | Prodrug | Prodrug | Prodrug | Prodrug | Prodrug | Prodrug | |
| 100 mg | 300 mg | 1000 mg | 100 mg | 300 mg | 1000 mg | ||
| N = 6 | N = 6 | N = 6 | N = 6 | N = 6 | N = 6 | ||
| C max (ng ml –1 ) | Fed / Fasted | 0.69 | 0.59 | 4.53 | 26.8 | 100.5 | 567.2 |
| 0.20 | 0.85 | 3.10 | 31.3 | 142.2 | 671.6 | ||
| AUC 0–24h (h × ng ml –1 ) | Fed / Fasted | 0.36a | 1.53 | 9.83 | 313.0 | 1079.1 | 6411.2 |
| 0.13b | 1.43 | 6.06 | 232.8 | 1052.7 | 5267.1 | ||
| AUC 0–t (h × ng ml –1 ) | Fed / Fasted | 0.36a | 1.53 | 9.83 | 335.9 | 1201.7 | 6874 |
| 0.13b | 1.43 | 6.06 | 232.8 | 1052.7 | 5267.1 | ||
| AUC 0–inf (h × ng ml –1 ) | Fed / Fasted | 1.47c | 2.38d | 10.67 | 402.5d | 1579.5d | 8059.3 |
| ‐ | 1.56b | 6.39 | 316.3 | 1559.1 | 7177.7 | ||
| B. Evaluation of food effect | |||||||
|---|---|---|---|---|---|---|---|
| Prodrug (fed vs. fasted) | OBE002 (fed vs. fasted) | ||||||
| Parameter | No. of subjects/observations | Estimate | 90% CI | No. of subjects/ observations | Estimate | 90% CI | |
| C max | 17/28 | 1.20 | 0.75–1.92 | 17/34 | 0.80 | 0.68–0.94 | |
| AUC 0–24h | 17/28 | 1.55 | 0.98–2.44 | 17/34 | 1.06 | 0.95–1.18 | |
| AUC 0–t | 17/28 | 1.55 | 0.98–2.44 | 17/34 | 1.06 | 0.95–1.18 | |
| AUC 0–inf | 17/20 | 1.84 | 1.55–2.17 | 17/32 | 1.15 | 1.07–1.24 | |
N = number of subjects in cohort;
Four missing values;
Two missing values;
Five missing values;
One missing value;
CI = confidence interval
The estimate for AUC0–inf of the prodrug was 1.836‐fold (90% CI: 1.552, 2.172) higher in the fed state than fasted (Table 5B). Evaluation of the effects of food on the other prodrug PK parameters was inconclusive due to high levels of variability. A food effect was observed for OBE002 in AUC0–inf, although less distinct than that seen with the prodrug and within bioequivalence limits: point estimate of 1.151 (90% CI: 1.067, 1.242) (Figure 3). The OBE002 Cmax was lower with food than fasting and was estimated as 0.796 (90% CI: 0.677, 0.937). No food effect was confirmed for OBE002 AUC0–24h or AUC0–t with their point estimates close to unity and the 90% CIs fully contained in the acceptance range.
Figure 3.
