

Abstract

Mutant isocitrate dehydrogenase 1 (IDH1) is an attractive therapeutic target for the treatment of various cancers such as AML, glioma, and glioblastoma. We have evaluated 3-pyrimidin-4-yl-oxazolidin-2-ones as mutant IDH1 inhibitors that bind to an allosteric, induced pocket of IDH1R132H. This Letter describes SAR exploration focused on improving both the in vitro and in vivo metabolic stability of the compounds, leading to the identification of 19 as a potent and selective mutant IDH1 inhibitor that has demonstrated brain penetration and excellent oral bioavailability in rodents. In a preclinical patient-derived IDH1 mutant xenograft tumor model study, 19 efficiently inhibited the production of the biomarker 2-HG.
Keywords: Mutant IDH1, inhibition of 2-HG production, in vivo anticancer activity, brain penetration, clinical candidate
As an isocitrate dehydrogenase, NADP+-dependent IDH1 catalyzes the oxidative decarboxylation of isocitrate to produce α-ketoglutarate (α-KG) as part of the citric acid cycle in aerobic metabolism,1−5 while NADP+ is reduced to NADPH.2−4 Heterozygous mutations in human IDH1 at Arg132 (R132*) have been identified in multiple cancer types, including acute myeloid leukemia (AML), glioblastoma, glioma, chondrosarcoma, and cholangiocarcinoma. The gain-of-function mutant loses normal enzymatic function and reduces α-KG to 2-hydroxyglutarate (2-HG), resulting in elevated intracellular levels of this metabolite.6 2-HG has been found to inhibit enzymatic function of many α-KG-dependent dioxygenases, including histone and DNA demethylases, causing widespread alterations in histone and DNA methylation which may potentially contribute to tumorigenesis.7 Recently, a first-in-class inhibitor of mutant IDH1 has shown efficacy against IDH1 mutant glioma and cholangiocarcinoma in clinical studies.8,9 A number of preclinical studies also provided evidence to support targeting mutant IDH1 tumors with small molecule inhibitors.10−15
We recently reported the identification of clinical candidate IDH305 (3) as a brain penetrant, potent, and selective mutant IDH1 inhibitor (Figure 1).16 A major part of the lead optimization efforts focused on improving intrinsic clearance without compromising physicochemical properties (e.g., solubility). In this Letter, we describe the rational design of fluoromethyl and fluoroethyl (as in 3) substituted oxazolidinones to enhance metabolic stability.
Figure 1.

SAR progression from hit-to-lead to clinical candidate.
As reported previously, our hit-to-lead optimization efforts from 1 produced a potent and selective lead compound, IDH889 (2).17 Oral dosing of 2 in a murine IDH1 mutant tumor xenograft model resulted in robust reduction of tumor-derived 2-HG, a PD biomarker of mutant IDH1R132* activity. However, in addition to low aqueous solubility (39 μM at pH 6.8), 2 also displayed poor rodent PK due to its high in vivo clearance (Table 1). This correlated with high in vitro intrinsic clearance (CLint) across different species (rat/mouse/dog/human 588/143/548/205 μL/min/mg). Thus, improving the CLint was identified as a top priority for the series.
Table 1. Cellular Potency, in vitro CLint, and Rat PK Data of 9, 10, and 2.

| 9 | 10 | 2 | |
|---|---|---|---|
| cellular HCT116-IDH1R132H/+ (IC50/μM) | 0.56 | 0.047 | 0.014 |
| in vitro liver microsomal CLint (μL/min/mg) | 34 | 101 | 588 |
| in vivo plasma clearance (mL/min/kg) | n.d.a | 17 | 41 |
| oral AUC (μM·h) | n.d. | 14 | 0.4 |
| oral bioavailability (%) | n.d. | 67 | 4 |
Not determined.
In silico and in vivo metabolic studies had identified debenzylation resulting from benzylic oxidation as a major metabolic pathway for 1 (Scheme 1). Common strategies to improve metabolic stability focus on reducing the lipophilicity of the molecule to affect binding to cytochrome P450 proteins (CYPs) and blocking metabolically labile sites.19
Scheme 1. Major Metabolite of 1 in Rat Liver Microsomes.

