

Abstract
Objectives
In a separate document, we have provided specific guidance on performing individual pharmacokinetic (PK) studies using limited samples in persons with hemophilia with the goal to optimize prophylaxis with clotting factor concentrates. This paper, intended for clinicians, aims to describe how to interpret and apply PK properties obtained in persons with hemophilia.
Methods
The members of the Working Party on population PK (PopPK) of the ISTH SSC Subcommittee on Factor VIII and IX and rare bleeding disorders, together with additional hemophilia and PK experts, completed a survey and ranking exercise whereby key areas of interest in the field were identified. The group had regular web conferences to refine the manuscript’s scope and structure, taking into account comments from the external feedback to the earlier document.
Results
Many clinical decisions in hemophilia are based on some form of explicit or implicit PK assessment. Individual patient PK profiles can be analyzed through traditional or PopPK methods, with the latter providing the advantage of fewer samples needing to be collected on any prophylaxis regimen, and without the need the for a washout period. The most useful presentation of PK results for clinical decision making are a curve of the factor activity level over time, the time to achieve a certain activity level, or related parameters like half‐life or exposure (AUC). Software platforms have been developed to deliver this information to clinicians at the point of care. Key characteristics of studies measuring average PK parameters were reviewed, outlining what makes a credible head‐to‐head comparison among different concentrates. Large data collections of PK and treatment outcomes currently ongoing will advance care in the future.
Conclusions
Traditionally used to compare different concentrates, PK can support tailoring of hemophilia treatment by individual profiling, which is greatly simplified by adopting a PopPK/Bayesian method and limited sampling protocol.
Keywords: factor IX, factor VIII, population pharmacokinetics, tailored prophylaxis, tailoring
Essentials.
The use of pharmacokinetics (PK) and population PK (PopPK) in tailoring hemophilia treatment is growing steadily.
We provide clinical guidance on uses and adoption of PK and PopPK in hemophilia.
We provide guidance on appraising PK reports, including studies and claims comparing different factor concentrates.
We discuss the importance of large PK data collection for advancement of hemophilia treatment approaches.
1. INTRODUCTION
The goal of hemophilia A and B treatment is the prevention of bleeding and thus to minimize the consequences of bleeding into joints and vital organs, consequently enhancing both the expected length and quality of life.1 This is usually achieved by regular preventive intravenous administration of the deficient coagulation factor, a treatment strategy called prophylaxis.2 The dose and frequency of factor concentrate infusions to improve important patient outcomes, such as a reduction in the number and severity of spontaneous or traumatic bleeding episodes or a reduction in the burden of care, vary largely among individuals, and may vary in the same individual over time.3 This variability is attributed to many factors, first of which is the individual’s tendency to bleed. This can be referred to as a pharmacodynamic (PD) component of the process, ie, the mechanisms linking the plasma activity level of clotting factor concentrate with the relevant outcome. Other sources of variability are: the bleeding history, including recent pattern of bleeding as a function of factor activity level and presence of target joints, level of physical activity, preferences with regard to infusion frequency, availability and affordability of clotting factor concentrates, targeted or tolerated annualized bleeding rate and the individual’s specific pharmacokinetic (PK) profile.4, 5 Accounting for each of these causes of variability is critical to individualizing treatment. While an understanding of an individual’s PK and PD are equally important in clinical decision making, knowledge of individual PK has slowly become a key driver of personalized hemophilia therapy. The variability of the disposition of the infused clotting factor concentrate (ie, the specific activity‐time curve after the infusion) is larger among different individuals than within an individual over time or across different concentrates of the same class.6, 7 Therefore, assessing the individual disposition of the infused concentrate for each specific patient should be considered as a primary objective in tailoring prophylaxis to individual needs.8 Whereas PK does not set optimal thresholds or define patient needs, tailoring treatment to individual characteristics, changes in lifestyle and response to clinical events using a “trial and error” approach without the knowledge of individual PK yields suboptimal results.
The primary aim of this article is to describe how PK analyses in persons with hemophilia, using a proposed common terminology, are currently interpreted and applied while considering the recommendations of the ISTH.
2. MANUSCRIPT DEVELOPMENT WORKFLOW
This manuscript is the result of the collaborative effort of the working party on Population Pharmacokinetics of the Scientific Standardization Committee (SSC) of the International Society for Thrombosis and Hemostasis (https://www.isth.org/members/group.aspx?id=100348). The group was established in July 2015 and met regularly through June 2017 to establish recommendations for performing individual PK assessments adopting a PopPK approach. These recommendations can be found in Iorio et al.9 The present document, although not an official communication of the SSC, elaborates on pharmacokinetics in hemophilia beyond what could be addressed in Iorio et al.9 Open comments from experts in the field of coagulation factor concentrates PK (independent investigators, pharmaceutical company PK experts, and members of regulatory bodies) were invited beyond the original Working Party membership.
3. THE EMPIRICAL APPROACH TO DOSING CLOTTING FACTOR CONCENTRATES IN PERSONS WITH HEMOPHILIA
Dosing guidance for clotting factor concentrate replacement tends to provide flexibility to the treater in response to the known PK variability amongst persons with hemophilia. Using prophylaxis with a standard half‐life factor VIII concentrate as an example, a typical dosing regimen would be 20 to 40 IU/kg administered every other day. Assuming a recovery of 0.02 IU/mL (ie. 2 IU/dL) for each 1 IU/kg of infused factor VIII and an average half‐life of 12 h, this regimen would provide the “average” persons with hemophilia a trough level at or above 0.01 IU/mL. This “one‐size‐fits‐all” dosing usually requires doses to be titrated within the dose range by use of blood sampling and empirical methods for individualization. This “trial and error” approach is commonly applied in practice.
When looking across classes of concentrates, the way that concentrate‐specific PK properties are accounted for is in the recommended starting regimens for the phase III studies. For example, 50 IU/kg twice a week or 100 IU/kg weekly for a standard half‐life recombinant factor IX concentrate (rFIX), or 100 IU/kg every 10 days for an extended half‐life (EHL) product, are all intended to target a given trough level. In practice, irrespective of which starting regimen is chosen, the range of doses and intervals that patients are ultimately on varies widely, implying that during titration, some patients will be under‐ or over‐dosed.
Furthermore, this “population average” approach does not account for patient variables such as age, Body Mass Index (BMI) or blood group that are already known to affect PK.10, 11, 12 Thus, the population average and subsequent “trial and error” approach to dosing does not incorporate current knowledge and available PK modeling and simulation tools.
4. ESTABLISHED USES OF PK MEASURES IN ROUTINE CLINICAL CARE OF PERSONS WITH HEMOPHILIA AND THEIR LIMITATIONS
The use of any measurement of postinfusion plasma activity level can be considered a basic application of PK to the treatment of hemophilia. The three most established measurements are: (i) the measurement of trough levels during prophylactic treatment, (ii) measuring peak and trough in a perioperative setting, or (iii) recovery and half‐life as guidance to wean off immune tolerance induction (ITI).
In routine prophylaxis, the classical approach to monitoring patients is to have their plasma factor activity levels measured just prior to the next infusion or, in other words, the trough level. This is to ensure that the plasma activity level of the infused factor is still above the level considered critical to prevent bleeding.13 This critical threshold is often assumed to be 0.01 IU/mL although different thresholds have been proposed for differing levels of physical activity or tendency to bleed.3 Dose adjustment based on measurement of pre‐dose (trough) levels is a simplified and empirical PK‐guided approach to prescribing prophylaxis.
