

Abstract
Aims
To determine the safety, tolerability, pharmacokinetics and pharmacodynamics of the Janus kinase 1‐selective inhibitor, PF‐04965842.
Methods
This was a phase 1, first‐in‐human, randomized, double‐blind, placebo‐controlled, combination single‐ and multiple‐dose escalation, parallel design study in healthy subjects (http://clinicaltrials.gov, NCT01835197). Subjects received a single dose of placebo or 3, 10, 30, 100, 200, 400 or 800 mg PF‐04965842 (single ascending dose phase) and placebo or 30 mg once daily (QD), 100 mg QD, 200 mg QD, 400 mg QD, 100 mg twice daily (BID) or 200 mg BID PF‐04965842 for 10 consecutive days (multiple ascending dose phase). The primary objective was to determine the safety and tolerability of PF‐04965842.
Results
Seventy‐nine subjects were randomized and received study treatments. There were no deaths or serious adverse events. The most frequent treatment‐emergent adverse events were headache (n = 13), diarrhoea (n = 11) and nausea (n = 11). PF‐04965842 was absorbed rapidly (median time at which maximum plasma concentration occurred generally ≤1 h following either single‐ or multiple‐dose administration) and eliminated rapidly (mean t ½ 2.8−5.2 h after 10 days of QD or BID administration in the multiple ascending dose phase). Increases in maximum plasma concentration and area under the concentration–time curve were dose proportional up to 200 mg (single or total daily doses) with an apparent trend towards greater than proportional increases with higher doses. Less than 4.4% of the dose was recovered unchanged in urine. Changes in pharmacodynamic biomarkers were consistent with the known effects of Janus kinase signalling inhibition.
Conclusions
These results support further evaluation of PF‐04965842 for clinical use in patients with inflammatory diseases.
Keywords: inflammatory diseases, JAK1 inhibitor, PF‐04965842, pharmacodynamics, pharmacokinetics, phase 1
What is Already Known about this Subject
There remains a need for alternative treatment options for patients with inflammatory diseases such lupus, psoriasis and atopic dermatitis.
Selective inhibition of Janus kinase (JAK)‐1 may provide a more favourable safety profile than pan‐JAK inhibitors by avoiding the effects of JAK2 inhibition.
What this Study Adds
This first‐in‐human study assessed the safety, tolerability, pharmacokinetics and pharmacodynamics of PF‐04965842, a potent JAK1‐selective inhibitor.
The favourable safety, tolerability, pharmacokinetics and pharmacodynamics profiles from this study support further evaluation of PF‐04965842 for clinical use.
Introduction
Disease‐modifying antirheumatic drugs are commonly used for the treatment of inflammatory diseases, including rheumatoid arthritis and psoriasis 1, 2, 3. Despite their proven efficacy and tolerability, there remains a need for alternative treatment options because some patients do not respond, stop responding after an initial positive response or are intolerant to these treatments 4, 5. Furthermore, an oral treatment may be preferable for some patients.
The Janus kinase (JAK) family of cytoplasmic tyrosine kinases [JAK1, JAK2, JAK3 and tyrosine kinase (TYK) 2] mediates signal transduction critical for leucocyte activation, proliferation, survival and function, via interactions with Type 1 and Type II cytokine receptors 6, 7. JAK1 pairs with JAK3 to mediate γ‐common cytokine signalling and also with JAK2 and TYK2 to transmit the signals of additional cytokines important in inflammation and immune responses. Key cytokines implicated in the pathophysiology of multiple inflammatory diseases, including interferon (IFN)‐α and IFN‐β (JAK1/TYK2 dependent), IFN‐γ (JAK1/JAK2 dependent), interleukin (IL)‐6 (JAK1/JAK2/TYK2 dependent) and IL‐21 (JAK1/JAK3 dependent), require JAK1 for signal transduction, suggesting that JAK1 inhibition could be efficacious in a broad range of inflammatory diseases 8, 9, 10.
Several inhibitors with reportedly greater selectivity for JAK1 than JAK2 or JAK3 are in clinical development, including: filgotinib 11, INCB039110 12, GSK2586184 13 and ABT‐494 14. Several other JAK inhibitors that are less selective for JAK1 are also currently approved or being tested for use in inflammatory diseases (e.g. ruxolitinib, baricitinib and tofacitinib) 8, 15, 16.
The aim of this study was to determine the safety and tolerability, as well as pharmacokinetics (PK) and pharmacodynamics (PD), of PF‐04965842, a JAK1‐selective inhibitor 17, in healthy adult subjects.
Methods
Study design
This was a phase 1, first‐in‐human, within‐cohort, randomized, double‐blind, placebo‐controlled, combination single‐ and multiple‐dose escalation, parallel design study in healthy subjects (http://clinicaltrials.gov, NCT01835197). The study was at a single centre in the USA and was subject blind, investigator blind and sponsor open (double‐blind with respect to within‐group assignments and single‐blind for between‐group assignments). Subjects were allocated randomization numbers and received treatment according to sponsor‐generated randomization codes. Study treatments were provided by the sponsor as bulk powders for preparation of suspensions at the study centre. An unblinded site pharmacist prepared the treatments and provided them to the blinded administrator. Treatments were given to subjects in individual dosing containers that were labelled with the randomization number. The study was approved by the Institutional Review Board IntegReview in Austin (TX, USA; Protocol B7451001, approved 25 April 2013) and was conducted in compliance with the ethical principles outlined in the Declaration of Helsinki (2008) and guidelines for Good Clinical Practice issued by the International Conference on Harmonisation (1996). All subjects gave written informed consent prior to study initiation.
The sample size was based on clinical and pharmacological considerations (no statistical considerations). There were nine cohorts comprising about eight subjects each (Figure 1). Subjects in each cohort were randomized in a 3:1 ratio of PF‐04965842:placebo in block sizes of 4. In the single ascending dose (SAD) phase, western subjects in Cohorts 1–7 received a single dose of placebo or 3, 10, 30, 100, 200, 400 or 800 mg PF‐04965842 in a dose escalation format. Subjects remained in the treatment facility from day –1 to day 5 in the SAD phase and returned for a follow‐up visit on day 8. The multiple ascending dose (MAD) phase was initiated at least 14 days after the start of the SAD phase, and subjects in Cohorts 3–7 received placebo or 30, 100 or 200 mg once daily (QD) or 100 or 200 mg twice daily (BID) PF‐04965842 for 10 consecutive days (the evening dose for the BID cohorts was not administered on day 10). In the MAD phase, subjects remained in the treatment facility until day 14 and returned for a follow‐up visit on day 28. Cohort 8 consisted of Japanese subjects who received a single dose of placebo or 800 mg PF‐04965842 in the SAD phase followed by 200 mg BID PF‐04965842 for 10 consecutive days in the MAD phase. Cohort 9 (western subjects) was added to expand the range of QD dosing; subjects received placebo or 400 mg QD PF‐04965842 for 10 consecutive days. During both the SAD and the MAD periods, escalation to subsequent dose levels occurred at a minimum of 7 days only if the last dose was well tolerated and after satisfactory review of the available safety and PK data. Dose escalation and stopping rules are described in Supplementary Methods 1.
Figure 1.
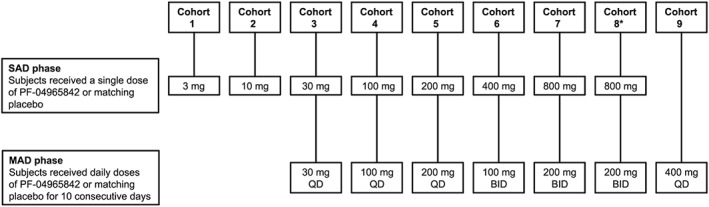
Study design. Subjects in each cohort (n = ~8 per cohort) were randomized 3:1 to PF‐04965842:placebo. BID, twice daily; MAD, multiple ascending dose; QD, once daily; SAD, single ascending dose. *Japanese subjects
The primary endpoint was to determine the safety and tolerability of PF‐04965842. The secondary endpoint was to characterize the PK and PD of PF‐04965842.