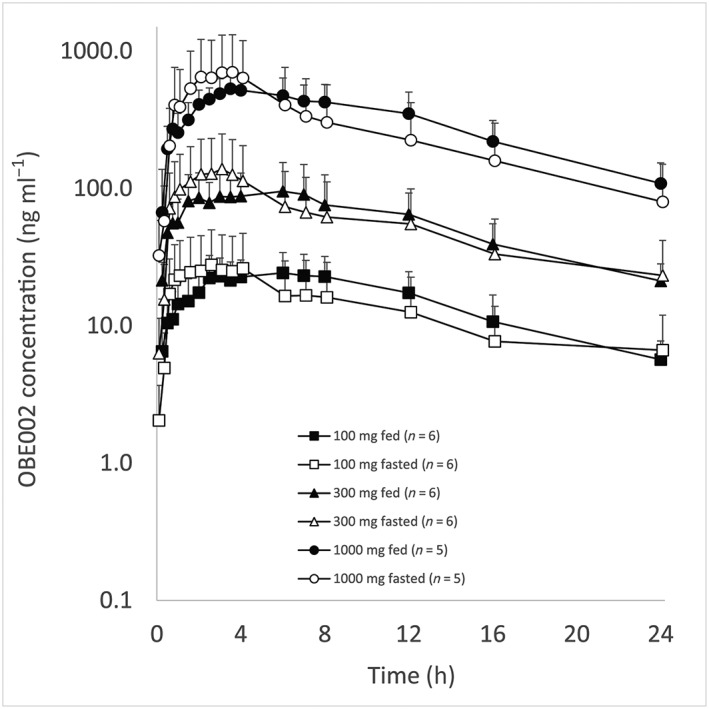
Mean (standard deviation) OBE002 plasma concentrations vs. nominal time – fed vs. fasted administration (log‐linear scale)
The food effect was not considered clinically relevant.
Safety
Following single doses, 10 (76.9%) subjects reported 19 treatment emergent adverse events (TEAE) following prodrug administration; eight (66.7%) subjects reported 11 TEAE following placebo (Table 6A). Two TEAEs (headache, ventricular extra‐systoles) in two subjects (15.4%) and four TEAEs (headache, constipation) in two subjects (16.7%) following placebo administration were judged – in blinded condition – to be related to IMP. These events were considered Grade 1 (mild) according to the Common Terminology Criteria for Adverse Events 23.
Table 6.
Summary of adverse events
| Part A. SAD | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Pooled | Pooled | 10 mg | 30 mg | 100 mg | 300 mg | 1000 mg | 1300 mg | 1300 mg | |
| Placebo | Prodrug | C1 P1 | C2 P1 | C1 P2 | C2 P2 | C1 P3 | C2 P3 | C1 P4 | |
| Adverse events | (N = 12) | (N = 13) | (N = 4) | (N = 4) | (N = 4) | (N = 4) | (N = 4) | (N = 4) | (N = 4) |
| Total, n (%) E | 8 (66.7) 11 | 10 (76.9) 19 | 2 (50.0) 6 | 2 (50.0) 2 | 3 (75.0) 5 | 2 (50.0) 4 | 1 (25.0) 1 | 1 (25.0) 1 | 0 (0.0) 0 |
| Preferred term, n (%) E | |||||||||
| Dizziness | 0 | 1 (25.0) 1 | 0 | 1 (25.0) 1 | 0 | 0 | 0 | 0 | |
| Presyncope | 0 | 0 | 1 (25.0) 1 | 0 | 0 | 0 | 0 | 0 | |
| Ventricular extra‐systoles | 0 | 0 | 0 | 0 | 1 (25.0) 1 | 0 | 0 | 0 | |
| Abdominal discomfort | 1 (8.3) 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Constipation | 2 (16.7) 2 | 1 (25.0) 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Dental discomfort | 0 | 0 | 0 | 0 | 1 (25.0) 1 | 0 | 0 | 0 | |
| Nausea | 0 | 0 | 0 | 1 (25.0) 1 | 0 | 0 | 0 | 0 | |
| Nasopharyngitis | 1 (8.3) 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Urinary tract infections | 0 | 1 (25.0) 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Back injury | 0 | 0 | 1 (25.0) 1 | 0 | 0 | 0 | 0 | 0 | |
| Decreased appetite | 0 | 1 (25.0) 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Muscle spasms | 0 | 0 | 0 | 0 | 1 (25.0) 1 | 0 | 0 | 0 | |
| Musculoskeletal pain | 0 | 0 | 0 | 0 | 1 (25.0) 1 | 0 | 0 | 0 | |
| Headache | 5 (41.7) 6 | 0 | 0 | 1 (25.0) 1 | 1 (25.0) 1 | 0 | 1 (25.0) 1 | 0 | |
| Endometrial hypertrophy | 1 (8.3) 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Postmenopausal haemorrhage | 0 | 0 | 0 | 1 (25.0) 1 | 0 | 0 | 0 | 0 | |
| Acne | 0 | 0 | 0 | 0 | 0 | 1 (25.0) 1 | 0 | 0 | |
