

Abstract

Protein arginine methyltransferase 5 (PRMT5) is a type II arginine methyltransferase that catalyzes the formation of symmetric dimethylarginine in a number of nuclear and cytoplasmic proteins. Although the cellular functions of PRMT5 have not been fully unraveled, it has been implicated in a number of cellular processes like RNA processing, signal transduction, and transcriptional regulation. PRMT5 is ubiquitously expressed in most tissues and its expression has been shown to be elevated in several cancers including breast cancer, gastric cancer, glioblastoma, and lymphoma. Here, we describe the identification and characterization of a novel and selective PRMT5 inhibitor with potent in vitro and in vivo activity. Compound 1 (also called LLY-283) inhibited PRMT5 enzymatic activity in vitro and in cells with IC50 of 22 ± 3 and 25 ± 1 nM, respectively, while its diastereomer, compound 2 (also called LLY-284), was much less active. Compound 1 also showed antitumor activity in mouse xenografts when dosed orally and can serve as an excellent probe molecule for understanding the biological function of PRMT5 in normal and cancer cells.
Keywords: PRMT5, methyltransferase, inhibitor, splicing, methylation
Arginine methylation of proteins is an important class of post-translational modification that is known to play a key functional role in crucial cellular pathways including cell growth and proliferation, apoptosis, angiogenesis, and metastasis by regulating both transcription and post-transcriptional RNA processing.1 This modification is catalyzed by the PRMT (Protein aRginine MethylTransferase) family of arginine methyltransferases, which transfer a methyl group from S-adenosyl methionine (AdoMet) to the terminal guanidino nitrogen atoms of arginine side-chains of histones and nonhistone proteins. Nine human PRMTs have been identified to date,2 and they are further subdivided into type I, type II, type III, and type IV enzymes based on the kind of arginine methylation catalyzed. PRMT5 is the predominant type II PRMT3 that catalyzes the formation of ω-NG-monomethyl and ω-NG,N′G-symmetric dimethyl arginine residues and can methylate arginine residues in both a GAR and PGM motif.
PRMT5 is involved in the regulation of diverse cellular processes including transcription,4 RNA metabolism,5 signal transduction,6 maintenance of Golgi apparatus architecture,7 and cellular differentiation.8 However, how PRMT5 impacts these biological processes is not yet clearly defined. In the nucleus, PRMT5 has been shown to be involved in transcriptional repression including that of tumor suppressor and cell cycle genes like ST7 (suppressor of tumorigenicity 7 protein),9 cyclin E1,10 and CDKN2A (cyclin-dependent kinase inhibitor 2A).11 PRMT5 brings about these epigenetic modifications through the symmetric dimethylation of histone H4 on the R3 residue (H4R3me2s) and on the R8 residue of histone H3 (H3R8me2s).12 H4R3me2s is generally associated with transcriptional repression,13 while H3R8me2s has been seen as a mark for both transcriptional activation and repression.14 Besides these marks, H2AR3me2s and H3R2me2s modifications have also been associated with PRMT5 activity. H3R2me2s is present in euchromatic regions and at several promoter regions like those of gluconeogenic genes; this methylation is followed by the recruitment of WDR5 and the subsequent expression of corresponding genes.15 The function of the cytoplasmic H2AR3me2s modification remains unclear.
An important facet of PRMT5 biology involves the regulation of RNA splicing and snRNP biogenesis in the cytoplasm where PRMT5 mainly exists in a 20S methylosome complex together with MEP50 and pICln.16,17 In this complex, PRMT5 catalyzes the symmetric dimethylation of the Sm proteins B/B′, D1, and D3 of U1, U2, U4, U5 snRNP and U6 snRNA-associated LSm418 thus enabling their binding to Survival Motor Neuron (SMN)19 and subsequent deposition onto U-snRNAs thereby forming U-snRNPs.20 It is believed that Sm protein methylation increases their binding affinity for SMN though the effect was apparent only in vitro.21 However, the spliceosomal role of PRMT5 appears to be conserved across species, and mutations in PRMT5 in Arabidopsis thaliana(22) and Drosophila melanogaster (Dart5)23 are believed to affect alternative splicing of critical genes involved in germ cell speciation in Drosophila and circardian rhythms in Arabidopsis. Similarly, PRMT5-null knockouts are embryonic lethal in mice showing that PRMT5 is essential for mammalian development.24 Remarkably, deletion of PRMT5 in neural stem/progenitor cells (NPCs) resulted in widespread effects on alternative splicing events including that of Mdm45 producing, in the latter case, a truncated splice variant lacking exon 6 that is thus targeted for degradation by nonsense-mediated decay. This causes a decrease in full-length MDM4 leading to induction of p53 pathway and, subsequently, cell cycle arrest and apoptosis. However, p53 activity is not solely responsible for the phenotype arising from the loss of PRMT5.5
The important role of PRMT5 in cancer including its interaction with pathways deregulated in cancer implicates it as an attractive therapeutic target in cancer.4,25 Several small molecule inhibitors of PRMTs including PRMT5 have been identified.26−28 However, most of these are low potency inhibitors or lack cellular and/or in vivo activity. Recently, a substrate competitive and cell active PRMT5 inhibitor, EPZ015666, was demonstrated to have antiproliferative effects in both in vitro and in vivo models of Mantle cell lymphoma (MCL).29,30 We had earlier reported the crystal structure of human PRMT5:MEP50 complex,31 which aided the synthesis of novel inhibitors of PRMT5. Here, we describe the identification and characterization of a potent and selective inhibitor of PRMT5, compound 1 (Figure 1), that specifically binds in the SAM pocket of PRMT5 in contrast to EPZ015666, which is histone peptide (substrate) competitive. The unique characteristics of compound 1 enable it to be highly potent and cell permeable with well-defined PK characteristics.