To ensure bleeding control during surgery, national and international guidelines recommend maintaining plasma activity levels of factor concentrates above specific thresholds for specific durations of time, both of which depend on the type of surgery.14 As a result, persons with hemophilia undergoing surgery often have one or more plasma factor activity levels measured to ensure optimal levels are maintained.15 Perisurgical dose adjustment based on these measurements can be considered a simplified and empirical PK guided approach to bleeding prevention. Similarly, when perisurgical hemostasis is obtained by using a continuous infusion of clotting factor concentrate, the initial infusion rate can be calculated based on the anticipated clearance of the concentrate itself. It has been observed, however, in a large surgery study using these methods that the majority of levels continue to be outside of the targeted range.16 Recently, a population PK (PopPK) approach to perisurgical dosing has been proposed,17 and a randomized controlled trial is currently ongoing to evaluate this approach to individualized dosing in the perisurgical setting.18
Defining tolerance in the context of an ITI regimen after the inhibitor is no longer detectable with the Bethesda assay (ideally the Nijmegen method), requires monitoring of the recovery of infused factor VIII and then its half‐life. Specific thresholds are suggested for both outcomes to define success or partial success.19, 20, 21, 22 Very recently, a more pragmatic application of PK to tailor the dose during ITI in children was suggested by the UK Haemophilia Centre Doctors’ Organisation (UKHCDO) that uses only trough level and mitigates the need to take multiple samples to assess both recovery and half‐life.23 Calculating the half‐life or measuring the recovery or trough level of the infused factor constitutes a (simplified) PK approach to tailoring individual treatment.
5. THE IMPORTANCE OF RELIABLE LABORATORY MEASUREMENTS
An important consideration when using plasma factor activity level measurements for clinical purposes is the precision and accuracy of the laboratory measurements. There is robust evidence that the choice of assay type (ie, one‐stage versus chromogenic), the choice of aPTT reagent, as well as the choice of reference standard (generic versus concentrate specific) impacts the measurement result in a significant way.14, 24 According to the general theory of measurements, the variability attributed to the measurement methods (eg, when using different assays on the same plasma sample) is due to random or systematic measurement errors.25, 26
The random error translates into imprecision or variation. A typical coefficient of variation of measurements for clotting assays is equal to or below 15% that results in, for example, a measurement of 0.50 IU/mL, if repeated multiple times, giving results between 0.43 IU/mL to 0.57 IU/mL two‐thirds of the time.
The systematic error translates into poor accuracy or significant deviation from the true value. For example, a test based on a specific reagent will systematically report a lower or higher result than another reagent. Systematic errors can also apply to a combination of specific concentrates and specific assays. A typical example is the finding that the original formulation of B‐Domain deleted factor VIII had a lower than expected recovery when measured with a one stage clotting assay using a full length factor VIII as a reference standard, but not when using a B‐Domain deleted specific standard.27, 28 This also seems to be relevant for some wild type and modified recombinant and plasma‐derived FVIII and FIX products,29, 30, 31 specifically where the one‐stage clotting assay result is influenced by the aPTT reagent selected.32 A review of the current evidence about the performance of different reagents for different factor concentrates has been performed by Young and colleagues.33 Manufacturers are responsible for providing information to clinical laboratories on appropriate assay and assay conditions for their product and can support efforts to ensure measurement accuracy when a single assay is used in the laboratory across a number of different products.32 While the one‐stage clotting assay is most commonly used for clinical monitoring, there is a move towards adoption of the chromogenic assay, which tends to be less prone to systematic errors.34
As recommended in the guidance,9 any measurement that is below the limit of quantification (BLQ) of the specific assay should be reported (eg, <0.01 IU/mL and not 0 IU/mL or 0.01 IU/mL). When BLQs are removed from the PK modeling process, the resulting half‐life will be overestimated (ie, longer than if the model used these values)35 resulting in a potentially unsafe reduction in dose or extension of frequency. Nevertheless, a number of methods are available for using BLQs in PopPK analyses.36 Avoiding sampling times where BLQ levels are expected is also good practice.
6. DOSE INDIVIDUALIZATION BASED ON ESTIMATION OF INDIVIDUAL PK PROFILES
Owing to the wide variability in factor concentrate PK between persons with hemophilia, assessing and using individual PK knowledge for dosing is an attractive option over the “trial and error” methods as described above and has been found to reduce factor concentrate usage and bleeding events as compared to standard prophylaxis.37, 38 In addition, utilization of a PopPK method to derive individual PK parameters will contribute significantly to individualized treatment of persons with hemophilia. To facilitate understanding this potential we will compare and contrast it to the traditional approach.
6.1. Traditional approach to obtaining individual PK information and its disadvantages
All non‐empirical (ie, non–”trial and error”) approaches to calculate an individual dose require some assessment of the individuals’ PK parameters. A publication of the International Society of Thrombosis and Hemostasis (ISTH) in 200139 recommends 10 to 11 postinfusion samples following a washout period with subsequent PK modeling to obtain PK parameter estimates. While the aim of the guideline was to understand the PK of a specific factor concentrate in a population of 12 to 15 persons with hemophilia, the suggested PK study can also be used as a means to generate individual PK estimates for use in dosing guidance. Some tailored prophylaxis programs based on a similar method are currently ongoing.37 However, all such previously published approaches share some common limitations: usually using a standard test dose (eg, 50 IU/kg) and requiring a wash‐out period, which is potentially risky for patients, as well as numerous postinfusion samples, over a period of days, which is burdensome and impractical for many patients, especially children.
6.2. PopPK approach and Bayesian estimation to obtaining individual PK information
Determination of individual PK parameters can be achieved with fewer samples than the traditional approach through integration of information from both a patient population and an individual. Limited‐sampling models (LSM) that rely on 1 to 3 blood samples have been primarily used for the estimation of area under the curve (AUC) and maximum plasma concentration (Cmax).40, 41 The Bayesian approach, which is based on Bayes’ theorem, has been used for a wide variety of drugs to predict individual PK parameters from 1 to 4 blood samples.42, 43, 44, 45 With these methods, there is an underlying assessment of the dose‐exposure relationship and the relevant covariates that modify this relationship such as age or weight from a patient population. Coupled with patient specific covariates and drug levels in blood, the models integrate population and individual level information to derive individual PK parameters that can be used to derive an individual PK profile.
Population level PK information can be analyzed and understood using PopPK methods that employ non‐linear mixed effect models. In hemophilia, PopPK uses both dense and sparse PK data from persons with hemophilia in either the presence or absence of a washout to derive a unique understanding of inter‐individual variability (IIV) and its predictors (eg, age, weight, BMI, blood group), inter‐occasion variability (IOV) that defines how an individual patients’ PK changes over time, and left‐over or residual variability.46 One goal of a PopPK model is to use the derived relationships between PK and patient characteristics (eg, age, weight, BMI, blood group) to predict PK in the next individual in the absence of individual factor activity levels. An extension of this and the method recommended on behalf of the ISTH SSC on FVIII and FIX,9 is to use an appropriately derived PopPK model and Bayesian estimation techniques to predict individual PK parameters using patient‐specific characteristics plus patient‐measured FVIII or FIX activity levels.
The characteristics of PK variability of clotting factor concentrates are especially suited to this dose individualization technique. In general, the variability in the dose‐exposure relationship is judged against the therapeutic window of a drug where the variability is considered large or clinically relevant when it places different patients (IIV), or the same patient over time (IOV), outside of the therapeutic window.47 When the therapeutic window is large, PK variability is less important to attaining target activity levels (Figure 1, panel A) than when there is a narrow therapeutic window (Figure 1, panel B). Indeed, when PK variability is small in relation to the therapeutic window (Figure 1, panel A), the disposition of a specific dosage of a drug (either as fixed dose or weight adjusted dose) can be predicted for most individuals in a population, or, more precisely, it can be predicted that most individuals will have their plasma activity levels within the therapeutic window. This is the case for many drugs, especially over‐the‐counter drugs, and is an ideal situation. Narrow therapeutic window drugs that have a relatively large IIV and IOV require continuous dose adjustments, as is the case of warfarin, and individual PK understanding is not needed because it is unable to solve this large variability issue (Figure 1, panel B). The intermediate scenario, and the one applying to clotting factor concentrates, is where, relative to the therapeutic window, the IIV is large and the IOV is small (Figure 1, panel C). In this case, assessing the individual PK, which changes minimally day over day (small IOV) but greatly between patients (IIV) allows for dose individualization. By adjusting the dose for each subject, the individual PK will be maintained within the therapeutic window over time (Figure 1, panel D).
