

Abstract
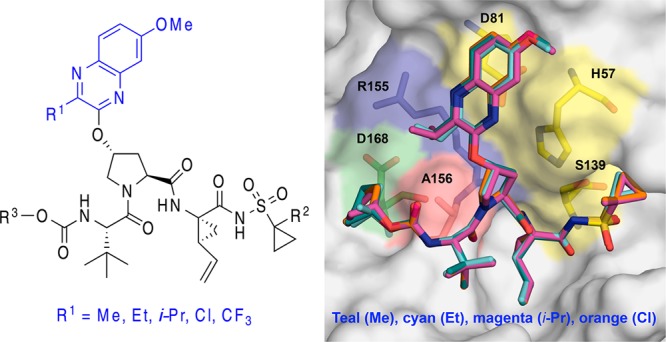
A series of linear HCV NS3/4A protease inhibitors was designed by eliminating the P2–P4 macrocyclic linker in grazoprevir, which, in addition to conferring conformational flexibility, allowed structure–activity relationship (SAR) exploration of diverse quinoxalines at the P2 position. Biochemical and replicon data indicated preference for small hydrophobic groups at the 3-position of P2 quinoxaline for maintaining potency against resistant variants R155K, A156T, and D168A/V. The linear inhibitors, though generally less potent than the corresponding macrocyclic analogues, were relatively easier to synthesize and less susceptible to drug resistance. Three inhibitor cocrystal structures bound to wild-type NS3/4A protease revealed a conformation with subtle changes in the binding of P2 quinoxaline, depending on the 3-position substituent, likely impacting both inhibitor potency and resistance profile. The SAR and structural analysis highlight inhibitor features that strengthen interactions of the P2 moiety with the catalytic triad residues, providing valuable insights to improve potency against resistant variants.
Keywords: HCV, NS3/4A protease, protease inhibitor, quinoxaline, crystal structure, drug resistance
Hepatitis C virus (HCV) is the leading cause of liver diseases such as cirrhosis, fibrosis, and liver cancer. The most recent estimates indicate that at least 70 million people worldwide have chronic HCV infection, and about 1.75 million new cases are recorded each year.1 The treatment of HCV infection has been challenging due to high genetic diversity of the virus and a lack of antiviral drugs that effectively target all HCV genotypes.2 Fortunately, recent advances in direct-acting antivirals (DAAs) targeting the HCV NS3/4A, NS5A, and NS5B proteins have significantly improved sustained virological response (SVR) in patients across genotypes.3 However, the high genetic diversity of HCV and the rapid emergence of drug resistance necessitate the use of combination therapies with two or three drugs from different classes to effectively treat infected patients.
HCV NS3/4A protease inhibitors (PIs) are a key component of new all-oral combination therapies.3 The five FDA approved PIs are all macrocyclic acylsulfonamides with different heterocyclic moieties at the P2 position.4 Of these, paritaprevir and grazoprevir are active against most HCV genotypes,5,6 while the recently approved glecaprevir and voxilaprevir exhibit pan-genotypic activity.7,8 These new PIs are also significantly more active against most polymorphic and drug resistant HCV variants.9 The development of NS3/4A PIs with pan-genotypic activity and improved resistance profiles marks a major milestone in anti-HCV drug discovery.
Despite remarkable improvements in potency and resistance profiles, the current NS3/4A PIs are still susceptible to drug resistance due to substitutions at one or more amino acid positions in the target protein.10,11 The most common resistance-associated substitutions (RASs) in the NS3/4A protease occur at residues Arg155, Ala156, and Asp168. Inhibitors show varying susceptibility to RASs at these and other positions depending on the heterocyclic moiety at the P2 position and the location of the macrocycle.12,13 Grazoprevir (1) (Figure 1) was the first NS3/4A PI to show activity against most HCV genotypes and resistant variants due to its unique binding conformation.12,14 The P2 quinoxaline moiety in 1 largely avoids direct interactions with residues Arg155 and Asp168, but instead interacts with the catalytic His57 and Asp81.12 As a result, 1 maintains potency against substitutions at Arg155 and is only moderately susceptible to substitutions at Asp168. However, the P2–P4 macrocycle in 1 causes steric clashes with larger substitutions at Ala156, resulting in drastically reduced potency.12 Both glecaprevir and voxilaprevir are also P2–P4 macrocyclic compounds structurally similar to 1 and are expected to bind in similar conformations, as indicated by their particularly high susceptibility to substitutions at Ala156.9,15 These three PIs currently in clinical use indeed exhibit similar resistance profiles and thus are prone to cross-resistance. Therefore, more robust NS3/4A PIs need to be developed, preferably incorporating diverse structural features.