Figure 1.

Synthesis of PRMT5 chemical probe 1 and negative control compound 2.
Deazapurine analogue 3 was prepared according to literature procedures.32,33 Hypervalent iodine oxidation of primary alcohol 3 gave acid 4, which was converted to morpholinoamide 5 without evidence of epimerization of the C4 center. Grignard addition proceeded smoothly at low temperatures to yield acetophenone 6. Chemical probe 1 and diastereomer 2 are readily prepared from this common intermediate. Asymmetric transfer hydrogenation of the ketone with (R,R)-Ts-DENEB34 delivered the hydride from the re-face, producing 7 with high diastereoselectivity. The relative configuration of the 5′-hydroxyl was confirmed from single crystal X-ray structure of the para-bromo benzoate (Supporting Information). The chloride was displaced by ammonia in MeOH, and the acetal 8 was hydrolyzed to produce 1. To obtain the 5′-OH diastereomer, l-selectride was used to enforce delivery of the hydride from the si-face of acetophenone 6, yielding 9 with high facial selectivity. Finally, treatment with ammonia and mild acid hydrolysis of the acetal delivered compound 2.
To understand the binding mode of compound 1 and to characterize its molecular interactions with PRMT5, we determined the crystal structure of the PRMT5:MEP50 complex bound to compound 1 (Figure 2). The crystal structure was determined to 2.9 Å resolution using published methods.31 Coordinates and structure factors have been deposited with the Protein Data Bank under the accession code 6CKC. Data quality and refinement statistics are presented in Table S1 (Supporting Information). In contrast to the previously published inhibitors of PRMT5 that bind in the peptide substrate pocket,29 compound 1 is the first potent inhibitor that binds in the AdoMet pocket, with the adenine and ribose moieties adopting very similar conformations and making similar interactions observed in other PRMT5:MEP50 structures with AdoMet analogs.31 The adenine makes hydrogen bonds to the side chain carboxylate of Asp419 and the main chain amino of Met420, while the ribose makes a pair of hydrogen bonds to the glutamate side chain of Glu392, as well as a hydrogen bond to the side chain of Tyr324. Comparison to the PRMT5:MEP50 complex with a substrate peptide in addition to an AdoMet analog reveals that the phenyl group of compound 1 displaces the side chain of Phe327, which adopts an alternate rotamer with minimal movement of the main chain.
Figure 2.
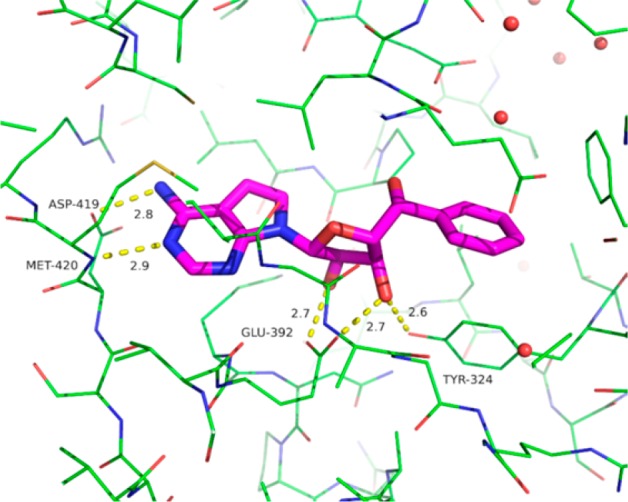
Crystal structure of the PRMT5:MEP50 complex (green) bound to compound 1 (purple). Key hydrogen bonds are indicated in dotted yellow lines as the ligand binds in the AdoMet pocket. Select residues are labeled.