Figure 1.
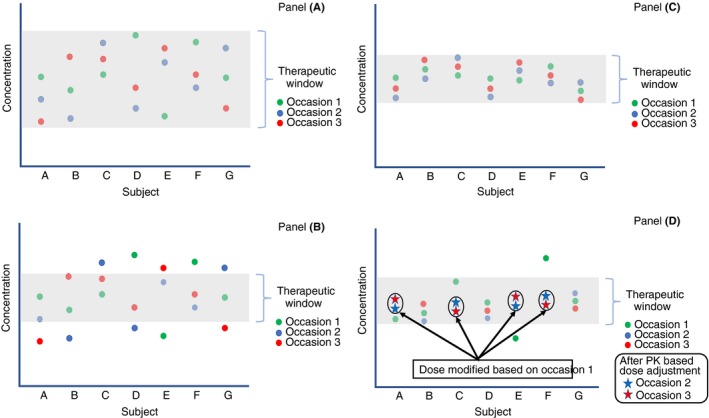
Impact of sources of variability in drug disposition and the impact of individualized dosing. The plot describes repeated measurements of drug concentrations in patients over time. The red, blue, and green dots for a given patient indicates three measurements for that patient at different times. The greyed‐out area represents the therapeutic window. Panel A describes that when the therapeutic window is larger than the variability among (IIV) and within (IOV) patients, patients have therapeutic concentrations most of the time. In this case, an average dose (either as a fixed dose or a weight‐adjusted dose) is expected to be therapeutic in most patients most of the time. Panel B describes a drug producing the same measurements as in Panel A but having a narrower therapeutic window. In this case, IIV and IOV are large relative to the therapeutic window and the relevant patient dose will need to differ amongst patients as well as within the same patient over time. Panel C describes the situation where, relative to the therapeutic window, the IIV is large and the IOV is small. In this case, deriving an individual dose from an assessment of individual PK will maintain the patient at therapeutic concentrations over time because their PK is stable (low IOV). This is the case for FVIII and FIX in persons with hemophilia. Panel D presents an example of adjusting the dose based on individual PK assessment following occasion 1 with subsequent occasions falling in the therapeutic range. This is the concept of individualized dosing of factor concentrates in persons with hemophilia
While IOV tends to be small when the patient is in a stable condition meaning that their PK assessment remains valid over time, non‐stable conditions will necessitate reassessment of PK to ensure that dosing is congruent with condition. Noteworthy examples include children where weight‐normalized clearance is higher in young children and gradually reaches adult levels with increasing age10 and, as a result, reassessment of PK profiles in young children is done every two to three years in some centers, and can be greatly facilitated by using a limited sampling PopPK approach. Other examples include the clearance changes associated with the immediate postsurgical period in patients receiving FVIII/FIX by bolus or continuous infusion and in patients with changing inhibitor titers to FVIII/FIX on ITI. The rate of change of PK within a patient in non‐stable conditions is unique to the patient and condition. Specially constructed sampling schedules and PopPK programs for these various clinical scenarios are an area of active research.18
6.3. Limitations to PopPK individual profiling
As with all regression models, the predictive accuracy of a model outside of the covariate space (eg, age, weight, inhibitor status) used for model development is uncertain. This was demonstrated when a previously derived FVIII model10 was used to predict PK in a cohort of persons with hemophilia undergoing surgery.17 Since the model was not built on patients during surgery, it was not an accurate predictor in that scenario and a surgery specific PopPK model was built. Other important scenarios in hemophilia where PopPK models could be built if enough data were available, includes patients with inhibitors, the obese, and children. Regardless of the scenario, we do not yet know how many patients are sufficient to build a predictive brand‐specific PopPK model best suited for Bayesian estimation. Large data collections, such as the Web‐Accessible Population Pharmacokinetics Service–Hemophilia (WAPPS‐Hemo),48 aim to gather FVIII and FIX data from thousands of patients on various brands in order to develop PopPK models that span the entirety of the covariate space, better representing persons with hemophilia than clinical trial participants. Prospective evaluation of the developed models is also possible and future research will address these limitations more robustly to further inform practice.
Given densely sampled profiles, traditional noncompartmental analysis produces PK estimates equivalent to PopPK estimates.49 Bayesian forecasting of individual PK having a set of limited patient activity levels has an uncertainty that is tied to the number and timing of those samples.50 Brekken et al.50 demonstrated that if only two samples were taken for plasma‐derived FIX, there is greater precision of the estimates when those two samples are taken at the end of the profile (day 4) vs at the beginning of the profile (day 2) with the caveat that imprecision increases when samples are BLQ, which tends to be at the end of the profile. The ISTH guidance aims to reduce this uncertainty by providing instruction to clinicians on timing and number of samples.9
A limitation to the use of PK and PopPK is in instances of a discordance between concentrate activity in blood and response (eg, bleeding), where PD plays a more important role. For example, plasma FIX activity levels may represent a suboptimal marker for clinical efficacy,51 and data for different FIX products may not be directly comparable.51 The techniques applied in order to extend the half‐life (EHL) of rFIX using pegylation, albumin fusion or Fc fusion, makes the EHL rFIX products substantially different on a molecular level, presumably affecting their extravascular distribution, which translates into differing PK characteristics as well as differences in the relationship between measured plasma FIX activity levels and clinical outcome. Knowledge of individual PK in isolation of the individual activity‐response relationship (PD) is unlikely to lead to optimal treatment.
Another limitation of a PK and PopPK tailored approach is patient and treater acceptance. A formal analysis of patient and treater attitudes towards PK‐tailored prophylaxis from both low‐ and high‐income countries was completed and showed that the majority of patients and, to a greater extent, treaters would be willing to switch to PK‐tailored dosing.52 This was not without hesitation where daily dosing was a barrier unless bleeding frequency was greatly reduced. It was interesting to note that number of blood samples and frequency of sampling for PK estimation were not barriers to acceptance52 suggesting that follow‐up samples for verification of a new regimen would be feasible. While resource rich countries using high dose prophylaxis (20‐40 IU/kg Q48 h) may use a PK‐tailored approach to reduce costs, resource poorer countries using low dose prophylaxis (6‐10 IU/kg twice weekly) may use PK‐tailoring to optimize their limited resources (eg, guide administration around high risk activities).
Finally, a practical limitation to the adoption of a PopPK based tailoring approach is the complexity of performing a post‐hoc Bayesian estimation. Indeed, this is beyond what most hemophilia treatment centers may accomplish and was the main driver for developing WAPPS‐Hemo. It is worth noting that, whereas other generic PopPK software (eg, Doseme LLC, Taringa Qld, Australia, doseme.com.au; InsightRX, Inc. San Francisco, CA, USA, insight‐rx.com; TDMx, University of Hamburg, Hamburg, Germany, http://www.tdmx.eu) and specialized dedicated software classified as a medical device (eg, my PKFit, Shire Pharmaceutical Holdings Ireland Limited, Dublin, Ireland, http://www.mypkfit.com) exist, the former requires a significant time commitment and expertise. The latter are product specific and heavily constrained in their estimation and simulation capacity by the need to adhere to the labelling specifications of the products they serve. Defining WAPPS as a collaborative research network was a decision taken after multiple informal and formal consultation with relevant regulatory agencies. This decision seems to have preserved the capacity of WAPPS to fully model the observed variability, empowering and not limiting the capacity of hemophilia doctors to exercise their clinical judgement.