Subjects
Eligible subjects included healthy males and postmenopausal/non‐childbearing females aged 18–55 years, with a body mass index of 17.5–30.5 kg m–2 and a total body weight >50 kg. Exclusion criteria included, but were not limited to: evidence or history of abnormal cardiovascular, pulmonary, renal or hepatic function; white blood cell count below 4.5 × 103 mm−3; haematocrit value below 38% (males) or 33% (females); platelet counts <150 × 103 mm−3; red blood cell, reticulocyte and lymphocyte counts that fell outside the reference range; any other laboratory parameter with an abnormal value and judged to be clinically significant; treatment with an investigational drug within 30 days or five half‐lives (whichever is longer) preceding the first dose of study medication; a history of tuberculosis or active/latent/inadequately treated infection; and history of cancer. Subjects were screened within 28 days before administration of the study drug. Concomitant medication was prohibited unless required for the treatment of adverse events (AEs) and the use of all concomitant medication was recorded.
Safety and tolerability
Subjects abstained from all food and drink (except water) at least 4 h prior to safety laboratory evaluations and 10 h prior to lipid evaluation. All AEs were monitored and recorded daily while the subjects were at the treatment facility, at follow‐up visits and as needed in addition to the scheduled timepoints. Vital signs were measured at screening, immediately prior to dosing and at regular intervals postdose. Blood samples for haematology and laboratory tests were taken during screening, on days 0, 2, 3, 5 and 8 during the SAD phase, and days 0, 4, 6, 8, 10 (at 0, 2 and 6 h postdose), 11, 14 and 28 during the MAD phase. Samples for lipid evaluations were taken during screening, on days 0, 2 and 5 during the SAD phase, and days 0, 4, 10, 11 and 28 during the MAD phase. Urine samples for creatinine clearance were collected over 24 h on days 0 and 1 during the SAD phase and on days 0 and 10 of the MAD phase.
PK
Subjects abstained from all food and drink 8 h prior to the collection of predose PK samples (water was permitted until 1 h prior to dosing). Blood samples to assess PF‐04965842 concentrations and for the PK endpoints were collected at 0 (predose), 0.5, 1, 2, 4, 6, 8, 12, 16, 24, 36 and 48 h postdose during the SAD phase. During the MAD phase, blood samples were collected prior to the morning dose on days 1, 2, 4, 6, 8, and then at 0, 0.5, 1, 2, 4, 6, 8, 12, 16, 24, 48 and 72 h after the last dose on day 10. These were collected in tubes containing dipotassium ethylenediamine tetra‐acetic acid, centrifuged at approximately 1700 g for about 10 min at 4°C, and stored at –70°C within 1 h of collection.
During the MAD phase, urine samples for PK analysis were collected on day 0 (predose blank) and day 10 (0–12 h for BID doses and 0–24 h for QD doses). The total volume was measured and recorded at the end of each 12‐h or 24‐h urine collection period. A 7–8 ml aliquot was withdrawn for measurement of drug concentrations and was frozen at –20°C within 1 h of the end of the collection interval.
Plasma and urine samples were analysed for PF‐04965842 concentrations using validated, sensitive and specific liquid chromatography tandem mass spectrometric methods. The lower limit of quantification for PF‐04965842 was 1 ng ml–1 for plasma concentrations and 10 ng ml–1 for urine concentrations. For plasma samples, the between‐day assay accuracy, expressed as percent relative error, for quality control (QC) concentrations ranged from –2.8% to 5.0% for the low, medium, high and diluted QC samples. Assay precision, expressed as the between‐day percent coefficient of variation (%CV) of the mean estimated concentrations of QC samples ranged from 6.2% to 13.0% for low (3 ng ml–1), medium (60 ng ml–1), high (750 ng ml–1) and diluted (5000 ng ml–1) concentrations. For urine samples, the between‐day assay accuracy ranged from –1.2% to 5.0% for the low, medium, high and diluted QC samples. Assay precision ranged from 1.3% to 6.5% for low (30 ng ml–1), medium (600 ng ml–1), high (7500 ng ml–1) and diluted (50 000 ng ml–1) concentrations.
PK parameters were determined with an internally developed and validated software system using standard non‐compartmental analysis of concentration–time data. Maximum plasma concentration (Cmax) was observed directly from data and time at which Cmax occurred (Tmax) was defined as the time of the first occurrence of Cmax. The t 1/2 was estimated using linear regression of the log‐linear concentration–time curve. Areas under the concentration–time curve to time τ (AUCτ), extrapolated to infinity (AUCinf) and to the time of the last quantifiable concentration (AUClast) were estimated using linear/log trapezoidal methods.
PD
PD biomarkers evaluated in this study included interferon gamma‐induced protein 10 (IP‐10) and high‐sensitivity C‐reactive protein (hsCRP) measurements from serum and neutrophils, platelets, reticulocytes, lymphocyte subset analysis [CD16+CD56+ natural killer (NK) cells, CD19+ B cells, and T cells including CD3+ (total), CD3+CD4+ helper T cells, CD3+CD8+ cytotoxic T cell subsets] from whole blood. Detailed information on sample collection and analysis of PD parameters are shown in Supplementary Methods 2.
Results
Subjects
Seventy‐nine subjects were randomized and received study treatments from 13 May 2013 to 09 June 2014 (Table 1). Eleven subjects discontinued from the study (n = 4 and n = 7 from the SAD and MAD phases, respectively). Reasons for discontinuations, as judged by the investigator, were AEs related to study drug (n = 3), an AE unrelated to study drug (n = 1), lost to follow‐up (n = 2) and other (n = 5). All subjects were included in the safety and PD analysis while only subjects treated with PF‐04965842 were included in the PK analysis.
Table 1.
Subject disposition
| Number of subjects | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SAD phase | MAD phase | ||||||||||||||
| PBO | PF‐04965842 | PBO | PF‐04965842 | ||||||||||||
| 0 mg | 3 mg | 10 mg | 30 mg | 100 mg | 200 mg | 400 mg | 800 mg | 0 mg | 30 mg QD | 100 mg QD | 200 mg QD | 400 mg QD | 100 mg BID | 200 mg BID | |
| Assigned to study treatment, n = 79 | |||||||||||||||
| Treated | 16 | 6 | 6 | 6 | 7 | 6 | 6 | 16 | 14 | 6 | 5 | 6 | 8 | 6 | 14 |
| Completed | 16 | 6 | 6 | 6 | 5 | 6 | 6 | 14 | 14 | 5 | 5 | 6 | 5 | 6 | 11 |
| Discontinued | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 2 | 0 | 1 | 0 | 0 | 3 | 0 | 3 |
| Relation to study drug not defined | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 3 |
| Lost to follow‐up | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| Other | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 3 |
| Relation to study drug | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 2 | 0 | 0 |
| AE | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 2 | 0 | 0 |
| Not related to study drug | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| AE | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Analysed for safety | |||||||||||||||
| AEs | 16 | 6 | 6 | 6 | 7 | 6 | 6 | 16 | 14 | 6 | 5 | 6 | 8 | 6 | 14 |
| Laboratory data | 16 | 6 | 6 | 6 | 7 | 6 | 6 | 16 | 14 | 6 | 5 | 6 | 8 | 6 | 14 |
| Analysed for PK | |||||||||||||||
| Parameter | 0 | 6 | 6 | 6 | 7 | 6 | 6 | 15 | 0 | 5 | 5 | 6 | 6 | 6 | 11 |
| Concentration | 0 | 6 | 6 | 6 | 7 | 6 | 6 | 15 | 0 | 6 | 5 | 6 | 7 | 6 | 13 |
| Analysed for PD | 16 | 6 | 6 | 6 | 7 | 6 | 6 | 16 | 14 | 6 | 5 | 6 | 8 | 6 | 14 |
Discontinuations were attributed to the last study treatment received. AE, adverse event; BID, twice daily; MAD, multiple ascending dose; PBO, placebo; PD, pharmacodynamics; PK, pharmacokinetics; QD, once daily; SAD, single ascending dose.
