

Abstract

Occurrence of acute myeloid leukemia (AML) results in abundant endogenous reactive oxygen species (ROS)/reactive nitrogen species (RNS) in AML cells and in disease-relevant microenvironments. Histone deacetylase inhibitor (HDACi) prodrug approach was designed accordingly by masking the hydroxamic acid zinc binding group with hydrogen peroxide (H2O2)/peroxynitrite (PNT)-sensitive, self-immolative aryl boronic acid moiety. Model prodrugs 5–82 and 5–23 were activated in AML cells to release cytotoxic HDACis, evidenced by inducing acetylation markers and reducing viability of AML cells. Intracellular activation and antileukemic activities of prodrug were increased or decreased by ROS/PNT inducers and scavengers, respectively. Prodrugs 5–82 and 5–23 also enhanced the potency of chemotherapy drug cytarabine, supporting the potentials of this prodrug class in combinatorial treatment.
Keywords: HDAC inhibitor, HDAC inhibitor prodrugs, acute myeloid leukemia, hydrogen peroxide, peroxynitrite, reactive oxygen species
Acute myeloid leukemia (AML) remains a challenging disease. The overall 5-year survival is less than 5% in patients over the age of 65.1 Histone deacetylase inhibitors (HDACis) target aberrant epigenetic modifications of cancer and have been studied as an attractive “targeted” treatment for AML.2 Although promising in preclinical models, HDACis as monotherapy only resulted in modest or poor clinical outcomes in AML trials.3 Multiple factors, such as undesired cardiovascular and gastrointestinal toxicity,4,5 fast elimination, and poor tissue penetration caused by metabolically labile and highly polar hydroxamic acid zinc binding group (ZBG),6,7 hindered the use of HDAC is in AML and other cancer indications. Several prodrug approaches, e.g., using carbamate6,8 and quinone7-based moieties to protect hydroxamic acid have been proposed to partially address these shortcomings.
Different from the reported approaches, we designed novel HDACi prodrugs by considering chemical properties of the “promoiety” and the biological characteristics of AML. The prodrugs were constructed by masking the hydroxamic acid ZBG with hydrogen peroxide (H2O2)/peroxynitrite (PNT)-sensitive, self-immolative aryl boronic acid promoiety (Figure 1a). Aromatic boronates/boronic acids specifically react with H2O2 (a ROS)9 or PNT (a RNS)10 under physiological conditions and have been used to design anticancer prodrugs and ROS probes.11 Similar to arylboronate-based nitrogen mustard12 and aminoferrocene13 prodrugs, boronic acid HDACi prodrugs could form phenol intermediates during activation and subsequently release free hydroxamic acid and a short-lived, chemically reactive quinone methide (QM) through 1,6-elimination (Figure 1a). In this study, two HDACis, SAHA (i.e., vorinostat), a FDA-approved drug to treat cutaneous T cell lymphoma,14 and 1–58, a piperlongumine (PL)-HDACi hybrid molecule with potent anti-AML activity,15 were converted to prodrugs 5–82 and 5–23, respectively (Figure 1a,b). We used boronic acid instead of boronic acid ester as a masking group to retain water solubility and, at the same time, to increase lipophilicity of the parental hydroxamic acid HDACis. For instance, cLogP of 5–82 is 3.26, compared with that of SAHA at 0.99 (calculated by using ChemDraw 12.0), predicting better tissue/tumor penetration. Hydroxamic acid is liable to O-glucoronidation16 and hydrolysis, and derivatizations with boronic acid or another “promoiety” could potentially increase metabolic stability of hydroxamic acid-based HDACis.7
Figure 1.
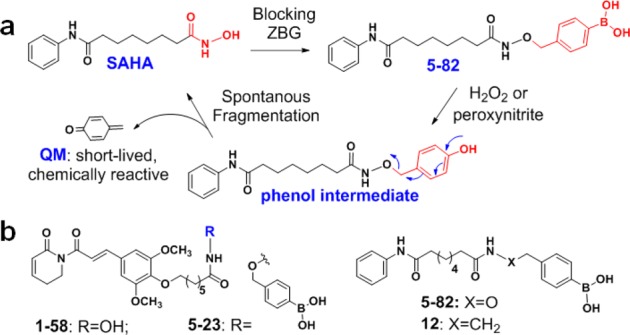
(a) Design of arylboronic acid-masked hydroxamic acid prodrug and the mechanism of activation, exemplified using SAHA and 5–82. (b) Chemical structures of HDACi 1–58, prodrug 5–23, and compound 12.