Figure 1.

Structures of the HCV NS3/4A protease inhibitors grazoprevir (1), P1–P3 macrocyclic analogue 5172-mcP1P3 (2), linear analogue 5172-linear (3), and SAR compounds.
We have shown that the unique binding conformation of 1 does not depend on the P2–P4 macrocycle, as the P1–P3 macrocyclic and linear analogues, 5172-mcP1P3 (2) and 5172-linear (3) (Figure 1), still bind in a similar conformation as the parent compound.16 In the absence of the P2–P4 macrocyclic constraint, the conformationally flexible P2 quinoxaline moiety can allow the inhibitor to better adapt to structural changes in the protease active site due to RASs, particularly at Ala156. Accordingly, compared to 1, the P1–P3 macrocyclic analogue 2 exhibits similar potency against RASs at Arg155 and Asp168 but is significantly less susceptible to substitutions at Ala156, resulting in an overall improved resistance profile.13
In addition to conferring conformational flexibility, elimination of the P2–P4 macrocycle in 1 provides opportunities to further improve potency and resistance profile by exploring structure–activity relationship (SAR) at the 3-position of P2 quinoxaline in both the P1–P3 macrocyclic (2) and linear (3) analogues. Toward this end, we recently developed a substrate envelope guided design strategy that aims to exploit interactions with the invariant catalytic triad, minimize interactions with the S2 subsite residues, and incorporate conformational flexibility at the P2 moiety, which led to the identification of P1–P3 macrocyclic analogues of 2 with exceptional potency and resistance profiles.17 These exciting findings encouraged us to explore the SAR of linear compound 3 to identify analogues with improved potency against drug resistant HCV variants. In this letter, we report the structure-guided design, synthesis, and SAR studies of a series of quinoxaline-based linear NS3/4A PIs. In addition, we report cocrystal structures of three linear inhibitors with different P2 quinoxaline moieties bound to NS3/4A protease, providing valuable structural insights to further optimize potency and resistance profiles.
The synthesis of linear NS3/4A PIs with diverse P2 quinoxaline moieties is outlined in Scheme 1. The key Boc-protected P2 intermediates 4a–e were prepared from the corresponding 3-substituted 7-methoxy-quinoxalin-2-ones by an SN2 displacement reaction with the activated cis-hydroxyproline derivative as described previously.17 Deprotection of the Boc group and reaction with N-Boc-l-tert-leucine under HATU coupling conditions gave the P2–P3 intermediates 5a–e. After ester hydrolysis, the resulting acids 6a–e were coupled with the P1–P1′ acylsulfonamide intermediates 7 and 8 to afford the target inhibitors 3, 9b–e, and 10a–e. The tert-butyl-capped compounds were converted to the corresponding cyclopentyl derivatives in two steps, involving Boc deprotection and reaction with N-(cyclopentyloxycarbonyloxy)-succinimide, to provide the desired compounds 11a–e and 12a–e.
Scheme 1. Synthesis of HCV NS3/4A Protease Inhibitors.

Reagents and conditions: (a) 4 N HCl in dioxane, CH2Cl2, RT, 3 h; (b) Boc-Tle-OH, HATU, DIEA, DMF, RT, 4 h; (c) LiOH·H2O, THF, H2O, RT, 24 h; (d) HATU, DIEA, DMF, RT, 2 h; (e) 4 N HCl in dioxane, RT, 3 h; (f) N-(cyclopentyloxy-carbonyloxy)-succinimide, DIEA, CH3CN, RT, 36 h.