Baseline demographic characteristics are shown in Table 2. All but one of the subjects were male, the mean age of the subjects was 37.9 years (range 21–55 years), the mean weight was 80.8 kg (range 51.0–102.7 kg), and the mean body mass index was 26.4 kg m–2 (range 17.6–30.5 kg m–2).
Table 2.
Baseline demographic characteristics
| PBO | PF‐04965842 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 mg (SAD) (n = 4) | 0 mg (SAD) then 0 mg QD or BID (MAD) (n = 12) | 0 mg QD (MAD) (n = 2) | 3 mg (SAD) (n = 6) | 10 mg (SAD) (n = 6) | 30 mg (SAD) then 30 mg QD (MAD) (n = 6) | 100 mg (SAD) then 100 mg QD (MAD) (n = 7) | 200 mg (SAD) then 200 mg QD (MAD) (n = 6) | 400 mg QD (MAD) (n = 8) | 400 mg (SAD) then 100 mg BID (MAD) (n = 6) | 800 mg (SAD) then 200 mg BID (MAD) (n = 16) | |
| Sex, n | |||||||||||
| Male | 4 | 12 | 2 | 6 | 6 | 6 | 7 | 6 | 8 | 6 | 15 |
| Female | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| Age, y | 34.8 (7.0) | 38.0 (6.2) | 35.0 (18.4) | 39.7 (9.9) | 39.0 (10.6) | 43.0 (12.3) | 32.0 (12.4) | 35.8 (5.7) | 36.0 (8.6) | 36.2 (7.1) | 41.2 (8.4) |
| Race, n | |||||||||||
| White | 1 | 3 | 0 | 3 | 5 | 2 | 4 | 0 | 2 | 1 | 0 |
| Black | 1 | 5 | 1 | 2 | 0 | 4 | 2 | 4 | 4 | 3 | 4 |
| Asian | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 10 |
| Other | 2 | 2 | 1 | 1 | 1 | 0 | 1 | 2 | 2 | 2 | 2 |
| Weight, kg | 87.7 (10.1) | 78.1 (11.3) | 94.5 (2.5) | 81.1 (6.4) | 84.1 (5.6) | 80.2 (9.3) | 78.4 (9.6) | 83.3 (14.0) | 85.1 (8.1) | 86.3 (5.1) | 74.2 (13.3) |
| BMI, kg m –2 | 26.1 (2.9) | 26.3 (3.6) | 29.0 (0.7) | 26.7 (3.4) | 26.6 (2.2) | 26.4 (2.3) | 25.3 (2.5) | 27.2 (2.4) | 28.1 (1.6) | 27.6 (2.5) | 24.9 (3.6) |
| Height, cm | 183.5 (9.1) | 172.3 (4.8) | 180.5 (4.9) | 175.0 (7.6) | 177.8 (3.9) | 174.2 (5.6) | 175.9 (7.6) | 174.3 (7.3) | 173.8 (4.8) | 177.0 (7.3) | 172.4 (6.8) |
Values are mean (standard deviation) unless otherwise stated. BID, twice daily; BMI, body mass index; MAD, multiple ascending dose; PBO, placebo; QD, once daily; SAD, single ascending dose.
Safety and tolerability
There were no deaths or serious AEs during this study. During the SAD phase, 24 subjects had a total of 64 treatment‐emergent AEs (TEAEs), 36 of which were considered to be treatment‐related. During the MAD phase, 35 subjects had a total of 97 TEAEs, 59 of which were considered to be treatment‐related. Most of the TEAEs were mild in severity and occurred at higher frequencies in subjects who received the highest doses in both the SAD (800 mg) and MAD phases (400 mg QD and 200 mg BID). The most frequent TEAEs (Table 3) were headache (n = 13), diarrhoea (n = 11) and nausea (n = 11). Seventeen mild/moderate infections were reported. Eight of these were considered to be treatment‐related by the investigator and occurred predominantly in the higher dose cohorts.
Table 3.
Treatment‐emergent adverse events occurring in two or more subjects in any dosing group
| Number of subjects with treatment‐emergent adverse events: all causalities (treatment‐related) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SAD phase | MAD phase | ||||||||||||||
| PBO | PF‐04965842 | PBO | PF‐04965842 | ||||||||||||
| AEs by SOC and MedRA* preferred term | 0 mg n = 16 | 3 mg n = 6 | 10 mg n = 6 | 30 mg n = 6 | 100 mg n = 7 | 200 mg n = 6 | 400 mg n = 6 | 800 mg n = 16 | 0 mg n = 14 | 30 mg QD n = 6 | 100 mg QD n = 5 | 200 mg QD n = 6 | 400 mg QD n = 8 | 100 mg BID n = 6 | 200 mg BID n = 14 |
| Gastrointestinal disorders | 0 | 0 | 0 | 0 | 0 | 1 (1) | 0 | 10 (10) | 4 (3) | 1 (0) | 1 (1) | 3 (3) | 5 (4) | 2 (1) | 7 (5) |
| Abdominal discomfort | 0 | 0 | 0 | 0 | 0 | 1 (1) | 0 | 2 (2) | 0 | 0 | 0 | 0 | 1 (1) | 0 | 2 (2) |
| Upper abdominal pain | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (2) | 0 | 1 (1) |
| Constipation | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (1) | 2 (1) | 0 | 0 | 1 (1) | 1 (1) | 1 (1) | 0 |
| Diarrhoea | 0 | 0 | 0 | 0 | 0 | 1 (1) | 0 | 1 (1) | 1 (1) | 0 | 0 | 1 (1) | 3 (2) | 1 (0) | 3 (3) |
| Flatulence | 0 | 0 | 0 | 0 | 0 | 1 (1) | 0 | 3 (3) | 0 | 0 | 0 | 0 | 0 | 0 | 2 (2) |
| Nausea | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 (3) | 1 (1) | 0 | 1 (1) | 2 (2) | 3 (3) | 0 | 1 (1) |
| Vomiting | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 (4) | 0 | 0 | 0 | 0 | 1 (1) | 0 | 0 |
| Infections and infestations | 1 (0) | 0 | 0 | 1 (0) | 0 | 1 (0) | 0 | 2 (2) | 1 (0) | 1 (0) | 0 | 1 (0) | 2 (2) | 1 (0) | 4 (4) |
| Upper respiratory tract infection | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1) | 1 (0) | 0 | 0 | 0 | 1 (1) | 1 (0) | 2 (2) |
| Musculoskeletal and connective tissue disorders | 1 (0) | 0 | 0 | 0 | 0 | 0 | 0 | 4 (1) | 0 | 0 | 0 | 0 | 1 (0) | 1 (0) | 1 (0) |
| Pain in extremity | 1 (0) | 0 | 0 | 0 | 0 | 0 | 0 | 3 (0) | 0 | 0 | 0 | 0 | 1 (0) | 0 | 1 (0) |
| Nervous system disorders | 0 | 0 | 0 | 0 | 1 (0) | 0 | 2 (2) | 5 (4) | 1 (0) | 0 | 1 (1) | 4 (3) | 2 (2) | 1 (1) | 3 (3) |
| Dysgeusia | 0 | 0 | 0 | 0 | 0 | 0 | 2 (2) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1) | 0 |
| Headache | 0 | 0 | 0 | 0 | 0 | 0 | 1 (0) | 4 (3) | 0 | 0 | 1 (1) | 3 (1) | 2 (2) | 0 | 2 (2) |
| Somnolence | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (2) | 0 | 0 | 0 | 2 (2) | 0 | 0 | 1 (1) |
| Respiratory, thoracic and mediastinal disorders | 1 (0) | 0 | 0 | 0 | 0 | 0 | 0 | 2 (0) | 0 | 0 | 0 | 1 (0) | 2 (0) | 1 (1) | 0 |
| Nasal congestion | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (0) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Skin and subcutaneous tissue disorders | 1 (0) | 1 (0) | 0 | 0 | 1 (0) | 1 (0) | 0 | 4 (1) | 2 (1) | 0 | 0 | 2 (0) | 1 (0) | 1 (1) | 6 (3) |
| Acne | 0 | 0 | 0 | 0 | 0 | 1 (0) | 0 | 1 (1) | 0 | 0 | 0 | 2 (0) | 0 | 1 (1) | 3 (3) |
| Ecchymosis | 0 | 0 | 0 | 0 | 1 (0) | 0 | 0 | 2 (0) | 0 | 0 | 0 | 0 | 0 | 0 | 3 (0) |
| Skin irritation | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (0) | 0 | 0 | 0 | 0 | 0 | 0 | 2 (0) |
Includes all data collected since the first dose of study drug. Subjects were counted only once per treatment in each row.