6.4. Characteristics of a clinically useful individual PK profile
Irrespective of the underlying PK method and assumptions, a clinically useful PK profile of an individual patient must provide at a minimum: (i) the predicted plasma activity level at any given time, and (ii) a measure of precision of the estimates. Coupled with patient‐specific thresholds, the individual plasma activity level vs. time profile contains the required information needed to identify when to reinfuse a patient (eg, target trough > 0.03 IU/mL) or when the risk of bleeding would be low (eg, level > 0.12 IU/mL53). It includes the predicted plasma activity level at any given time, or, as alternate display, the time elapsed from the infusion to any level of interest with associated uncertainty (Figure 2, panel A). The time to critical activity level is increasingly reported as a relevant outcome measure in PopPK papers of factor concentrates.54, 55, 56 In the event of a change in dose or frequency, a PK profile presenting the new regimen can be calculated using the individuals’ PK estimates (Figure 2, panel B and C), and again provide all of the information needed for clinical decision making. Indeed, whereas an individual’s primary PK parameters such as clearance and volume of distribution are important for derivation of a PK profile, they are usually not meaningful to clinicians. Even secondary PK parameters, like individual terminal half‐life and AUC, are more translatable to clinical practice, but still too complicated for many clinicians. Independent interpretation and use of relevant PK outcomes is beyond reach for most clinics, and there is a move towards embracing software that both calculates an individual’s PK profile using Bayesian methods and allows for individualized dose regimen design.48 Mobile applications that extend the software scope and allow the patient access to their predicted activities in real time are currently under development.
Figure 2.
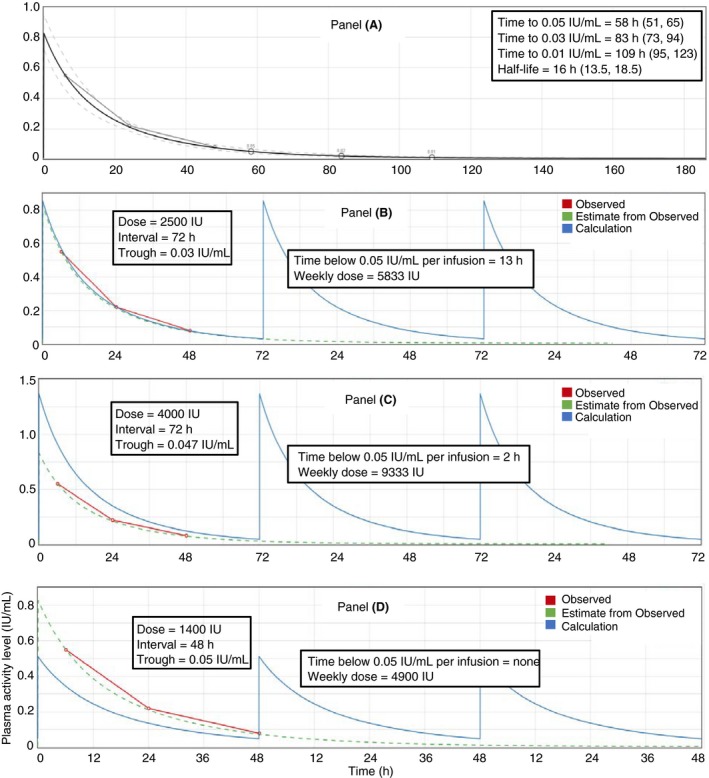
Characteristics and information content of an individual PK profile. The individual plasma activity level vs. time profile contains most of the information needed to identify the dose and interval for the optimal regimen for a specific patient. We are using as an example plots produced with WAPPS‐Hemo (http://www.wapps-hemo.ca). Panel A represents a profile from a simulated patient dosed with 2500 IU FVIII and plasma activity levels measured at 4, 24, and 48 h post‐administration (small hollow circles and interpolated line). Using a PopPK model and a Bayesian approach the fitted plasma activity level vs time profile is produced (solid black line) with its associated uncertainty (prediction intervals as derived from the underlying PopPK model—dashed grey lines). Estimates of terminal half‐life and time to threshold levels (95% prediction intervals) are clinically actionable outcomes. Panel B presents the process of simulation using patient specific PK. The original measured plasma activity levels (red) and model fit (green) for the 2500‐IU dose are presented for reference. For the patient in Panel A, Panel B shows the weekly profile (solid blue line) on their current regimen of 2500 IU infused every third day. The trough was estimated at 0.03 IU/mL with a weekly consumption of 5833 IU. Assuming a safety threshold of 0.05 IU/mL for the intended level of activity, the time spent below 0.05 IU/mL is estimated to be 13 hours per interval. Panel C shows the calculated curve obtained by keeping the interval at every third day, and increasing the dose at 4000 IU. This would increase the trough level to 0.047 IU/mL and the weekly consumption to 9333 IU. The time spent below 0.05 IU/mL would be 2 hours. Panel D shows the calculated curve obtained by reducing the frequency to every second day and the dose to 1400 IU. This would increase the trough level to 0.05 IU/mL with no time spent below and the weekly consumption would be 4900 IU
7. PK AND POPPK CONTRIBUTION TO CHOOSING A SPECIFIC FACTOR CONCENTRATE
We have demonstrated that knowledge of one’s PK profile is needed to optimize an individual dosing regimen. But is there value in knowing concentrate specific “average” PK characteristics? The theoretical answer is yes. A concentrate with lower average clearance, higher average exposure (ie, AUC/Dose) and longer average terminal half‐life, is more likely to yield favorable profiles, on average, in the population. Practically, a robust comparison of PK across different concentrates is not trivial, and requires certain critical considerations in appraising scientific evidence in the field (Table 1). The single most important concept is that there is more variability among individuals (the population) than among concentrates (the treatment).6 There are two important consequences to this concept: the first is the need to check if the studies providing the PK estimates have been performed on populations comparable to the patients we are planning to apply those results to (ie, external validity).57 The second is that when we compare the average PK characteristics of two or more concentrates, we need to make sure the tested populations and the study designs are comparable and robust enough. The most efficient study design to ensure comparability is the crossover study, where each individual receives each concentrate and they therefore act as their own control.39 Of critical importance is comparing only PK data generated with comparable methods: too often, and sometimes even in crossover studies, different assumptions and methods (including sampling schedules) are used for the two concentrates under comparison, and the method more than the concentrates is responsible for the observed difference.58, 59, 60, 61 Irrespective of the goodness of the decision‐making process and quality of the supportive evidence, generic choices at the population level cannot substitute for individual PK profiling, as they do not account for inter‐patient variability.
Table 1.
