

Abstract
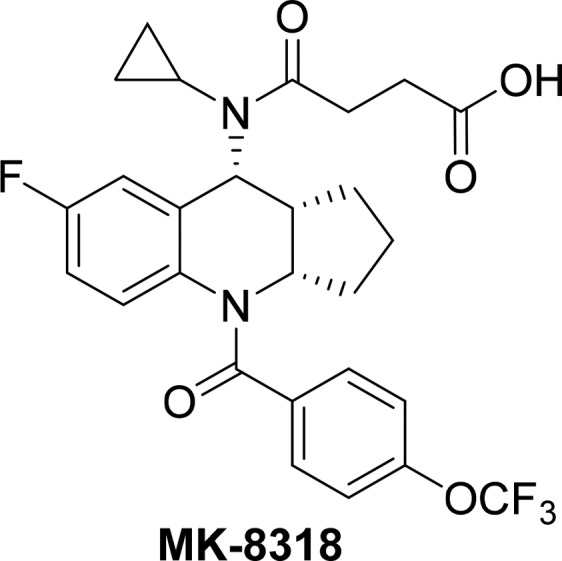
A novel series of tricyclic tetrahydroquinolines were identified as potent and selective CRTh2 receptor antagonists. The agonism and antagonism switch was achieved through structure-based drug design (SBDD) using a CRTh2 receptor homologue model. The challenge of very low exposures in pharmacokinetic studies was overcome by exhaustive medicinal chemistry lead optimization through focused SAR studies on the tricyclic core. Further optimization resulted in the identification of the preclinical candidate 4-(cyclopropyl((3aS,9R,9aR)-7-fluoro-4-(4-(trifluoromethoxy)benzoyl)-2,3,3a,4,9,9a-hexahydro-1H-cyclopenta[b]quinolin-9-yl)amino)-4-oxobutanoic acid (15c, MK-8318) with potent and selective CRTh2 antagonist activity and a favorable PK profile suitable for once daily oral dosing for potential treatment of asthma.
Keywords: MK-8318, CRTh2 receptor, DP2 receptor, receptor antagonist, homology model, PK lead optimization, pharmacokinetics, asthma, tetrahydroquinoline
Asthma is a common chronic lung disease that inflames and narrows the airways and affects millions of people of all ages in all parts of the world. It is a cause of substantial burden to society and often results in a reduced quality of life. It was estimated in 2014 that about 334 million people worldwide had asthma with a potential increase of incidences by 50% every decade.1,2 For mildly asthmatic patients, the disease can be well controlled by multiple treatment options such as inhaled formulations including inhaled corticosteroids (ICS), both short-acting beta agonists (SABAs) and long-acting beta agonists (LABAs), long-acting muscarinic antagonists (LAMAs), and LABA/inhaled corticosteroid ICS fixed-dose combinations (FDCs).3,4 However, convenience and patient compliance continue to be an issue with these treatments even though nonsteroid oral asthma medications such as leukotriene receptor antagonists (e.g., montelukast) are available but are used as a second-line treatment due to lower clinical efficacy. For severe and refractory asthmatic patients, the treatment options are more limited and biologic-based treatments became available just recently.5,6 With such a global disease burden and cause of suffering, unmet medical needs remain in finding efficient treatments and control of asthma and other allergic diseases. In recent years, the chemoattractant receptor homologous molecule expressed on T-helper type 2 (CRTh2) receptor (alternatively DP2 receptor) antagonists have attracted much attention from the pharmaceutical industry7,8 due to their significant role in modulating allergic inflammation via stimulation of Th2 cells, eosinophils and basophils. It has been shown that the PGD2 induced chemotaxis, degranulation, and cytokine production are exclusively exerted through CRTh2 receptors, and modulation of CRTh2 receptors should result in reducing excessive allergic responses and could be used for the treatment of asthma.9−11 TP receptor antagonist (thromboxane receptor) ramatroban is marketed in Japan for the treatment of allergic rhinitis and was considered as a potential treatment of asthma. Its antiallergic efficacy was believed to be related to its moderate CRTh2 activity.12 Market research indicates physicians and patients would welcome an improved oral therapy. An oral CRTh2 antagonist has potential utility for the mild asthmatics who prefer oral nonsteroid control over an inhaled option and/or who have not had an adequate response to leukotriene receptor antagonists. This can be used for patients who need greater control as add-on to ICS or ICS/LABA or leukotrienes. Due to these potential clinical benefits, many companies have since identified potent CRTh2 antagonists in recent years with about 20 of them having progressed into clinical development.13−17 At MRL, as an addition to the Singulair franchise, a CRTh2 antagonist was progressed to clinical trials (Figure 1, MK-7246) targeting asthma.13,18 To continue the effort in this area, a backup program was established in house with the goal to identify a CRTh2 antagonist suitable for once daily dosing, which would be structurally distinct from current clinical candidates.19 Herein, we report our effort in identifying structurally novel MK-8318 as a potent and selective CRTh2 receptor antagonist suitable for once-daily oral dosing.