Compounds 1 and 2 were screened for their in vitro inhibitory activity toward PRMT5:MEP50 complex (Figure 3). Using a radioactivity-based assay monitoring the transfer of the methyl group from 3H-SAM to peptide substrate, we confirmed that compound 1, potently inhibited PRMT5:MEP50 activity in vitro with an IC50 of 22 ± 3 nM, while its 5′-stereoisomer, compound 2, was almost 50-fold less active (IC50 of 1074 ± 53 nM). Binding of compound 1 to PRMT5:MEP50 complex was confirmed by surface plasmon resonance (SPR; Supporting Information) with a KD (equilibrium dissociation constant) value of 6 ± 2 nM (n = 3) and dissociation- (kon) and association- (koff) rate constants of 3.9 ± 0.4 × 105 M–1 s–1 and 2.2 ± 0.8 × 10–3 s–1, respectively. The mechanism of action (MOA) of compound 1 was assessed by determining the IC50 values in the presence of various concentrations of peptide and SAM. No dramatic change was observed in IC50 values upon increasing the concentration of each substrate to as high as 25× of the respective Km (Michaelis–Menten constant) values. Even though binding of compound 1 in the SAM pocket was clearly seen in cocrystal structures, the SAM competition assay did not reveal competitive inhibition with respect to SAM; rather the MOA data supports apparent noncompetitive patterns with respect to both substrates. The reason(s) for noncompetiveness is (are) unknown at this time. We have seen this earlier in other cases, e.g., PRMT4 and PRMT6 for MS04935 (a dual PRMT4 and 6 inhibitor) and PRMT6 for MS02336 (a pan-Type 1 PRMT inhibitor). However, in both these cases the ligand is binding in the peptide pocket.
Figure 3.
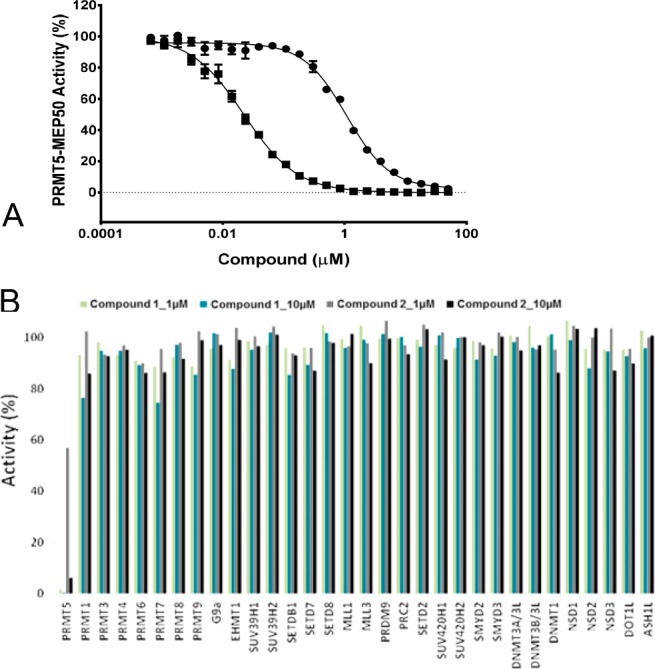
(A) Enzyme inhibition assay for compound 1 and its negative control (compound 2). The IC50 values of 22 ± 3 nM (Hill Slope of 1) and 1074 ± 53 nM (Hill Slope of 1.2) were determined for compound 1 (■) and compound 2 (●), respectively. The experiments were performed in triplicate. (B) Selectivity of compounds 1 and 2 at 1 and 10 μM (as indicated on the plot) were assessed against a collection of 32 methyltransferases.
Compounds 1 and 2 were also assessed for their activity against a panel of 32 methyltransferases using radioactive assays as described in Supporting Information. In spite of being a close analogue of SAM, compound 1 showed remarkable specificity for PRMT5 with the activity dropping significantly even against other arginine methyltransferases including the closely related arginine methyltransferase PRMT7 and the Type I PRMTs such as PRMT4 and 6.
The cellular activity of the compounds was evaluated by monitoring SmBB′ symmetric dimethylation in MCF7 cells in Western blot (Figure 4) and alternative splicing of MDM4 in A375 cells by qPCR (Figure 5). It has been reported previously that the various SMN complex proteins, including SmBB′ are symmetrically dimethylated by PRMT5.17 We found that knocking down of PRMT5, but not PRMT1, 3, 4, 6, 7, and 9 resulted in decreased basal SmBB′ symmetric dimethylation levels in MCF7 cells (Supporting Information). Similar to PRMT5 knockdown, two day treatment of MCF7 cells with compound 1 decreased SmBB′-Rme2s levels in a dose-dependent manner (IC50 = 25 ± 1 nM) (Figure 4). At 1 μM, compound 1 decreased the levels of SmBB′-Rme2s to the same extent as previously published peptide-competitive PRMT5 inhibitor, GSK591.29 At the same concentration, negative control compound, compound 2, caused only 20% decrease in SmBB′-Rme2s levels.
Figure 4.