Our early optimization efforts focused on the exploration of 5-membered heterocyclic aromatic rings on the amine side chain, which were expected to reduce benzylic oxidation20 through the inductive effect of the electronegative heteroatoms and reduction of the overall lipophilicity of the molecule. We elected to maintain the isopropyl group on the oxazolidinone ring throughout our initial studies because it enhanced potency relative to compounds lacking this moiety. This is likely due both to its interaction with the hydrophobic protein pocket and, more importantly, its van der Waals contact with the aryl group of the amine side chain. Indeed, both the bound and small molecule X-ray structures of 1 overlay closely, suggesting a prearrangement of the molecule into the active binding conformation that is driven by hydrophobic collapse.17,18 We believe this ligand preorganization is critical for maintaining mutant IDH1 potency. Additionally, a cocrystal structure of 2(17) with the homodimer of IDH1R132H showed a hydrogen bond between Ser278 and one of the ortho nitrogen atoms in the pyrimidine ring. Therefore, in our optimization efforts, at least one hydrogen bond acceptor, either a nitrogen or oxygen atom, was maintained in ring A (Figure 1).
Most of the heterocyclic analogs thus prepared did indeed attenuate the aforementioned benzylic oxidation observed in Met ID studies; however, the rat in vitro microsomal CLint remained high. For example, benzylic oxidation of oxadiazole 4 was reduced during incubation with rat liver microsomes (Scheme 2), but the major observed metabolites now resulted from oxazolidinone ring opening and oxidation. The change in the site of metabolism observed with 4 was not predicted by in silico modeling, but we were nonetheless encouraged by the reduced benzylic oxidation observed with 4 and para-chloro analog 5, which is more potent and has improved CLint. The tetramethyl substituted analog of 5 (6) (Figure 2) was designed and synthesized to block oxazolidinone ring oxidation. Unfortunately, this resulted in a 20-fold loss in potency, and only a slight improvement in CLint was observed.
Scheme 2. Major Metabolites of 4 in Rat Liver Microsomes.

Figure 2.

Biochemical potency and rat liver microsomal CLint for 5 and 6.
In parallel to the work on the heteroaromatic analogs, compounds that displayed acceptable CLint (although were insufficiently potent) were evaluated to understand what structural characteristics were important for metabolic stability. For instance, unsubstituted oxazolidinone 7 (Figure 3) had low clearance, while the corresponding isopropyl analog 8 displayed high clearance. It seemed counterintuitive that analogs containing sterically encumbered oxazolidinone rings would undergo ring metabolism more readily than their unsubstituted congeners. We hypothesized that the substituents on the oxazolidinone might play a role in CYP recognition and binding that could be important for the metabolic stability of the molecules.
Figure 3.

Biochemical potency and rat liver microsomal CLint for 7 and 8.
The cocrystal structures of both IDH125 and IDH889(17) showed van der Waals interactions between the isopropyl group and ring A. Thus, the decreased potency of 7 relative to isopropyl analog 8 was not surprising because of the ligand preorganization resulting from the aforementioned hydrophobic collapse.17 However, the relative importance of the isopropyl group to inducing the hydrophobic collapse was unclear. Therefore, the unsubstituted oxazolidinone was viewed as providing a starting point for building in potency while maintaining good metabolic stability. In addition to high clearance, isopropyl analogs also generally displayed low aqueous solubility and high plasma protein binding across species. Substituents that would reduce lipophilicity as compared to isopropyl would likely improve the overall physicochemical profile of the series and lead to better druglike properties.
Toward this goal, an effort was initiated to evaluate various substituents on both the oxazolidinone and the biaryl amine side chain to identify a suitable combination of metabolic stability and potency. A number of 5,6- and 6,6-membered biaryl analogs were synthesized, and the imidazole analog 9 was identified as a promising lead, with a cellular IC50 of 0.5 μM and much improved in vitro CLint across species (mouse/rat/human 73/34/59 μL/min/mg). The addition of a methyl group onto the oxazolidinone ring (10) increased cellular potency 10-fold, while only moderately increasing the rat CLint (103 μL/min/mg) as compared to 2 and other isopropyl analogs (Table 1). Compound 10 also displayed significantly lower in vivo clearance vs 2 (17 vs 41 mL/min/kg) during pharmacokinetic profiling in rat (IV/PO 1/10 mg/kg). This translated to a 35-fold increase in oral AUC and a 17-fold improvement in bioavailability vs 2 at the same oral dosage.