Figure 1.

Structures of MK-7246, in-house CRTh2 antagonist, and indomethacin.
In addition to the leads such as compound 1(19) generated through in-house compound screening, we decided to pursue a literature-based drug design strategy to generate a structurally different lead. In this regard, tetrahydroquinoline CRTh2 antagonists attracted our attention due to their unique structure and potent binding and functional antagonist activity.20 Starting from known compound 2(20) (Figure 2), extensive structural overlay through conformational analysis identified fused tricyclic compound 3 as a potent CRTh2 ligand in the binding assay. However, to our surprise, when it was tested in the functional cAMP assay with the agonist mode, this compound turned out to be a functional partial agonist. It was known that subtle structural changes might impact the properties of the molecule to serve as an antagonist or agonist.19,21 To overcome this problem, we decided to compare the structure with a known CRTh2 agonist indomethacin22 in the CRTh2 homologue model. It suggested that the aniline phenyl group in 3 was overlapping with the methoxybenzene in indomethacin, which was occupying the agonist pocket of the receptor (Figure 3). From the modeling study, we also noticed two key hydrogen bond interactions (between side chain acid and Lys210, and between benzocarbonyl and Tyr262), which were crucial for activity based on future SAR studies. With this information in mind, a smaller ethyl group was introduced (4) to minimize the interaction in this region. To our delight, compound 4 indeed lacked agonist activity and was a potent CRTh2 antagonist in both binding and functional assays.
Figure 2.

Evolution of initial leads.
Figure 3.
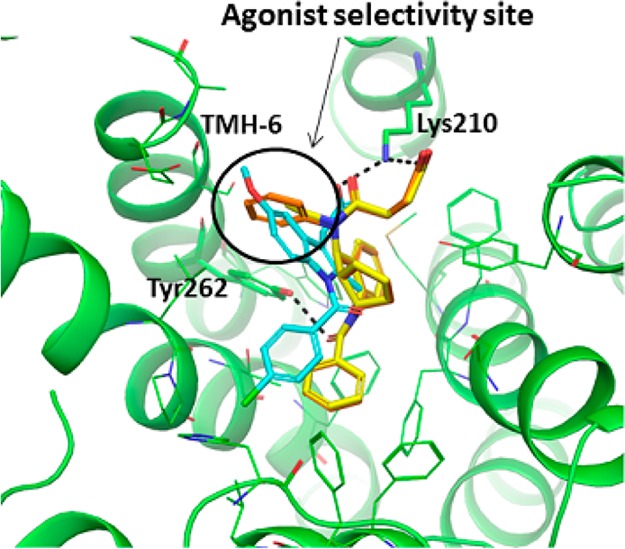
Overlay of homology models of human CRTh2 in complex with indomethacin, 3, or 4 (stereochemistry as drawn: 3aS,9R,9aR). The acid of three ligands forms a salt-bridge with the Lys210 side chain amino group; benzocarbonyl of 3 and 4 makes a hydrogen bond with the Tyr262 side chain hydroxy group, shown with dashed lines. The area occupied by methoxybenzene of indomethacin was defined as an agonist selectivity pocket (circled), which is pointed toward TMH-6. Carbon atoms of indomethacin are colored in cyan, 3 in orange, and 4 in yellow.
With multiple positions for SAR modifications in compound 4, a systematic optimization strategy was taken to quickly determine the essential pharmacophores at each portion of the molecule (Figure 2, north, middle, and south portions) before focused SAR to optimize overall profiles. Considering the profound effect of the exocyclic alkylamine on the functional activity, we first started the optimization of the northern portion of the molecule (Table 1, 5a–5h). Small lipophilic alkyl groups were acceptable at this position with cyclobutyl and cyclopropyl derivatives being the most potent. Polar substitutions were not well tolerated (data not shown), which may be because this portion of the molecule occupies the lipophilic region of the receptor. Based on the better binding activity of the cyclopropyl group, it was chosen as the optimum substitution at this position for further SAR development. The length of the acid side chain is important for activity, and a two carbon spacer was proven to be optimal length. When the acid is three carbons away from the amide carbonyl (Figure 4, 6 vs 9), the activity decreased. However, carbamates at the northern side chain were acceptable with a two-atom spacer (7 vs 8).