Besides chemical properties, the design of H2O2/PNT-activated HDACi prodrugs is also reasoned by abundant endogenous ROS/RNS in AML cells and in disease-relevant microenvironments. Due to malfunctioned redox homeostasis pathways, such as alteration of mitochondrial electron transfer chain, FLT3-ITD-caused upregulation of NADPH oxidase, and the elevated xanthine oxidoreductase activity,17 AML blasts display higher levels of ROS compared to normal leukocytes.18−20 The occurrence of AML also abnormally upregulates and activates NOX4 (an superoxide-generating enzyme) and nitric oxide synthase 3 (i.e., eNOS) in bone marrow (BM) endothelial cells and results in overproduction of nitric oxide (NO) and ROS.21 This event could eventually produce higher levels of PNT from the reaction of NO and superoxide (O2–•) in AML BM microenvironment. PNT reacts with boronic acid up to a million-fold faster than H2O210,22 and could be another activator of boronic acid-based HDACi prodrugs. Compared with healthy tissue, the elevated H2O2 and PNT may activate prodrug and release higher concentrations of cytotoxic HDACi in AML cells and in AML-relevant, ROS/RNS-enriched microenvironments.
Starting from pinacol ester 1, hydroxylamine 3 was made by deprotecting Mitsunobu reaction product 2 with methylhydrazine. Coupling of 3 with carboxylic acids 4(15) and 7,23 followed by the treatment of NaIO4/NH4OAc, gave prodrugs 5–23 and 5–82, respectively (Scheme 1). To study the potential biological effects of aryl boronic acid, we prepared a close structural analogue of 5–82, i.e., compound 12 (Figure 1b), that does not convert to HDACi due to replacing the oxygen of ZBG with a methylene group. 12 was made by condensing 7 with amine 11 which was synthesized from 8 via sequential amine protection, borylation and acidic deprotection (Scheme 1).
Scheme 1. Synthesis of Prodrugs 5-23 and 5-82 and the 5-82 Analogue 12.

Reagents and conditions: (a) N-hydroxyphthalimide, PPh3, DEAD, THF, 82%; (b) methylhydrazine, THF, 0 °C, 77%; (c) aniline, BOP, DIPEA, DCM, 70%; (d) LiOH, THF/H2O (1:1), 80%; (e) 1. HBTU, DIPEA, THF; 2. NaIO4, NH4OAc, acetone/H2O (1:1); 57% for 5–23, 60% for 5–82; 75% for 12; (f) (BOC)2O, Et3N, 95%; (g) Bis(pinacolato)diboron, AcOK, Pd(dppf)Cl2, 1,4-dioxane, 80 °C, 80%; (h) trifluoroacetic acid, DCM, 91%.
By using SAHA prodrug 5–82 as a model compound, we first investigated if the prodrug can be activated by H2O2 or PNT in cell-free incubations. Prodrug 5–82 was incubated with H2O2 in PBS buffer (pH 7.4) at 37 °C. As revealed by HPLC analysis (Figure 2a), it reacted with H2O2 to form two new peaks: the retention time of A was identical to that of SAHA, and peak B was assigned as the phenol intermediate (Figure 1a) as the measured m/z value matched F.W. of the proposed structure in a high resolution mass spectrometry-based LC-MS analysis (Figure S1a). Peak B eventually disappeared, and the disappearance was accompanied by the increased peak area of SAHA. At a given time point (e.g., at 30 min, Figure 2a), the formation of phenol intermediate and SAHA were increased along with the elevated H2O2 concentrations (Figure 2b). Prodrug 5–82 (100 μM) was completely converted to SAHA by equivalent (100 μM) or excessive (500 μM) H2O2, whereas only half of the 5–82 was consumed by 50 μM of H2O2 as determined by comparisons of the peak area of SAHA with the standard sample (Figures 2b and S1b).
Figure 2.