Given these encouraging results, we continued to explore the oxazolidinone substitutions to further improve the stability and potency of the imidazole series. We anticipated that the oxazolidinone SAR would translate beyond the imidazole series described here and that the findings could prove useful for the optimization of the other biaryl series as well. The data summarized in Table 2 highlights both steric and electronic effects of the substituents on cellular IC50 and microsomal CLint. Ethyl analog 11 was about 3-fold more potent than methyl analog 10; however, the CLint of 11 was significantly higher. Introduction of a fluorine atom generated a pair of fluoroethyl diastereomers, 12 and 13. While both isomers demonstrated a trend toward improved stability over ethyl analog 11, S-isomer 13 was shown to be the more potent diastereomer by 10-fold over 12. A significant improvement in stability was obtained with fluoromethyl analog 14 with maintenance of cellular potency.
Table 2. Cellular Potency and CLint in Rat Liver Microsomes.

| Cmpd | X | Y | R | Cellular HCT116-IDH1R132H/+ (IC50/μM) | Rat liver microsomal CLint (μL/min/mg) |
|---|---|---|---|---|---|
| 10 | Cl | F | Me | 0.047 | 103 |
| 11 | Cl | F | Et | 0.013 | 448 |
| 12 | Cl | F | (R)-1-F-Et | 0.24 | 237 |
| 13 | Cl | F | (S)-1- F-Et | 0.022 | 272 |
| 14 | Cl | F | F-Me | 0.024 | 28 |
| 15 | Cl | H | i-Pr | 0.015 | 489 |
| 16 | Cl | H | (S)-1- F-Et | 0.004 | 148 |
| 17 | Cl | H | F-Me | 0.030 | 75 |
| 18 | CF2H | H | (S)-1- F-Et | 0.015 | 72 |
| 19 | CF3 | H | F-Me | 0.039 | 7 |
Throughout our medicinal chemistry campaign, both the 5-fluoro and 5-hydrogen pyrimidine cores had been explored, and the 5-substituent was identified as a handle for the fine-tuning of potency, lipophilicity, and physicochemical properties. As observed in the other biaryl series, isopropyl analog 15 displayed good potency and high CLint. The (S)-fluoroethyl analog of 15 (16) demonstrated an improvement in potency (4-fold) and stability vs 15. Once again, lower CLint was observed with fluoromethyl analog 17 relative to 16. However, unlike with the 5-fluoro pyrimidines (13–14), the cellular potency dropped over 7-fold from 16 to 17.
Encouraged by the improvement in CLint with the fluoromethyl and (S)-1-fluoroethyl oxazolidinones, we decided to modify the phenyl substitutions of the biaryl amine side chain and identified a number of potent compounds with low to medium CLint, exemplified by 18 and 19.
Both 18 and 19 demonstrated a good balance of cellular potency, in vitro clearance, permeability and aqueous solubility (Table 3). Although 19 was about 2-fold less potent than 18, its superior microsomal CLint correlated with significantly improved in vivo clearance, AUC, and oral bioavailability in rat vs 18. When rat plasma protein binding data were used to approximate unbound plasma AUC, 10 mg/kg oral dosing of 18 and 19 provided cellular IC50 coverage for roughly the same amount of time (Figure SI-1). The AUC brain to plasma ratio of 19 in rat at 10 mg/kg was 0.17, suggesting 19 is brain penetrant. The limited exposure of 18 in rat brain is presumably due to the p-glycoprotein driven efflux mechanism (MDR1-MDCK efflux ratio >9).