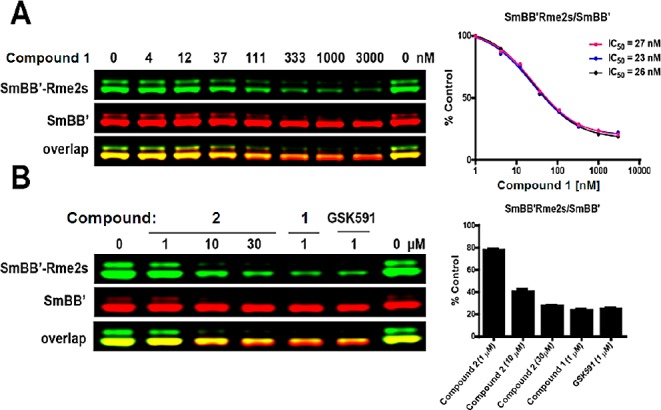
Compound 1 inhibits PRMT5 activity in MCF7 cells. MCF7 cells were treated with compounds at indicated concentrations for 48 h and SmBB′-Rme2s levels were determined by Western blot. (A) SmBB′-Rme2s dose response to compound 1. The graph represents nonlinear fits of SmBB′-Rme2s signal intensities normalized to total SmBB′. (B) Effect of compound 1, GSK3203591, and compound 2 on SmBB′-Rme2s levels. The graph represents SmBB′-Rme2s signal intensities normalized to total SmBB′ represented as % DMSO control. The results are mean ± SEM of three replicates.
Figure 5.
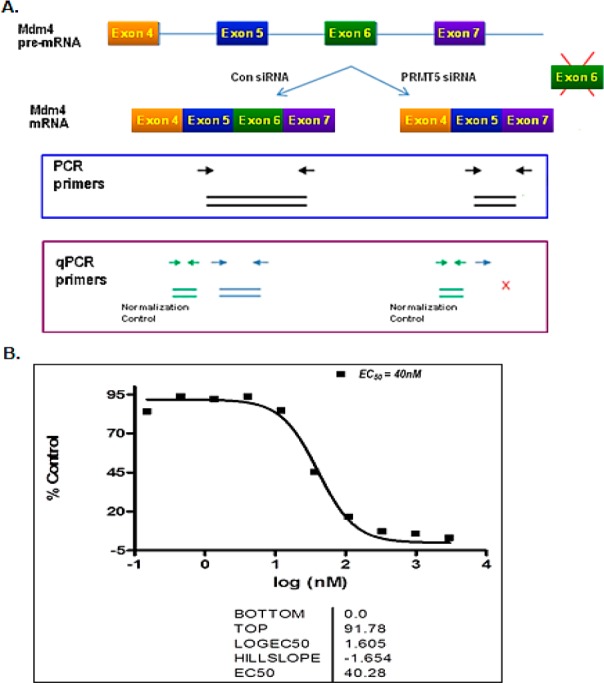
Compound 1 inhibits PRMT5 activity in A375 cells. (A) Scheme depicting the qPCR assay strategy. (B) Mdm4 exon 6 splicing (normalized to exon 5) dose response to compound 1 (run in duplicate; Supporting Information). Subsequent repeats of the assay (n > 3) gave an EC50 of 37 ± 3 nM (mean ± SEM).
Reduced methylation of Sm proteins caused by the depletion of PRMT5 has been shown to lead to aberrant alternative splicing of specific mRNAs with weak 5′ donor sites.5 Mdm4 was seen to be one of these key mRNAs that is sensitive to the defects in the spliceosomal machinery. Similarly, compound 1 also inhibited the PRMT5 mediated regulation of the splicing of MDM4, as indicated by the ratio of MDM4 mRNA bearing exons 5 and 6 to that bearing solely exon 5 (Figure 5). Treatment of A375 melanoma cells with compound 1 for 72 h caused skipping of the exon 6 (cassette exon) of Mdm4 with an EC50 of 40 nM when normalized to the levels of exon 5.
The effects of PRMT5 target engagement manifested as an antiproliferative phenotype in many cancer cell lines when tested in a 7-day proliferation assay (Cell titer Glo assay; Supporting Information). Though hematological tumors were especially sensitive, potent antiproliferative effects were observed in many solid tumor lines including those from breast, lung, skin ovarian, and gastric cancer (representative examples from some of the common cancer types are shown in Table 1). In the cell lines where proliferation inhibition assay was repeated, reproducible IC50 values were obtained, for example, A375 (n = 3) gave an average IC50 of 46 ± 5 nM (mean ± SEM).
Table 1. Cancer Cell Line Response to Compound 1 in 7-Day Proliferation Assay (Supporting Information).