(a) Prodrug 5–82 was activated by H2O2. Prodrug 5–82 (100 μM) was incubated with H2O2 in PBS (pH 7.4) at 37 °C for 30 min. Aliquots were analyzed by using HPLC. (b) Prodrug 5–82 (100 μM) time-dependently released SAHA in the presence of H2O2 in 4 h in PBS (pH 7.4) at 37 °C. (c) Prodrug 5–82 was activated by PNT. Prodrug 5–82 (100 μM) was incubated with indicated reagents in PBS (pH 7.4) at room temperature for 15 min. Aliquots were analyzed by using HPLC. Benzophenone was used as an internal standard (IS). X, xanthine; XO, xanthine oxidase; Cat., catalase; PAPA/NO, PAPA-NONOate.
Prodrug 5–82 was also activated by PNT (Figure 2c). PNT was generated by a mixture of xanthine (X, 200 mM), xanthine oxidase (XO, 10 mU/mL), and NO donor PAPA-NONOate (100 μM).10 X and XO produce fluxes of superoxide anion (O2–•), which further reacts with NO to form PNT. Even at room temperature, 5–82 was completely converted to the phenol intermediate by X/XO/PAPA-NONOate within 15 min. In sharp contrast, PAPA-NONOate itself barely induced any change of 5–82, emphasizing the specific reaction between boronic acid and PNT. X/XO alone partially consumed 5–82 to generate the phenol, and this conversion was abrogated by the addition of catalase (100 U/mL), indicating that 5–82 was specifically oxidized by H2O2, a disproportionate product of O2–• yielded from the X/XO system. The addition of catalase also reduced consumption of 5–82 in the X/XO/PAPA-NONOate system, suggesting that both H2O2 and PNT were generated and were responsible for the oxidation of aryl boronic acid in this incubation. Since catalase removes H2O2, 5–82 was mainly consumed by PNT in the X/XO/PAPA-NONOate/catalase system to generate the phenol intermediate that gradually released SAHA (Figure S2). Taken together, we conclude that 5–82 can rapidly react with either H2O2 or PTN and subsequently releases SAHA in PBS buffer via a phenol intermediate.
We next tested antileukemic activities of prodrugs 5–82 and 5–23 in AML cells. Both prodrugs concentration-dependently reduced the viability of U937 and MV4–11 cells, with IC50s in sub to low μM range (Figure 3a; Table S1). MV4–11 is more sensitive to prodrug treatments than U937 cells, consistent with their sensitivity to parental HDACis SAHA and 1–58 (Table S1).15 Since boronic acid is a structural component of several approved (e.g., bortezomib) or clinically tested drugs,24 we used 12 (Figure 1b, Scheme 1), an analogue of 5–82, to assess biological effects of aryl boronic acid of the prodrugs. Analogue 12 did not affect the viability of MV4–11 cells at the concentrations up to 10 μM (Figure S3a), suggesting that aryl boronic acid does not contribute to 5–82-induced growth inhibition. We also compared prodrug 5–23 and the methyl ester of 4 (i.e., 27(15) in our previous research, Figure S3b) in U937 cells. Although sharing the same PL hybrid drug chemical scaffold, 27 was significantly less effective than 5–23 in MTT assay (Figure S3b), providing another evidence supporting the importance of HDACi release for the antileukemic activities of prodrugs.
Figure 3.
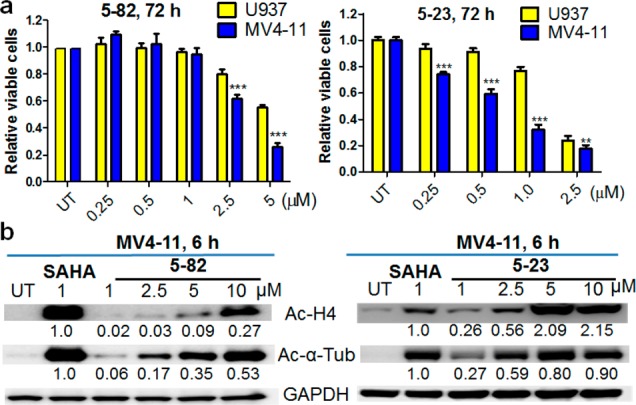
(a) Prodrugs 5–82 and 5–23 reduced viability of both U937 and MV4–11 AML cells. Viability were measured using MTT assay. Bars represent mean ± SD, n = 8. ** indicates P < 0.01; *** indicates P < 0.001. (b) Prodrugs 5–82 and 5–23 concentration-dependently increased acetylation of histone H4 (Ac–H4) and α-tubulin (Tub) in MV4–11 cells determined by Western Blotting. MV4–11 cells were treated with 5–82 or 5–23 at indicated concentrations for 6 h. The fold changes for the Ac–H4 and Ac-α-Tub were obtained using densitometry measurements, normalized to GAPDH, and then compared with SAHA treatment. One representative blot from three experiments is shown.