Appraisal of the characteristics of PK studies that affect the comparability of results among factor concentrates. Presented are the domains of a study to be considered when assessing if a study reporting a PK analysis can be trusted, applied to a given clinical situation, or its results compared to those from another study. The same criteria apply when assessing comparative studies
| Domain | Cueing question | Characteristic assessed | Notes |
|---|---|---|---|
| Population | Are the populations used to assess the PK characteristics of the concentrates similar to each other and to the population of interest? | ||
| Did the study design and conduct control for baseline imbalance of participant characteristics? | Study design | Crossover design (each participant acts as its own control); randomized trial (the two arms are practically identical) . | |
| Did participants represent the full, or at least similar, spectrum of the population? Were the demographics and clinical characteristics of the population(s) at baseline described? | Population composition | The baseline characteristics of the participants are usually described in a table.The range of observed participant characteristics (eg, age, weight) is similar to the population of interest. | |
| Was a sufficiently large sample enrolled in the study? | Study size | The number of subjects is sufficient to capture the variability. For a conventional study, 12‐15 subjects are deemed sufficient; for a population PK study around 20‐30 subjects with dense data or 100 with sparse data are suggested. | |
| Is the precision of the findings appropriate? | Observed variability | The range of observed PK values around the average is typical for the population; smaller or larger variability may require careful consideration. | |
| Is (are) the population(s) in the studies representative of the one I plan to apply the results to? | External validity | Would the patient(s) I am planning to apply the results of the study to have been enrolled in the study(ies)? | |
| Intervention | Did the administration of the concentrates under assessment happen in a similar way across the comparators and with respect to the intended use? | ||
| Was the study performed under routine clinical conditions? | Study setting | Usually patients studied during regular prophylaxis, in non‐bleeding conditions, with exclusion of the surgical setting. | |
| Were participants subject to a wash‐out? | Study design | If no washout then comparisons should be in steady‐state conditions. | |
| Were the doses of the concentrates tested comparable? | Study design | PK of factor concentrates is supposed to be dose independent, but use of extreme doses may require specific considerations. | |
| Measurements | Were the sampling strategies sound and similar across the comparison? | ||
| Were samples drawn over comparable time periods across the comparison? | PK assessment method | PK estimates can change depending on how many samples are used in the analysis, and for how long they are collected. | |
| Were samples measured with the same laboratory test and reference standard? | Laboratory method | Using different laboratory tests and/or reference standard may imbalance the comparison. | |
| Were samples below the limit of quantitation (BLQ) recorded? | Laboratory method | Results for measurement below the level of detection must be reported as “BLQ” followed by the minimum detectable concentration. | |
| Analysis | Were the analysis plans similar, sound, and clearly reported across the comparison? | ||
| Was the PK and/or PopPK analytical approach described in sufficient detail to be reproduced? | PK analysis | Details of the modelling approach must be provided and discussed, particularly when different for different concentrates. | |
| Were the structural models (non‐compartmental, one or multiple compartment) assumptions similar across the comparison? If not, was the case for the difference explained? | PK analysis | Justification for the modelling approach must be provided and discussed, particularly when different for different concentrates. | |
| Were reasonable assumptions used for PopPK analysis? | PopPK analysis | Justification for the endogenous activity, choice of covariates, number of samples, and subjects, modelling approach must be provided and discussed. | |
| Were BLQs accounted for in the analysis? | PopPK analysis | BLQs must be modeled as other post‐infusion measures. The M3 method is often used, but others may be acceptable. | |
| Results | What are the results? Are they similar, sound and clearly reported across the comparison(s)? | ||
| Were all expected results reported with their variability? | PK/PopPK analysis | Are there any incomplete data reporting or any selective outcome reporting? | |
| Were results comparable with previous/contemporary analyses on the same concentrate? | PK/PopPK analysis | Differences in the results that cannot be explained by differences in the population, intervention or analysis should be carefully considered. | |
| Were results comparable with those obtained with other concentrates in the same class? | PK/PopPK analysis | Differences in the results that cannot be explained by differences in the population, intervention, or analysis should be carefully considered. | |
| Are clinical outcomes presented in addition to the PK? | Study Design | PK/PopPK studies are often performed as part of a larger efficacy/safety study. Reporting (or referencing) clinical outcomes might be of help in interpreting, comparing, and applying the PK results. | |
PK, pharmacokinetic; PopPK, population pharmacokinetic.
8. PARTICIPATING IN LARGE PRAGMATIC POPPK DATA COLLECTIONS
Until recently, the vast majority of PK and PopPK studies have been performed by drug manufacturers to support the filing of regulatory applications or by a few specialized research centers keen in using PK to tailor treatment.6, 10, 50, 54, 62 These studies have also been completed to control or compare cost of different concentrates or regimens63, 64, 65 and to develop new PK applications to hemophilia.6, 50 PK is now becoming more often considered in decision making in hemophilia. This has been precipitated by a higher usage and capability of web‐based applications, more intense international research collaboration, larger number of concentrates competing on the market, the advent of EHL products and the continuous pressure on fair use of resources, including tendering processes. In this era of large web‐based databases used to support day‐to‐day management of hemophilia including the UKHCDO database (http://www.ukhcdo.org), the American Thrombosis & Hemostasis Network (ATHN) (http://www.athn.org), the Australian‐Canadian Bleeding Disorders Registry (ABDR [http://www.blood.gov.au/abdr]/CBDR [http://www.cbdr.ca]) family of products, the FranceCoag database (http://www.francecoag.org), and the newly launched World Federation of Haemophilia (WFH) Patient Registry (http://www.wfh.org/en/wbdr) there is an opportunity to perform large population based data collection of postinfusion plasma samples. Coupled with PK approaches, this large‐scale data could provide a valuable contribution to clinical decision making at the patient and policy levels. Many centers have adopted, as routine clinical practice, a PopPK based individual estimation method using one of the available PopPK applications.48 This is feeding a large international database and has been integrated into the hemophilia management software used in the Czech Republic, the US, and Canada. One of the important advancements provided by these large data collections involves the simultaneous consideration of clinical information, such as bleeding and treatment logs, adherence information, and activity levels. It is important for reliable PK information to be stored, centralized, and analyzed to enhance our collective capacity to understand how to best individualize and optimize the treatment of persons with hemophilia.8 The hemophilia community is producing an impressive capacity of data collection. For example, in less than 2 years, the 180 centers of the WAPPS research network have collected from >2000 unique patients over 3500 individual PK profiles.
9. CONCLUSIONS
The use of PK in the treatment of hemophilia continues to increase in importance and studies have demonstrated its utility. Along with providing a means to compare and contrast different concentrates, PK can also be used to aid in local clinical decision making. One such use is in deriving individual PK for persons with hemophilia to help with dose tailoring and this can be achieved through a number of methods. PopPK methods that integrate information from the population of persons with hemophilia along with individual PK information and characteristics are poised to provide a convenient and accessible means of individualizing dose tailoring; especially when made available to treaters and patients through dedicated software, albeit raising further questions about appropriate thresholds for troughs and/or peaks for participation in activities with varying trauma/bleed risk. Large data collection efforts are ongoing in the hemophilia community and this has the potential to further advance care.
ACKNOWLEDGMENTS
The authors would like to acknowledge the contribution of the following external experts who provided comments on the manuscript: H. Agersø, Novo Nordisk A/S, Denmark; B. Beaufils, LFB, France; E. Berntorp, Malmö, Sweden; J. Feddern, Octapharma, Germany; M. Germer, Biotest, Germany; D. Moj, Biotest, Germany; S. Jönsson, Uppsala University, M. Ragni, Pittsburgh, USA; J. Roberts, CSL Behring, US; A. Shah, Bayer, US; and G. Spotts, Shire, Ireland.
RELATIONSHIP DISCLOSURES
V. Blanchette has received honoraria for participating in Educational events and Advisory Boards funded by Bayer, Biogen, Novartis, Novo Nordisk, Octapharma, Pfizer and Shire. He is a member of Data Safety Monitoring Boards for Octapharma and Shire. He is Chair of the International Prophylaxis Study Group, a collaborative study group that receives funding support from Bayer, Biogen, CSL‐Behring, Novo Nordisk, Pfizer and Shire. J. Blatny has received speaker’s fees from Bayer, Baxter, CSL Behring, Octapharma, Pfizer, Novo Nordisk and LFB; performed consultancy for Shire, Baxter, Novo Nordisk, Pfizer and Roche. A. Boban has no conflict to declare. M. Cnossen has received grants from Baxter, Bayer Schering Pharma, CSL Behring and Novo Nordisk, personal fees and travel grants from Roche, and is a member of advisory boards for Roche and Bayer Schering Pharma. P. Collins has received research support from CSL Behring, consultancy fees from Novo Nordisk, Sobi, Shire, CSL Behring and sponsorship for meeting Novo Nordisk, CSL Behring, and Shire. S.E. Croteau has received consultant/advisory board participant fees from Aptevo, Bayer, CSL‐Behring, Octapharma, Genentech, Novo Nordisk, honoraria from Octapharma and research support from Octapharma, Pfizer, Shire, and Spark Therapeutics. A.N. Edginton has received speaker fees from Bayer. K. Fischer has performed consultancy for Baxter, Biogen, CSL Behring, Freeline, Novo Nordisk, Pfizer, Roche, and Sobi; has received research support from Bayer, Wyeth/Pfizer, Baxter, and Novo Nordisk. D.P. Hart has received speaker or consultancy fees from Bayer, Biomarin, Uniqure, Roche, Shire, Pfizer, Biotest, Octapharma, and Sobi. He has received research grant awards from Bayer, Shire, and Octapharma. A. Iorio’s institution has received project based funding via research or service agreements with Bayer, Baxalta/Shire, Bioverativ, Grifols, Novo Nordisk, Pfizer, Octapharma, and Roche. S. Ito has received grants from UBC Pharma GmbH, Novartis Pharma AG; donation support from Shoppers Drug Mart and personal fees from the Canadian Pharmacy Association. Joan Korth‐Bradley is an employee and shareholder of Pfizer, Inc. Stefan Lethagen is an employee of Sobi. D. Lillicrap has no conflict to declare. M. Makris has performed consultancy for CSL Behring, Grifols, and has attended advisory board meetings for Novo Nordisk and Shire. Ron Mathôt has received grants and non‐financial support from Bayer, grants from Merck and non‐financial support from Shire. Massimo Morfini has received personal fees from Novo Nordisk, Octapharma, Kedrion, Sobi, Pfizer, Freeline Therapeutics, and CSL Behring. E. Neufeld has received consultant/advisory board fees from CSL Behring, Octapharma, Grifols, Genentech, Kedrion, Novo Nordisk, Pfizer, Shire, and research support from Octapharma. He is a member of the Data Safety Monitoring Board for Bayer. Jeffrey Spears is an employee of Grifols.