| cell line | histology | IC50 (μM) | max inhibition (%) |
|---|---|---|---|
| HCC1937 | breast | 0.030 | 83 |
| T-42D | breast | 0.003 | 84.0 |
| UACC-812 | breast | >2.5 | 21.0 |
| MX-1 cells | breast | >2.5 | 36.9 |
| NUGC-3 | gastric | 0.017 | 92 |
| KE-97 | gastric | 0.024 | 95.7 |
| MV4–11 | hematological | 0.016 | 93 |
| Daudi | hematological | 0.017 | 99.3 |
| Karpas-422 | hematological | 0.085 | 99.9 |
| HEL 92.1.7 | hematological | 0.003 | 91.2 |
| ARH-77 | hematological | 0.010 | 97.2 |
| REC-1 | hematological | 0.022 | 99.7 |
| IM-9 | hematological | 0.046 | 93 |
| KMS-12-BM | hematological | 0.006 | 99 |
| RPMI-8226 | hematological | 0.009 | 88.5 |
| OPM-2 | hematological | >2.5 | 22.5 |
| NCI-H2171 | lung | 0 010 | 97.7 |
| H-2122 | lung | 0.027 | 90 |
| NCI-H1930 | lung | >2.5 | 49.6 |
| NCI-H1651 | lung | >2.5 | 32 |
| A375 | skin | 0.037 | 91.1 |
| A2058 | skin | 0.013 | 96 |
| GAK | skin | 0.016 | 99.9 |
| Colo829 | skin | 0.013 | 51 |
| OVCAR-3 | ovarian | 0.032 | 95 |
Further in vitro and in vivo characterization of compound 1 revealed that it possesses desirable physical and ADME properties that will enable broad exploration of the PRMT5 mechanism in animals. In brief, the compound has excellent physicochemical properties, high unbound levels in plasma, moderate to slow plasma clearance, low potential for competitive CYP inhibition, and dynamic range of oral bioavailability (Supporting Information) that result in high exposure in rodents when given once daily. As seen in Figure 6, unbound Cmax (@10 mg/kg PO dose) exceeds the cellular MDM4 IC50 value for compound 1.
Figure 6.

Compound 1 mouse PO PK: Oral dose response.
Since compound 1 is amenable to in vivo studies, we tested its efficacy in A375 derived xenografts grown subcutaneously in severe combined immunodeficiency (SCID) mice in a 28-day study, with an oral once a day (QD) dose of compound 1 at 20 mg per kilogram of body weight (mg/kg).
After 28 days of dosing, compound 1 (20 mg/kg; QD) induced statistically significant tumor growth inhibition (TGI) compared to vehicle-treated tumors (Figure 7). A more pronounced in vivo response to compound 1 is predicted in cell lines from hematological cancers like DLBCL, MCL, ALL, etc., since many such lines are highly sensitive to compound 1in vitro. Further, at the doses given, compound 1 was well tolerated with moderate body weight loss (<10%) and no apparent toxicological observations.
Figure 7.
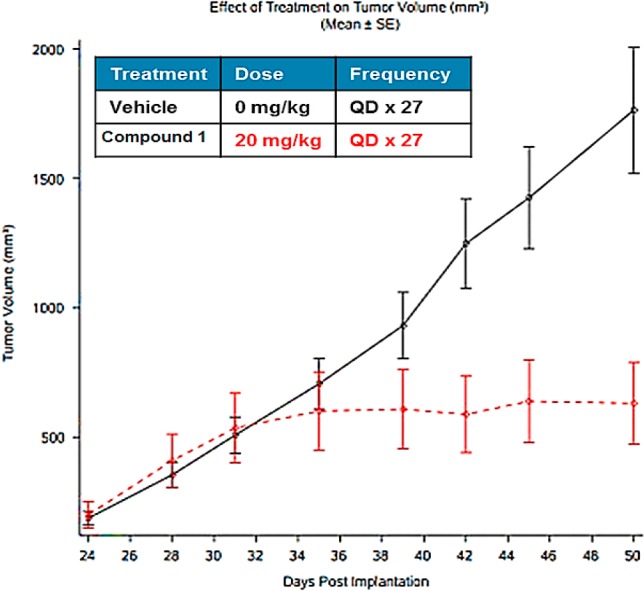
Compound 1 inhibits tumor growth in mouse xenograft (A375).
In summary, we have discovered and characterized a potent and selective PRMT5 inhibitor that can be used as an in vivo tool compound to evaluate the pharmacology of PRMT5 inhibition. Unlike earlier PRMT inhibitors such as EPZ015666, which is mainly substrate competitive, compound 1 directly binds to and occupies the SAM pocket and is, to our knowledge, the first such PRMT5 inhibitor that has been reported. We have shown that compound 1 is a low nanomolar enzymatic inhibitor of the PRMT5:MEP50 complex in biochemical assays and is selective for PRMT5 over other methyltransferases including related family members. The binding mode in the SAM pocket of PRMT5 was confirmed from a 2.9 Å resolution crystal structure. In addition, compound 1 has submicromolar cell-based potency as revealed by its ability to inhibit the arginine methylation of spliceosomal protein SmB/B′ and, consequently, affects the alternative splicing of Mdm4. We have shown that compound 1 has broad in vitro antiproliferative activity across several tumor types. Further, well-behaved and predictable pharmacokinetic characteristics of compound 1 enable investigation of its antitumor effects in vivo as seen in A375 mouse xenografts. Although PRMT5 is also known as an epigenetic modulator through its regulation of histone arginine methylation especially that of histones H3 (H3R8) and H4 (H4R3), we did not observe much evidence of such effects in our cell-based assays with compound 1 using corresponding commercially available and proprietary methylated histone antibodies. However, at this stage we cannot rule out the epigenetic role of PRMT5. Further characterization of the mechanism of antitumor effects of PRMT5 inhibition will be critical in defining the therapeutic potential of PRMT5 across different tumor types.
Acknowledgments
The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, Merck & Co., Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and the Wellcome Trust. The help provided by John M. Strelow, Eli Lilly and Company, on enzyme kinetics is gratefully acknowledged.