Table 1. Alkyl Side Chain Modification of the Northern Portion.
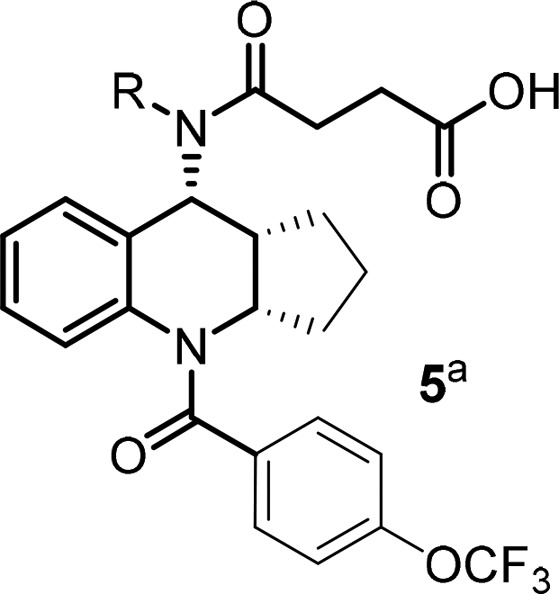

OCF3 benzamide was chosen to establish this SAR due to improved rat PK comparing to simple benzamide from initial screening SAR.
The data is the mean of at least two determinations.
Figure 4.

SAR of the acid side chain and tricyclic core. aThese compounds were all cis racemic and were generated during initial SAR screening. All other compounds in the Letter are enantiopure as drawn except those indicated as all-cis racemic. Trans isomers were less active and not discussed. Cbz and benzamide gave similar binding potency, but amides showed better PK.
For the middle portion, both cyclobutyl- and cyclohexyl-fused rings provided compounds having comparable binding activity to the compound having a cyclopentyl-fused ring (Figure 4, 10, 11 vs 9). However, the SAR is very narrow. When small substitutions such as methyl and gem-dimethyl were introduced to the cyclic alkyl rings in an effort to improve the PK properties of the compounds, such substitutions resulted in less active antagonists (data not shown). From the point of view of improving the metabolic profile, we felt the smaller cyclobutyl-fused ring might offer some advantages and decided to fix it as the core structure while developing SAR of the southern portion of the molecule.
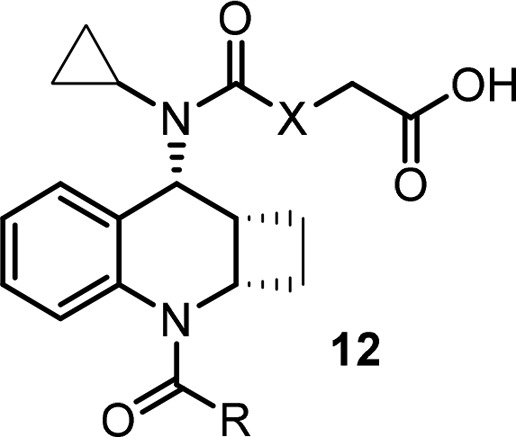
Initial SAR efforts with amide substitutions at the southern portion were promising. Aryl amides with different groups were well tolerated and showed potent CRTh2 binding and functional activity (Table 2, 12a–12d). Southern arylamides also worked well with a northern carbamate acid side chain (12e–12g) and had potent activity with all analogs showing single digit nanomolar binding potency. Compounds 12e and 12f also met program activity target profile of single digit nanomolar potency in functional assay (cAMP and beta-arrestin) and EOS whole blood assay (eosinophil shape change assay in human whole blood). Unfortunately, when these compounds were tested in rat PK studies,23 most of them had very poor exposure (AUC) with the carbamate analogs being the worst (12e, 12f). The analysis of in vitro hepatocyte stability and PK parameters correlations unfortunately failed to predict exposure in rat (data not shown).
Table 2. SAR of Southern Portion of the Cyclobutyl-Fused Tricyclic Core.

In nM; the data is the mean of at least two determinations.
0.5 mpk IV.