Table 3. Physicochemical Properties and Rodent PK Data for 18 and 19 (IV/PO 1/10 mg/kg for rat and mouse).
| 18 | 19 | |
|---|---|---|
| cellular HCT116-IDH1R132H/+ IC50 (μM) | 0.015 | 0.039 |
| solubility at pH 6.8 (μM) | 422 | 124 |
| Caco-2 Papp (AB/BA) | 21/31 | 20/24 |
| MDR1-MDCK (AB/BA) | 6/57 | 11/39 |
| plasma protein binding (rat/mouse) | 95/92 | 98/98 |
| in vitro liver microsomal CLint (μL/min/mg) (rat/mouse) | 72/63 | 7.7/11 |
| in vivo plasma clearance (mL/min/kg) (rat/mouse) | 20/n.d.a | 2.6/5 |
| oral AUC (μM·h) (rat/mouse) | 15/n.d. | 180/172 |
| oral bioavailability (%) (rat/mouse) | 82/n.d. | 127/149 |
| brain to plasma (total) AUC ratio (rat) | <0.1 | 0.17 |
Not determined.
Compound 19 was evaluated for specificity against mutant IDH1 relative to wild-type (WT) IDH1 in a biochemical assay measuring consumption (mutant) or production (WT) of NADPH.21 Compound 19 exhibited over 50- to 100-fold selectivity for mutant IDH1 isoforms vs WT (IDH1R132H IC50 0.021 μM; IDH1R132C IC50 0.045 μM; IDH1WT IC50 2.52 μM). Compound 19 was also profiled in a patient-derived IDH1 mutant HCT116-IDH1R132H/+ mechanistic xenograft tumor model in mice to evaluate in vivo inhibition of 2-HG production. Compound 19, dosed orally at 150 mg/kg, inhibited new 2-HG production. Given the high baseline tissue concentration of 2-HG, it takes several hours for it to be cleared from tissues, as observed previously in the pharmacodynamic (PD) study with 2.17 Unbound plasma concentration of 19, approximated from mouse plasma protein binding of 98%, stayed above the HCT116-IDH1R132H/+ cellular IC50 for 24 h. This resulted in reduction of tumor 2-HG concentration (Figure 4), with maximal 2-HG inhibition of over 90% relative to the vehicle-treated tumors at 16–24 h post-treatment. As a comparison, when 3 was dosed orally at 200 mg/kg in the same PK/PD study,16 2-HG levels began to rebound after 16 h post-treatment as the free concentration of 3 dropped below the in vitro cellular IC50.
Figure 4.
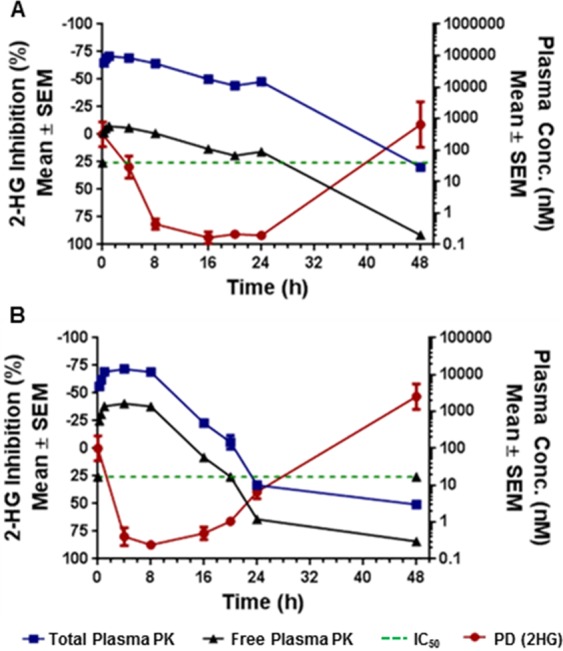
Percent 2-HG inhibition in HCT116-IDH1R132H/+ xenograft tumor tissue in correlation with total and calculated unbound plasma concentration following (a) 150 mg/kg dose of 19 and (b) 200 mg/kg dose of 3.
The synthesis of 19 is described in Scheme 3. Condensation of (S)-2-methylpropane-2-sulfinamide22 and tert-butyl 4-formyl-1H-imidazole-1-carboxylate provided 20 in 74% yield. The addition of methylmagnesium bromide to tert-butanesulfinimine 20,23 followed by Boc deprotection gave 21 in 86% yield over two steps. Palladium-catalyzed cross-coupling24 of 21 with 1-bromo-4-(trifluoromethyl)benzene, followed by removal of the tert-butanesulfinyl group, provided amine 22 efficiently in 81% yield over two steps. Silyl protection of (S)-4-(hydroxymethyl)oxazolidin-2-one (23) produced 24 in 86% yield, and the subsequent SNAr reaction of 24 with 2,4-difluoropyrimidine afforded compound 25 in 81% yield. The removal of the silyl protecting group and subsequent conversion to fluoromethyl compound 26 was achieved efficiently in one-pot using perfluorobutanesulfonyl fluoride (PFBSF) and triethylamine trihydrofluoride.25 Coupling of 22 and 26 in DMSO then provided 19 in 74% yield.