AUTHOR CONTRIBUTIONS
V. Blanchette, J. Blatny, P. Collins, A.N. Edginton, K. Fischer, D.P. Hart, A. Iorio (Chair), S. Ito, D. Lillicrap, M. Makris, and E.J. Neufeld were appointed as working group on Population Pharmacokinetic of the ISTH SSC on Factor VIII and IX. All members of the group equally contributed to the group discussion. A. Boban, M. Cnossen, S.E. Croteau, J. Korth‐Bradley, S. Lethagen, R. Mathot, M. Morfini, and J. Spears joined the writing group and provided substantial comments on early draft of the manuscript. A. Iorio conceived and lead the project. A.N. Edginton drafted and finalized the manuscript. All authors commented on the draft and approved the final version of the manuscript.
GLOSSARY OF TERMS
Area under the curve (AUC): Surface beneath the activity vs time profile; it measures “exposure” to the concentrate.
Baseline factor level: The level of factor activity measured in plasma in absence of therapeutically administered factor concentrate. It is the level of factor activity, if any, endogenously produced by the individual. It is also the factor level used to classify the patient as severe (<0.01 IU/mL), moderate (0.01‐0.05 IU/mL) or mild (>0.05 IU/mL).
Below limit of quantitation (BLQ): Indicates a measurement of factor activity below the minimum amount detected by the laboratory assay. Most often BLQ values are reported as “undetectable”, or “not measurable”, or <0.01 IU/mL.
Clearance (eg, L/h): Volume of blood that is completely removed of factor activity in a specified unit of time.
Extended half‐life (EHL): Recombinant factor concentrates engineered to obtain a prolonged exposure of the active substance in the plasma. Extension of the half‐life is obtained by conjugation (to the Fc fragment of Ig, albumin, or PEG) or other techniques.
Half‐life: Time required for the plasma activity to decrease by half. It is qualified as terminal half‐life when estimated on the last portion of the activity versus time profile.
Recovery: Amount of factor activity measured in the plasma directly following an infusion as a proportion of the amount of concentrate infused.
International Units (IU): The unit used to define plasma factor activity level. The normal range for factor VIII and factor IX is from 0.5 IU/mL (50 IU/dL) to 1.5 IU/mL (150 IU/dL).
Immune tolerance induction treatment (ITI): Administration of factor VIII or IX meant to induce tolerance in patients with inhibitory antibodies.
Inter‐individual variability (IIV): The variability of PK between different individuals
Inter‐occasion variability (IOV): The variability of PK over time within the same individual
Lean body weight: Residual body weight after subtraction of the fat component (equal or more often inferior to the total body weight)
Maximum plasma concentration ( C max ): The plasma factor activity measured after a concentrate infusion. For bolus infusions it should theoretically be the concentration measured at the end of the infusion (C0).
Mean residence time (MRT): The average amount of time that a single molecule of factor VIII or factor IX stays in the body
Pharmacodynamics (PD): The study of the exposure‐ response relationship of a drug (ie, what the drug does to the body).
Pharmacokinetics (PK): The study of the absorption, distribution, metabolism, and excretion of drugs (ie, what the body does to the drug).
Population pharmacokinetics (PopPK): The study of the sources and correlates of variability in drug concentrations among individuals of the target patient population receiving clinically relevant doses of a drug of interest.
Prediction intervals: Probabilistic limits around a Bayesian predicted value.
Sparse data: Sampling technique by which few blood samples are drawn at any time after a drug infusion.
Therapeutic window: The interval between the lowest effective and the highest tolerable (safe) plasma concentration of a drug in the plasma/body.
Trough level: The lowest plasma level reached by a drug between two infusions (usually reached immediately before the subsequent infusion, and also called pre‐dose level).
Volume of distribution: The theoretical volume that would be necessary to contain the total amount of a factor concentrate to generate the same activity level that it is observed in the plasma. The link between the total amount of factor concentrate in the body and the plasma activity.
WAPPS‐Hemo: Web Accessible Population Pharmacokinetic Service–Hemophilia. A web‐based solution dedicated to individual pharmacokinetic profiling of patients with hemophilia treated with factor concentrates.
Wash‐out: Time spent off‐treatment before a conventional PK study to ensure no residual factor activity level generated by the factor concentrate is present in the blood. Usually equal or longer than 5 times the anticipated half‐life. The residual measurable activity level after an appropriate wash‐out is the baseline factor level.
Bayesian modelling: Probabilistic approach to forecasting individual PK profiles based on limited/sparse samples from one individual and previous knowledge from a population study.
Iorio A, Edginton AN, Blanchette V, et al. Performing and interpreting individual pharmacokinetic profiles in patients with Hemophilia A or B: Rationale and general considerations. Res Pract Thromb Haemost. 2018;2:535–548. 10.1002/rth2.12106
Contributor Information
Alfonso Iorio, Email: iorioa@mcmaster.ca, https://twitter.com/AlfonsoIorio.
Ron Mathôt, https://twitter.com/ProfMakris.