MedRA version 17.0 coding was applied. AE, adverse event; BID, twice daily; MAD, multiple ascending dose; MedDRA, Medical Dictionary for Regulatory Activities; PBO, placebo; QD, once daily; SAD, single ascending dose; SOC, system organ class.
There were no temporary discontinuations or dose reductions due to AEs reported in this study. Four subjects discontinued from the study due to AEs. In the SAD phase, one subject in the PF‐04965842 100 mg group experienced a second‐degree atrioventricular block (attributed to a pre‐existing condition and not considered treatment‐related by the investigator) and one subject in the PF‐04965842 800 mg group experienced an AE of vomiting (considered to be treatment‐related by the investigator). In the MAD phase, one subject in the PF‐04965842 400 mg QD group experienced an AE of respiratory syncytial virus infection and another experienced vomiting (both AEs were moderate in severity and were reported as treatment‐related by the investigator).
Overall, 25 and 40 subjects had laboratory abnormalities during the SAD and MAD phases, respectively, none of which were considered clinically relevant. The most frequently reported laboratory abnormalities were decreased mean platelet volume (<0.9× lower limit of normal, n = 10) and positive urine blood tests (≥1, n = 5) in the SAD phase, and decreased reticulocyte counts (<0.5× lower limit of normal, n = 10), increased lymphocyte counts (>1.2× upper limit of normal, n = 8) and decreased mean platelet volume (n = 7) in the MAD phase. Some subjects had decreases in haematological laboratory values, such as haemoglobin (Grade 1 criteria for haemoglobin, n = 7; Figure S1A), platelet counts (Grade 1 criteria for thrombocytopenia, n = 9; Figure S1B) or white blood cell counts (Grade 2 criteria for leukopenia, n = 4), that met Grade 1 or Grade 2 Common Terminology Criteria for Adverse Events (version 4.0) severity, but these were not associated with any clinical symptoms. All subjects had normal bilirubin and aspartate aminotransferase/alanine aminotransaminase levels.
During the SAD phase, there were increases and decreases in median percent change from baseline in total cholesterol and high‐density lipoprotein (HDL) cholesterol levels; the greatest increase at 24 h postdose was in the PF‐04965842 800 mg group (10% for both) while the greatest decrease was in the PF‐04965842 10 mg group (2% and 9%, respectively). At 24 h postdose, the PF‐04965842 100 mg group had the greatest increase (17%) in low‐density lipoprotein (LDL) cholesterol level. During the MAD phase, the PF‐04965842 200 mg BID group had the highest increase in median percent change from baseline in total cholesterol (22%) and HDL cholesterol (27%), and the 100 mg BID group had the highest increase in LDL cholesterol (42%) at day 10 predose (i.e., day 10, 0 h), as shown in Figure S2.
At 24 h postdose on day 1, median changes from baseline in creatinine clearance ranged between –13 and 10 ml min–1 during the SAD phase and between –53 and 8 ml min–1 during the MAD phase, with no subjects outside the normal range.
In the SAD phase, four subjects had sporadic measurements of absolute supine systolic blood pressure values <90 mmHg (placebo, n = 1; PF‐04965842 800 mg, n = 3) and three subjects had absolute supine diastolic blood pressure values <50 mmHg (placebo, n = 1; PF‐04965842 800 mg, n = 2). In the MAD phase, three subjects had occasional absolute supine systolic blood pressure values <90 mmHg (n = 1 each in the PF‐04965842 100 mg QD, 400 mg QD and 200 mg BID treatment groups) and seven subjects had absolute supine diastolic blood pressure values <50 mmHg (PF‐04965842 100 mg QD, n = 1; PF‐04965842 400 mg QD, n = 1; PF‐04965842 200 mg BID, n = 1; PF‐04965842 200 mg QD, n = 2; placebo, n = 2). One subject in the SAD phase (placebo group) and three subjects in the MAD phase (one each in the placebo, PF‐04965842 30 mg QD, and 100 mg QD treatment groups) had a maximum QT corrected for heart rate using Fridericia's method (QTcF) interval of 450–<480 ms. These changes were sporadic and not significant clinically.
PK
PF‐04965842 PK parameters and plasma concentration vs. time profiles are shown in Table 4 and Figure 2, respectively. In the SAD phase (Table 4a, Figure 2A) PF‐04965842 was absorbed rapidly following single doses of 3 mg to 200 mg (median Tmax < 1 h), and more slowly at the higher doses (median Tmax 1.5−4.0 h for 400 mg and 800 mg). Following attainment of Cmax, a monophasic decline was observed at the lower doses of 3 mg to 30 mg (mean t ½ 2.0−2.5 h) while a biphasic decline was observed at doses of 100 mg to 800 mg (mean t ½ 3.6−5.3 h). Plasma PF‐04965842 Cmax appeared to increase proportionally across the entire dose range, while increases in AUCinf were greater than proportional at single doses of 400 and 800 mg. The difference was most notable in western subjects who received 800 mg. While Cmax was similar in western and Japanese subjects who received 800 mg, the geometric mean AUCinf was 26% higher in western subjects than that observed in Japanese subjects.
Table 4.