QM is formed along with HDACi release (Figure 1a) and is antiproliferative to cancer cells.25 4-(Chloromethyl)phenyl acetate (4-CPA) is a QM precursor:26 the acetate is hydrolyzed within cells by esterases, and the resulting 4-(chloromethyl)phenol simultaneously converts to QM (Figure S4a). 4-CPA decreased viability of MV4–11 cells with IC50 value (1.13 μM, Figure S4b) higher than those of SAHA (0.50 μM) and 1–58 (0.23 μM). Because the inferior potency vs SAHA and the quick trapping of QM by water and GSH, combinations of 4-CPA (up to 500 nM) and SAHA did not outperform SAHA alone when used in 1:1 ratio to mimic prodrug setting (Figure S4c). However, in treatments where the mole ratio of 4-CPA was in excess of SAHA/1–58 and/or concentrations of 4-CPA was increased beyond the cellular buffering capacity (Figure S4d,e), 4-CPA indeed enhanced the potency of HDACis, suggesting that although the QM may not be critical for the activities of 5–82 or 5–23 in current work, it could become a positive contributor to the overall cytotoxicity especially when the released HDACis are less cytotoxic than QM.
We also measured cellular HDAC inhibition markers, acetylated histone H4 and α-tubulin, to further confirm intracellular activation of prodrugs. Both 5–82 and 5–23 concentration-dependently increased acetylation of histone H4 and α-tubulin within 6 h in MV4–11 (Figure 3b) and U937 cells (Figure S5). Compared to the same standard SAHA, densitometry analysis revealed that 1–58 prodrug 5–23 caused more prominent global acetylation than SAHA prodrug 5–82 in MV4–11 (Figure 3b) and U937 cells (Figure S5). To understand this observation, we measured intrinsic HDAC inhibition activity of parental HDACis using an enzymatic assay against nuclear HDACs extracted from Hela cells. Interestingly, 1 μM 1–58 and SAHA treatment resulted in 32% and 65% inhibition, respectively (Figure S6), demonstrating that 1–58 is weaker than SAHA at nuclear HDAC inhibition. Therefore, compared to 5–82, the increased H4 acetylation induced by 5–23 was likely attributed to the higher levels of intracellular free HDACi (but not higher intrinsic HDACi activity), potentially caused by (1) increased intracellular prodrug concentration since 5–23 is more lipophilic than 5–82 (clogP 4.73 vs 3.26); and (2) the presence of ROS-inducing piperlongumine functionality27 may enhance oxidative stress in AML cells, which facilitates prodrug activation. Prodrug 5–23 was also more potent than 5–82 in reducing cell viability, reflected by the IC50s of the two prodrugs: 0.70 μM vs 3.07 μM in MV4–11 cells and 1.61 μM vs 6.13 μM in U937 cells (Table S1). At least two factors, the higher intracellular free HDACi concentration and the stronger antiproliferative activity of the released HDACi (1–58 vs SAHA),15 may have contributed to the greater in vitro activities of 5–23 than 5–82.