Scheme 3. Synthesis of 19.

Examples can be found in the literature demonstrating that metabolism can be reduced for some molecules when a metabolically labile C–H is replaced by C–F. These include the cholesterol-absorption inhibitor Ezetimibe26 and the cathepsin K inhibitor Odanacatib27 for the treatment of osteoporosis. Multiple factors may contribute to the improvement of CLint of the fluoroalkyl-substituted analogs (12–14 and 16–19), including a reduction in potential for oxazolidinone ring oxidation by an inductive effect of the electronegative fluorine atom. Another factor could be the effect of fluorine on molecular lipophilicity. Examples of a hydrogen-to-fluorine substitution decreasing lipophilicity have been reported by Roche scientists.28 Those compounds contained a common motif whereby an oxygen atom was located either two or three carbons away from a fluorine. The decreased lipophilicity was attributed to the polarization of the oxygen atom by the neighboring fluorine. Interestingly, this structural pattern is also observed in our fluoroalkyl analogs. It is noted that some modifications to the biaryl amine side chain modestly improved CLint of the isopropyl-substituted analogs, but not as substantially as the replacement of the isopropyl group with methyl, fluoromethyl, or fluoroethyl groups. This suggests that the compounds may be recognized differently within the CYP enzyme active site and that the substituents on the oxazolidinone ring can affect the binding of the compounds in the active site.
In conclusion, our optimization of lead compound 2 with a focus on improving in vitro CLint led to the discoveries of both the imidazole subseries and fluoroalkyl-substituted oxazolidinones that demonstrated improved metabolic stability. Potent, orally bioavailable and brain-penetrant mutant IDH1 inhibitor 19 effectively inhibited 2-HG production in a preclinical xenograft tumor model. The fluoroalkyl oxazolidinone SAR was also applied to other biaryl series, and a number of compounds with good potency and desirable in vivo properties, including the clinical candidate IDH305, were subsequently identified.
Acknowledgments
The authors would like to thank Dazhi Tang and Weiping Jia for analytical support and Bill Sellers, Karin Briner, Tim Ramsey, Michael Dillon, Juliet Williams, and Travis Stams for project support.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00182.
Synthetic procedures, analytical data, assay protocols, PK, PK/PD, and efficacy study protocols and data (PDF)
Author Present Address
§ (J.S.) Ideaya Biosciences, 280 Utah Avenue, Suite 250, South San Francisco, CA 94080.
Author Present Address
∥ (A.C.) Chevron Oronite LLC, 100 Chevron Way, Richmond, CA 94802.
Author Present Address
⊥ (J.R.L.) Constellation Pharmaceuticals, 215 First Street, Suite 200, Cambridge, MA 02142.
The authors declare the following competing financial interest(s): The authors are all current or former employees of Novartis and some own shares in Novartis.