The SAR exploration of 3 was focused on optimizing interactions of the P2 quinoxaline moiety and minimizing direct interactions with S2 subsite residues. In addition, modifications at the P1′ (R2) and the N-terminal capping groups (R3) were investigated (Figure 1). The potency and resistance profiles of the resulting linear PIs were evaluated using biochemical and replicon assays. The enzyme inhibition constants (Ki) were determined against WT GT1a NS3/4A protease and resistant variants R155K and D168A (Table 1). For a subset of compounds, cellular antiviral potencies (EC50) were determined using replicon-based antiviral assays against WT HCV and resistant variants R155K, A156T, D168A, and D168V (Table 2). Grazoprevir (1) was used as a control in all assays.
Table 1. Inhibitory Activity against Wild-type HCV NS3/4A Protease and Drug Resistant Variants.

|
Ki (nM) |
|||||
|---|---|---|---|---|---|
| compd | R1 | R2 | WT | R155K | D168A |
| 3 | Et | H | 19 ± 2.7 | 17 ± 2.3 | 642 ± 101 |
| 10a | Et | Me | 16 ± 1.3 | 14 ± 1.1 | 385 ± 31 |
| 11a | Et | H | 9.8 ± 2.0 | 15 ± 2.2 | 350 ± 30 |
| 12a | Et | Me | 6.9 ± 0.5 | 13 ± 2.7 | 145 ± 14 |
| 9b | Me | H | 18 ± 1.6 | 8.5 ± 2.1 | 290 ± 24 |
| 10b | Me | Me | 14 ± 2.1 | 14 ± 1.7 | 265 ± 26 |
| 11b | Me | H | 9.2 ± 0.9 | 9.6 ± 0.9 | 144 ± 23 |
| 12b | Me | Me | 7.1 ± 1.1 | 10 ± 1.3 | 140 ± 13 |
| 9c | i-Pr | H | 32 ± 5.1 | 49 ± 11 | 1086 ± 137 |
| 10c | i-Pr | Me | 29 ± 9.4 | 27 ± 5.6 | 1179 ± 170 |
| 11c | i-Pr | H | 17 ± 3.2 | 55 ± 11 | 985 ± 106 |
| 12c | i-Pr | Me | 21 ± 2.6 | 43 ± 11 | 1000 ± 80 |
| 9d | Cl | H | 7.8 ± 1.1 | 2.2 ± 0.4 | 128 ± 16 |
| 10d | Cl | Me | 6.1 ± 1.1 | 3.8 ± 0.6 | 119 ± 16 |
| 11d | Cl | H | 3.8 ± 0.6 | 4.1 ± 0.5 | 99 ± 10 |
| 12d | Cl | Me | 3.9 ± 0.7 | 5.2 ± 0.8 | 51 ± 6.0 |
| 9e | CF3 | H | 87 ± 18 | 24 ± 3.3 | 723 ± 80 |
| 10e | CF3 | Me | 46 ± 9.6 | 12 ± 1.7 | 513 ± 50 |
| 11e | CF3 | H | 34 ± 8.3 | 26 ± 7.7 | 703 ± 63 |
| 12e | CF3 | Me | 22 ± 3.4 | 22 ± 6.9 | 516 ± 61 |
| 2 | 2.0 ± 0.1 | 3.1 ± 0.34 | 91 ± 10 | ||
| 1 | 0.20 ± 0.1 | 0.80 ± 0.3 | 40 ± 5.0 | ||
Table 2. Antiviral Activity against Wild-type HCV and Drug Resistant Variants.