Pharmacokinetic parameters: (a) single‐dose phase; (b) multiple‐dose phase
| (a) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Parametera, units | 3 mg | 10 mg | 30 mg | 100 mg | 200 mg | 400 mg | 800 mg | 800 mg | 800 mg |
| Cohort 1 | Cohort 2 | Cohort 3 | Cohort 4 | Cohort 5 | Cohort 6 | Cohort 7 | Cohort 8 | Cohorts 7 + 8 | |
| N = 6 | N = 6 | N = 6 | N = 7 | N = 6 | N = 6 | N = 6 | N = 10 | N = 16 | |
| n 1 , n 2 | 6, 5 | 6, 5 | 6, 6 | 7, 5 | 6, 6 | 6, 6 | 5, 5 | 10, 9 | 15, 14 |
| AUC last , ng h ml –1 | 36.8 (84) | 135.1 (40) | 386.6 (44) | 1226 (60) | 2878 (29) | 10 150 (28) | 27 480 (35) | 19 810 (53) | 22 090 (49) |
| AUC inf , ng h ml –1 | 48.8 (64) | 142.8 (44) | 392.3 (43) | 1465 (41) | 2886 (29) | 10 180 (28) | 27 540 (35) | 21 860 (43) | 23 740 (40) |
| Cmax, ng ml–1 | 17.1 (27) | 47.0 (21) | 142.5 (50) | 459.7 (53) | 1072 (35) | 2291 (42) | 3819 (26) | 3660 (48) | 3712 (41) |
| T max , h | 0.634 (0.517–1.27) | 0.750 (0.500–1.00) | 0.792 (0.500–1.08) | 0.550 (0.500–1.03) | 0.767 (0.500–1.17) | 1.50 (0.517–4.03) | 4.03 (2.00–4.30) | 2.02 (1.00–4.00) | 3.92 (1.00–4.30) |
| t ½ , h | 2.09 ± 0.87 | 1.94 ± 0.55 | 2.53 ± 1.32 | 3.59 ± 2.08 | 3.89 ± 2.20 | 3.55 ± 1.11 | 5.27 ± 2.92 | 4.67 ± 1.57 | 4.88 ± 2.06 |
| CL/F, l h–1 | 61.44 (65) | 70.02 (44) | 76.41 (43) | 68.34 (41) | 69.31 (29) | 39.32 (28) | 29.00 (35) | 36.61 (43) | 33.69 (41) |
| V z /F, l | 172.5 (26) | 190.1 (18) | 256.2 (63) | 318.5 (66) | 343.0 (46) | 193.3 (39) | 197.1 (77) | 235.0 (64) | 220.7 (66) |
| (b) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Parameterb, units | 30 mg QD | 100 mg QD | 200 mg QD | 100 mg BID | 200 mg BID | 200 mg BID | 200 mg BID | 400 mg QD |
| Cohort 3 | Cohort 4 | Cohort 5 | Cohort 6 | Cohort 7 | Cohort 8 | Cohorts 7 + 8 | Cohort 9 | |
| N = 6 | N = 5 | N = 6 | N = 6 | N = 5 | N = 9 | N = 14 | N = 8 | |
| n 1 , n 2 | 5, 5 | 5, 5 | 6, 6 | 6, 6 | 5, 5 | 6, 6 | 11, 11 | 6, 6 |
| Plasma | ||||||||
| AUC τ , ng h ml –1 | 500 (38) | 1977 (47) | 4277 (34) | 3107 (31) | 13 680 (27) | 8754 (30) | 10 720 (36) | 18 230 (24) |
| C max , ng ml –1 | 161 (50) | 700 (31) | 1199 (23) | 773.0 (36) | 2691 (8) | 2294 (27) | 2467 (22) | 3334 (17) |
| T max , h | 0.55 (0.50–1.03) | 0.55 (0.50–2.02) | 1.05 (1.02–1.05) | 0.759 (0.50–1.02) | 1.02 (0.50–1.07) | 0.50 (0.50–1.00) | 0.50 (0.50–1.07) | 0.767(0.50–2.05) |
| t ½ , h | 2.76 ± 1.11 | 2.99 ± 0.76 | 3.06 ± 0.24 | 5.03 ± 2.34 | 5.17 ± 1.60 | 3.64 ± 0.83 | 4.34 ± 1.42 | 4.85 ± 1.86 |
| CL/F, l h –1 | 59.93 (38) | 50.56 (47) | 46.76 (34) | 32.16 (31) | 14.61 (27) | 22.86 (29) | 18.65 (36) | 21.99 (24) |
| V z /F, l | 223.0 (43) | 212.9 (43) | 206.0 (29) | 212.3 (65) | 104.7 (42) | 117.8 (31) | 111.6 (35) | 145.4 (34) |
| Urine | ||||||||
| Ae τ % | 1.03 (74) | 1.17 (99) | 1.42 (49) | NCc | 4.38 (51) | 2.60 (25) | 3.30 (47) | 2.64 (37) |
| CL r , l h –1 | 0.618 (33) | 0.590 (41) | 0.665 (20) | NCc | 0.639 (40) | 0.596 (13) | 0.615 (27) | 0.579 (23) |
Notes (a): Data for one subject in Cohort 7 (800 mg) were excluded due to vomiting.
Values are geometric mean (geometric percent coefficient of variation) for all except: median (range) for Tmax; arithmetic mean ± standard deviation for t ½.
AUClast, area under the concentration–time profile from time zero to the time of the last quantifiable concentration; AUCinf, area under the concentration–time profile from time zero extrapolated to infinite time; CL/F, apparent clearance; Cmax, maximum plasma concentration; N, number of subjects in the treatment group; n1, number of subjects contributing to the mean; n2, number of subjects where t ½, AUCinf, CL/F and Vz/F were determined; t ½, terminal half‐life; Tmax, time at which Cmax occurred; Vz/F, apparent volume of distribution.
Notes (b): Data for one subject in the 400 mg QD (Cohort 9) group were excluded due to vomiting.
Values are geometric mean (percent coefficient of variation) for all except: median (range) for Tmax; arithmetic mean ± SD for t ½.
Protocol deviation in urine collection for Cohort 6.
Aeτ %, cumulative percent of dose recovered unchanged in urine over the dosing interval τ; AUCτ, area under the concentration–time curve from time zero to time τ, the dosing interval, where τ = 24 h for QD dosing and 12 h for BID dosing; BID, twice daily; CL/F, apparent clearance; CLr, renal clearance; Cmax, maximum plasma concentration; N, number of subjects in the treatment group (start of multiple dosing on day 1); n1, number of subjects contributing to the mean (completed multiple dosing through day 10); n2, number of subjects where t ½ and Vz/F were determined; NC, not calculated; QD, once daily; SD, standard deviation; t ½, terminal half‐life; Tmax, time at which Cmax occurred; Vz/F, apparent volume of distribution.
Figure 2.
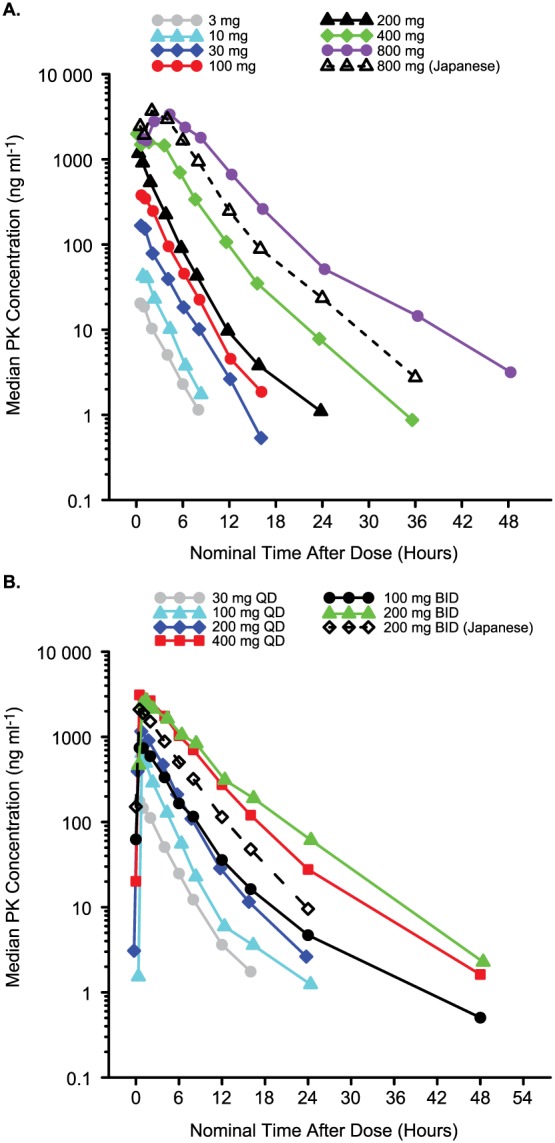
Plasma PF‐04965842 concentration vs. time profiles. (A) SAD phase. Data for 1 subject in the 800 mg (western) group were excluded due to vomiting. (B) MAD phase. Data shown are for day 10 predose (0 h) up to 72 h postdose. Data for 1 subject in the 400 mg QD group were excluded due to vomiting. BID, twice daily; MAD, multiple ascending dose; PK, pharmacokinetic; QD, once daily; SAD, single ascending dose
Multiple‐dose PK parameters are shown in Table 4b and concentration–time profiles are illustrated in Figure 2B. On day 10, PF‐04965842 was absorbed rapidly (median Tmax ≤ 1 h) across the entire range of doses, from a total daily dose of 30 mg (30 mg QD) up to 400 mg (200 mg BID or 400 mg QD). Following attainment of Cmax, a biphasic decline was observed following all but the lowest dose and the mean t ½ ranged from 2.8 h to 5.2 h. Plasma PF‐04965842 Cmax and AUCτ both showed a trend towards greater than proportional increases with increasing daily dose. Western subjects who received 200 mg BID had the highest dose‐normalized AUCτ and Cmax of any dose level (data not shown), including Japanese subjects who received the same dosing regimen. Geometric mean Cmax and AUCτ following multiple‐dose administration were 17% and 56% higher, respectively, in western subjects than Japanese subjects. Similarly, t ½ was also longer in western vs. Japanese subjects (5.2 h vs. 3.6 h, respectively). However, dose normalized AUCτ for western subjects with the same total daily dose (400 mg QD) was similar to that observed for the Japanese subjects who received 200 mg BID, with geometric mean values of 45.51 ng h ml–1 mg–1and 43.74 ng h ml–1 mg–1, respectively. Urinary recovery of PF‐04965842 was low (1.0−4.4%) and renal clearance averaged about 0.6 l h–1.