We further evaluated selectivity of prodrugs using three human peripheral blood mononuclear cell (PBMNC) samples obtained from healthy donors (two for MTT and one for Western blot assays). Compared to vehicle control, prodrugs 5–82 and 5–23 induced little to no acetylation of histone H4 and α-tubulin in PBMNCs at 5 and 10 μM (Figure S7a), which is in contrast to the substantial increase of HDAC inhibition markers in AML cells (Figures 3b and S5). In MTT assays, the IC50 of 5–82 was greater than 10 μM in both PBMNC samples, and the IC50 of 5–23 was 7.6 μM in one sample and was greater than 10 μM in another sample (Figure S7b). The higher IC50 values (Table S1) and lower induction of HDAC inhibition markers in PBMNC samples vs AML cells support the selectivity of 5–82 and 5–23 to AML cells over normal cells. HDACi SAHA also showed selectivity; however, SAHA was more potent against normal cells than the prodrugs (Table S1 and Figure S7b). Besides cancer cells, the cancer microenvironment may also generate higher levels of ROS/RNS21 that promote the release of HDACis. Selectivity of this HDACi prodrug class can be further tested in in vitro and in vivo models containing microenvironmental components.
We then investigated if antileukemic activities of prodrug could be affected by exogenous redox reagents. Pretreatment with an antioxidant, such as GSH, GSH ethyl ester (GSHEE), or catalase, significantly rescued viability of MV4–11 cells from 5–82 treatment (Figure 4a). Prodrug 5–82 was stable in GSH coincubation (Figure S8), suggesting GSH or GSHEE rescued cells via scavenging intracellular ROS/RNS instead of their reactions with the electrophilic boron center. On the contrary, H2O2 and/or PNT inducers, such as superoxide dismutase (SOD) and a combination of lipopolysaccharide (LPS) and buthionine-(S,R)-sulfoximine (BSO), notably enhanced inhibitory effect of 5–82 in MV4–11 cells (Figure 4b). Besides potentially affecting biological activities of the released HDACis, redox agents could modulate prodrug activation. Indeed, SOD, the combination of LPS and BSO, increased and catalase decreased acetylation of histone H4 and α-tubulin (Figure 4c). Catalase suppresses whereas SOD promotes intracellular H2O2 production, consistent with their opposite effects on HDAC inhibition markers. LPS induces iNOS and BSO depletes GSH; their combination could boost ROS (e.g., H2O2) and PNT levels and generate oxidative environment favoring prodrug activation. Collectively, antileukemic activities of HDACi prodrugs could be enhanced by exogenous ROS/RNS inducers, whereas being weakened by antioxidants, modulating prodrug activation could be one of the factors responsible for the observed cellular effects.
Figure 4.

Antileukemic activities of prodrug 5–82 were influenced by H2O2/PNT inducers and scavengers. (a,b) MV4–11 cells were pretreated with GSH (5 mM), GSHEE (5 mM), catalase (0.125 mg/mL), SOD (0.05 mg/mL), or the combination of LPS (10 μg/mL) and BSO (2.5 μM) for 2 h, respectively, followed by 5–82 (2.5 μM) treatment for 72 h. Viable cells were measured using MTT assay. Bars represent mean ± SD, n = 6. *** indicates P < 0.001. (c) MV4–11 cells were pretreated with catalase (Cat, 0.5 mg/mL), SOD (0.1 mg/mL), LPS (10 μg/mL), or BSO (10 μM) for 2 h, followed by 5–82 (2.5 μM) treatment for 24 h. Acetylation of histone H4 and α-tubulin were analyzed using Western blotting. The fold changes for the Ac–H4 and Ac-α-Tub were obtained by densitometry measurements, normalized to GAPDH, and then compared to 5–82 treatment. One representative blot from three experiments is shown.
Because of pleiotropic anticancer mechanisms, HDACi is a versatile component of combinatorial AML treatments.28 For instance, SAHA was combined with idarubicin and cytarabine in an AML clinical trial.29 To explore the feasibility of HDACi prodrugs in combinatorial therapy, MV4–11 cells were treated with 5–82 or 5–23 in combination with cytarabine. Both prodrugs significantly enhanced the potency of cytarabine (0.05–0.5 μM) (Figure 5a), and the inhibitory effects of drug combination were accompanied by the substantially increased PARP-1 and caspase-3 cleavage compared to the individual drug treatments (Figure 5b).
Figure 5.

Prodrugs 5–82 (2.5 μM) and 5–23 (0.5 μM) enhanced the antileukemic effects of cytarabine in MV4–11 cells. (a) MV4–11 cells were treated with drugs at indicated concentrations for 72 h, and cell viability was measured using MTT assay. Bars represent mean ± SD, n = 6. *** indicates P < 0.001. (b) MV4–11 cells were treated with prodrugs and/or cytarabine (0.5 μM) for 24 h, and expression of cleaved caspase-3 and PARP-1 was analyzed using Western blotting. One representative blot from three experiments is shown.