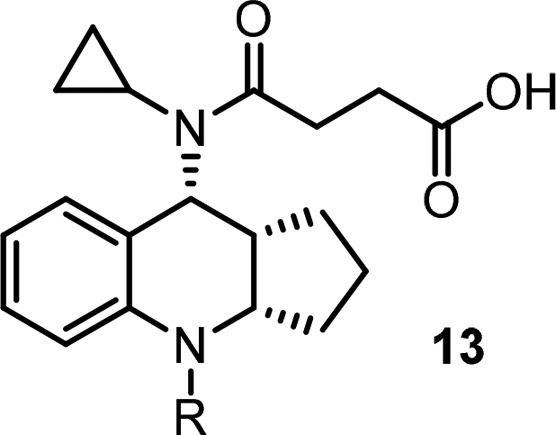
Presented with the poor PK results of the cyclobutyl fused analogs, we decided to investigate the cyclopentyl fused core to determine whether any improvement could be achieved and hope a compound with improved PK exposure can be identified through the empirical study. In this regard, different substitutions were introduced at the southern part (Table 3, selected examples.) Amides and carbamates were tolerated in terms of activity (5d, 13a, 13d–13e), but rat PK exposure was still poor. Elongated amide (13b) was not tolerated, which confirmed the importance of the carbonyl group as indicated in the homologue model. Urea 13c lost the binding activity by about 10-fold. Heterocycle 13f gave reasonable binding activity, but functional activity decreased dramatically. Based on these observations, an amide functionality off the tricyclic core at the southern portion was necessary in order to achieve the desired binding and functional activity, but exposure in rat PK still needed to be improved.
Table 3. Modifications of Southern Portion of the Cyclopentyl-Fused Tricyclic Core.

The data is the mean of at least two determinations.
0.5 mpk IV.
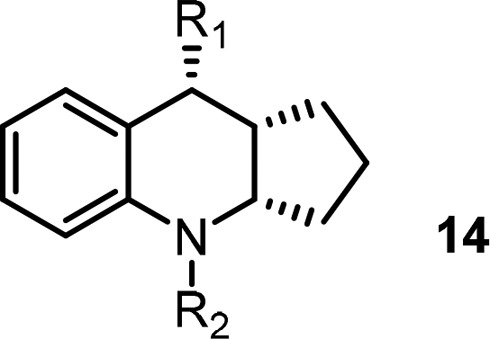
To understand the cause of the poor PK properties even with good solubility and permeability profiles, in vitro metabolite ID studies in rat were carried out. The studies showed that the major metabolic pathway was glucuronidation of the side chain acid in addition to oxidative metabolism at the middle core and southern region. In order to block or minimize acid metabolism, different side chains were introduced to increase steric hindrance and impact pKa of the acid (Table 4). However, these modifications proved to be fruitless: removal of the acid from the side chain resulted in much reduced functional activity (14a; cAMP IC50 = 1840 nM); bulky side chains resulted in loss of binding potency (14b-14d); aryl and heteroaryl linker analogs either lost binding activity (14e) or resulted in partial agonism (14f). An acid bioisostere provided no advantage and reduced activity was observed (14g). To this point, the SAR development had led us to a very narrow space for improvement in terms of PK exposure profile. In reviewing the PK data, a slight exposure (AUC) improvement in compound 5d (Table 3) caught our attention and left us some hope that there might be opportunity to identify compounds with increased plasma exposure.
Table 4. Acid Side Chain Modifications.

EOS shape IC50 = 4930 nM, racemic.
cAMP IC50 = 2920 nM; cAMP agonist EC50 = 12 nM, racemic.
All-cis racemic.
The data is the mean of at least two determinations.
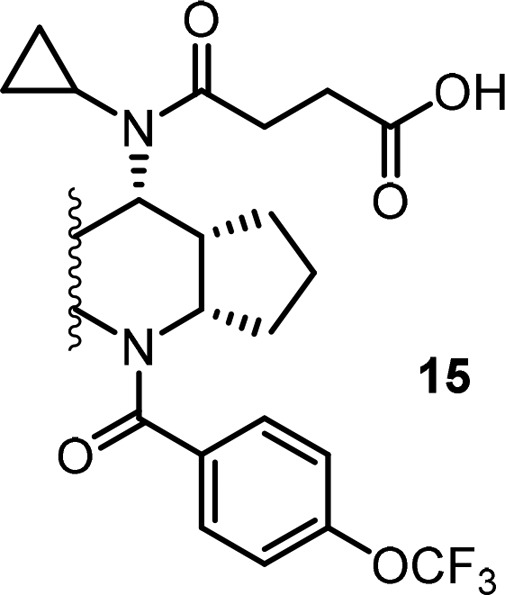
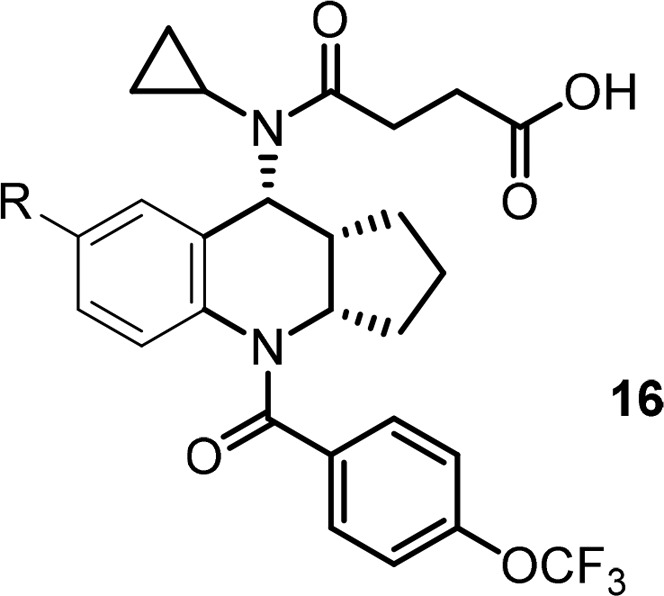
Based on this observation, we decided to explore the modification of the phenyl ring in the middle core portion, an area we have not explored yet. In this regard, small electron withdrawing groups such as F and Cl atoms were introduced first to block a potential metabolic hot spot and to affect overall electronic properties of the core, which might impact PK (Table 5). To our delight, most of these compounds retained very good biological activity and a few of them provided improved exposure when dosed in rat, with compound 15c is the best (Table 5, 15c–15e). To capitalize on this effort, a few close analogs of compound 15c were prepared to see whether further PK improvement could be achieved. Disappointingly, these modifications again resulted in loss of binding activity (Table 6, 16a–16f) and did not offer much improvement in PK profile (data not shown), which again suggested a very narrow SAR in this series.
Table 5. Focused SAR of the Phenyl Ring of the Cyclopentyl-Fused Tricyclic Tetrahydroquinoline.

In nM; the data is the mean of at least two determinations.
0.5 mpk IV.
3 mpk IV.
2 mpk IV.
Table 6. Targeted Modifications Based on Compound 15c.

In nM; the data is the mean of at least two determinations.
To this end, compound 15c stood out as one of the best molecules from the rat PK evaluation. It was further profiled in rat, dog, and monkey. The compound demonstrated overall good pharmacokinetic characteristics including oral bioavailability and half-life across the species (Table 7), which allowed once daily oral dosing in human. In addition to single digit nanomolar potency in all assays meeting program activity target profile (Ki, cAMP, beta-arrestin, and EOS shape change), compound 15c also showed clean profiles in ancillary studies (such as P450 enzyme inhibition, TDI, and PXR) and excellent selectivity against other related prostanoid receptors (please see Supporting Information for ancillary profiles summary). Based on the good overall profiles, we selected 15c for in vivo efficacy studies. In a sheep model, compound 15c blocked late airway response (LAR) and airway hyperresponsiveness (AHR) after i.v. infusion at 1 mpk.24 It also displayed good efficacy on BAL cells/pulmonary function from ovalbumin sensitized and challenged Brown Norway rats at 3, 10, and 30 mpk oral dosing (Table 8). With these favorable profiles, compound 15c was selected as a preclinical candidate (MK-8318) for further development.
Table 7. Pharmacokinetics of 15c in Preclinical Species.
| intravenous
PK parameters (IV, n = 3) | |||||
|---|---|---|---|---|---|
| species | dose (mg/kg)a | Clpb | Vd (L/kg) | T1/2 (h) | AUC (μM·h) |
| rat | 3 | 29.1 ± 7.7 | 5.8 ± 3.1 | 7.5 ± 1.0 | 3.4 ± 0.8 |
| dog | 1 | 9.4 ± 2.3 | 2.4 ± 0.63 | 10.4 ± 2.4 | 3.46 ± 0.80 |
| monkey | 1c | 16.4 ± 2.30 | 5.80 ± 0.76 | 12.9 ± 2.05 | 1.92 ± 0.27 |
| oral
PK parameters (PO, n = 3) | |||||
|---|---|---|---|---|---|
| species | dose (mg/kg) | Cmax (μM) | Tmax (h) | AUC (μM·h) | Foral |
| rat | 10d | 2.07 ± 1.59 | 0.58 ± 0.38 | 6.77 ± 3.19 | 61 ± 29 |
| dog | 2e | 2.47 ± 0.67 | 0.42 ± 0.14 | 8.51 ± 3.09 | 121 ± 44 |
| rhesus | 10e | 5.69 ± 2.02 | 0.42 ± 0.14 | 6.76 ± 1.78 | 28 ± 7.6 |
20% hydroxypropyl-beta-cyclodextrin with 100 mM sodium phosphate (pH 8).
mL/min/kg.
5% dimethyl sulfoxide/30% propylene glycol/30% polyethylene glycol 300/35% (40% hydroxypropyl-beta-cyclodextrin).
0.5% methyl cellulose/0.2% sodium laurel sulfate/5 mM HCI.
0.4% hydroxypropyl methylcellulose.
Table 8. Effect of Compound 15c with Oral Dosing on Recoverable BAL Cells from Ovalbumin Sensitized and Challenged Brown Norway Ratsa.
| cell type | % inhibition @ 3 mg/kg | % inhibition @ 10 mg/kg | % inhibition @ 30 mg/kg |
|---|---|---|---|
| eosinophils | 36 | 65 | 58 |
| neutrophils | 56 | 49 | 77 |
| total cells | 52 | 68 | 78 |
The effect of 15c (3–30 m/kg, p.o.; QD for 3 days) on differential BAL recoverable inflammatory cells measured 24 h post-antigen (ovalbumin 1%; 30 min) challenge. p < 0.05 compared to negative and positive controls.
The typical synthetic scheme for the preparation of the tetrahydroquinoline analogs is illustrated in Scheme 1. The synthesis started from ethyl 2-oxocyclopentanecarboxylate 17. It was treated with 4-fluoroaniline in the presence of Zn to give an aminoester, which was hydrolyzed to acid 18. The acid was converted to ketone 19 upon treatment with Eaton’s reagent25 followed by Boc protection. At this stage, the racemic intermediate 19 was resolved with chiral SFC to provide enantiomerically pure isomer. The cyclopropyl amine was introduced through reductive amination to give compound 20, which was acylated resulting in amide 21. The Boc moiety was removed with TFA, and the intermediate was coupled with 4-trifluoromethoxy benzoyl chloride to give compound 22, which was hydrolyzed to give final product 15c. This synthetic sequence was very efficient, and analogs could be prepared readily.
Scheme 1. Representative Synthetic Process for the Preparation of Tetrahydroquinolines as CRTh2 Antagonist.

Conditions: (a) Zn, 4-fluoroaniline, acetic acid/water, 80 °C 100%; NaOH (5 N), dioxane, 100%. (b) Eaton’s reagent, 70 °C, 40%; Boc2O, NEt3, DMAP, dioxane, 58%. (c) Titanium(IV) ethoxide, cyclopropylamine, THF, then NaBH4, CH2Cl2/MeOH, 90%. (d) Ethyl 4-chloro-4-oxobutanoate, Hunig’s base, dioxane, 90%. (e) TFA, CH2Cl2, 96%; 4-trifluoromethoxy benzoyl chloride, NEt3, DMAP, CH2Cl2, 87%. (f) NaOH (1 N), THF/MeOH, 91%.
In conclusion, a novel series of tricyclic tetrahydroquinolines were identified as potent and selective CRTh2 antagonists employing literature-based drug design. The agonism and antagonism switch was achieved through structure-based drug design (SBDD) using a CRTh2 receptor homologue model. A systematic SAR study was carried out, and the key pharmacophores were quickly identified to guide the SAR effort. The challenge of poor exposure seen in rat was overcome by dosing a number of compounds orally and metabolite ID studies through focused SAR studies on the middle tricyclic core. A balance between rational drug design and extensive SAR study resulted in the identification of compound 15c as a potent CRTh2 receptor antagonist with good in vitro and in vivo activity. This compound was selected as a preclinical candidate (MK-8318) due to its favorable PK profile suitable for once daily oral dosing and clean ancillary profiles. The detailed pharmacological and clinical evaluation of 15c as a potential treatment of asthma will be reported in due course.
Acknowledgments
We thank Dr. Eric Meade for his help editing the manuscript, Dr. Hong Mei for helpful discussion, the MS and NMR groups for spectroscopic data, and Drs. William Greenlee and Joseph Vacca for support of the program.
Glossary
ABBREVIATIONS
- CRTh2
chemoattractant receptor homologous molecule expressed on T-Helper type 2
- SBDD
structure-based drug design
- ICS
inhaled corticosteroids
- SABAs
short-acting beta agonists
- LABAs
long-acting beta agonists
- LAMAs
long-acting muscarinic antagonists
- FDCs
fixed-dose combinations
- DP2
prostaglandin D2 receptor 2
- TP receptor
thromboxane A2-prostanoid receptor
- PGD2
prostaglandin D2
- MRL
Merck Research Laboratory
- cAMP
cyclic adenosine monophosphate
- SAR
structure–activity relationship
- PK
pharmacokinetics
- AUC
area under curve
- TDI
time-dependent inhibition
- PXR
pregnane X receptor
- LAR
late airway response
- AHR
airway hyperresponsiveness
- TFA
trifluoroacetic acid
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00145.
Biological protocol, in vitro activity profile and ancillary profiles of compound 15c, in vivo metabolism of 15c, homology modeling of CRTh2 receptor, representative experimental procedures, and NMR and MS data for key compounds (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Dedication
Dedicated to Professor E. J. Corey on the occasion of his 90th birthday.
Supplementary Material
References
- The Global Asthma Network. The Global Asthma Report 2014. http://globalasthmareport.org/.
- Braman S. S. The Global Burden of Asthma. Chest 2006, 130, 4S–12S. 10.1378/chest.130.1_suppl.4S. [DOI] [PubMed] [Google Scholar]
- Xia Y.; Kelton C. M. L.; Xue L.; Guo J. J.; Bian B.; Wigle P. R. Safety of Long-acting beta Agonists and Inhaled Corticosteroids in Children and Adolescents with Asthma. Ther. Adv. Drug Saf. 2013, 4, 254–263. 10.1177/2042098613504124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kew K. M.; Dahri K. Long-acting Muscarinic Antagonists (LAMA) Added to Combination Long-acting beta2-agonists and Inhaled Corticosteroids (LABA/ICS) versus LABA/ICS for Adults with Asthma. Cochrane Database Syst. Rev. 2016, CD011721. 10.1002/14651858.CD011721.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasa M. Management of Severe Asthma: A Transition from Inhalations to Biologics. https://decisionresourcesgroup.com/drg-blog/management-severe-asthma-transition-inhalations-biologics/.
- Barnes P. J. Drugs for Asthma. Br. J. Pharmacol. 2006, 147, S297–S303. 10.1038/sj.bjp.0706437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Budelsky A.; Elsevier B. V.; Lawton G.; Witty D. R. Prostaglandin D2 Receptor CRTH2 Antagonists for the Treatment of Inflammatory Diseases. Prog. Med. Chem. 2011, 50, 49–107. 10.1016/B978-0-12-381290-2.00002-1. [DOI] [PubMed] [Google Scholar]
- There were about 60 discovery research publications from the pharmaceutical industry. We apologize that these were not cited here due to space limits.
- Schuligoi R.; Sturm E.; Luschnig P.; Konya V.; Philipose S.; Sedej M.; Waldhoer M.; Peskar B. A.; Heinemann A. CRTh2 and D-type Prostanoid Receptor Antagonists as Novel Therapeutic Agents for Inflammatory Diseases. Pharmacology 2010, 85, 372–382. 10.1159/000313836. [DOI] [PubMed] [Google Scholar]
- Pettipher R.; Hansel T. T. Antagonists of the Prostaglandin D2 Receptor CRTh2. Prog. Respir. Res. 2010, 39, 193–200. 10.1159/000320819. [DOI] [PubMed] [Google Scholar]
- Xue L.; Salimi M.; Panse I.; Mjösberg J. M.; McKenzie A. N.; Spits H.; Klenerman P.; Ogg G. Prostaglandin D2 activates group 2 innate lymphoid cells through chemoattractant receptor-homologous molecule expressed on TH2 cells. J. Allergy Clin. Immunol. 2014, 133, 1184–1194. 10.1016/j.jaci.2013.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto H.; Shichijo M.; Iino T.; Manabe Y.; Watanabe A.; Shimazaki M.; Gantner F.; Bacon K. B. An Orally Bioavailable Small Molecule Antagonist of CRTh2, Ramatroban (BAY u3405), Inhibits Prostaglandin D2-induced Eosinophil Migration in vitro. J. Pharmacol. Exp. Ther. 2003, 305, 347–352. 10.1124/jpet.102.046748. [DOI] [PubMed] [Google Scholar]
- Norman P. Update on the Status of DP2 Receptor Antagonists; from Proof of Concept Through Clinical Failures to Promising New Drugs. Expert Opin. Invest. Drugs 2014, 23, 55–66. 10.1517/13543784.2013.839658. [DOI] [PubMed] [Google Scholar]
- Pettipher R.; Whittaker M. Update on the Development of Antagonists of Chemoattractant Receptor-Homologous Molecule Expressed on Th2 Cells (CRTH2). From Lead Optimization to Clinical Proof-of-Concept in Asthma and Allergic Rhinitis. J. Med. Chem. 2012, 55, 2915–2931. And references cited therein. 10.1021/jm2013997. [DOI] [PubMed] [Google Scholar]
- Norman P. Indole-based CRTH2 Antagonists. Expert Opin. Ther. Pat. 2005, 15, 1817–1823. 10.1517/13543776.15.12.1817. [DOI] [Google Scholar]
- Ulven T.; Kostenis E. Novel CRTh2 Antagonists: a Review of Patents from 2006 to 2009. Expert Opin. Ther. Pat. 2010, 20, 1505–1530. 10.1517/13543776.2010.525506. [DOI] [PubMed] [Google Scholar]
- Lamers C.; Flesch D.; Schubert-Zsilavecz M.; Merk D. Novel Prostaglandin Receptor Modulators: a Patent Review (2002–2012) - part I: non-EP Receptor Modulators. Expert Opin. Ther. Pat. 2013, 23, 47–77. 10.1517/13543776.2013.736495. [DOI] [PubMed] [Google Scholar]
- Gallant M.; Beaulieu C.; Berthelette C.; Colucci J.; Crackower M. A.; Dalton C.; Denis D.; Ducharme Y.; Friesen R. W.; Guay D.; Gervais F. G.; Hamel M.; Houle R.; Krawczyk C. M.; Kosjek B.; Lau S.; Leblanc Y.; Lee E. E.; Levesque J.-F.; Mellon C.; Molinaro C.; Mullet W.; O’Neill G. P.; O’Shea P.; Sawyer N.; Sillaots S.; Simard D.; Slipetz D.; Stocco R.; Sørensen D.; Truong V. L.; Wong E.; Wud J.; Zaghdane H.; Wang Z. Discovery of MK-7246, a Selective CRTh2 Antagonist for the Treatment of Respiratory Diseases. Bioorg. Med. Chem. Lett. 2011, 21, 288–293. 10.1016/j.bmcl.2010.11.015. [DOI] [PubMed] [Google Scholar]
- Xiao D.; Zhu Z.; Yu Y.; Shao N.; Wu J.; McCormick K. D.; Dhondi P.; Qin J.; Mazzola R.; Tang H.; Rao A.; Siliphaivanh P.; Qiu H.; Yang X.; Rivelli M.; Garlisi C. G.; Eckel S.; Mukhopadhyay G.; Correll C.; Rindgen D.; Aslanian R.; Palani A. Quality by Design (QbD) of Amide Isosteres: 5,5-Disubstituted Isoxazolines as Potent CRTh2 Antagonists with Favorable Pharmacokinetic and Drug-like Properties. Bioorg. Med. Chem. Lett. 2014, 24, 1615–1620. 10.1016/j.bmcl.2014.01.043. [DOI] [PubMed] [Google Scholar]
- Liu J.; Wang Y.; Sun Y.; Marshall D.; Miao S.; Tonn G.; Anders P.; Tocker J.; Tang H. L.; Medina J. Tetrahydroquinoline Derivatives as CRTh2 Antagonists. Bioorg. Med. Chem. Lett. 2009, 19, 6840–6844. 10.1016/j.bmcl.2009.10.094. [DOI] [PubMed] [Google Scholar]
- Luker T.; Bonnert R.; Schmidt J.; Sargent C.; Paine S. W.; Thom S.; Pairaudeau G.; Patel A.; Mohammed R.; Akam E.; Dougall I.; Davis A. M.; Abbott P.; Brough S.; Millichip I.; McInally T. Switching between Agonists and Antagonists at CRTh2 in a Series of Highly Potent and Selective Biaryl Phenoxyacetic Acids. Bioorg. Med. Chem. Lett. 2011, 21, 3616–3621. 10.1016/j.bmcl.2011.04.101. [DOI] [PubMed] [Google Scholar]
- Hirai H.; Tanaka K.; Takano S.; Ichimasa M.; Nakamura M.; Nagata K. Cutting Edge: Agonistic Effect of Indomethacin on a Prostaglandin D2 Receptor, CRTh2. J. Immunol. 2002, 168, 981–985. 10.4049/jimmunol.168.3.981. [DOI] [PubMed] [Google Scholar]
- Cox K. A.; Dunn-Meynell K.; Korfmacher W.; Broske L.; Nomeir A. A.; Lin C. C.; Cayen M. N.; Barr W. H. Novel in vivo Procedure for Rapid Pharmacokinetic Screening of Discovery Compounds in Rats. Drug Discovery Today 1999, 4, 232–237. 10.1016/S1359-6446(98)01299-9. [DOI] [PubMed] [Google Scholar]
- Smith N.; Broadley K. J. Optimisation of the Sensitisation Conditions for an Ovalbumin Challenge Model of Asthma. Int. Immunopharmacol. 2007, 7, 183–190. 10.1016/j.intimp.2006.09.007. [DOI] [PubMed] [Google Scholar]
- Eaton P. E.; Carlson G. R.; Lee J. T. Phosphorus Pentoxide-methanesulfonic Acid. Convenient Alternative to Polyphosphoric Acid. J. Org. Chem. 1973, 38, 4071–4073. 10.1021/jo00987a028. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.