| Replicon
EC50 (nM) (fold change) |
|||||
|---|---|---|---|---|---|
| compd | WT | R155K | A156T | D168A | D168V |
| 3 | 24 | 50 (2.1) | 73 (3.0) | >500 (>21) | >500 (>21) |
| 11b | 10 | 48 (4.8) | 164 (16) | 101 (10) | 150 (15) |
| 12b | 4.5 | 28 (6.2) | 87 (19) | 45 (10) | 59 (13) |
| 9d | 6.6 | 10 (1.5) | 107 (16) | 38 (5.8) | 12 (1.8) |
| 10d | 6.3 | 10 (1.6) | 100 (16) | 30 (4.8) | 12 (1.9) |
| 11d | 7.4 | 40 (5.4) | 292 (40) | 50 (6.8) | 54 (7.3) |
| 12d | 3.1 | 27 (8.7) | 163 (53) | 25 (8.1) | 23 (7.4) |
| 2 | 0.33 | 1.8 (5.5) | 9.7 (29) | 6.3 (19) | 9.1 (28) |
| 1 | 0.12 | 1.9 (16) | 200 (1667) | 11 (92) | 5.3 (44) |
Compared to the macrocyclic PIs 1 and 2, the linear analogue 3 exhibited significantly lower potency against WT protease (Ki = 19 nM) and experienced an even larger reduction in antiviral potency (EC50 = 24 nM), as reported previously.13 Compound 3 was also less potent than 1 and 2 against the resistant variants R155K, D168A, and D168V in both enzyme inhibition and replicon assays. The significant potency losses for the linear inhibitor 3 are likely due to the increase in conformational flexibility and associated entropic penalty of binding to the protease. However, close examination of the overall resistance profile revealed that fold losses in potency were generally lower for compound 3 than 1 in both enzyme inhibition and replicon assays (Tables S1 and 2). Moreover, while 1 was highly susceptible to the A156T variant (EC50 = 200 nM), with >1600-fold loss in potency compared to WT, compound 3 showed better antiviral potency against this variant (EC50 = 73 nM). The reduced susceptibility to RASs, particularly at Ala156, observed for 3 demonstrates that removal of the macrocyclic linker and the resulting conformational flexibility allows the inhibitor to adapt to substitutions in the S2 subsite.
To improve potency, analogues of inhibitor 3 with modifications at the P1′ and P4 capping groups were prepared and tested. Replacement of the P1′ cyclopropylsulfonamide with a more hydrophobic 1-methylcyclopropylsulfonamide moiety generally improved potency of the resulting analogues. Thus, compared to 3, analogue 10a afforded a slight increase in enzyme potency against WT protease and resistant variants R155K and D168A. Similarly, replacing the tert-butyl P4 capping group with a bulkier cyclopentyl moiety in 11a provided compounds with improved potency. Analogue 12a with the 1-methylcyclopropylsulfonamide moiety at P1′ and cyclopentyl P4 capping group was 2- and 4-fold more potent than 3 against the WT protease (Ki = 6.9 nM) and D168A variant (Ki = 145 nM), respectively. Thus, minor modifications at the P1′ and P4 moieties of inhibitor 3 provided analogues with improved potency against WT protease and the D168A variant.
We have shown that minimizing inhibitor interactions in the S2 subsite resulted in an overall improvement in potency and resistance profiles.17 Although the P2 quinoxaline in 3 largely avoids direct interactions with residues in the S2 subsite, the ethyl group at the 3-position of this moiety makes hydrophobic interactions with the hydrocarbon portion of the Arg155 side chain as well as with Ala156. Thus, in an effort to optimize interactions with these residues, changes at the 3-position of the P2 quinoxaline moiety were explored. Based on the cocrystal structures and SAR results from the P1–P3 macrocyclic series,17 replacing the ethyl group with a smaller methyl group at this position while reducing overall inhibitor interactions in the S2 subsite was expected to maintain key hydrophobic interactions with side chains of Arg155 and Ala156. As anticipated, compound 9b incorporating the 3-methylquinoxaline was 2-fold more potent than 3 against the R155K and D168A protease variants in enzyme inhibition assays. However, analogue 10b with the 1-methylcyclopropylsulfonamide moiety at the P1′ position did not show much improvement in enzyme potency compared to 9b. Compounds 11b and 12b with the cyclopentyl P4 capping group were slightly more potent than the corresponding tert-butyl analogues 9b and 10b against WT and D168A. Furthermore, compounds 11b and 12b showed nanomolar potency in replicon assays against WT HCV, and despite losing about 5–20-fold potency, 12b maintained significant potency against all variants tested. Although both the 3-ethyl and 3-methyl-quinoxaline compounds showed similar potencies against WT and R155K protease variants, PIs with the smaller methyl substituent were generally more potent against the D168A variant. Together, the enzyme inhibition and replicon data indicate a preference for smaller substituents at the 3-position of the P2 quinoxaline to maintain potency against resistant variants.