In summary, we report new HDACi prodrugs designed by masking the hydroxamic acid ZBG with an aryl boronic acid-based promoiety. Model prodrugs were activated by H2O2 or PNT, leading to intracellular release of active HDACi and reduction of viable AML cells. Supported by enhancing the potency of cytarbine, this HDACi prodrug class could be combined with conventional chemotherapy drugs as well as other “targeted” anti-AML agents. Given the broad pharmacological effects of HDACi30 and the critical roles of ROS31 and PNT32 in human diseases, H2O2 and PNT-activated HDACi prodrugs may have great potential in the treatment of cancer and noncancer indications.
Acknowledgments
This study was supported by a Wayne State University startup fund (to Z.Q.) and pilot grant from Karmanos Cancer Institute Cancer Epigenetics Strategic Research Funding (to Z.Q. and Y.G.). Children's Hospital of Michigan Foundation and the LaFontaine Family/U Can-cer Vive Foundation (to Y.G.). The authors would like to thank Dr. Maozi Liu (Agilent, Inc.) for characterizing the phenol intermediate using high resolution mass spectrometry.
Glossary
ABBREVIATIONS
- AML
acute myeloid leukemia
- SAHA (vorinostat)
suberanilohydroxamic acid
- HDACis
histone deacetylase inhibitors
- H2O2
hydrogen peroxide
- PNT
peroxynitrite
- NOX
NADPH oxidase
- X
xanthine
- XO
xanthine oxidase
- PAPA/NO
PAPA-NONOate
- GSHEE
glutathione ethyl ester
- LPS
lipopolysaccharide
- BSO
buthionine-(S,R)-sulfoximine
- SOD
superoxide dismutase
- PBS
phosphate-buffered saline
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00057.
General experimental methods, HPLC analysis of incubation mixtures, detailed synthetic procedures, and compound characterization (1H, 13C NMR and MS) (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Thein M. S.; Ershler W. B.; Jemal A.; Yates J. W.; Baer M. R. Outcome of older patients with acute myeloid leukemia: an analysis of SEER data over 3 decades. Cancer 2013, 119 (15), 2720–2727. 10.1002/cncr.28129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintas-Cardama A.; Santos F. P.; Garcia-Manero G. Histone deacetylase inhibitors for the treatment of myelodysplastic syndrome and acute myeloid leukemia. Leukemia 2011, 25 (2), 226–235. 10.1038/leu.2010.276. [DOI] [PubMed] [Google Scholar]
- Kirschbaum M. H.; Foon K. A.; Frankel P.; Ruel C.; Pulone B.; Tuscano J. M.; Newman E. M. A phase 2 study of belinostat (PXD101) in patients with relapsed or refractory acute myeloid leukemia or patients over the age of 60 with newly diagnosed acute myeloid leukemia: a California Cancer Consortium Study. Leuk. Lymphoma 2014, 55 (10), 2301–2304. 10.3109/10428194.2013.877134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gryder B. E.; Sodji Q. H.; Oyelere A. K. Targeted cancer therapy: giving histone deacetylase inhibitors all they need to succeed. Future Med. Chem. 2012, 4 (4), 505–524. 10.4155/fmc.12.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian S.; Bates S. E.; Wright J. J.; Espinoza-Delgado I.; Piekarz R. L. Clinical toxicities of histone deacetylase inhibitors. Pharmaceuticals 2010, 3 (9), 2751–2767. 10.3390/ph3092751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silhar P.; Eubanks L. M.; Seki H.; Pellett S.; Javor S.; Tepp W. H.; Johnson E. A.; Janda K. D. Targeting botulinum A cellular toxicity: a prodrug approach. J. Med. Chem. 2013, 56 (20), 7870–7879. 10.1021/jm400873n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel K. B.; Sullivan E. D.; Chen Y.; Chan J. C.; Jennings P. A.; Fierke C. A.; Cohen S. M. Dual-Mode HDAC prodrug for covalent modification and subsequent inhibitor release. J. Med. Chem. 2015, 58 (11), 4812–4821. 10.1021/acs.jmedchem.5b00539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlimme S.; Hauser A. T.; Carafa V.; Heinke R.; Kannan S.; Stolfa D. A.; Cellamare S.; Carotti A.; Altucci L.; Jung M.; Sippl W. Carbamate prodrug concept for hydroxamate HDAC inhibitors. ChemMedChem 2011, 6 (7), 1193–1198. 10.1002/cmdc.201100007. [DOI] [PubMed] [Google Scholar]