REFERENCES
- 1. Berntorp E, Astermark J, Baghaei F, et al. Treatment of haemophilia A and B and von Willebrand’s disease: summary and conclusions of a systematic review as part of a Swedish health‐technology assessment. Haemophilia. 2012;18:158–65. [DOI] [PubMed] [Google Scholar]
- 2. Iorio A, Marchesini E, Marcucci M, Stobart K, Chan AK. Clotting factor concentrates given to prevent bleeding and bleeding‐related complications in people with hemophilia A or B. Cochrane Database Syst Rev. 2011(9):CD003429. [DOI] [PubMed] [Google Scholar]
- 3. Iorio A, Iserman E, Blanchette V, et al. Target plasma factor levels for personalized treatment in haemophilia: a Delphi consensus statement. Haemophilia. 2017;23:e170–9. [DOI] [PubMed] [Google Scholar]
- 4. Ljung R, Fischer K, Carcao M, et al. Practical considerations in choosing a factor VIII prophylaxis regimen: role of clinical phenotype and trough levels. Thromb Haemost. 2016;115:913–20. [DOI] [PubMed] [Google Scholar]
- 5. Carcao MD, Iorio A. Individualizing factor replacement therapy in severe hemophilia. Semin Thromb Hemost. 2015;41:864–71. [DOI] [PubMed] [Google Scholar]
- 6. McEneny‐King A, Iorio A, Foster G, Edginton AN. The use of pharmacokinetics in dose individualization of factor VIII in the treatment of hemophilia A. Expert Opin Drug Metab Toxicol. 2016;12;11:1313–21. [DOI] [PubMed] [Google Scholar]
- 7. Morfini M, Marchesini E, Paladino E, Santoro C, Zanon E, Iorio A. Pharmacokinetics of plasma‐derived vs. recombinant FVIII concentrates: a comparative study. Haemophilia. 2015;21:204–9. [DOI] [PubMed] [Google Scholar]
- 8. Iorio A, McEneny‐King A, Keepanasseril A, Foster G, Edginton A. What is the role for population pharmacokinetics in hemophilia? Int J Pharmacokinetics. 2017;2;2:125–36. [Google Scholar]
- 9. Iorio A, Blanchette V, Blatny J, Collins P, Fischer K, Neufeld E. Estimating and interpreting the pharmacokinetic profiles of individual patients with hemophilia A or B using a population pharmacokinetic approach: communication from the SSC of the ISTH. J Thromb Haemost. 2017;15:2461–5. [DOI] [PubMed] [Google Scholar]
- 10. Bjorkman S, Oh M, Spotts G, Schroth P, et al. Population pharmacokinetics of recombinant factor VIII: the relationships of pharmacokinetics to age and body weight. Blood. 2012;119:612–8. [DOI] [PubMed] [Google Scholar]
- 11. McEneny‐King A, Chelle P, Henrard S, Hermans C, Iorio A, Edginton AN. Modeling of body weight metrics for effective and cost‐efficient conventional factor VIII dosing in hemophilia A prophylaxis. Pharmaceutics. 2017;9:E47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vlot AJ, Mauser‐Bunschoten EP, Zarkova AG, et al. The half‐life of infused factor VIII is shorter in hemophiliac patients with blood group O than in those with blood group A. Thromb Haemost. 2000;83:65–9. [PubMed] [Google Scholar]
- 13. Nilsson IM, Hedner U, Ahlberg A. Haemophilia prophylaxis in Sweden. Acta Paediatr Scand. 1976;65:129–35. [DOI] [PubMed] [Google Scholar]
- 14. Srivastava A, Brewer AK, Mauser‐Bunschoten EP, et al. Guidelines for the management of hemophilia. Haemophilia. 2013;19:e1–47. [DOI] [PubMed] [Google Scholar]
- 15. de Moerloose P, Fischer K, Lambert T, et al. Recommendations for assessment, monitoring and follow‐up of patients with haemophilia. Haemophilia. 2012;18:319–25. [DOI] [PubMed] [Google Scholar]
- 16. Hazendonk HC, Lock J, Mathot RA, et al. Perioperative treatment of hemophilia A patients: blood group O patients are at risk of bleeding complications. J Thromb Haemost. 2016;14:468–78. [DOI] [PubMed] [Google Scholar]
- 17. Hazendonk H, Fijnvandraat K, Lock J, et al. A population pharmacokinetic model for perioperative dosing of factor VIII in hemophilia A patients. Haematologica. 2016;101:1159–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hazendonk HC, van Moort I, Fijnvandraat K, et al. The “OPTI‐CLOT” trial. A randomised controlled trial on periOperative PharmacokineTIc‐guided dosing of CLOTting factor concentrate in haemophilia A. Thromb Haemost. 2015;114:639–44. [DOI] [PubMed] [Google Scholar]
- 19. Berntorp E, Shapiro A, Astermark J, et al. Inhibitor treatment in haemophilias A and B: summary statement for the 2006 international consensus conference. Haemophilia. 2006;12(Suppl 6):1–7. [DOI] [PubMed] [Google Scholar]
- 20. Blanchette VS, Key NS, Ljung LR, et al. Definitions in hemophilia: communication from the SSC of the ISTH. J Thromb Haemost. 2014;12:1935–9. [DOI] [PubMed] [Google Scholar]
- 21. Hay CR, DiMichele DM; International Immune Tolerance Study . The principal results of the International Immune Tolerance Study: a randomized dose comparison. Blood. 2012;119(6):1335–44. [DOI] [PubMed] [Google Scholar]
- 22. Collins PW, Chalmers E, Hart DP, et al. Diagnosis and treatment of factor VIII and IX inhibitors in congenital haemophilia: (4th edition). UK Haemophilia Centre Doctors Organization. Br J Haematol. 2013;160:153–70. [DOI] [PubMed] [Google Scholar]
- 23. Collins P, Chalmers E, Alamelu J, et al. First‐line immune tolerance induction for children with severe haemophilia A: a protocol from the UK Haemophilia Centre Doctors’ Organisation Inhibitor and Paediatric Working Parties. Haemophilia. 2017;23:654–9. [DOI] [PubMed] [Google Scholar]
- 24. Preston FE, Kitchen S. Quality control and factor VIII assays. Haemophilia. 1998;4:651–3. [DOI] [PubMed] [Google Scholar]
- 25. Bland JM, Altman DG. Applying the right statistics: analyses of measurement studies. Ultrasound Obstet Gynecol. 2003;22:85–93. [DOI] [PubMed] [Google Scholar]
- 26. Hulley SB, Newman TB, Cummings SR. Planning the measurements: precision and accuracy In: Hulley SB, Cummings SR, Browner WS, Grady DG, Newman TB, editors. Designing Clinical Research, 3rd ed. Philadelphia: Lippincott Williams & Wilkins; 2007: 37–50. [Google Scholar]
- 27. Gruppo RA, Brown D, Wilkes MM, Navickis RJ. Comparative effectiveness of full‐length and B‐domain deleted factor VIII for prophylaxis–a meta‐analysis. Haemophilia. 2003;9:251–60. [DOI] [PubMed] [Google Scholar]
- 28. Santoro C, Iorio A, Ferrante F, et al. Performance of recalibrated ReFacto laboratory standard in the measurement of FVIII plasma concentration via the chromogenic and one‐stage assays after infusion of recalibrated ReFacto (B‐domain deleted recombinant factor VIII). Haemophilia. 2009;15:779–87. [DOI] [PubMed] [Google Scholar]
- 29. Hubbard AR. Potency labeling of novel factor VIII and factor IX concentrates: past experience and current strategy. Semin Thromb Hemost. 2015;41:849–54. [DOI] [PubMed] [Google Scholar]
- 30. Dodt J, Hubbard AR, Wicks SJ, et al. Potency determination of factor VIII and factor IX for new product labelling and postinfusion testing: challenges for caregivers and regulators. Haemophilia. 2015;21:543–9. [DOI] [PubMed] [Google Scholar]
- 31. Wilmot HV, Hogwood J, Gray E. Recombinant factor IX: discrepancies between one‐stage clotting and chromogenic assays. Haemophilia. 2014;20:891–7. [DOI] [PubMed] [Google Scholar]
- 32. Sommer JM, Buyue Y, Bardan S, et al. Comparative field study: impact of laboratory assay variability on the assessment of recombinant factor IX Fc fusion protein (rFIXFc) activity. Thromb Haemost. 2014;112:932–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Young GB, Fischer K, Iorio A, et al.; on behalf of the International Prophylaxis Study Group (IPSG) . Laboratory assay measurement of modified clotting factor concentrates: a review of the literature and recommendations for practice, (Submitted). [DOI] [PubMed]
- 34. Peyvandi F, Oldenburg J, Friedman KD. A critical appraisal of one‐stage and chromogenic assays of factor VIII activity. J Thromb Haemost. 2016;14:248–61. [DOI] [PubMed] [Google Scholar]
- 35. Garmann D, McLeay S, Shah A, Vis P, Maas Enriquez M, Ploeger BA. Population pharmacokinetic characterization of BAY 81‐8973, a full‐length recombinant factor VIII: lessons learned—importance of including samples with factor VIII levels below the quantitation limit. Haemophilia. 2017;23:528–37. [DOI] [PubMed] [Google Scholar]
- 36. Beal SL. Ways to fit a PK model with some data below the quantification limit. J Pharmacokinet Pharmacodyn. 2001;28:481–504. [DOI] [PubMed] [Google Scholar]
- 37. Lissitchkov T, Rusen L, Georgiev P, et al. PK‐guided personalized prophylaxis with Nuwiq((R)) (human‐cl rhFVIII) in adults with severe haemophilia A. Haemophilia. 2017;23:697–704. [DOI] [PubMed] [Google Scholar]
- 38. Pasca S, Milan M, Sarolo L, Zanon E. PK‐driven prophylaxis versus standard prophylaxis: When a tailored treatment may be a real and achievable cost‐saving approach in children with severe hemophilia A. Thromb Res. 2017;157:58–63. [DOI] [PubMed] [Google Scholar]
- 39. Lee M, Schulman S, Ingerslev J. The design and analysis of pharmacokinetic studies of coagulation factors. ISTH Website. Available from https://c.ymcdn.com/sites/www.isth.org/resource/group/d4a6f49a-f4ec-450f-9e0f-7be9f0c2ab2e/official_communications/fviiipharmaco.pdf.