| Rash papular | 0 | 1 (25.0) 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Hot flush | 0 | 1 (25.0) 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Part B. MAD | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Pooled | Pooled | 100 mg | 300 mg | 1000 mg | |||||
| Placebo | Prodrug | Cohort 1 | Cohort 2 | Cohort 3 | |||||
| Adverse events | (N = 6) | (N = 17) | (N = 6) | (N = 6) | (N = 5) | ||||
| Total, n (%) E | 6 (100.0) 16 | 16 (94.1) 40 | 6 (100.0) 24 | 6 (100.0) 8 | 4 (80.0) 8 | ||||
| Preferred Term, n (%) E | |||||||||
| Vision blurred | 0 | 1 (5.9) 1 | 0 | 0 | 1 (20.0) 1 | ||||
| Abdominal distension | 0 | 1 (5.9) 1 | 1 (16.7) 1 | 0 | 0 | ||||
| Abdominal pain | 0 | 1 (5.9) 2 | 1 (16.7) 2 | 0 | 0 | ||||
| Constipation | 5 (83.3) 5 | 8 (47.1) 9 | 6 (100.0) 6 | 0 | 2 (40.0) 3 | ||||
| Diarrhoea | 1 (16.7) 1 | 0 | 0 | 0 | 0 | ||||
| Dyspepsia | 0 | 1 (5.9) 1 | 1 (16.7) 1 | 0 | 0 | ||||
| Nausea | 1 (16.7) 2 | 1 (5.9) 1 | 0 | 1 (16.7) 1 | 0 | ||||
| Rectal haemorrhage | 1 (16.7) 1 | 0 | 0 | 0 | 0 | ||||
| Vomiting | 0 | 1 (5.9) 1 | 1 (16.7) 1 | 0 | 0 | ||||
| Chest discomfort | 1 (16.7) 1 | 0 | 0 | 0 | 0 | ||||
| Gastroenteritis | 1 (16.7) 1 | 0 | 0 | 0 | 0 | ||||
| Nasopharyngitis | 0 | 3 (17.6) 3 | 1 (16.7) 1 | 1 (16.7) 1 | 1 (20.0) 1 | ||||
| Oral herpes | 0 | 1 (5.9) 1 | 1 (16.7) 1 | 0 | 0 | ||||
| Ligament sprain | 0 | 1 (5.9) 1 | 1 (16.7) 1 | 0 | 0 | ||||
| Arthralgia | 0 | 1 (5.9) 1 | 1 (16.7) 1 | 0 | 0 | ||||
| Myalgia | 0 | 1 (5.9) 1 | 0 | 0 | 1 (20.0) 1 | ||||
| Cervicogenic headache | 0 | 1 (5.9) 1 | 0 | 1 (16.7) 1 | 0 | ||||
| Dizziness | 2 (33.3) 2 | 0 | 0 | 0 | 0 | ||||
| Headache | 2 (33.3) 2 | 9 (52.9) 9 | 4 (66.7) 4 | 3 (50.0) 3 | 2 (40.0) 2 | ||||
| Lethargy | 0 | 1 (5.9) 1 | 1 (16.7) 1 | 0 | 0 | ||||
| Urine odour abnormal | 0 | 1 (5.9) 1 | 1 (16.7) 1 | 0 | 0 | ||||
| Varicose veins vaginal | 0 | 1 (5.9) 1 | 0 | 1 (16.7) 1 | 0 | ||||
| Epistaxis | 0 | 1 (5.9) 1 | 1 (16.7) 1 | 0 | 0 | ||||
| Productive cough | 0 | 1 (5.9) 1 | 0 | 1 (16.7) 1 | 0 | ||||
| Hot flush | 1 (16.7) 1 | 2 (11.8) 2 | 2 (33.3) 2 | 0 | 0 | ||||
SAD = single ascending dose; C = Cohort; P = Period; N = number of subjects at risk; n = number of subjects with at least one event; E = number of events; MAD = multiple ascending dose
Among subjects treated with the prodrug, TEAEs were less frequent with increasing doses. No serious adverse events were reported. One subject experienced urinary tract infection after receiving 10 mg of the prodrug, leading to her withdrawal from the trial but this was not considered to be related to prodrug exposure. Headache was the most frequently reported TEAE (prodrug: n = 3; placebo: n = 5).
Following multiple doses, 16 (94.1%) subjects reported 40 TEAE following the prodrug doses and six (100%) subjects reported 16 TEAE following placebo (Table 6B). Headache (prodrug: n = 9; placebo: n = 2) and constipation (prodrug: n = 8; placebo: n = 5) were the most frequently reported adverse events. Three TEAEs (constipation) in two subjects (11.8%) following prodrug administration and two TEAEs (headache, constipation) in one subject (16.7%) following placebo administration were judged – in blinded condition – to be related to IMP. These events were considered CTCAE Grade 1. Among subjects treated with the prodrug, TEAEs were less frequent with increasing doses.
No serious adverse events were reported in the trial. No clinically significant changes were detected in the laboratory parameters or vital signs. The effects on ECGs were reported elsewhere 24.
Discussion
Preterm birth remains a significant risk for later disability. Current treatment strategies attempt to delay delivery as long as possible when indicated. A delay of 1 week in extremely preterm infants results in a decrease in neonatal morbidity in the region of 30% 25. A delay of 48 h allows administration of antenatal corticosteroids to accelerate fetal lung maturation and magnesium sulfate for fetal neuroprotection. It gives time to transfer the mother to a centre with neonatal intensive care facilities. Both measures reduce neonatal mortality and morbidity 26.
Short‐term tocolytic therapy has been demonstrated to be superior to placebo in prolonging pregnancy for at least 48 h 27. Currently available tocolytic therapies include betamimetics, calcium channel blockers, magnesium sulfate, oxytocin antagonists and prostaglandin synthesis inhibitors. Tocolytics decrease uterine contractions by acting on the uterine muscle through various mechanisms of action but have limited efficacy and some have restrictive safety issues. For example, betamimetics cause cardiac arrhythmias, hypotension, hyperglycaemia and pulmonary oedema; calcium channel blockers cause maternal hypotension and dizziness; magnesium sulfate causes flushing, respiratory suppression and cardiac arrest; oxytocin receptor blockers cause gastrointestinal disturbances; and prostaglandin inhibitors cause maternal gastrointestinal disturbances, oligohydramnios, fetal kidney dysfunction and premature constriction of the ductus arteriosus 28. None of the currently available treatments is recommended for dosing beyond 48 h.
A recent network meta‐analysis indicated that prostaglandin inhibitors have the highest probability of being the best therapy for preterm labour 27, which may be due to this pathway's potential not only to suppress uterine contractility but also to prevent cervical changes resulting from preterm labour and to inhibit inflammatory responses.