- Zielonka J.; Sikora A.; Hardy M.; Joseph J.; Dranka B. P.; Kalyanaraman B. Boronate probes as diagnostic tools for real time monitoring of peroxynitrite and hydroperoxides. Chem. Res. Toxicol. 2012, 25 (9), 1793–1799. 10.1021/tx300164j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikora A.; Zielonka J.; Lopez M.; Joseph J.; Kalyanaraman B. Direct oxidation of boronates by peroxynitrite: mechanism and implications in fluorescence imaging of peroxynitrite. Free Radical Biol. Med. 2009, 47 (10), 1401–1407. 10.1016/j.freeradbiomed.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X.; Gandhi V. ROS-activated anticancer prodrugs: a new strategy for tumor-specific damage. Ther. Delivery 2012, 3 (7), 823–833. 10.4155/tde.12.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang Y.; Balakrishnan K.; Gandhi V.; Peng X. Hydrogen peroxide inducible DNA cross-linking agents: targeted anticancer prodrugs. J. Am. Chem. Soc. 2011, 133 (48), 19278–19281. 10.1021/ja2073824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagen H.; Marzenell P.; Jentzsch E.; Wenz F.; Veldwijk M. R.; Mokhir A. Aminoferrocene-based prodrugs activated by reactive oxygen species. J. Med. Chem. 2012, 55 (2), 924–934. 10.1021/jm2014937. [DOI] [PubMed] [Google Scholar]
- Mann B. S.; Johnson J. R.; Cohen M. H.; Justice R.; Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12 (10), 1247–1252. 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- Liao Y.; Niu X.; Chen B.; Edwards H.; Xu L.; Xie C.; Lin H.; Polin L.; Taub J. W.; Ge Y.; Qin Z. Synthesis and antileukemic activities of piperlongumine and HDAC inhibitor hybrids against Acute myeloid leukemia cells. J. Med. Chem. 2016, 59 (17), 7974–7990. 10.1021/acs.jmedchem.6b00772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balliet R. M.; Chen G.; Gallagher C. J.; Dellinger R. W.; Sun D.; Lazarus P. Characterization of UGTs active against SAHA and association between SAHA glucuronidation activity phenotype with UGT genotype. Cancer Res. 2009, 69 (7), 2981–2989. 10.1158/0008-5472.CAN-08-4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin M. E.; Rivera-Del Valle N.; Chandra J. Redox control of leukemia: from molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signaling 2013, 18 (11), 1349–1383. 10.1089/ars.2011.4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Er T. K.; Tsai S. M.; Wu S. H.; Chiang W.; Lin H. C.; Lin S. F.; Wu S. H.; Tsai L. Y.; Liu T. Z. Antioxidant status and superoxide anion radical generation in acute myeloid leukemia. Clin. Biochem. 2007, 40 (13–14), 1015–1019. 10.1016/j.clinbiochem.2007.05.013. [DOI] [PubMed] [Google Scholar]
- Zhou F. L.; Zhang W. G.; Wei Y. C.; Meng S.; Bai G. G.; Wang B. Y.; Yang H. Y.; Tian W.; Meng X.; Zhang H.; Chen S. P. Involvement of oxidative stress in the relapse of acute myeloid leukemia. J. Biol. Chem. 2010, 285 (20), 15010–15015. 10.1074/jbc.M110.103713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hole P. S.; Zabkiewicz J.; Munje C.; Newton Z.; Pearn L.; White P.; Marquez N.; Hills R. K.; Burnett A. K.; Tonks A.; Darley R. L. Overproduction of NOX-derived ROS in AML promotes proliferation and is associated with defective oxidative stress signaling. Blood 2013, 122 (19), 3322–3330. 10.1182/blood-2013-04-491944. [DOI] [PubMed] [Google Scholar]