Glossary
ABBREVIATIONS
- SAM
S-adenosylmethionine
- PRMT
protein arginine methyl transferase
- SmBB′me2s
small ribonucleoprotein particle protein BB′ symmetric dimethylation
- PK
pharmacokinetic
- MEP50
methylosome protein 50
- TEMPO
2,2,6,6-tetramethylpiperidine 1-oxyl
- (R,R)-Ts-DENEB
chloro{N-[(1R,2R)-1,2-diphenyl-2-(2-(4-methylbenzyloxy)ethylamino)-ethyl]-4-methylbenzenesulfonamide(chloro)ruthenium(II) (R,R)
- CYP
cytochrome P450 enzyme
- DLCBL
diffused large B-cell lymphoma
- MCL
mantle cell lymphoma
- ALL
acute lymphoblastic leukemia
- l-selectride
lithium tri-sec-butyl borohydride
- GAR
glycine and arginine-rich motif
- PGM
proline, glycine and methionine-rich motif
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00014.
In vitro and in vivo biological methods describing biochemical activity assays, surface plasmon resonance (SPR), cellular PRMT5 assay, Western blot, MDM4 Exon5/6 PCR assay in A375 tumor cells, 7-day proliferation assay, A375 xenograft tumor model, and a table listing crystal structure data quality and refinement statistics; experimental and characterization data for 1 and 2, and copies of 1H NMR spectra; SPR and MOA data; PRMT knockdown data; summary table for ADME and PK properties (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Blanc R. S.; Richard S. Arginine methylation: the coming of age. Mol. Cell 2017, 65, 8–24. 10.1016/j.molcel.2016.11.003. [DOI] [PubMed] [Google Scholar]
- Wolf S. S. The protein arginine methyltransferase family: an update about function, new perspectives and the physiological role in humans. Cell. Mol. Life Sci. 2009, 66, 2109–2121. 10.1007/s00018-009-0010-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branscombe T. L.; Frankel A.; Lee J. H.; Cook J. R.; Yang Z.; Pestka S.; Clarke S. PRMT5 (Janus kinase-binding protein 1) catalyzes the formation of symmetric dimethylarginine residues in proteins. J. Biol. Chem. 2001, 276, 32971–32976. 10.1074/jbc.M105412200. [DOI] [PubMed] [Google Scholar]
- Chen H.; Lorton B.; Gupta V.; Shechter D. A TGFβ-PRMT5-MEP50 axis regulates cancer cell invasion through histone H3 and H4 arginine methylation coupled transcriptional activation and repression. Oncogene 2017, 36, 373–386. 10.1038/onc.2016.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezzi M.; Teo S. X.; Muller J.; Mok W. C.; Sahu S. K.; Vardy L. A.; Bonday Z. Q.; Guccione E. Regulation of constitutive and alternative splicing by PRMT5 reveals a role for Mdm4 pre-mRNA in sensing defects in the spliceosomal machinery. Genes Dev. 2013, 27, 1903–1916. 10.1101/gad.219899.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson M.; Durant S. T.; Cho E. C.; Sheahan S.; Edelmann M.; Kessler B.; La Thangue N. B. Arginine methylation regulates the p53 response. Nat. Cell Biol. 2008, 10, 1431–1439. 10.1038/ncb1802. [DOI] [PubMed] [Google Scholar]
- Zhou Z.; Sun X.; Zou Z.; Sun L.; Zhang T.; Guo S.; Wen Y.; Liu L.; Wang Y.; Qin J.; Li L.; Gong W.; Bao S. PRMT5 regulates Golgi apparatus structure through methylation of the golgin GM130. Cell Res. 2010, 20, 1023–1033. 10.1038/cr.2010.56. [DOI] [PubMed] [Google Scholar]
- Dacwag C. S.; Ohkawa Y.; Pal S.; Sif S.; Imbalzano A. N. The protein arginine methyltransferase Prmt5 is required for myogenesis because it facilitates ATP-dependent chromatin remodeling. Mol. Cell. Biol. 2007, 27, 384–394. 10.1128/MCB.01528-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S.; Vishwanath S. N.; Erdjument-Bromage H.; Tempst P.; Sif S. Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol. Cell. Biol. 2004, 24, 9630–9645. 10.1128/MCB.24.21.9630-9645.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbrizio E.; El Messaoudi S.; Polanowska J.; Paul C.; Cook J. R.; Lee J. H.; Negre V.; Rousset M.; Pestka S.; Le Cam A.; Sardet C. Negative regulation of transcription by the type II arginine methyltransferase PRMT5. EMBO Rep. 2002, 3, 641–645. 10.1093/embo-reports/kvf136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung J.; Karkhanis V.; Tae S.; Yan F.; Smith P.; Ayers L. W.; Agostinelli C.; Pileri S.; Denis G. V.; Baiocchi R. A.; Sif S. Protein arginine methyltransferase 5 (PRMT5) inhibition induces lymphoma cell death through reactivation of the retinoblastoma tumor suppressor pathway and polycomb repressor complex 2 (PRC2) silencing. J. Biol. Chem. 2013, 288, 35534–35547. 