Supplementary Material
References
- Dimitrov L.; Hong C. S.; Yang C.; Zhuang Z.; Heiss J. D. New developments in the pathogenesis and therapeutic targeting of the IDH1 mutation in glioma. Int. J. Med. Sci. 2015, 12 (3), 201–213. 10.7150/ijms.11047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molenaar R. J.; Radivoyevitch T.; Maciejewski J. P.; van Noorden C. J. F.; Bleeker F. E. The driver and passenger effects of isocitrate dehydrogenase 1 and 2 mutations in oncogenesis and survival prolongation. Biochim. Biophys. Acta, Rev. Cancer 2014, 1846 (2), 326–341. 10.1016/j.bbcan.2014.05.004. [DOI] [PubMed] [Google Scholar]
- Kim H.–J.; Fei X.; Cho S.–C.; Choi B. Y.; Ahn H.–C.; Lee K.; Seo S.–Y.; Keum Y.–S. Discovery of α-mangostin as a novel competitive inhibitor against mutant isocitrate dehydrogenase-1. Bioorg. Med. Chem. Lett. 2015, 25, 5625–5631. 10.1016/j.bmcl.2015.10.034. [DOI] [PubMed] [Google Scholar]
- Guo C.; Pirozzi C. J.; Lopez G. Y.; Yan H. Isocitrate dehydrogenase mutations in gliomas: mechanisms, biomarkers and therapeutic target. Curr. Opin. Neurol. 2011, 24 (6), 648–652. 10.1097/WCO.0b013e32834cd415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X.; Zhao J.; Xu Z.; Peng B.; Huang Q.; Arnold E.; Ding J. Structures of human cytosolic NADP-dependent isocitrate dehydrogenase reveal a novel self-regulatory mechanism of activity. J. Biol. Chem. 2004, 279, 33946–33957. 10.1074/jbc.M404298200. [DOI] [PubMed] [Google Scholar]
- Turkalp Z.; Karamchandani J.; Das S. IDH mutation in glioma: new insights and promises for the future. JAMA Neurology 2014, 71, 1319–1325. 10.1001/jamaneurol.2014.1205. [DOI] [PubMed] [Google Scholar]
- Liu X.; Ling Z. Q. Role of isocitrate dehydrogenase 1/2 (IDH 1/2) gene mutations in human tumors. Histology and Histopathology 2015, 30, 1155–1160. [DOI] [PubMed] [Google Scholar]
- Hansen E.; Quivoron C.; Straley K.; Lemieux R. M.; Popovici-Muller J.; Sadrzadeh H.; Fathi A. T.; Gliser C.; David M.; Saada V.; Micol J.–B.; Bernard O.; Dorsch M.; Yang H.; Su M.; Agresta S.; de Botton S.; Penard-Lacronique V.; Yen K. AG-120, an oral, selective, first-in-class, potent inhibitor of mutant IDH1, reduces intracellular 2HG and induces cellular differentiation in TF-1 R132H cells and primary human IDH1 mutant AML patient samples treated ex vivo. Blood 2014, 124, 3734. [Google Scholar]
- Mondesir J.; Willekens C.; de Botton S. IDH1 and IDH2 mutations as novel therapeutic targets: current perspectives. J. Blood Med. 2016, 7, 171–180. 10.2147/JBM.S70716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovici-Muller J.; Saunders J. O.; Salituro F. G.; Travins J. M.; Yan S.; Zhao F.; Gross S.; Dang L.; Yen K. E.; Yang H.; Straley K. S.; Jin S.; Kunii K.; Fantin V. R.; Zhang S.; Pan Q.; Shi D.; Biller S. A.; Su S. M. Discovery of the first potent inhibitors of mutant IDH1 that lower tumor 2-HG in vivo. ACS Med. Chem. Lett. 2012, 3, 850–855. 10.1021/ml300225h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; Yao Y.; Kogiso M.; Zheng B.; Deng L.; Qiu J. J.; Dong S.; Lv H.; Gallo J. M.; Li X.–N.; Song Y. Inhibition of cancer-associated mutant isocitrate dehydrogenases: synthesis, structure–activity relationship, and selective antitumor activity. J. Med. Chem. 2014, 57, 8307–8318. 10.1021/jm500660f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks E.; Wu X.; Hanel A.; Nguyen S.; Wang J.; Zhang J. H.; Harrison A.; Zhang W. Identification and characterization of small-molecule inhibitors of the R132H/R132H mutant isocitrate dehydrogenase 1 homodimer and R132H/wild-type heterodimer. J. Biomol. Screening 2014, 19, 1193–1200. 10.1177/1087057114541148. [DOI] [PubMed] [Google Scholar]
- Deng G.; Shen J.; Yin M.; McManus J.; Mathieu M.; Gee P.; He T.; Shi C.; Bedel O.; McLean L. R.; Le-Strat F.; Zhang Y.; Marquette J.–P.; Gao Q.; Zhang B.; Rak A.; Hoffmann D.; Rooney E.; Vassort A.; Englaro W.; Li Y.; Patel V.; Adrian F.; Gross S.; Wiederschain D.; Cheng H.; Licht S. Selective inhibition of mutant isocitrate dehydrogenase 1 (IDH1) via disruption of a metal binding network by an allosteric small molecule. J. Biol. Chem. 2015, 290, 762–774. 10.1074/jbc.M114.608497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohle D.; Popovici-Muller J.; Palaskas N.; Turcan S.; Grommes C.; Campos C.; Tsoi J.; Clark O.; Oldrini B.; Komisopoulou E.; Kunii K.; Pedraza A.; Schalm S.; Silverman L.; Miller A.; Wang F.; Yang H.; Chen Y.; Kernytsky A.; Rosenblum M. K.; Liu W.; Biller S. A.; Su S. M.; Brennan C. W.; Chan T. A.; Graeber T. G.; Yen K. E.; Mellinghoff I. K. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013, 340, 626–630. 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B.; Yao Y.; Liu Z.; Deng L.; Anglin J. L.; Jiang H.; Prasad B. V. V.; Song Y. Crystallographic investigation and selective inhibition of mutant isocitrate dehydrogenase. ACS Med. Chem. Lett. 2013, 4, 542–546. 10.1021/ml400036z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y. S.; Levell J. R.; Liu G.; Caferro T.; Sutton J.; Shafer C. M.; Costales A.; Manning J. R.; Zhao Q.; Sendzik M.; Shultz M.; Chenail G.; Dooley J.; Villalba B.; Farsidjani A.; Chen J.; Kulathila R.; Xie X.; Dodd S.; Gould T.; Liang G.; Heimbach T.; Slocum K.; Firestone B.; Pu M.; Pagliarini R.; Growney J. D. Discovery and evaluation of clinical candidate IDH305, a brain penetrant mutant IDH1 inhibitor. ACS Med. Chem. Lett. 2017, 8, 1116–1121. 10.1021/acsmedchemlett.7b00342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levell J. R.; Caferro T.; Chenail G.; Dix I.; Dooley J.; Firestone B.; Fortin P. D.; Giraldes J.; Gould T.; Growney J. D.; Jones M. D.; Kulathila R.; Lin F.; Liu G.; Mueller A.; van der Plas S.; Slocum K.; Smith T.; Terranova R.; Touré B. B.; Tyagi V.; Wagner T.; Xie X.; Xu M.; Yang F. S.; Zhou L. X.; Pagliarini R.; Cho Y. S. Optimization of 3-pyrimidin-4-yl-oxazolidin-2-ones as allosteric and mutant specific inhibitors of IDH1. ACS Med. Chem. Lett. 2017, 8, 151–156. 10.1021/acsmedchemlett.6b00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X.; Baird D.; Bowen K.; Capka V.; Chen J.; Chenail G.; Cho Y. S.; Dooley J.; Farsidjani A.; Fortin P.; Kohls D.; Kulathila R.; Lin F.; McKay D.; Rodrigues L.; Sage D.; Touré B. B.; van der Plas S.; Wright K.; Xu M.; Yin H.; Levell J.; Pagliarini R. A. Allosteric mutant IDH1 inhibitors reveal mechanisms for IDH1 mutant and isoform selectivity. Structure 2017, 25, 506–513. 10.1016/j.str.2016.12.017. [DOI] [PubMed] [Google Scholar]
- Nassar A.–E. F.; Kamel A. M.; Clarimont C. Improving the decision-making process in the structural modification of drug candidates: enhancing metabolic stability. Drug Discovery Today 2004, 9, 1020–1028. 10.1016/S1359-6446(04)03280-5. [DOI] [PubMed] [Google Scholar]