Next, a larger isopropyl group was incorporated in compounds 9c and 10c to further explore the optimal size of the substituent at the 3-position of P2 quinoxaline that can be accommodated in the S2 subsite without causing unfavorable interactions. These compounds displayed considerably lower potency compared to the 3-methyl- and 3-ethyl-quinoxaline compounds across all variants in enzyme inhibition assays. Moreover, compounds with a larger isopropyl group at this position were highly susceptible to RASs at Arg155 and Asp168, with Ki values in the millimolar range against the D168A protease variant. Analogues 11c and 12c, with a cyclopentyl P4 capping group, showed similar trends to the corresponding tert-butyl analogues across all protease variants tested. These findings further support our hypothesis that large substituents at the 3-position of the P2 quinoxaline are detrimental to potency against resistant variants.
After determining the optimal size of the substituent at the 3-position of the P2 quinoxaline, isosteric replacements of the alkyl group with different electronic properties were explored. Thus, a set of compounds bearing a 3-chloroquinoxaline P2 moiety, with comparable size to the 3-methylquinoxaline, was analyzed. In general, compounds with the 3-chloroquinoxaline were significantly more potent than the corresponding 3-ethyl- and 3-methyl-quinoxaline analogues. Compounds 9d and 10d, with a tert-butyl P4 capping group, showed about 2-fold better potency than the corresponding 3-methylquinoxaline analogues 9b and 10b against WT, R155K, and D168A proteases. Similarly, the cyclopentyl-capped compounds 11d and 12d (WT Ki = 3.8 and 3.9 nM, respectively) were more potent than the corresponding 11b and 12b, showing excellent potency against WT protease and resistant variants. In fact, both 11d and 12d exhibited Ki values against WT protease and resistant variants in the same range as the macrocyclic inhibitor 2 (WT Ki = 2.0 nM), indicating that potency of the quinoxaline-based linear PIs could be improved significantly by SAR exploration. In replicon assays, the 3-chloroquinoxaline compounds exhibited the best overall potency profile among the linear compounds, with PIs 9d, 10d, and 12d showing significant improvement in replicon potency against the multidrug resistant HCV variants D168A/V (EC50 = 12–38 nM). However, these compounds were more susceptible to the A156T substitution than the corresponding macrocyclic analogues.17 The improved potency profiles of the 3-chloroquinoxaline compounds compared to the corresponding 3-methylquinoxaline analogues indicate that the chloro group likely renders more favorable electronic properties to the quinoxaline moiety, which improves the critical π–π stacking interactions with the catalytic residue His57.
To further investigate the effect of electron-withdrawing groups on the activity of the inhibitors, derivatives with a more electronegative, although relatively larger, 3-trifluoromethylquinoxaline were examined. In contrast to the 3-chloroquinoxaline inhibitors, compounds 9e and 10e showed considerable loss in potency against WT protease. However, despite relatively lower potency against WT, the 3-trifluoromethylquinoxaline analogues were slightly more potent than the corresponding 3-isopropylquinoxaline PIs against the resistant variants R155K and D168A. Similar trends were observed for inhibitors 11e and 12e with the cyclopentyl P4 capping group. While it is difficult to separate the effects of electronic properties of the chloro- and trifluoromethyl-quinoxaline moieties, it is likely that size played a more important role in determining the overall potency profile of these inhibitors.
In an effort to explain the observed potency and resistance profiles, crystal structures of three linear PIs incorporating different P2 quinoxaline moieties were determined in complex with WT NS3/4A protease (Table S2). The crystal structures of WT-12b, -12c, and -12d were compared with our previously determined structures of 1 and 3 (PDB IDs 3SUD and 5EQQ, respectively).12,16 These high-resolution (1.78–1.80 Å) structures provided details of protein–inhibitor interactions to elucidate the structural differences that underlie varied potency and susceptibility to resistant variants.