10.1074/jbc.M113.510669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karkhanis V.; Hu Y. J.; Baiocchi R. A.; Imbalzano A. N.; Sif S. Versatility of PRMT5-induced methylation in growth control and development. Trends Biochem. Sci. 2011, 36, 633–641. 10.1016/j.tibs.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X.; Hoang S.; Mayo M. W.; Bekiranov S. Application of machine learning methods to histone methylation ChIP-Seq data reveals H4R3me2 globally represses gene expression. BMC Bioinformatics 2010, 11, 396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S.; Baiocchi R. A.; Byrd J. C.; Grever M. R.; Jacob S. T.; Sif S. Low levels of miR-92b/96 induce PRMT5 translation and H3R8/H4R3 methylation in mantle cell lymphoma. EMBO J. 2007, 26, 3558–3569. 10.1038/sj.emboj.7601794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirmizis A.; Santos-Rosa H.; Penkett C. J.; Singer M. A.; Vermeulen M.; Mann M.; Bähler J.; Green R. D.; Kouzarides T. Arginine methylation at histone H3R2 controls deposition of H3K4 trimethylation. Nature 2007, 449, 928–932. 10.1038/nature06160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesen W. J.; Paushkin S.; Wyce A.; Massenet S.; Pesiridis G. S.; Van Duyne G.; Rappsilber J.; Mann M.; Dreyfuss G. The methylosome, a 20S complex containing JBP1 and pICln, produces dimethylarginine-modified Sm proteins. Mol. Cell. Biol. 2001, 21, 8289–8300. 10.1128/MCB.21.24.8289-8300.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister G.; Eggert C.; Bühler D.; Brahms H.; Kambach C.; Fischer U. Methylation of Sm proteins by a complex containing PRMT5 and the putative U snRNP assembly factor pICln. Curr. Biol. 2001, 11, 1990–1994. 10.1016/S0960-9822(01)00592-9. [DOI] [PubMed] [Google Scholar]
- Brahms H.; Meheus L.; de Brabandere V.; Fischer U.; Lührmann R. Symmetrical dimethylation of arginine residues in spliceosomal Sm protein B/B′ and the Sm-like protein LSm4, and their interaction with the SMN protein. RNA 2001, 7, 1531–1542. 10.1017/S135583820101442X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesen W. J.; Massenet S.; Paushkin S.; Wyce A.; Dreyfuss G. SMN, the product of the spinal muscular atrophy gene, binds preferentially to dimethylarginine-containing protein targets. Mol. Cell 2001, 7, 1111–1117. 10.1016/S1097-2765(01)00244-1. [DOI] [PubMed] [Google Scholar]
- Meister G.; Fischer U. Assisted RNP assembly: SMN and PRMT5 complexes cooperate in the formation of spliceosomal UsnRNPs. EMBO J. 2002, 21, 5853–5863. 10.1093/emboj/cdf585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisvert F. M.; Cote J.; Boulanger M. C.; Cleroux P.; Bachand F.; Autexier C.; Richard S. Symmetrical dimethylarginine methylation is required for the localization of SMN in Cajal bodies and pre-mRNA splicing. J. Cell Biol. 2002, 159, 957–969. 10.1083/jcb.200207028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez S. E.; Petrillo E.; Beckwith E. J.; Zhang X.; Rugnone M. L.; Hernando C. E.; Cuevas J. C.; Godoy Herz M. A.; Depetris-Chauvin A.; Simpson C. G.; Brown J. W.; Cerdán P. D.; Borevitz J. O.; Mas P.; Ceriani M. F.; Kornblihtt A. R.; Yanovsky M. J. A methyl transferase links the circadian clock to the regulation of alternative splicing. Nature 2010, 468, 112–116. 10.1038/nature09470. [DOI] [PubMed] [Google Scholar]
- Gonsalvez G. B.; Praveen K.; Hicks A. J.; Tian L.; Matera A. G. Sm protein methylation is dispensable for snRNP assembly in Drosophila melanogaster. RNA 2008, 14, 878–887. 10.1261/rna.940708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tee W. W.; Pardo M.; Theunissen T. W.; Yu L.; Choudhary J. S.; Hajkova P.; Surani M. A. Prmt5 is essential for early mouse development and acts in the cytoplasm to maintain ES cell pluripotency. Genes Dev. 2010, 24, 2772–2777. 10.1101/gad.606110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng X.; Wang Z. Protein arginine methyltransferase 5 regulates multiple signaling pathways to promote lung cancer cell proliferation. BMC Cancer 2016, 16, 567. 