Steady state was reached by day 4 for QD dosing and day 6 for BID dosing, based on comparison of predose trough concentrations across days. Geometric mean values for the observed accumulation ratio (Rac: multiple‐dose AUCτ/single‐dose AUCτ) were 1.3−1.5 for QD dosing and 1.3−2.3 for BID dosing. Geometric mean values for the steady‐state accumulation ratio (Rss: multiple‐dose AUCτ/single‐dose AUCinf) were consistently >1 (1.3−1.5 for QD dosing and 1.2−2.0 for BID dosing), suggesting that there may be greater than linear increase in PF‐04965842 exposure with multiple‐dose administration. However, based on Rac, the increase in exposure after 10 days of multiple dosing is less than three‐fold for any of the dosing regimens studied.
PD
In the MAD phase, dose‐dependent decreases from baseline in circulating IP‐10 (Figure 3A) and hsCRP (Figure 3B) levels were observed as early as day 2 with all PF‐04965842 doses. The maximal decreases were at predose on day 10, with 56% decrease in the 100 mg BID group and 54% in the 200 mg BID group in IP‐10 and 85% decrease in hsCRP in the 100 mg BID group.
Figure 3.
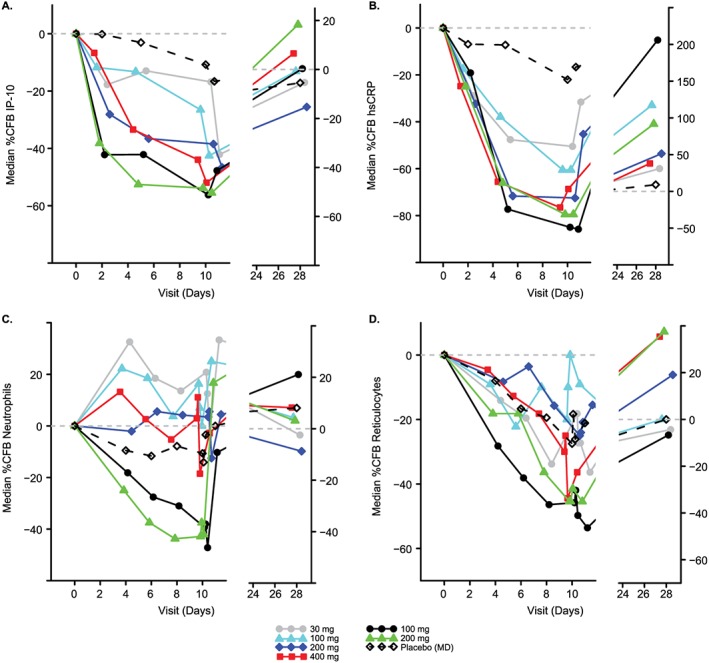
Median percent change from baseline in pharmacodynamic parameters over time (A) IP‐10 levels; (B) hsCRP levels; (C) neutrophil counts; (D) reticulocyte counts. BID, twice daily; hsCRP, high‐sensitivity C‐reactive protein; IP‐10, interferon gamma‐induced protein 10; MAD, multiple ascending dose; MD, multiple dose; QD, once daily; SAD, single ascending dose
PF‐04965842 100 mg BID and 200 mg BID doses led to decreases from baseline in neutrophil counts from day 4 through day 10, but quickly recovered to or above the baseline levels following the termination of dosing on day 10, and this recovery was observed as early as day 11 (24 h post last dose of PF‐04965842; Figure 3C). The maximum observed decrease of –47% occurred on Day 10, 6 h postdose in the 100 mg BID treatment group. Decreases from baseline in reticulocytes were observed in the 100 mg BID and 200 mg BID PF‐04965842 doses relative to placebo; the maximal reduction was observed at 24 h postdose on day 10 in the 100 mg BID group (54%; Figure 3D).
Fluorescence‐activated cell sorting (FACS) assessment of lymphocyte subsets showed varying effects on NK, T and B cells following PF‐04965842 doses compared with placebo in the MAD phase. Generally, there were decreases in CD16+CD56+ NK cells, minimal changes in total CD3+ T cells, small increases in CD3+CD4+ T cells, small decreases in CD3+CD8+ T cells, and increases in CD19+ B cells (data not shown).
Discussion
This study in healthy subjects investigated the safety, tolerability, PK and PD properties of PF‐04965842, a JAK1‐selective inhibitor. There were no deaths, serious AEs, temporary discontinuations or dose reductions due to AEs. The most frequent TEAEs were headache, diarrhoea and nausea, which have previously been reported in healthy volunteers treated with other JAK inhibitors 18, 19, 20. TEAEs were more common with the higher doses (single dose 800 mg and multiple dose 200 mg BID and 400 mg QD), but most were mild in severity. Occurrence of anaemia, thrombocytopenia and infections is of special interest in studies of JAK inhibitors 8, 13, 21. There were no incidences of anaemia or thrombocytopenia during this study and the incidence of infections was low. Changes in lipid parameters were observed in this study, as has been reported in some studies with other JAK inhibitors 12, 13, 14, 22, 23. The most dramatic effects on HDL and LDL cholesterol were observed in subjects receiving PF‐04965842 BID.
Following single doses of PF‐04965842, peak plasma concentrations occurred within 1 h for doses up to 200 mg, but were delayed following the 400 mg and 800 mg doses (1.5–4.0 h). Plasma PF‐04965842 Cmax concentrations increased proportionally across the entire dose range, while exposure in terms of the AUC was greater than proportional at doses of 400 mg and 800 mg. A monophasic decline was observed in the disposition of PF‐04965842 at the lower doses (3–30 mg) and a biphasic decline was observed at the higher doses, probably due to concentrations for the lower doses declining below the lower limit of quantification before the slower terminal phase was observed. In comparison, a median Tmax of 0.5–1 h followed by monophasic elimination was observed with single doses of tofacitinib (0.1–100 mg) in healthy volunteers 18. Dose proportional systemic exposures and a mean t 1/2 of 2.5 h were observed for tofacitinib ≥3 mg 18. Ruxolitinib was absorbed rapidly in healthy volunteers, typically attaining peak plasma concentrations within 2 h for single doses of 5–200 mg, and plasma concentration declined in a multiphasic manner with a mean terminal disposition t 1/2 of 3 h for the five lowest doses and 5 h for 200 mg 20. Cmax and AUC increased in a linear, dose‐proportional manner for all the ruxolitinib doses 20.
On day 10 following multiple‐dose PF‐04965842 administration, peak plasma concentrations occurred within 1 h across the entire range of doses and plasma Cmax and AUCτ both appeared to show a trend towards greater than proportional increases with increasing daily dose. The PK profiles of PF‐04965842 were comparable with those of baricitinib in healthy volunteers, with fast absorption (within 1.5 h) followed by biphasic decline in plasma concentrations 24. For PF‐04965842, steady state plasma concentrations generally appeared to have been reached by day 4 for QD dosing and day 6 for BID dosing, while ruxolitinib and baricitinib reached steady state by day 2 for all regimens 20, 24. Urinary recovery of PF‐04965842 was low, with 1.0−4.4% of the dose recovered unchanged on day 10 of multiple‐dose administration, which was higher than the 0.11% urinary recovery reported on day 10 following 100 mg QD dosing of ruxolitinib 20, and lower than the 64.1% reported for baricitinib 2–20 mg QD doses 24. The PK profiles of the western and Japanese subjects showed that dose adjustment would not be needed for Japanese subjects. However, some variability in geometric mean Cmax, AUCτ and t ½ between western and Japanese subjects was observed, suggesting differences in absorption as well as metabolism between the ethnic groups.
Decreases in neutrophil and reticulocyte counts consistent with the known effects of JAK signalling inhibition were greater with BID than with QD doses of PF‐04965842. Reduced neutrophil and reticulocyte counts have been reported for healthy volunteers who received ruxolitinib BID 20 and reduced neutrophil counts have also been observed in patients with rheumatoid arthritis and psoriasis who were treated with tofacitinib 25, 26. The decreases in reticulocyte counts, which were most prominent with BID PF‐04965842 administration, may indicate some modulation of JAK2, leading to inhibition of erythropoietin (EPO) signalling 27, 28. Transient and reversible dose‐dependent decline in reticulocyte counts has been reported in patients with psoriasis who received tofacitinib BID 26. The variability in baseline neutrophil counts observed in this study is not unusual. This variability shows difference by race with the African American subjects having lower neutrophil counts than Caucasians 29.
During the MAD phase, dose‐dependent reductions occurred for IP‐10 (downstream of IFNγ) 9, 30, 31 and hsCRP (downstream of IL‐6) 9, 32, 33, which were included in this study as biomarkers of JAK1 inhibition. Decreases in NK cells and increases in B cells, minimal changes in total T cells, small increases in CD4+ T cells and small decreases in CD8+ T cells were observed. Similar effects have been observed with the JAK inhibitor tofacitinib 31, 34, 35.
In terms of study limitations, the current study only evaluated the effect of PF‐04965842 administration over 10 days in healthy subjects. Patients with inflammatory diseases could react differently and additional adverse effects may emerge with prolonged dosing.
The pharmacokinetic and haematological safety data from this Phase 1 study provided upper and lower dose limits for the completed Phase 2 studies in psoriasis (NCT02201524) and atopic dermatitis (NCT02780167), which guided the doses implemented in the ongoing Phase 3 clinical trial for atopic dermatitis (JADE Mono‐1; NCT03349060). The PK/PD modelling and simulation supporting dose selection for Phase 3 will be the subject of a separate publication.
In conclusion, the study results indicate that the JAK1 inhibitor PF‐04965842 has a favourable safety profile and is well tolerated in healthy subjects, supporting further evaluation of PF‐04965842 in patients with inflammatory diseases such as psoriasis and atopic dermatitis.
Competing Interests
E.P., M.R.H., E.K., S.T. and C.B. are employees of Pfizer and M.L.V., K.G. and C.W.A. were Pfizer employees when this study was conducted. All authors own stock in Pfizer.
We wish to thank all subjects who participated in this study and medical staff at the New Haven Pfizer Clinical Research Unit. We would also like to thank all the laboratory analysts and both internal and external members of operational teams who provided essential services during this study, including Jenny Zhang who managed the IP‐10 assays, Shan Mei Liao who provided statistical support, and Joyce Van Winkle who sadly passed away in June 2014. Medical writing support was provided by Rina Vekaria Passmore, PhD, of Engage and was funded by Pfizer.
This study was funded by Pfizer.
Supporting information
Figure S1 Median percent change from baseline over time (A) haemoglobin; (B) platelet counts. BID, twice daily; %CFB, percent change from baseline; QD, once daily
Figure S2 Median percent change from baseline over time (A) cholesterol; (B) high‐density lipoprotein; (C) low‐density lipoprotein. BID, twice daily; %CFB, percent change from baseline; QD, once daily
Peeva, E. , Hodge, M. R. , Kieras, E. , Vazquez, M. L. , Goteti, K. , Tarabar, S. G. , Alvey, C. W. , and Banfield, C. (2018) Evaluation of a Janus kinase 1 inhibitor, PF‐04965842, in healthy subjects: A phase 1, randomized, placebo‐controlled, dose‐escalation study. Br J Clin Pharmacol, 84: 1776–1788. 10.1111/bcp.13612.
Clinical trial registration: ClinicalTrials.gov, NCT01835197.
References
- 1. Nast A, Gisondi P, Ormerod AD, Saiag P, Smith C, Spuls PI, et al European S3‐guidelines on the systemic treatment of psoriasis vulgaris – update 2015 – short version – EDF in cooperation with EADV and IPC. J Eur Acad Dermatol Venereol 2015; 29: 2277–2294. [DOI] [PubMed] [Google Scholar]
- 2. Singh JA, Saag KG, Bridges SL Jr, Akl EA, Bannuru RR, Sullivan MC, et al 2015 American College of Rheumatology guideline for the treatment of rheumatoid arthritis. Arthritis Rheumatol 2016; 68: 1–26. [DOI] [PubMed] [Google Scholar]
- 3. Smolen JS, Landewé R, Breedveld FC, Buch M, Burmester G, Dougados M, et al EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease‐modifying antirheumatic drugs: 2013 update. Ann Rheum Dis 2014; 73: 492–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hsu L, Armstrong AW. JAK inhibitors: treatment efficacy and safety profile in patients with psoriasis. J Immunol Res 2014; 2014: 283617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pope JE, Haraoui B, Thorne JC, Vieira A, Poulin‐Costello M, Keystone EC. The Canadian methotrexate and etanercept outcome study: a randomised trial of discontinuing versus continuing methotrexate after 6 months of etanercept and methotrexate therapy in rheumatoid arthritis. Ann Rheum Dis 2014; 73: 2144–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Murray PJ. The JAK‐STAT signaling pathway: input and output integration. J Immunol 2007; 178: 2623–2629. [DOI] [PubMed] [Google Scholar]
- 7. O'Sullivan LA, Liongue C, Lewis RS, Stephenson SE, Ward AC. Cytokine receptor signaling through the Jak‐Stat‐Socs pathway in disease. Mol Immunol 2007; 44: 2497–2506. [DOI] [PubMed] [Google Scholar]