The overall binding mode of linear inhibitors 12b–d is similar to that of macrocyclic inhibitors 1–2 and the parent compound 3, where the P2 quinoxaline predominately interacts with the catalytic triad residues (Figure 2). These structures confirm that the quinoxaline moiety maintains this unique binding conformation irrespective of macrocyclization and the substituent at the 3-position. Inhibitors 12b–d span the S1′–S4 pockets in the active site, with a conserved hydrogen bond network present in all WT NS3/4A protease structures. The hydrogen bonds between the P1 amide and the backbone carbonyl of Arg155 as well as the P3 amide and Ala157 backbone are maintained. The P1′ acylsulfonamide moiety is positioned in the oxyanion hole, stabilized by hydrogen bonds with His57, Gly137, Ser138, and Ser139. Although the overall binding mode of linear analogues is similar to that of compound 1, there are subtle changes in the binding of P2 quinoxaline that may impact inhibitor potency.
Figure 2.

Cocrystal structures of WT1a HCV NS3/4A protease in complex with linear inhibitors (a) 12b, (b) 12c, and (c) 12d. The protease active site is presented as a light gray surface with bound inhibitors depicted as orange sticks. The catalytic triad is highlighted in yellow, and drug resistance residues Arg155, Ala156, and Asp168 are shown as sticks.
The differences between the WT cocrystal structures of 1 and 3-methylquinoxaline inhibitor 12b occur predominantly in the S2 subsite. Relative to WT-1, the Asp168 side chain in the WT-12b structure is shifted to allow additional hydrogen bonding with the side chain of Arg155 (Figures 2A and 3A). This conformation of Asp168, which allows the P4 cyclopentyl capping group to occupy the S4 pocket, is observed in most WT protease–inhibitor complexes.12,17 The 3-methylquinoxaline moiety is shifted away from the catalytic residues toward the S2 subsite relative to the conformation of P2 quinoxaline in the WT-1 structure. This shift was also observed in the parent compound 3, though to a lesser extent, likely to accommodate the larger ethyl group at the 3-position of quinoxaline (Figure S1). However, despite a larger shift of the 3-methylquinoxaline moiety, inhibitor 12b has an improved potency profile against resistant variants compared to 3, likely due to weaker contacts of the smaller methyl group with residues in the S2 subsite that mutate to confer resistance. Thus, while a slight shift of the P2 quinoxaline toward the S2 subsite does not appear to affect the overall potency profile, the substituent at the 3-position of this moiety significantly impacts inhibitor potency against resistant variants.
Figure 3.

Superposition of WT-1 and (a) WT-12b, (b) WT-12c, and (c) WT-12d complexes, focusing on the differences at the P2 quinoxaline. The protease is in a ribbon representation (light gray), with bound inhibitors 1 (blue) and 12b–d (orange) depicted as sticks. The side chains of the catalytic triad and drug resistance residues Arg155, Ala156, and Asp168 are shown as balls and sticks.
The shift of the quinoxaline moiety toward the S2 subsite residues was also observed in the WT-12c and WT-12d complexes (Figure 3B,C), as well as in WT-2 and other structures of the P1–P3 macrocyclic analogues.17 However, the crystal structure of inhibitor 12c with the 3-isopropylquinoxaline revealed an additional rearrangement of the P2 moiety. Compared to 1, the P2 quinoxaline in 12c, with a larger isopropyl substituent, packs less against the catalytic His57 and moves away from the binding surface toward the solvent (Figure 3B). This movement of the quinoxaline away from the catalytic His57 is not observed in the inhibitor complexes with smaller substituents at the 3-position (Figure 3A,C). Interestingly, this conformation is reminiscent of the conformation of 1 when bound to the A156T protease variant (PDB ID 3SUG) (Figure S2), where the larger threonine residue causes steric clash with the P2–P4 macrocycle.12,16 To accommodate the larger side chain in the A156T protease, inhibitor 1 undergoes a rearrangement resulting in the shift of the P2 quinoxaline moiety away from the binding surface toward the solvent, weakening the critical π–π interactions with the catalytic His57 (Figure S2A). This altered conformation of 1 results in dramatic potency losses against RASs at Ala156.12
Similar to the A156T-1 complex, the altered conformation of the 3-isopropylquinoxaline results in significantly reduced interactions with the catalytic His57. Any perturbation to the protease active site due to RASs may further reduce interactions with His57. Indeed, the 3-isopropylquinoxaline compounds exhibit reduced potency against WT relative to the parent compound 3 and are the most susceptible to resistant variants. These results suggest that modifications of the inhibitor scaffold that cause movement of the quinoxaline away from His57 toward the solvent are highly detrimental to potency, whereas movement of the quinoxaline away from His57 toward the S2 subsite residues has less of an effect on inhibitor potency when a smaller substituent is present at the 3-position of P2 quinoxaline.
The 3-chloroquinoxaline series exhibited an excellent potency profile against WT and resistant variants even with a shift of the P2 quinoxaline moiety toward the S2 subsite. Interestingly, comparison of the WT-12b and WT-12d structures (Figure 3C) did not reveal any noticeable difference in binding poses for the 3-methyl- and 3-chloro-quinoxaline analogues that could explain the disparity in inhibitory activity. The electronic effects of the chloro group appear to improve stacking interactions of the quinoxaline moiety with the catalytic residue His57, which are crucial for the binding of inhibitors with a P2 quinoxaline moiety. Thus, enhancing interactions with the catalytic triad residues by modifying the inhibitor P2 quinoxaline moiety is likely to improve the overall binding energy and potency profiles.
Acknowledgments
This work was supported by a grant from the National Institute of Allergy and Infectious Diseases of the NIH (R01 AI085051). A.N.M. was also supported by the National Institute of General Medical Sciences of the NIH (F31 GM119345). M.J. was supported by a Postdoctoral Fellowship from the Higher Education Commission (HEC) of Pakistan.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00150.
Tables S1–S2, Figures S1–S2, experimental procedures and characterization data for intermediates and final compounds, and details of protein expression, purification, X-ray crystallography, enzyme inhibition, and antiviral assays (PDF)
Accession Codes
The PDB accession codes for X-ray crystal structures of WT NS3/4A protease in complex with inhibitors 12b, 12c, and 12d are 6CVW, 6CVX, and 6CVY.
Author Present Address
§ Department of Chemistry, GC University Lahore, Katchery Road, Lahore 54000, Pakistan.
The authors declare no competing financial interest.
Supplementary Material
References
- World Health Organization (WHO). Hepatitis C, Fact Sheet (Updated October 2017). http://www.who.int/mediacentre/factsheets/fs164/en/ (March 12, 2018).
- Fried M. W.; Shiffman M. L.; Reddy K. R.; Smith C.; Marinos G.; Goncales F. L. Jr.; Haussinger D.; Diago M.; Carosi G.; Dhumeaux D.; Craxi A.; Lin A.; Hoffman J.; Yu J. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N. Engl. J. Med. 2002, 347, 975–982. 10.1056/NEJMoa020047. [DOI] [PubMed] [Google Scholar]
- Falade-Nwulia O.; Suarez-Cuervo C.; Nelson D. R.; Fried M. W.; Segal J. B.; Sulkowski M. S. Oral direct-acting agent therapy for hepatitis C virus infection: a systematic review. Ann. Intern. Med. 2017, 166, 637–648. 10.7326/M16-2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCauley J. A.; Rudd M. T. Hepatitis C virus NS3/4A protease inhibitors. Curr. Opin. Pharmacol. 2016, 30, 84–92. 10.1016/j.coph.2016.07.015. [DOI] [PubMed] [Google Scholar]
- Pilot-Matias T.; Tripathi R.; Cohen D.; Gaultier I.; Dekhtyar T.; Lu L.; Reisch T.; Irvin M.; Hopkins T.; Pithawalla R.; Middleton T.; Ng T.; McDaniel K.; Or Y. S.; Menon R.; Kempf D.; Molla A.; Collins C. In vitro and in vivo antiviral activity and resistance profile of the hepatitis C virus NS3/4A protease inhibitor ABT-450. Antimicrob. Agents Chemother. 2015, 59, 988–997. 10.1128/AAC.04227-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summa V.; Ludmerer S. W.; McCauley J. A.; Fandozzi C.; Burlein C.; Claudio G.; Coleman P. J.; DiMuzio J. M.; Ferrara M.; Di Filippo M.; Gates A. T.; Graham D. J.; Harper S.; Hazuda D. J.; McHale C.; Monteagudo E.; Pucci V.; Rowley M.; Rudd M. T.; Soriano A.; Stahlhut M. W.; Vacca J. P.; Olsen D. B.; Liverton N. J.; Carroll S. S. MK-5172, a selective inhibitor of hepatitis C virus NS3/4A protease with broad activity across genotypes and resistant variants. Antimicrob. Agents Chemother. 2012, 56, 4161–4167. 10.1128/AAC.00324-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwo P. Y.; Poordad F.; Asatryan A.; Wang S.; Wyles D. L.; Hassanein T.; Felizarta F.; Sulkowski M. S.; Gane E.; Maliakkal B.; Overcash J. S.; Gordon S. C.; Muir A. J.; Aguilar H.; Agarwal K.; Dore G. J.; Lin C. W.; Liu R.; Lovell S. S.; Ng T. I.; Kort J.; Mensa F. J. Glecaprevir and pibrentasvir yield high response rates in patients with HCV genotype 1–6 without cirrhosis. J. Hepatol. 2017, 67, 263–271. 10.1016/j.jhep.2017.03.039. [DOI] [PubMed] [Google Scholar]
- Bourliere M.; Gordon S. C.; Flamm S. L.; Cooper C. L.; Ramji A.; Tong M.; Ravendhran N.; Vierling J. M.; Tran T. T.; Pianko S.; Bansal M. B.; de Ledinghen V.; Hyland R. H.; Stamm L. M.; Dvory-Sobol H.; Svarovskaia E.; Zhang J.; Huang K. C.; Subramanian G. M.; Brainard D. M.; McHutchison J. G.; Verna E. C.; Buggisch P.; Landis C. S.; Younes Z. H.; Curry M. P.; Strasser S. I.; Schiff E. R.; Reddy K. R.; Manns M. P.; Kowdley K. V.; Zeuzem S. Polaris; Investigators, P.-. Sofosbuvir, velpatasvir, and voxilaprevir for previously treated HCV infection. N. Engl. J. Med. 2017, 376, 2134–2146. 10.1056/NEJMoa1613512. [DOI] [PubMed] [Google Scholar]
- Ng T. I.; Tripathi R.; Reisch T.; Lu L.; Middleton T.; Hopkins T. A.; Pithawalla R.; Irvin M.; Dekhtyar T.; Krishnan P.; Schnell G.; Beyer J.; McDaniel K. F.; Ma J.; Wang G.; Jiang L. J.; Or Y. S.; Kempf D.; Pilot-Matias T.; Collins C. In vitro antiviral activity and resistance profile of the next-generation hepatitis C virus NS3/4A protease inhibitor glecaprevir. Antimicrob. Agents Chemother. 2017, 62, e01620-17. 10.1128/AAC.01620-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlotsky J. M. Hepatitis C virus resistance to direct-acting antiviral drugs in interferon-free regimens. Gastroenterology 2016, 151, 70–86. 10.1053/j.gastro.2016.04.003. [DOI] [PubMed] [Google Scholar]
- Sarrazin C. The importance of resistance to direct antiviral drugs in HCV infection in clinical practice. J. Hepatol. 2016, 64, 486–504. 10.1016/j.jhep.2015.09.011. [DOI] [PubMed] [Google Scholar]
- Romano K. P.; Ali A.; Aydin C.; Soumana D.; Özen A.; Deveau L. M.; Silver C.; Cao H.; Newton A.; Petropoulos C. J.; Huang W.; Schiffer C. A. The molecular basis of drug resistance against hepatitis C virus NS3/4A protease inhibitors. PLoS Pathog. 2012, 8, e1002832. 10.1371/journal.ppat.1002832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali A.; Aydin C.; Gildemeister R.; Romano K. P.; Cao H.; Özen A.; Soumana D.; Newton A.; Petropoulos C. J.; Huang W.; Schiffer C. A. Evaluating the role of macrocycles in the susceptibility of hepatitis C virus NS3/4A protease inhibitors to drug resistance. ACS Chem. Biol. 2013, 8, 1469–1478. 10.1021/cb400100g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper S.; McCauley J. A.; Rudd M. T.; Ferrara M.; DiFilippo M.; Crescenzi B.; Koch U.; Petrocchi A.; Holloway M. K.; Butcher J. W.; Romano J. J.; Bush K. J.; