

Abstract
Aims
To characterize the pharmacokinetics, pharmacodynamics and safety of esaxerenone, a mineralocorticoid receptor antagonist, in healthy adult Japanese men.
Methods
Double‐blind, placebo‐controlled, sequential, dose‐escalation studies were conducted in subjects randomized to receive oral once‐daily esaxerenone (ranges: 5–200 mg [single‐dose]; 10–100 mg over 10 days [multiple‐dose]) or placebo under fasting conditions. Plasma concentrations were analysed by liquid chromatograph–tandem mass spectrometry. Pharmacokinetic parameters were determined by noncompartment analysis. Plasma/urine levels of pharmacodynamic biomarkers for mineralocorticoid receptor activity were evaluated.
Results
In total, 48/48 and 39/40 subjects completed the single‐ and multiple‐dose studies, respectively. Exposures were generally dose‐proportional. The tmax, t1/2 and CL/F remained unchanged, independent of dose; the respective ranges were 1.5–4.0 h, 22.3–25.1 h, and 4.0–5.2 l h–1 (multiple‐dose study). Vz/F ranged from 136.5 to 283.7 l in the multiple‐dose study, and exposure reached steady state by day 4. The mean observed accumulation ratio, by dose, ranged from 1.36–1.98. The urinary Na+/K+ ratio increased after single‐dose administration; however, its relationship to the doses tested remains unclear. Plasma renin activity, active renin concentration and aldosterone concentration increased dose‐dependently. Although blood potassium levels increased dose‐dependently in the multiple‐dose study (reaching a maximum mean ± standard deviation of 4.63 ± 0.354 mmol l–1 in the 100‐mg group), no safety/tolerability‐related problems were detected in either study.
Conclusions
Exposure levels in healthy adults receiving esaxerenone were generally dose‐proportional. Dose‐dependent changes in plasma pharmacodynamic biomarkers for the mineralocorticoid receptor were identified during multiple‐dose treatment and support the pharmacological activity of esaxerenone. No important safety concerns were identified.
Keywords: esaxerenone, mineralocorticoid receptor antagonist, pharmacodynamics, pharmacokinetics, phase I, safety
What is Already Known about this Subject
Mineralocorticoid receptor (MR) antagonists are the drug of choice for primary hyperaldosteronism, and they elicit an antihypertensive effect in patients with low‐renin hypertension or refractory hypertension.
The currently licensed MR antagonist spironolactone is associated with sex hormone‐related effects such as gynaecomastia, whereas eplerenone is contraindicated for patients with diabetes, hyperkalaemia, microalbuminuria or proteinuria.
Esaxerenone is a nonsteroidal MR antagonist that has been shown to exert antihypertensive and cardiorenal protective effects in nonclinical studies.
What this Study Adds
Esaxerenone has a favourable pharmacokinetic profile for daily oral treatment, with rapid absorption, a long elimination half‐life (~20 h), generally dose‐proportional exposure (up to 200 mg in the single‐dose study and 100 mg day–1 in the multiple‐dose study), and steady state reached on day 4.
Esaxerenone is pharmacologically effective in humans, as demonstrated by the dose‐dependent increase in plasma renin activity.
No clinically significant safety concerns were associated with esaxerenone up to 200 mg (single dose), or 100 mg (multiple doses, once daily).
Introduction
Hypertension is the single most important risk for cerebrocardiovascular events. A well‐recognized public health problem, hypertension was estimated in 2010 to affect approximately 43 million Japanese individuals 1, 2. Management of hypertension aims to prevent or limit organ damage and cardiovascular disease by controlling blood pressure (BP). However, many people do not receive treatment for hypertension, or have poorly controlled BP where stricter control is required 1, 3. The causes of hypertension are multifactorial, and treatment often entails a combination of different antihypertensive drugs when monotherapy proves inadequate in controlling BP.
http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2872 acts on the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=626 (MR), a nuclear receptor expressed in tubular epithelial cells, to promote reabsorption of Na+ and excretion of urinary K+, regulating fluid volume and electrolytes in the blood 4, 5. MR antagonists are considered potassium‐sparing diuretics, exerting their antihypertensive effect by antagonizing the effect of aldosterone. The 2014 Japanese Society of Hypertension Guidelines for the Management of Hypertension recommend the addition of an aldosterone antagonist when BP is not adequately controlled with triple therapy (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=35&familyId=6&familyType=GPCR blocker or http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1613 inhibitor plus calcium antagonist plus thiazide diuretics) 1.
MR antagonists are the drug of choice for primary hyperaldosteronism and yield a satisfactory antihypertensive effect in patients with low‐http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2413 hypertension or refractory hypertension 1. In addition to their antihypertensive effects, the already marketed MR antagonists http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2875 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2876 have been shown to improve the prognosis of cardiac failure 6, 7, 8. However, because spironolactone is a nonselective MR antagonist with moderate affinity for http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=627 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=628, it is associated with sex hormone‐related effects such as gynaecomastia 9. Eplerenone has improved MR selectivity; however, it is contraindicated in patients with diabetes and microalbuminuria or proteinuria and patients with renal impairment owing to the incidence of hyperkalaemia 1, 10.
http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9894 (CS‐3150) is a nonsteroidal MR antagonist under development. Results from an in vitro study showed that esaxerenone inhibited binding of aldosterone to the MR, with no agonistic or antagonistic effects on http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=625, androgen or progesterone receptors, even at high concentrations 11. Owing to this high selectivity for the MR, esaxerenone is likely to be less associated with sex hormone‐related adverse effects than other drugs in the same class.
Here, we report findings from two phase I studies evaluating the pharmacokinetics (PK), pharmacodynamics (PD) and safety of esaxerenone in Japanese healthy adult male subjects after single and multiple oral administration.
Methods
Study participants
Participants were healthy Japanese adult volunteers meeting the following criteria (for both the single‐ and multiple‐dose studies): male subjects aged ≥20 and ≤45 years at the time of informed consent; body mass index of ≥18.5 and <25.0 kg m–2 at screening; willing to quit smoking during hospitalization; having systolic BP <140 mmHg and diastolic BP <90 mmHg (measured after the subject had rested for ≥3 min in a sitting position); and having pulse rate ≤99 beats min–1. The exclusion criteria (similar for both studies reported here) were as follows: any prior, serious disease considered to affect the study by the investigator or subinvestigator, including central nervous system, cardiovascular, respiratory, blood/haematopoietic, gastrointestinal, hepatic/renal, thyroid, pituitary gland or adrenal disease; hypersensitivity to a drug or idiosyncratic reactions (such as penicillin allergy); drug or alcohol dependence; positive infection test result (hepatitis B surface antigen, hepatitis C virus antibody, syphilis, HIV antibody); having had a total of ≥1200 ml of whole blood collected within 1 year, a total of ≥400 ml collected within 84 days, or a total of ≥200 ml collected within 28 days before screening, or having undergone plasmapheresis or platelet apheresis within 14 days before screening; having participated in another clinical study and received a study drug within 120 days before screening; having already participated in a clinical study of esaxerenone and received esaxerenone (i.e. in the case of the single‐dose study, participants could only participate in one step of the dose escalation); any finding considered clinically significant at screening, including headache, dizziness, weakness, abnormal electrocardiography finding, or a deviation in laboratory data from the reference range of the study centre (excluding those not considered to affect the study by the investigators); and being judged ineligible for participation in the study (for example, if visiting the study centre or complying with the administration instructions seemed difficult).
Study design and treatments
Both studies had a randomized, double‐blind, placebo‐controlled, sequential, dose‐escalation design. The studies received approval from the Osaka Pharmacology Clinical Research Hospital institutional review board. The single‐dose study protocol was approved on 14 December 2010 (control No. of the study site: 830PB), and the multiple‐dose study protocol was approved on 20 July 2011 (control No. of the study site: 830PC). Both studies were conducted in accordance with the ethical principles of the Declaration of Helsinki, Pharmaceutical Affairs Law, and the Ordinances Regarding Good Clinical Practice of the Ministry of Health, Labour, and Welfare, Japan. All subjects in the single‐dose study (JapicCTI No. 163473) and the multiple‐dose study (JapicCTI No. 163476) provided written informed consent ahead of enrolment in the study.
In both studies, subjects were required to fast for at least 10 h starting on the day before study drug administration and for 4 h following administration (the fasting following administration was restricted to days 1 and 10 in the multiple‐dose study). Study drugs were administered orally with 200 ml of water. No other beverages were permitted for 1 h before or 2 h after administration. Subjects rested in a sitting position for 3.5 h following drug administration (this was restricted to days 1 and 10 in the multiple‐dose study). Caffeinated drinks were prohibited during hospitalization, and subjects were only permitted food prepared by the study centre, which was provided at a predetermined time. Subjects were required to visit the study centre 6–8 days after the last study drug administration for a follow‐up examination.
Single‐dose study
Methodological details of the single‐dose study are provided in the Supplementary Methods.
Multiple‐dose study
The multiple‐dose study comprised four steps in which esaxerenone 10 mg, 20 mg, 50 mg, 100 mg or placebo was orally administered once daily as multiple doses under fasting conditions for 10 days. At each step, subjects were randomized to receive either esaxerenone or placebo in an 8:2 ratio (permuted block method). Study drugs were administered 1 h before breakfast, and subjects were asked not to have breakfast on days 1 and 10. At each step of the study, safety was evaluated before proceeding to the next step. The initial dose (10 mg) was selected based on the tolerated dose range of 5–200 mg in the single‐dose study, and the minimum dose at which PD biomarker changes were observed.
PK
Samples in the multiple‐dose study were collected before drug administration and at 1, 1.5, 2, 2.5, 3, 3.5, 4, 8 and 12 h on day 1 and before drug administration, and at 1, 1.5, 2, 2.5, 3, 3.5, 4, 8, 12, 24, 36, 48, 60 and 72 h after drug administration on day 10, with trough concentrations from day 2 to day 6 and day 8. Blood was collected from the antebrachial cutaneous vein of each subject into EDTA‐2K vacuum blood collection tubes, gently mixed by inversion and placed on ice. The plasma obtained from centrifugation at 1700 g for 10 min at 4°C was divided into two storage containers and kept frozen at –20°C until delivered to the laboratory for analysis. Esaxerenone and its internal standard (d7‐form) were extracted from plasma samples (50 μl) using a 96‐well solid‐phase extraction plate conditioned with 250 μl of acetonitrile and 250 μl of water. After washing with 500 μl of washing solution (acetonitrile/water, 10:90, v/v), the plate was eluted with 200 μl of a diluent (acetonitrile/water 50:50, v/v). A 20‐μl aliquot of the eluate was injected into the liquid chromatograph–tandem mass spectrometer. Chromatographic separation was performed using a CAPCELL PAK C18 MGIII (Shiseido Co., Ltd.; Tokyo, Japan) column (2.0 × 150 mm, 5 μm). Detection was performed using a Sciex API 4000 (AB SCIEX, Framingham, Massachusetts, USA) tandem mass spectrometer with TurboIonSpray source by electrospray ionization in the negative ion mode and multiple‐reaction monitoring of esaxerenone (m/z 465–365) and its internal standard (m/z 472–370). The within‐study assay precision was 3.7%, 3.7% and 3.5% for quality control samples of esaxerenone at 0.3, 4 and 80 ng ml–1, respectively. The assay accuracy was in the range of –1.0% to 0.3%, and the lower limit of quantification was 0.1 ng ml–1.
In the multiple‐dose study, the area under the concentration–time curve (AUC) over the dosing interval (AUCτ), maximum plasma concentration (Cmax), and time to reach maximum plasma concentration (tmax) were determined on days 1 and 10; terminal elimination half‐life (t1/2), apparent total body clearance at steady state (CLss/F), apparent volume of distribution based on the terminal phase (Vz/F), and observed accumulation ratio (Robs) were determined on day 10 only. Robs was calculated using AUC values by the following formula: Robs = AUCτ (Day 10) / AUCτ (Day 1).
PD
In the multiple‐dose study, PD biomarker measurements were performed on blood samples collected at 4 and 8 h after drug administration on day 1, and at 4, 8, and 24 h after drug administration on day 10 (after the final dose) with trough concentrations determined on days 2–5 and 10. Subjects rested in a supine position for 30 minutes before blood collection. PD biomarker measurements included plasma renin activity (PRA), active renin concentration (ARC), angiotensin II (AII) concentrations and plasma aldosterone concentrations (PAC), all of which were determined in plasma. Urine specimens were collected at specific serial intervals after administration on days 1 and 10 and between 0 and 24 h after administration on days 2 to 4. PD measurements in urine included volume, electrolyte (Na+ and K+) excretion, and Na+/K+ ratio. In the multiple‐dose study, the transtubular potassium gradient was also calculated using the following formula: [urine K+ concentration/serum K+ concentration]/[urine osmotic pressure/serum osmotic pressure]. Blood pressure was monitored throughout the study period.
Clinical safety and tolerability
The safety endpoints were adverse events (AEs), laboratory tests, 12‐lead electrocardiogram, physical measurements and vital signs. AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA/J Ver. 14.0) System Organ Class and Preferred Terms for the individual AEs.
Statistical analysis
The PK set for analysis comprised data from all enrolled subjects administered the study drug, for whom no major protocol violations were documented. Subjects given placebo were excluded. The PD sets for analysis matched those for the PK analysis, except in that subjects given placebo were not excluded. The safety analysis sets comprised data from all enrolled patients given the study drug. Data from subjects given placebo were pooled for each study and analysed. The following statistical software programs were used: WinNonlin Enterprise Version 5.2.1; Pharsight Knowledgebase Server™ Version 3.1, and WinNonlin Autopilot™ Ver 1.2.2 (all Pharsight Corporation); and SAS Version 9.1.3 (SAS Institute Japan Ltd.).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 12, and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/1 8 13, 14, 15.
Results
Subject disposition and baseline characteristics
In the single‐dose study, 48 subjects were randomized to receive the study drug, and all 48 completed the study. There were no withdrawals related to an AE. The safety and PD analysis sets comprised data from 48 subjects, whilst the PK analysis set comprised data from 36 subjects (the 12 subjects given placebo were excluded from the PK analysis). In the multiple‐dose study, 40 subjects were randomized to receive the study drug. One subject from the 10‐mg group was withdrawn after experiencing infectious gastroenteritis on day 7, and 39 subjects completed the study. The PK, PD and safety analysis sets comprised 32, 39 and 40 subjects, respectively; the eight subjects given placebo were excluded from the PK analysis set. The treatment and placebo groups were relatively well balanced in terms of demographic and PD characteristics at baseline across the administration groups in both studies (Tables S1 and S2).
PK
PK parameters and the time course of plasma esaxerenone concentrations for the single‐dose study are shown in Table S3 and Figure S1, respectively. Systemic exposure (based on both AUC and Cmax) increased generally in proportion to dose in the 5–200‐mg range, and tmax and t1/2 did not change across dose levels (ranges of 2.5–3.5 h and 18.7–22.9 h, respectively). The apparent total body clearance (CL/F) and Vz/F also remained constant regardless of dose.
Table 1 shows PK parameters on days 1 and 10 for the multiple‐dose study. It was observed that tmax (2.5–3.5 h) and t1/2 (22.32–25.05 h) on day 10 were similar to those of the single‐dose study (Table S3). Furthermore, CLss/F and Vz/F remained constant regardless of the dose and resembled those obtained for the single‐dose study. Systemic exposure to esaxerenone was greater on day 10 than day 1, yet plasma esaxerenone concentrations remained almost constant from day 4 onward (Figure 1). The mean observed accumulation ratio (Robs) ranged from 1.36 (10‐mg dose group) to 1.98 (20‐mg dose group). Drug accumulation during multiple‐dose treatment was consistent with predicted values based on the single‐dose PK profile of esaxerenone.
Table 1.
Pharmacokinetic parameters of esaxerenone on days 1 and 10 in the multiple‐dose study
| Dose (mg day –1 ) | 10 | 20 | 50 | 100 | |
|---|---|---|---|---|---|
| Day 1 | n | 8 | 8 | 8 | 8 |
| Cmax (ng ml–1) | 161.9 (25.9) | 220.5 (34.8) | 489.5 (134.8) | 858.6 (196.3) | |
| tmax (h) | 2.5 (1.5, 4.0) | 2.5 (1.5, 3.0) | 3.8 (2.0, 4.0) | 2.5 (1.5, 4.0) | |
| AUCτ (ng h ml–1) | 1722 (210) | 2651 (455) | 6294 (1168) | 11 300 (1817) | |
| Day 10 | n | 7 | 8 | 8 | 8 |
| Cmax (ng ml–1) | 174.4 (27.7) | 393.4 (78.8) | 744.8 (125.9) | 1416 (326.6) | |
| tmax (h) | 2.5 (1.5, 4.0) | 3.0 (1.0, 4.0) | 2.8 (1.5, 4.0) | 3.5 (1.5, 4.0) | |
| AUCτ (ng h ml–1) | 2353 (511) | 5224 (1117) | 10 520 (1259) | 20 170 (3967) | |
| t1/2 (h) | 25.1 (5.4) | 24.2 (6.3) | 22.4 (2.5) | 22.3 (4.4)a | |
| CLss/F (l h–1) | 4.41 (0.90) | 3.99 (0.87) | 4.81 (0.58) | 5.21 (1.2)a | |
| Vz/F (l) | 154.6 (15.8) | 136.5 (32.4) | 283.6 (146.1) | 168.3 (54.7)a | |
| Robs | 1.36 (0.23) | 1.98 (0.36) | 1.71 (0.29) | 1.82 (0.44) |
Values are expressed as the arithmetic mean (standard deviation). For tmax, the median (minimum, maximum) is provided.
Data for seven subjects; one subject was excluded for use of concomitant medication to treat an adverse event.
Cmax, maximum plasma concentration; tmax, time to reach maximum plasma concentration; AUCτ, area under the concentration‐time curve over the dosing interval; t1/2, terminal elimination half‐life; CLss/F, apparent total body clearance at steady state; Vz/F, volume of distribution based on the terminal phase; Robs, observed accumulation ratio calculated by AUCτ (Day 10) / AUCτ (Day 1).
Figure 1.
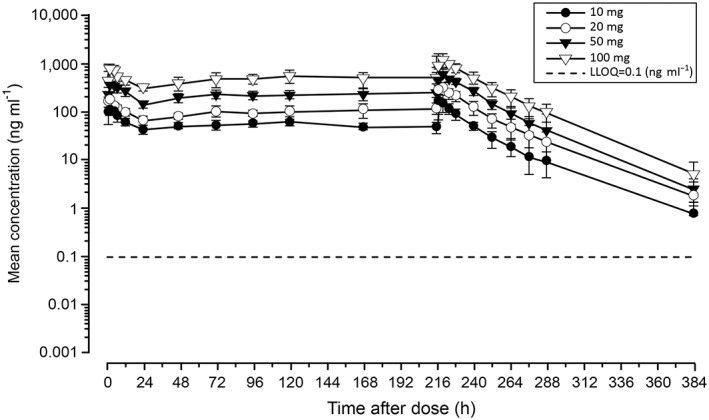
Plasma concentration–time profiles of esaxerenone for the multiple‐dose study. Data are presented as the arithmetic mean ± standard deviation. LLOQ, lower limit of quantitation
PD
In the single‐dose study, PRA, ARC and PAC increased after the administration of esaxerenone, whereas AII concentrations remained unchanged (Figures S2–S5). Both urinary Na+ excretion and Na+/K+ ratio increased after the administration of esaxerenone: the maximum mean changes from baseline in the Na+/K+ ratio were 5.041 (50 mg esaxerenone) and 5.041 (20 mg esaxerenone), compared with 0.508 and 1.530 in the placebo group during the 4–8 h and 8–12 h, respectively, after dosing. Of note, urinary K+ excretion decreased after the administration of esaxerenone. Figure S6 depicts the time course of change in urinary Na+/K+ ratio.
In the multiple‐dose study, PRA, ARC and PAC increased in a dose‐dependent manner (Figure 2A–C), whereas AII concentrations increased in the 50‐mg and 100‐mg groups (Figure 2D). The urinary Na+/K+ ratio increased from baseline by 1.078 and 1.430 in the 100‐mg esaxerenone group compared with –0.331 and –0.458 in the placebo group on days 1 and 2, respectively, returning to baseline levels or lower by day 10 (Figure 3). No obvious relationship was found between these changes and any of the esaxerenone doses. The time course of transtubular potassium gradient is plotted in Figure 4.
Figure 2.
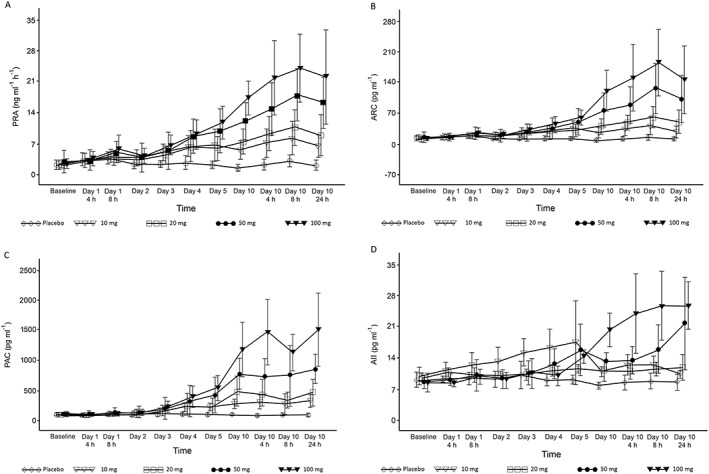
Time course changes in (A) plasma renin activity (PRA), (B) active renin concentration (ARC), (C) plasma aldosterone concentration (PAC), and (D) angiotensin II concentration (AII) in the multiple‐dose esaxerenone study. Data are presented as the arithmetic mean ± standard deviation
Figure 3.
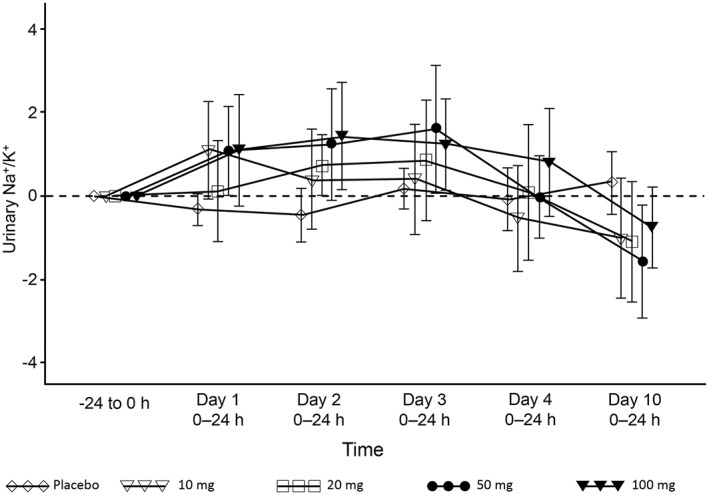
Time course of urinary Na+/K+ ratio (change from baseline) in the multiple‐dose esaxerenone study. Data are presented as the arithmetic mean ± standard deviation
Figure 4.
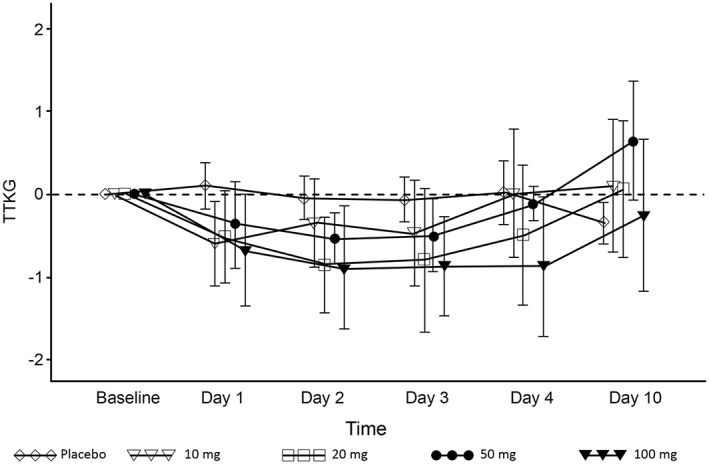
Time course of transtubular potassium gradient (TTKG; change from baseline) in the multiple‐dose esaxerenone study. Data are presented as the arithmetic mean ± standard deviation
No obvious relationship was found between changes in blood pressure (systolic or diastolic), and the esaxerenone dose in the single‐ or multiple‐dose studies.
Safety
The AEs occurring in the single‐dose study are shown in Table S4, and they affected two subjects in the esaxerenone group (n = 36) and another two in the placebo group (n = 12). For the esaxerenone group, AEs were noted as an increase in C‐reactive protein and creatine phosphokinase plasma levels. The latter occurred in the 20‐mg group and was considered related to the study drug. There were no serious AEs or AEs leading to withdrawal from the study; all four AEs were mild and resolved without treatment. Furthermore, no clinically significant changes in electrolytes or renal function biomarkers or clinically relevant findings for any other safety endpoint were noted.
In the multiple‐dose study, AEs occurred in 12 of the 32 subjects in the esaxerenone group, nine of which were considered to be related to the study drug (Table 2). No AEs were reported in the placebo group. The number of subjects experiencing AEs was highest in the 100‐mg group, although no obvious relationship was found between the incidence of AEs and the doses tested. Two of the affected subjects showed an increase in their alanine aminotransferase, aspartate aminotransferase and C‐reactive protein levels, besides gastroenteritis. All events reported, except for gastroenteritis (two subjects) and the increase in alanine aminotransferase (one subject in the 100‐mg group), were considered related to the study drug. The events gastroenteritis (one subject in the 10‐mg group) and rash (one subject in the 100‐mg group) were moderate, and both subjects recovered after treatment. All other events were mild and subsided without treatment. The subject experiencing moderate gastroenteritis was eventually withdrawn from the study, but the AE was considered unrelated to the study drug. No other withdrawals or serious AEs were reported.
Table 2.