- Passaro D.; Di Tullio A.; Abarrategi A.; Rouault-Pierre K.; Foster K.; Ariza-McNaughton L.; Montaner B.; Chakravarty P.; Bhaw L.; Diana G.; Lassailly F.; Gribben J.; Bonnet D. Increased Vascular Permeability in the Bone Marrow Microenvironment Contributes to Disease Progression and Drug Response in Acute Myeloid Leukemia. Cancer Cell 2017, 32 (3), 324–341. 10.1016/j.ccell.2017.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieracki N. A.; Gantner B. N.; Mao M.; Horner J. H.; Ye R. D.; Malik A. B.; Newcomb M. E.; Bonini M. G. Bioluminescent detection of peroxynitrite with a boronic acid-caged luciferin. Free Radical Biol. Med. 2013, 61, 40–50. 10.1016/j.freeradbiomed.2013.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gediya L. K.; Chopra P.; Purushottamachar P.; Maheshwari N.; Njar V. C. A new simple and high-yield synthesis of suberoylanilide hydroxamic acid and its inhibitory effect alone or in combination with retinoids on proliferation of human prostate cancer cells. J. Med. Chem. 2005, 48 (15), 5047–5051. 10.1021/jm058214k. [DOI] [PubMed] [Google Scholar]
- Das B. C.; Thapa P.; Karki R.; Schinke C.; Das S.; Kambhampati S.; Banerjee S. K.; Van Veldhuizen P.; Verma A.; Weiss L. M.; Evans T. Boron chemicals in diagnosis and therapeutics. Future Med. Chem. 2013, 5 (6), 653–676. 10.4155/fmc.13.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noh J.; Kwon B.; Han E.; Park M.; Yang W.; Cho W.; Yoo W.; Khang G.; Lee D. Amplification of oxidative stress by a dual stimuli-responsive hybrid drug enhances cancer cell death. Nat. Commun. 2015, 6, 6907. 10.1038/ncomms7907. [DOI] [PubMed] [Google Scholar]
- Hulsman N.; Medema J. P.; Bos C.; Jongejan A.; Leurs R.; Smit M. J.; de Esch I. J.; Richel D.; Wijtmans M. Chemical insights in the concept of hybrid drugs: the antitumor effect of nitric oxide-donating aspirin involves a quinone methide but not nitric oxide nor aspirin. J. Med. Chem. 2007, 50 (10), 2424–2431. 10.1021/jm061371e. [DOI] [PubMed] [Google Scholar]
- Raj L.; Ide T.; Gurkar A. U.; Foley M.; Schenone M.; Li X.; Tolliday N. J.; Golub T. R.; Carr S. A.; Shamji A. F.; Stern A. M.; Mandinova A.; Schreiber S. L.; Lee S. W. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature 2011, 475 (7355), 231–234. 10.1038/nature10167. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Bose P.; Grant S. Rational combinations of targeted agents in AML. J. Clin. Med. 2015, 4 (4), 634–664. 10.3390/jcm4040634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Manero G.; Tambaro F. P.; Bekele N. B.; Yang H.; Ravandi F.; Jabbour E.; Borthakur G.; Kadia T. M.; Konopleva M. Y.; Faderl S.; Cortes J. E.; Brandt M.; Hu Y.; McCue D.; Newsome W. M.; Pierce S. R.; de Lima M.; Kantarjian H. M. Phase II trial of vorinostat with idarubicin and cytarabine for patients with newly diagnosed acute myelogenous leukemia or myelodysplastic syndrome. J. Clin. Oncol. 2012, 30 (18), 2204–2210. 10.1200/JCO.2011.38.3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkenberg K. J.; Johnstone R. W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discovery 2014, 13 (9), 673–691. 10.1038/nrd4360. [DOI] [PubMed] [Google Scholar]
- Brieger K.; Schiavone S.; Miller F. J. Jr.; Krause K. H. Reactive oxygen species: from health to disease. Swiss Med. Wkly. 2012, 142, w13659. 10.4414/smw.2012.13659. [DOI] [PubMed] [Google Scholar]
- Pacher P.; Beckman J. S.; Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87 (1), 315–424. 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.