Prostaglandins have long been known to play a major role during pregnancy and parturition. Prostaglandins act directly or through modulation of other endocrine or paracrine factors involved in the preparation, activation and stimulation of uterine tissues that preclude the onset of labour 29, 30, 31, 32, 33. In uterine tissues, the levels of prostaglandins vary greatly and are under the control of their synthesis by cyclooxygenases isozymes (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1375&familyId=269&familyType=ENZYME and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1376&familyId=269&familyType=ENZYME) and specific http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=270 and their breakdown by http://www.guidetopharmacology.org/GRAC/FamilyIntroductionForward?familyId=58. They exert their effects through a number of http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=694 subtypes. In vitro and in vivo studies have demonstrated that prostaglandin F2α (PGF2α) causes contraction of the myometrium through activation of FP. This is known to result in raised intracellular calcium concentrations, which, in turn, leads to contraction of the uterine smooth cell muscle 34. Expression of the FP receptor has been shown to increase in rat myometrium with gestational ageing, thus enhancing myometrium sensitivity to the contractile actions PGF2α 8. Similarly, FP is expressed in term human myometrium 34, 35, 36. Upregulation of the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1628, an enzyme that breaks down collagen in cervical fibroblasts leading to cervical ripening, is also upregulated by PGF2α 37. Metalloproteinases can also downregulate their naturally‐occurring inhibitors in human term decidua, thus accelerating the breakdown of collagen and the rupture of membranes 38.
Tocolytic activity has been confirmed previously with other prostaglandin F2α receptor antagonists in rats, mice and sheep 10, 11. The prostaglandin F2α receptor antagonist OBE022 (prodrug) was designed to safely delay preterm birth. We confirmed that the prodrug markedly reduced spontaneous uterine contractions in pregnant rats without causing adverse effects on ductus arteriosus, kidneys or coagulation 39, 40.
The prodrug dosing regimens for this trial were selected based on nonclinical and emerging human data and covered the anticipated therapeutic dose range. The prodrug was safe and well tolerated across all dosing regimens. Significant changes in vital signs were not observed in this trial, despite the previously reported blood pressure elevation in response to topical application of FP agonists 41 and the association of tocolytic medication with hypotensive effects 42, 43. After oral administration, the prodrug was readily transformed into its metabolite OBE002. Plasma half‐life of OBE002 was compatible with once daily dosing. Dose proportionality for the prodrug was not confirmed. OBE002 levels were dose‐proportional following single doses, but not following multiple dosing. Comparison of dose accumulation ratios may indicate a limitation in metabolite formation with increasing prodrug doses. Due to the at least bi‐exponential elimination of OBE002, a rapid initial decline was followed by a much longer period with low concentrations. This may be explained by distribution‐redistribution of OBE002 from a small compartment.
The trial demonstrated no clinically relevant effect of food. Whilst the analyses for the prodrug and OBE002 were conducted across all three MAD levels, the number of possible comparisons for the prodrug was small due to plasma levels being below the level of quantification at the 100 mg dose level.
This early phase trial has some inherent limitations. Preterm labour is a critical situation that requires urgent medical intervention. The efficacy of a new medicine for this indication can only be tested in the target population once data supporting a positive benefit/risk are available. The data collected in this trial therefore originate from nonpregnant women. Postmenopausal women were chosen because at the time of trial initiation, the reproduction toxicity studies required to characterize the inherent risk of the prodrug during exposure of women of childbearing potential were not available. While significant PK differences between postmenopausal and nonpregnant women of child bearing potential are not expected, pregnancy‐associated changes in PK may occur. PK studies in pregnant women will be required to elucidate potential changes due to alterations in distribution and metabolism. This trial enables future trials in pregnant women.
Sample sizes of early phase trials are relatively small. Further investigation of the PK of the prodrug/OBE002 may be useful in light of the small sample size and the variability of PK parameters seen in this trial.
In conclusion, the novel, orally active, selective FP receptor antagonist OBE022 (prodrug) was safe and well tolerated in healthy postmenopausal women following administration for 7 days. Exposure to the prodrug and its active and stable metabolite OBE002 increased with dose and was compatible with once daily dosing, aiding administration in clinical practice. The results of this trial have enabled evaluation of this drug candidate in preterm labour patients and a clinical trial to characterize its safety, efficacy and PK profile in pregnant women with spontaneous preterm labour is ongoing (http://clinicaltrials.gov Identifier: NCT03369262).