- 40. van Warmerdam LJ, ten Bokkel Huinink WW, Maes RA, Beijnen JH. Limited‐sampling models for anticancer agents. J Cancer Res Clin Oncol. 1994;120:427–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sorensen BT, Stromgren A, Jakobsen P, Jakobsen A. A limited sampling method for estimation of the carboplatin area under the curve. Cancer Chemother Pharmacol. 1993;31:324–7. [DOI] [PubMed] [Google Scholar]
- 42. Garraffo R, Iliadis A, Cano JP, Dellamonica P, Lapalus P. Application of Bayesian estimation for the prediction of an appropriate dosage regimen of amikacin. J Pharm Sci. 1989;78:753–7. [DOI] [PubMed] [Google Scholar]
- 43. Gaulier JM, Boulieu R, Fischer C, Mauguiere . Evaluation of a bayesian pharmacokinetic program for phenytoin concentration predictions in outpatient population. Eur J Drug Metab Pharmacokinet. 1998;23:295–300. [DOI] [PubMed] [Google Scholar]
- 44. Denaro CP, Ravenscroft PJ. Comparison of Sawchuk‐Zaske and bayesian forecasting for aminoglycosides in seriously ill patients. Br J Clin Pharmacol. 1989;28:37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bjorkman S, Collins P. Project on Factor VIIIFIXPotFVFIXS, Standardization Committee of The I. Measurement of factor VIII pharmacokinetics in routine clinical practice. J Thromb Haemost. 2013;11:180–2. [DOI] [PubMed] [Google Scholar]
- 46. Ette EI, Williams PJ. Population pharmacokinetics I: background, concepts, and models. Ann Pharmacother. 2004;38:1702–6. [DOI] [PubMed] [Google Scholar]
- 47. Holford NH, Buclin T. Safe and effective variability‐a criterion for dose individualization. Ther Drug Monit. 2012;34:565–8. [DOI] [PubMed] [Google Scholar]
- 48. Iorio A, Keepanasseril A, Foster G, et al. Development of a Web‐Accessible Population Pharmacokinetic Service‐Hemophilia (WAPPS‐Hemo): Study Protocol. JMIR Res Protoc. 2016;5:e239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dubois A, Gsteiger S, Balser S, et al. Pharmacokinetic similarity of biologics: analysis using nonlinear mixed‐effects modeling. Clin Pharmacol Ther. 2012;91:234–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Brekkan A, Berntorp E, Jensen K, Nielsen EI, Jonsson S. Population pharmacokinetics of plasma‐derived factor IX: procedures for dose individualization. J Thromb Haemost. 2016;14:724–32. [DOI] [PubMed] [Google Scholar]
- 51. Iorio A, Fischer K, Blanchette V, et al. Tailoring treatment of haemophilia B: accounting for the distribution and clearance of standard and extended half‐life FIX concentrates. Thromb Haemost. 2017;117:1023–30. [DOI] [PubMed] [Google Scholar]
- 52. Lock J, de Bekker‐Grob EW, Urhan G, et al. Facilitating the implementation of pharmacokinetic‐guided dosing of prophylaxis in haemophilia care by discrete choice experiment. Haemophilia. 2016;22:e1–10. [DOI] [PubMed] [Google Scholar]
- 53. Den Uijl IE, Mauser Bunschoten EP, Roosendaal G, et al. Clinical severity of haemophilia A: does the classification of the 1950s still stand? Haemophilia. 2011;17:849–53. [DOI] [PubMed] [Google Scholar]
- 54. Nestorov I, Neelakantan S, Ludden TM, Li S, Jiang H, Rogge M. Population pharmacokinetics of recombinant factor VIII Fc fusion protein. Clin Pharmacol Drug Dev. 2015;4:163–74. [DOI] [PubMed] [Google Scholar]
- 55. Zhang Y, Roberts J, Bensen‐Kennedy D, et al. Population pharmacokinetics of a new long‐acting recombinant coagulation factor IX albumin fusion protein for patients with severe hemophilia B. J Thromb Haemost. 2016;14:2132–40. [DOI] [PubMed] [Google Scholar]
- 56. Diao L, Li S, Ludden T, Gobburu J, Nestorov I, Jiang H. Population pharmacokinetic modelling of recombinant factor IX Fc fusion protein (rFIXFc) in patients with haemophilia B. Clin Pharmacokinet. 2014;53:467–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Guyatt GH, Oxman AD, Kunz R, et al. GRADE guidelines: 8. Rating the quality of evidence—indirectness. J Clin Epidemiol. 2011;64:1303–10. [DOI] [PubMed] [Google Scholar]
- 58. Goudemand J, Peynet J, Chambost H, et al. A cross‐over pharmacokinetic study of a double viral inactivated factor IX concentrate (15 nm filtration and SD) compared to a SD factor IX concentrate. Thromb Haemost. 1998;80:919–24. [PubMed] [Google Scholar]
- 59. Thomas DP, Hampton KK, Dasani H, et al. A cross‐over pharmacokinetic and thrombogenicity study of a prothrombin complex concentrate and a purified factor IX concentrate. Br J Haematol. 1994;87:782–8. [DOI] [PubMed] [Google Scholar]
- 60. Ragni MV, Yabes JG, Fogarty PF, et al. Pilot randomized, non‐inferiority, cross‐over trial of once‐weekly vs. three times‐weekly recombinant factor VIII prophylaxis in adults with severe haemophilia A. Haemophilia. 2017;23:e43–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Spira J, Plyushch O, Zozulya N, et al. Safety, pharmacokinetics and efficacy of factor VIIa formulated with PEGylated liposomes in haemophilia A patients with inhibitors to factor VIII—an open label, exploratory, cross‐over, phase I/II study. Haemophilia. 2010;16:910–8. [DOI] [PubMed] [Google Scholar]
- 62. Suzuki A, Tomono Y, Korth‐Bradley JM. Population pharmacokinetic modelling of factor IX activity after administration of recombinant factor IX in patients with haemophilia B. Haemophilia. 2016;22:e359–66. [DOI] [PubMed] [Google Scholar]
- 63. Bjorkman S, Berntorp E. Pharmacokinetics of coagulation factors: clinical relevance for patients with haemophilia. Clin Pharmacokinet. 2001;40:815–32. [DOI] [PubMed] [Google Scholar]
- 64. Collins PW, Fischer K, Morfini M, Blanchette VS, Bjorkman S; International Prophylaxis Study Group Pharmacokinetics Expert Working Group . Implications of coagulation factor VIII and IX pharmacokinetics in the prophylactic treatment of haemophilia. Haemophilia. 2011;17:2–10. [DOI] [PubMed] [Google Scholar]
- 65. Carlsson M, Bjorkman S, Berntorp E. Multidose pharmacokinetics of factor IX: implications for dosing in prophylaxis. Haemophilia. 1998;4:83–8. [DOI] [PubMed] [Google Scholar]