Adverse events related to the study drug in the multiple‐dose study
| SOC | Placebo | 10 mg | 20 mg | 50 mg | 100 mg | Overall | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PT | n | 8 | 8 | 8 | 8 | 8 | 32 | |||||||
| Adverse event | 0 | (0.0%) | 1 | (12.5%) | 3 | (37.5%) | 1 | (12.5%) | 4 | (50.0%) | 9 | (28.1%) | ||
| Investigations | 0 | (0.0%) | 1 | (12.5%) | 3 | (37.5%) | 1 | (12.5%) | 3 | (37.5%) | 8 | (25.0%) | ||
| ALT increased | 0 | (0.0%) | 1 | (12.5%) | 2 | (25.0%) | 1 | (12.5%) | 0 | (0.0%) | 4 | (12.5%) | ||
| AST increased | 0 | (0.0%) | 1 | (12.5%) | 1 | (12.5%) | 1 | (12.5%) | 0 | (0.0%) | 3 | (9.4%) | ||
| Blood K + increased | 0 | (0.0%) | 0 | (0.0%) | 0 | (0.0%) | 0 | (0.0%) | 1 | (12.5%) | 1 | (3.1%) | ||
| Blood uric acid increased | 0 | (0.0%) | 0 | (0.0%) | 0 | (0.0%) | 0 | (0.0%) | 1 | (12.5%) | 1 | (3.1%) | ||
| CRP increased | 0 | (0.0%) | 0 | (0.0%) | 1 | (12.5%) | 0 | (0.0%) | 1 | (12.5%) | 2 | (6.3%) | ||
| Nervous system disorders | 0 | (0.0%) | 0 | (0.0%) | 0 | (0.0%) | 0 | (0.0%) | 1 | (12.5%) | 1 | (3.1%) | ||
| Dizziness | 0 | (0.0%) | 0 | (0.0%) | 0 | (0.0%) | 0 | (0.0%) | 1 | (12.5%) | 1 | (3.1%) | ||
| Skin and subcutaneous tissue disorders | 0 | (0.0%) | 0 | (0.0%) | 0 | (0.0%) | 0 | (0.0%) | 1 | (12.5%) | 1 | (3.1%) | ||
| Rash | 0 | (0.0%) | 0 | (0.0%) | 0 | (0.0%) | 0 | (0.0%) | 1 | (12.5%) | 1 | (3.1%) | ||
Adverse events were categorized using the Medical Dictionary for Regulatory Activities (MedDRA/J Ver. 14.0).
ALT, alanine aminotransferase; AST, aspartate aminotransferase; CRP, C‐reactive protein; n, number of subjects (incidence); PT, preferred term; SOC, system organ class
One subject in the 100‐mg group experienced an increase in serum K+ 1 day after the completion of study drug administration. However, this was mild and resolved by the following day. Serum K+ levels increased in a dose‐dependent manner from study day 2 to 72 h after the end of study drug administration (Figure 5), but decreased to similar levels as those in the placebo group over the 6–8 days following the last study drug administration. Mean serum K+ reached the highest level in the 100‐mg group (4.63 ± 0.354 mmol l–1). No particular concerns were observed in electrocardiography findings.
Figure 5.
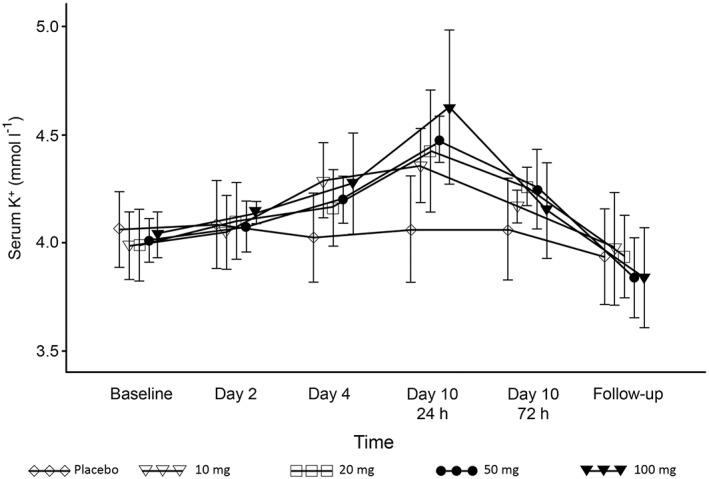
Time course of serum K+ concentration in the multiple‐dose esaxerenone study. Data are presented as mean ± standard deviation
Discussion
This report describes the PK, PD and safety characteristics of the MR antagonist esaxerenone administered in single and multiple doses to healthy Japanese males. Esaxerenone plasma concentrations increased generally in a dose‐proportional manner, and steady state was considered to have been reached by day 4 in the multiple‐dose study. Results for tmax, t1/2, and CL/F were comparable between the studies. Systemic exposure to esaxerenone in the multiple‐dose treatment was greater on day 10 compared with day 1. The t1/2 of esaxerenone is approximately 20 h, and it appears to be suitable for once‐daily dosing because efficacy is expected to be sustained throughout the day.
Certain biomarkers in blood and urine are helpful in assessing the potency of a novel MR antagonist and can be used to support appropriate dose regimens for future clinical studies. For blood markers of MR antagonism, a time‐ and dose‐dependent increase in aldosterone level was observed with chronic MR blockade by eplerenone in humans 16. Multiple administrations of eplerenone also resulted in dose‐dependent elevations in serum aldosterone in patients with mild‐to‐moderate hypertension 17. Both eplerenone (25–100 mg daily) and spironolactone (12.5–100 mg daily) for 12 months significantly increased PRA and PAC in patients with primary aldosteronism 18. Multiple administrations of esaxerenone for 10 days increased PRA, ARC and PAC in a time‐ and dose‐dependent manner in healthy male subjects (Figure 2A–C).
The urinary Na+/K+ ratio was also reported in humans as a translatable biomarker of MR antagonism 16. An increased urinary Na+/K+ ratio was observed following a single oral administration of 100, 300 or 1000 mg of eplerenone in healthy male subjects, although the effect of eplerenone on the urinary Na+/K+ ratio was not sustained with chronic dosing. A similar increase in urinary Na+/K+ ratio was observed from days 1 to 4 following multiple doses of esaxerenone, but the ratio returned to baseline levels or lower by day 10 (Figure 3). Although the reason for this phenomenon is not clear, potential compensatory mechanisms could account for this finding, as mentioned previously 16. A recent postmarketing surveillance study in Japan revealed that patients with essential hypertension who received 50–100 mg of eplerenone once daily experienced a clinically significant antihypertensive effect leading to favourable blood pressure control 19. The results of urinary Na+/K+ ratio from a single dose of eplerenone at 100 mg were almost comparable with those obtained from a single dose of esaxerenone 5 mg. Considering the plasma t1/2 and observed accumulation ratio (range, 1.36–1.98), esaxerenone may be sufficiently potent to elicit an adequate antihypertensive effect from a once‐daily dose lower than 5 mg in multiple administration. Furthermore, results from phase IIa and IIb studies (registered at Japic; JapicCTI‐121921 and JapicCTI‐152772, respectively) support the hypothesis that esaxerenone can elicit an antihypertensive effect at doses of ≤5 mg. Given the above, the maximum dose selected for the phase III study was 5 mg (registered at http://clinicaltrials.gov; NCT02890173).
In terms of the safety profile of esaxerenone, nine AEs were considered to be related to the study drug. These were, however, of mild or moderate intensity and subsided or resolved following treatment, and no relationship was established between the incidence of AEs and the doses tested in the two studies. Dose‐dependent increases in serum K+ levels were observed from day 2 up to 72 h following the end of drug administration, resuming to normal levels 6–8 days after the last administration. One subject in the 100‐mg group in the multiple‐dose study experienced an increase in serum K+ that exceeded the reference range. The reported AE was mild and transient, and the K+ level recovered to within the reference range. Nevertheless, this observation, along with reports of hyperkalaemia, electrolyte abnormality, and renal failure with eplerenone and spironolactone 10, 20, indicate that serum K+ should be carefully monitored in future longer‐term studies.
It should also be emphasized that, because of the subjects' characteristics (healthy young men) and the relatively short duration of the study, the antihypertensive effects of esaxerenone could not be appropriately measured; in fact, no changes in BP were observed in subjects with normal BP. Furthermore, a study with longer duration of intervention will be necessary to address any potential AEs or toxicities that may be time‐dependent. Finally, it should be noted that effects on K+ levels may be even more pronounced in patients with impaired renal function or those at increased risk of hyperkalaemia; this could not be evaluated in studies enrolling only healthy volunteers.
A phase III randomized trial to evaluate the safety and antihypertensive effects of esaxerenone 2.5 and 5 mg compared with eplerenone in essential hypertension has recently been completed (registered at http://clinicaltrials.gov; NCT02890173). The current study has demonstrated safety at considerably higher doses than those used in the phase III study.
Competing Interests
H.F. received research funding for this study from Daiichi Sankyo Co., Ltd. M.K., T.S., A.M., F.K. and H.I. are employees of Daiichi Sankyo.
The studies described in this report were funded by Daiichi Sankyo Co., Ltd. The authors would like to thank Rita Moreira da Silva, PhD, of Edanz Medical Writing for providing medical writing services.
Supporting information
Table S1 Demographics and baseline characteristics of participants across the safety, pharmacokinetic and pharmacodynamic analysis sets in the single‐dose study
Table S2 Demographics and baseline characteristics of participants in the safety analysis set in the multiple‐dose study
Table S3 Pharmacokinetic parameters of esaxerenone in the single‐dose study
Table S4 Adverse events reported in the single‐dose study
Figure S1 Plasma concentration–time profiles of esaxerenone for the single‐dose study. Data are presented as the arithmetic mean ± standard deviation. LLOQ, lower limit of quantification
Figure S2 Time course of plasma renin activity (PRA) in the single‐dose esaxerenone study. Data are presented as the arithmetic mean ± standard deviation
Figure S3 Time course of active renin concentration (ARC) in the single‐dose esaxerenone study. Data are presented as the arithmetic mean ± standard deviation
Figure S4 Time course of plasma aldosterone concentration (PAC) in the single‐dose esaxerenone study. Data are presented as the arithmetic mean ± standard deviation
Figure S5 Time course of angiotensin II concentration (AII) in the single‐dose esaxerenone study. Data are presented as the arithmetic mean ± standard deviation
Figure S6 Time course of urinary Na+/K+ ratio (change from baseline) in the single‐dose esaxerenone study. Data are presented as the arithmetic mean ± standard deviation
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Kato, M. , Furuie, H. , Shimizu, T. , Miyazaki, A. , Kobayashi, F. , and Ishizuka, H. (2018) Single‐ and multiple‐dose escalation study to assess pharmacokinetics, pharmacodynamics and safety of oral esaxerenone in healthy Japanese subjects. Br J Clin Pharmacol, 84: 1821–1829. 10.1111/bcp.13616.
Clinical Trial registration: Single‐dose study: JapicCTI No. 163473; multiple‐dose study: JapicCTI No. 163476.
References
- 1. Shimamoto K, Ando K, Fujita T, Hasebe N, Higaki J, Horiuchi M, et al The Japanese Society of Hypertension guidelines for the management of hypertension (JSH 2014). Hypertens Res 2014; 37: 253–390. [DOI] [PubMed] [Google Scholar]
- 2. Health Service Bureau, Ministry of Health, Labour and Welfare . Outline for the results of the National Health and Nutrition Survey Japan, 2006. Tokyo: Ministry of Health, Labour and Welfare 2008. Available at: http://www.mhlw.go.jp/houdou/2008/04/dl/h0430-2c.pdf [in Japanese] (last accessed 23 May 2018).
- 3. Hozawa A, Ohkubo T, Kikuya M, Yamaguchi J, Ohmori K, Fujiwara T, et al Blood pressure control assessed by home, ambulatory and conventional blood pressure measurements in the Japanese general population: the Ohasama study. Hypertens Res 2002; 25: 57–63. [DOI] [PubMed] [Google Scholar]
- 4. Shibata S, Nagase M, Fujita T. Activation mechanism of aldosterone and its receptor In: Annual Review, Diabetes/Metabolism/Endocrine 2010, eds Terauchi Y, Ito H, Ishibasi S. Shinjuku‐Ku: Chugai Igakusha, 2010; 196–205 [in Japanese]. [Google Scholar]
- 5. Young MJ, Funder JW. Mineralocorticoid receptors and pathophysiological roles for aldosterone in the cardiovascular system. J Hypertens 2002; 20: 1465–1468. [DOI] [PubMed] [Google Scholar]
- 6. RALES Investigators . Effectiveness of spironolactone added to an angiotensin‐converting enzyme inhibitor and a loop diuretic for severe chronic congestive heart failure (the Randomized Aldactone Evaluation Study [RALES]). Am J Cardiol 1996; 78: 902–907. [DOI] [PubMed] [Google Scholar]
- 7. Pitt B, Remme W, Zannad F, Neaton J, Martinez F, Roniker B, et al Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 2003; 348: 1309–1321. [DOI] [PubMed] [Google Scholar]
- 8. Zannad F, McMurray JJ, Krum H, van Veldhuisen DJ, Swedberg K, Shi H, et al Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011; 364: 11–21. [DOI] [PubMed] [Google Scholar]
- 9. Sica DA. Pharmacokinetics and pharmacodynamics of mineralocorticoid blocking agents and their effects on potassium homeostasis. Heart Fail Rev 2005; 10: 23–29. [DOI] [PubMed] [Google Scholar]
- 10. Selara tablets 25 mg/ Selara tablets 50 mg/ Selara tablets 100 mg [package insert]. Pfizer Japan Inc., Tokyo, Japan 2011. [in Japanese].
- 11. Arai K, Homma T, Morikawa Y, Ubukata N, Tsuruoka H, Aoki K, et al Pharmacological profile of CS‐3150, a novel, highly potent and selective non‐steroidal mineralocorticoid receptor antagonist. Eur J Pharmacol 2015; 761: 226–234. [DOI] [PubMed] [Google Scholar]
- 12. Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S, et al The IUPHAR/BPS guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 2018; 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alexander SPH, Cidlowski JA, Kelly E, Marrion NV, Peters JA, Faccenda E, et al The Concise Guide to PHARMACOLOGY 2017/18: Nuclear hormone receptors. Br J Pharmacol 2017; 174: S208–S224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA, et al The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 2017; 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E, et al The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 2017; 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Eudy RJ, Sahasrabudhe V, Sweeney K, Tugnait M, King‐Ahmad A, Near K, et al The use of plasma aldosterone and urinary sodium to potassium ratio as translatable quantitative biomarkers of mineralocorticoid receptor antagonism. J Transl Med 2011; 9: 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Weinberger MH, Roniker B, Krause SL, Weiss RJ. Eplerenone, a selective aldosterone blocker, in mild‐to‐moderate hypertension. Am J Hypertens 2002; 15: 709–716. [DOI] [PubMed] [Google Scholar]
- 18. Karashima S, Yoneda T, Kometani M, Ohe M, Mori S, Sawamura T, et al Comparison of eplerenone and spironolactone for the treatment of primary aldosteronism. Hypertens Res 2016; 39: 133–137. [DOI] [PubMed] [Google Scholar]
- 19. Takahashi S, Hiramatsu M, Hotta S, Watanabe Y, Suga O, Endo Y, et al Safety and antihypertensive effect of Selara (eplerenone): results from a postmarketing surveillance in Japan. Int J Hypertens 2016; 2016: 5091951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vukadinović D, Lavall D, Vukadinović AN, Pitt B, Wagenpfeil S, Böhm M. True rate of mineralocorticoid receptor antagonists‐related hyperkalemia in placebo‐controlled trials: a meta‐analysis. Am Heart J 2017; 188: 99–108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Demographics and baseline characteristics of participants across the safety, pharmacokinetic and pharmacodynamic analysis sets in the single‐dose study
Table S2 Demographics and baseline characteristics of participants in the safety analysis set in the multiple‐dose study
Table S3 Pharmacokinetic parameters of esaxerenone in the single‐dose study
Table S4 Adverse events reported in the single‐dose study
Figure S1 Plasma concentration–time profiles of esaxerenone for the single‐dose study. Data are presented as the arithmetic mean ± standard deviation. LLOQ, lower limit of quantification
Figure S2 Time course of plasma renin activity (PRA) in the single‐dose esaxerenone study. Data are presented as the arithmetic mean ± standard deviation
Figure S3 Time course of active renin concentration (ARC) in the single‐dose esaxerenone study. Data are presented as the arithmetic mean ± standard deviation
Figure S4 Time course of plasma aldosterone concentration (PAC) in the single‐dose esaxerenone study. Data are presented as the arithmetic mean ± standard deviation
Figure S5 Time course of angiotensin II concentration (AII) in the single‐dose esaxerenone study. Data are presented as the arithmetic mean ± standard deviation
Figure S6 Time course of urinary Na+/K+ ratio (change from baseline) in the single‐dose esaxerenone study. Data are presented as the arithmetic mean ± standard deviation
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item