Competing Interests
S.C., J.T. and U.L., are employees of Richmond Pharmacology Ltd. J.‐P.G., L.M. and O.P. are employees of ObsEva SA. OBE022 is being developed by ObsEva SA. ObsEva SA funded this clinical trial investigating OBE022.
The authors thank the bioanalytical site SGS Cephac Europe, in particular Nathalie Plaud and Gaëlle Remaud for their support and contributions to the trial.
Contributors
All authors conceived and designed the experiments. S.C., U.L. and J.T. performed the experiments. All authors read and revised the article and approved the final version.
Pohl, O. , Marchand, L. , Gotteland, J.‐P. , Coates, S. , Täubel, J. , and Lorch, U. (2018) Pharmacokinetics, safety and tolerability of OBE022, a selective prostaglandin F2α receptor antagonist tocolytic: A first‐in‐human trial in healthy postmenopausal women. Br J Clin Pharmacol, 84: 1839–1855. 10.1111/bcp.13622.
PI Statement: Ulrike Lorch was the Principal Investigator for this trial. Clinical trial registration number: EU Clinical Trials Register 2016–001957‐42
References
- 1. Liu L, Oza S, Hogan D, Chu Y, Perin J, Zhu J, et al Global, regional, and national causes of under‐5 mortality in 2000‐15: an updated systematic analysis with implications for the Sustainable Development Goals. Lancet 2016; 388: 3027–3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mwaniki MK, Atieno M, Lawn JE, Newton CRJC. Long‐term neurodevelopmental outcomes after intrauterine and neonatal insults: a systematic review. Lancet 2012; 379: 445–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Petrou S. The economic consequences of preterm birth during the first 10 years of life. BJOG 2005; 112 (Suppl 1): 10–15. [DOI] [PubMed] [Google Scholar]
- 4. Institute of Medicine . Preterm Birth: Causes, Consequences, and Prevention. Washington, DC, USA: National Academies Press, 2006. [Google Scholar]
- 5. Romero R, Dey SK, Fisher SJ. Preterm labor: one syndrome, many causes. Science 2014; 345: 760–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Crankshaw DJ, Gaspar V. Effects of prostanoids on the rat's myometrium in vitro during pregnancy. Biol Reprod 1992; 46: 392–400. [DOI] [PubMed] [Google Scholar]
- 7. Phillippe M, Saunders T, Basa A. Intracellular mechanisms underlying prostaglandin F2α‐stimulated phasic myometrial contractions. Am J Physiol 1997; 273: E665–E673. [DOI] [PubMed] [Google Scholar]
- 8. Al‐Matubsi HY, Eis ALW, Brodt‐Eppley J, MacPhee DJ, Lye S, Myatt L. Expression and localization of the contractile prostaglandin F receptor in pregnant rat myometrium in late gestation, labor, and postpartum . Biol Reprod 2001; 65: 1029–1037. [DOI] [PubMed] [Google Scholar]
- 9. Norton ME, Merrill J, Cooper BA, Kuller JA, Clyman RI. Neonatal complications after the administration of indomethacin for preterm labour. N Engl J Med 1993; 329: 1601–1607. [DOI] [PubMed] [Google Scholar]
- 10. Hirst JJ, Parkington HC, Young IR, Palliser HK, Peri KG, Olson DM. Delay of preterm birth in sheep by THG113.31, a prostaglandin F2α receptor antagonist. Am J Obstet Gynecol 2005; 193: 256–266. [DOI] [PubMed] [Google Scholar]
- 11. Chollet A, Tos EG, Cirillo R. Tocolytic effect of a selective FP receptor antagonist in rodent models reveals an innovative approach to the treatment of preterm labor. BMC Pregnancy Childbirth 2007; 7 (Suppl. 1): S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Moher D, Hopewell S, Schulz KF, Montori V, Gøtzsche PC, Devereaux PJ, et al CONSORT 2010 explanation and elaboration: updated guidelines for reporting parallel group randomised trials. BMJ 2010; 340: c869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. European Medicines Agency. Committee for Medicinal Products for Human Use . Guideline on strategies to identify and mitigate risks for first‐in‐human clinical trials with investigational medicinal products. July 2007. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002988.pdf (last accessed 19 February 2018).