10.1186/s12885-016-2632-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaniskan H. Ü.; Jin J. Recent progress in developing selective inhibitors of protein methyltransferases. Curr. Opin. Chem. Biol. 2017, 39, 100–108. 10.1016/j.cbpa.2017.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H.; Qian K.; Ho M. C.; Zheng Y. G. Small molecule inhibitors of protein arginine methyltransferases. Expert Opin. Invest. Drugs 2016, 25, 335–358. 10.1517/13543784.2016.1144747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao R.; Shao J.; Zhu K.; Zhang Y.; Ding H.; Zhang C.; Shi Z.; Jiang H.; Sun D.; Duan W.; Luo C. Potent, selective, and cell active protein arginine methyltransferase 5 (PRMT5) inhibitor developed by structure-based virtual screening and hit optimization. J. Med. Chem. 2017, 60, 6289–6304. 10.1021/acs.jmedchem.7b00587. [DOI] [PubMed] [Google Scholar]
- Chan-Penebre E.; Kuplast K. G.; Majer C. R.; Boriack-Sjodin P. A.; Wigle T. J.; Johnston L. D.; Rioux N.; Munchhof M. J.; Jin L.; Jacques S. L.; West K. A.; Lingaraj T.; Stickland K.; Ribich S. A.; Raimondi A.; Scott M. P.; Waters N. J.; Pollock R. M.; Smith J. J.; Barbash O.; Pappalardi M.; Ho T. F.; Nurse K.; Oza K. P.; Gallagher K. T.; Kruger R.; Moyer M. P.; Copeland R. A.; Chesworth R.; Duncan K. W. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat. Chem. Biol. 2015, 11, 432–437. 10.1038/nchembio.1810. [DOI] [PubMed] [Google Scholar]
- Duncan K. W.; Rioux N.; Boriack-Sjodin P. A.; Munchhof M. J.; Reiter L. A.; Majer C. R.; Jin L.; Johnston L. D.; Chan-Penebre E.; Kuplast K. G.; Porter Scott M.; Pollock R. M.; Waters N. J.; Smith J. J.; Moyer M. P.; Copeland R. A.; Chesworth R. Structure and property guided design in the identification of PRMT5 tool compound EPZ015666. ACS Med. Chem. Lett. 2016, 7, 162–166. 10.1021/acsmedchemlett.5b00380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonysamy S.; Bonday Z.; Campbell R. M.; Doyle B.; Druzina Z.; Gheyi T.; Han B.; Jungheim L. N.; Qian Y.; Rauch C.; Russell M.; Sauder J. M.; Wasserman S. R.; Weichert K.; Willard F. S.; Zhang A.; Emtage S. Crystal structure of the human PRMT5:MEP50 complex. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 17960–17965. 10.1073/pnas.1209814109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonday Z. Q.; Cortez G. S.; Dahnke K. R.; Grogan M. J.; Rodriguez Hergueta A.; Jamison J. A.; Watson B. M.; Woods T. A.. 5-Substituted nucleoside analogues. WO 2016/178870.
- Yu W.; Chory E. J.; Wernimont A. K.; Tempel W.; Scopton A.; Alexander Federation A.; Marineau J. J.; Qi J.; Barsyte-Lovejoy D.; Yi J.; Marcellus R.; Iacob R. E.; Engen J. R.; Griffin C.; Aman A.; Wienholds E.; Li F.; Javier Pineda J.; Estiu G.; Shatseva T.; Hajian T.; Al-awar R.; Dick J. E.; Vedadi M.; Brown P. J.; Arrowsmith C. H.; Bradner J. E.; Schapira M. Catalytic site remodeling of the DOT1L methyltransferase by selective inhibitors. Nat. Commun. 2012, 3, 1288. 10.1038/ncomms2304. [DOI] [PubMed] [Google Scholar]
- Touge T.; Hakamata T.; Nara H.; Kobayashi T.; Sayo N.; Saito T.; Kayaki Y.; Ikariya T. Oxo-Tethered Ruthenium (II) complex as a bifunctional catalyst for asymmetric transfer hydrogenation and H2 hydrogenation. J. Am. Chem. Soc. 2011, 133, 14960–14963. 10.1021/ja207283t. [DOI] [PubMed] [Google Scholar]
- Shen Y.; Szewczyk M. M.; Eram M. S.; Smil D.; Kaniskan H. Ü.; Ferreira de Freitas R. F.; Senisterra G.; Li F.; Schapira M.; Brown P. J.; Arrowsmith C. H.; Barsyte-Lovejoy D.; Liu J.; Vedadi M.; Jin J. Discovery of a potent, selective, and cell-active dual inhibitor of protein arginine methyltransferase 4 and protein arginine methyltransferase 6. J. Med. Chem. 2016, 59, 9124–9139. 10.1021/acs.jmedchem.6b01033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eram M. S.; Shen Y.; Szewczyk M.; Wu H.; Senisterra G.; Li F.; Butler K. V.; Kaniskan H. Ü; Speed B. A.; Dela Seña C.; Dong A.; Zeng H.; Schapira M.; Brown P. J.; Arrowsmith C. H.; Barsyte-Lovejoy D.; Liu J.; Vedadi M.; Jin J. A Potent, selective, and cell-active inhibitor of human Type I protein arginine methyltransferases. ACS Chem. Biol. 2016, 11, 772–781. 10.1021/acschembio.5b00839. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.