- 8. Kontzias A, Kotlyar A, Laurence A, Changelian P, O'Shea JJ. Jakinibs: a new class of kinase inhibitors in cancer and autoimmune disease. Curr Opin Pharmacol 2012; 12: 464–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. O'Shea JJ, Holland SM, Staudt LM. JAKs and STATs in immunity, immunodeficiency, and cancer. N Engl J Med 2013; 368: 161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. van Vollenhoven RF. Small molecular compounds in development for rheumatoid arthritis. Curr Opin Rheumatol 2013; 25: 391–397. [DOI] [PubMed] [Google Scholar]
- 11. Namour F, Diderichsen PM, Cox E, Vayssière B, Van der Aa A, Tasset C, et al Pharmacokinetics and pharmacokinetic/pharmacodynamic modeling of filgotinib (GLPG0634), a selective JAK1 inhibitor, in support of phase IIB dose selection. Clin Pharmacokinet 2015; 54: 859–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bissonnette R, Luchi M, Fidelus‐Gort R, Jackson S, Zhang H, Flores R, et al A randomized, double‐blind, placebo‐controlled, dose‐escalation study of the safety and efficacy of INCB039110, an oral janus kinase 1 inhibitor, in patients with stable, chronic plaque psoriasis. J Dermatolog Treat 2016; 27: 332–338. [DOI] [PubMed] [Google Scholar]
- 13. Ludbrook VJ, Hicks KJ, Hanrott KE, Patel JS, Binks MH, Wyres MR, et al Investigation of selective JAK1 inhibitor GSK2586184 for the treatment of psoriasis in a randomized placebo‐controlled phase IIa study. Br J Dermatol 2016; 174: 985–995. [DOI] [PubMed] [Google Scholar]
- 14. Kremer JM, Emery P, Camp HS, Friedman A, Wang L, Othman AA, et al A phase IIb study of ABT‐494, a selective JAK‐1 inhibitor, in patients with rheumatoid arthritis and an inadequate response to anti‐tumor necrosis factor therapy. Arthritis Rheumatol 2016; 68: 2867–2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. O'Shea JJ, Kontzias A, Yamaoka K, Tanaka Y, Laurence A. Janus kinase inhibitors in autoimmune diseases. Ann Rheum Dis 2013; 72 (Suppl 2): ii111–ii115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Norman P. Selective JAK inhibitors in development for rheumatoid arthritis. Expert Opin Investig Drugs 2014; 23: 1067–1077. [DOI] [PubMed] [Google Scholar]
- 17. Vazquez ML, Kaila N, Strohbach JW, Trzupek JD, Brown MF, Flanagan ME, et al Identification of N‐{cis‐3‐[methyl(7H‐pyrrolo[2,3‐d]pyrimidin‐4‐yl)amino]cyclobutyl}propane‐1‐sulfonamide (PF‐04965842): a selective JAK1 clinical candidate for the treatment of autoimmune diseases. J Med Chem 2018; 61: 1130–1152. [DOI] [PubMed] [Google Scholar]
- 18. Krishnaswami S, Boy M, Chow V, Chan G. Safety, tolerability, and pharmacokinetics of single oral doses of tofacitinib, a Janus kinase inhibitor, in healthy volunteers. Clin Pharmacol Drug Dev 2015; 4: 83–88. [DOI] [PubMed] [Google Scholar]
- 19. Mohamed MF, Camp HS, Jiang P, Padley RJ, Asatryan A, Othman AA. Pharmacokinetics, safety and tolerability of ABT‐494, a novel selective JAK 1 inhibitor, in healthy volunteers and subjects with rheumatoid arthritis. Clin Pharmacokinet 2016; 55: 1547–1558. [DOI] [PubMed] [Google Scholar]
- 20. Shi JG, Chen X, McGee RF, Landman RR, Emm T, Lo Y, et al The pharmacokinetics, pharmacodynamics, and safety of orally dosed INCB018424 phosphate in healthy volunteers. J Clin Pharmacol 2011; 51: 1644–1654. [DOI] [PubMed] [Google Scholar]
- 21. Neubauer H, Cumano A, Müller M, Wu H, Huffstadt U, Pfeffer K. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell 1998; 93: 397–409. [DOI] [PubMed] [Google Scholar]
- 22. Genovese MC, Smolen JS, Weinblatt ME, Burmester GR, Meerwein S, Camp HS, et al Efficacy and safety of ABT‐494, a selective JAK‐1 inhibitor, in a phase IIb study in patients with rheumatoid arthritis and an inadequate response to methotrexate. Arthritis Rheumatol 2016; 68: 2857–2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kahl L, Patel J, Layton M, Binks M, Hicks K, Leon G, et al Safety, tolerability, efficacy and pharmacodynamics of the selective JAK1 inhibitor GSK2586184 in patients with systemic lupus erythematosus. Lupus 2016; 25: 1420–1430. [DOI] [PubMed] [Google Scholar]
- 24. Shi JG, Chen X, Lee F, Emm T, Scherle PA, Lo Y, et al The pharmacokinetics, pharmacodynamics, and safety of baricitinib, an oral JAK 1/2 inhibitor, in healthy volunteers. J Clin Pharmacol 2014; 54: 1354–1361. [DOI] [PubMed] [Google Scholar]
- 25. Kremer J, Li ZG, Hall S, Fleischmann R, Genovese M, Martin‐Mola E, et al Tofacitinib in combination with nonbiologic disease‐modifying antirheumatic drugs in patients with active rheumatoid arthritis: a randomized trial. Ann Intern Med 2013; 159: 253–261. [DOI] [PubMed] [Google Scholar]
- 26. Strober B, Buonanno M, Clark JD, Kawabata T, Tan H, Wolk R, et al Effect of tofacitinib, a Janus kinase inhibitor, on haematological parameters during 12 weeks of psoriasis treatment. Br J Dermatol 2013; 169: 992–999. [DOI] [PubMed] [Google Scholar]
- 27. Shi J, Yuan B, Hu W, Lodish H. JAK2 V617F stimulates proliferation of erythropoietin‐dependent erythroid progenitors and delays their differentiation by activating Stat1 and other nonerythroid signaling pathways. Exp Hematol 2016; 44: 1044–1058 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wojchowski DM, Gregory RC, Miller CP, Pandit AK, Pircher TJ. Signal transduction in the erythropoietin receptor system. Exp Cell Res 1999; 253: 143–156. [DOI] [PubMed] [Google Scholar]
- 29. Reich D, Nalls MA, Kao WH, Akylbekova EL, Tandon A, Patterson N, et al Reduced neutrophil count in people of African descent is due to a regulatory variant in the Duffy antigen receptor for chemokines gene. PLoS Genet 2009; 5: e1000360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Luster AD, Unkeless JC, Ravetch JV. Gamma‐interferon transcriptionally regulates an early‐response gene containing homology to platelet proteins. Nature 1985; 315: 672–676. [DOI] [PubMed] [Google Scholar]
- 31. Hodge JA, Kawabata TT, Krishnaswami S, Clark JD, Telliez JB, Dowty ME, et al The mechanism of action of tofacitinib – an oral Janus kinase inhibitor for the treatment of rheumatoid arthritis. Clin Exp Rheumatol 2016; 34: 318–328. [PubMed] [Google Scholar]
- 32. Castell JV, Gómez‐Lechón MJ, David M, Andus T, Geiger T, Trullenque R, et al Interleukin‐6 is the major regulator of acute phase protein synthesis in adult human hepatocytes. FEBS Lett 1989; 242: 237–239. [DOI] [PubMed] [Google Scholar]
- 33. Li SP, Goldman ND. Regulation of human C‐reactive protein gene expression by two synergistic IL‐6 responsive elements. Biochemistry (Mosc) 1996; 35: 9060–9068. [DOI] [PubMed] [Google Scholar]
- 34. Conklyn M, Andresen C, Changelian P, Kudlacz E. The JAK3 inhibitor CP‐690550 selectively reduces NK and CD8+ cell numbers in cynomolgus monkey blood following chronic oral dosing. J Leukoc Biol 2004; 76: 1248–1255. [DOI] [PubMed] [Google Scholar]
- 35. van Gurp E, Weimar W, Gaston R, Brennan D, Mendez R, Pirsch J, et al Phase 1 dose‐escalation study of CP‐690 550 in stable renal allograft recipients: preliminary findings of safety, tolerability, effects on lymphocyte subsets and pharmacokinetics. Am J Transplant 2008; 8: 1711–1718. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Median percent change from baseline over time (A) haemoglobin; (B) platelet counts. BID, twice daily; %CFB, percent change from baseline; QD, once daily
Figure S2 Median percent change from baseline over time (A) cholesterol; (B) high‐density lipoprotein; (C) low‐density lipoprotein. BID, twice daily; %CFB, percent change from baseline; QD, once daily