- 14. US Department of Health and Human Services. Food and Drug Administration. Centre for Drug Evaluation and Research . Guidance for industry: estimating the maximum safe starting dose in initial clinical trials for therapeutics in adult healthy volunteers. July 2005. Available at: https://www.fda.gov/downloads/drugs/guidances/ucm078932.pdf (last accessed 19 February 2018).
- 15. US Department of Health and Human Services. Food and Drug Administration. Centre for Drug Evaluation and Research . Guidance for industry: food‐effect bioavailability and fed bioequivalence studies. December 2002. Available at https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm070241.pdf (last accessed 19 February 2018).
- 16. European Medicine Agency . Guideline on Bioanalytical Method Validation, EMEA, CHMP, EWP, July 2011. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/08/WC500109686.pdf (last accessed 19 February 2018).
- 17. Food and Drug Administration . Guidance for Industry. Bioanalytical Method Validation. May 2001. Available at https://www.fda.gov/downloads/Drugs/Guidance/ucm070107.pdf (last accessed 19 February 2018).
- 18. European Commission . Communication from the commission – detailed guidance on the collection, verification and presentation of adverse event/reaction reports arising from clinical trials on medicinal products for human use (CT‐3). 2011. Available at https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-10/2011_c172_01/2011_c172_01_en.pdf (last accessed 19 February 2018).
- 19. International conference on harmonisation guideline E2F “note for guidance on development safety update report”. Issued as EMA/CHMP/ICH/309348/2008. September 2011. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/09/WC500097061.pdf (last accessed 19 February 2018).
- 20. Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S, et al The IUPHAR/BPS guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 2018; 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA, et al The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 2017; 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E, et al The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 2017; 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. National Cancer Institute: common terminology criteria for adverse events (CTCAE) v4.03. Available at https://ctep.cancer.gov/protocoldevelopment/electronic_applications/ctc.htm#ctc_50 (last accessed 19 February 2018).
- 24. Täubel J, Lorch U, Coates S, Fernandes S, Foley P, Ferber G, et al Confirmation of the cardiac safety of PGF2α receptor antagonist OBE022 in a first‐in‐human study in healthy subjects, using intensive ECG assessments. Clin Pharmacol Drug Dev 2018; 10.1002/cpdd.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Haas DM, Imperiale TF, Kirkpatrick PR, Klein RW, Zollinger TW, Golichowski AM. Tocolytic therapy: a meta‐analysis and decision analysis. Obstet Gynecol 2009; 113: 585–594. [DOI] [PubMed] [Google Scholar]
- 26. van Vliet EOG, Boormans EM, de Lange TS, Mol BW, Oudijk MA. Preterm labor: current pharmacotherapy options for tocolysis. Expert Opin Pharmacother 2014; 15: 787–797. [DOI] [PubMed] [Google Scholar]
- 27. Haas DM, Caldwell DM, Kirkpatrick P, McIntosh JJ, Welton NJ. Tocolytic therapy for preterm delivery: systematic review and network meta‐analysis. BMJ 2012; 345: e6226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Haas DM, Benjamin T, Sawyer R, Quinney SK. Short‐term tocolytics for preterm delivery – current perspectives. Int J Womens Health 2014; 6: 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Challis JRG, Sloboda DM, Alfaidy N, Lye SJ, Gibb W, Patel FA, et al Prostaglandins and mechanisms of preterm birth. Reproduction 2002; 124: 1–17. [DOI] [PubMed] [Google Scholar]
- 30. Challis JRG, Matthews SG, Gibb W, Lye SJ. Endocrine and paracrine regulation of birth at term and preterm. Endocr Rev 2000; 21: 514–550. [DOI] [PubMed] [Google Scholar]
- 31. Olson DM. The promise of prostaglandins: have they fulfilled their potential as therapeutic targets for the delay of preterm birth? J Soc Gynecol Investig 2005; 12: 466–478. [DOI] [PubMed] [Google Scholar]
- 32. Slater DM, Zervou S, Thornton S. Prostaglandins and prostanoid receptors in human pregnancy and parturition. J Soc Gynecol Investig 2002; 9: 118–124. [PubMed] [Google Scholar]
- 33. Nathanielsz PW, Smith G, Wu W. Topographical specialization of prostaglandin function in late pregnancy and at parturition in the baboon. Prostaglandins Leukot Essent Fatty Acids 2004; 70: 199–206. [DOI] [PubMed] [Google Scholar]
- 34. Senior J, Marshall K, Sangha R, Clayton JK. In vitro characterization of prostanoid receptors on human myometrium at term pregnancy. Br J Pharmacol 1993; 108: 501–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Leonhardt A, Glaser A, Wegmann M, Hackenberg R, Nusing RM. Expression of prostanoid receptors in human lower segment pregnant myometrium. Prostaglandins Leukot Essent Fatty Acids 2003; 69: 307–313. [DOI] [PubMed] [Google Scholar]
- 36. Grigsby PL, Sooranna SR, Adu‐Amankwa B, Pitzer B, Brokman DE, Johnson MR, et al Regional expression of prostaglandin E2 and F2alpha receptors in human myometrium, amnion and choriodecidua with advancing gestation and labor. Biol Reprod 2006; 75: 297–305. [DOI] [PubMed] [Google Scholar]
- 37. Yoshida M, Nagawa N, Itoh H, Yura S, Takemura M, Wada Y, et al Prostaglandin F2α, cytokines and cyclic mechanical stretch augment matrix metalloproteinase‐1 secretion from cultured human uterine cervical fibroblast cells. Mol Hum Reprod 2002; 8: 681–687. [DOI] [PubMed] [Google Scholar]
- 38. Ulug U, Goldman S, Ben‐Shlomo I, Shalev E. Matrix metalloproteinase MMP‐2 and MMP‐9 and their inhibitor TIMP‐1, in human term decidua and fetal membranes: the effect of prostaglandin F2α and indomethacin. Mol Hum Reprod 2001; 7: 1187–1193. [DOI] [PubMed] [Google Scholar]
- 39. Pohl O, Méen M, Lluel P, Chollet A, Gotteland J‐P. Effect of OBE022, an oral and selective non‐prostanoid PGF2α receptor antagonist in combination with nifedipine for preterm labor: a study on RU486‐induced pregnant mice. Reprod Sci 2017; 24: 40A, S‐002. [Google Scholar]
- 40. Pohl O, Guillaume P, Bennett P, Legrand P, Bézivin S, Chollet A, et al OBE002, a prostaglandin F2α antagonist for the treatment of preterm labor, does not impair renal function in the newborn rabbit. 12th World Congress of Perinatal Medicine 03‐06 Nov. 2015. J Perinat Med 2015; 43: 1198. [Google Scholar]
- 41. Ohyama K, Kawakami H, Inuoue M. Blood pressure elevation associated with topical prostaglandin F2α analogs: an analysis of the different spontaneous adverse events report databases. Biol Pharm Bull 2017; 40: 616–620. [DOI] [PubMed] [Google Scholar]
- 42. Fabry I, De Paepe P, Kips J, Vermeersch S, Van Bortel L. Different effects of tocolytic medication on blood pressure and blood pressure amplification. Eur J Clin Pharmacol 2011; 67: 11–17. [DOI] [PubMed] [Google Scholar]
- 43. Luewan S, Mahathep R, Tongsong T. Hypotension in normotensive pregnant women treated with nifedipine as a tocolytic drug. Arch Gynecol Obstet 2011; 284: 527–530. [DOI] [PubMed] [Google Scholar]