- Ioannidis S.; Lamb M. L.; Wang T.; Almeida L.; Block M. H.; Davies A. M.; Peng B.; Su M.; Zhang H.–J.; Hoffmann E.; Rivard C.; Green I.; Howard T.; Pollard H.; Read J.; Alimzhanov M.; Bebernitz G.; Bell K.; Ye M.; Huszar D.; Zinda M. Discovery of 5-Chloro-N2-[(1S)-1-(5-fluoropyrimidin-2-yl)ethyl]-N4-(5-methyl-1H-pyrazol-3-yl)pyrimidine-2,4-diamine (AZD1480) as a Novel Inhibitor of the Jak/Stat Pathway. J. Med. Chem. 2011, 54, 262–276. 10.1021/jm1011319. [DOI] [PubMed] [Google Scholar]
- Gao H.; Korn J. M.; Ferretti S.; Monahan J. E.; Wang Y.; Singh M.; Zhang C.; Schnell C.; Yang G.; Zhang Y.; Balbin O. A.; Barbe S.; Cai H.; Casey F.; Chatterjee S.; Chiang D. Y.; Chuai S.; Cogan S. M.; Collins S. D.; Dammassa E.; Ebel N.; Embry M.; Green J.; Kauffmann A.; Kowal C.; Leary R. J.; Lehar J.; Liang Y.; Loo A.; Lorenzana E.; McDonald E. R. III; McLaughlin M. E.; Merkin J.; Meyer R.; Naylor T. L.; Patawaran M.; Reddy A.; Röelli C.; Ruddy D. A.; Salangsang F.; Santacroce F.; Singh A. P.; Tang Y.; Tinetto W.; Tobler S.; Velazquez R.; Venkatesan K.; Von Arx F.; Wang H. Q.; Wang Z.; Wiesmann M.; Wyss D.; Xu F.; Bitter H.; Atadja P.; Lees E.; Hofmann F.; Li E.; Keen N.; Cozens R.; Jensen M. R.; Pryer N. K.; Williams J. A.; Sellers W. R. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat. Med. 2015, 21, 1318–1325. 10.1038/nm.3954. [DOI] [PubMed] [Google Scholar]
- Collados J. F.; Toledano E.; Guijarro D.; Yus M. Microwave-assisted solvent-free synthesis of enantiomerically pure N-(tert-butylsulfinyl)imines. J. Org. Chem. 2012, 77, 5744–5750. 10.1021/jo300919x. [DOI] [PubMed] [Google Scholar]
- Liu G.; Cogan D. A.; Ellman J. A. Catalytic asymmetric synthesis of tert-butanesulfinamide. Application to the asymmetric synthesis of amines. J. Am. Chem. Soc. 1997, 119, 9913–9914. 10.1021/ja972012z. [DOI] [Google Scholar]
- Ueda S.; Ali S.; Fors B. P.; Buchwald S. L. Me3(OMe)tBuXPhos: A surrogate ligand for Me4tBuXPhos in palladium-catalyzed C–N and C–O bond-forming reactions. J. Org. Chem. 2012, 77, 2543–2547. 10.1021/jo202537e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J.; Zarkowsky D. S.; Thomas D. W.; Zhao M. M.; Huffman M. A. Direct and convenient conversion of alcohols to fluorides. Org. Lett. 2004, 6, 1465–1468. 10.1021/ol049672a. [DOI] [PubMed] [Google Scholar]
- Van Heek M.; France C. F.; Compton D. S.; Mcleod R. L.; Yumibe N. P.; Alton K. B.; Sybertz E. J.; Davis H. R. Jr In vivo metabolism-based discovery of a potent cholesterol absorption inhibitor, SCH58235, in the rat and rhesus monkey through the identification of the active metabolites of SCH48461. J. Pharmacol. Exp. Ther. 1997, 283, 157–163. [PubMed] [Google Scholar]
- Gauthier J. Y.; Chauret N.; Cromlish W.; Desmarais S.; Duong L. T.; Falgueyret J.–P.; Kimmel D. B.; Lamontagne S.; Léger S.; LeRiche T.; Li C. S.; Massé F.; McKay D. J.; Nicoll-Griffith D. A.; Oballa R. M.; Palmer J. T.; Percival M. D.; Riendeau D.; Robichaud J.; Rodan G. A.; Rodan S. B.; Seto C.; Thérien M.; Truong V.–L.; Venuti M. C.; Wesolowski G.; Young R. N.; Zamboni R.; Black W. C. The discovery of odanacatib (MK-0822), a selective inhibitor of cathepsin K. Bioorg. Med. Chem. Lett. 2008, 18, 923–928. 10.1016/j.bmcl.2007.12.047. [DOI] [PubMed] [Google Scholar]
- Böhm H.–J.; Banner D.; Bendels S.; Kansy M.; Kuhn B.; Müller K.; Obst-Sander U.; Stahl M. Fluorine in medicinal chemistry. ChemBioChem 2004, 5, 637–643. 10.1002/cbic.200301023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.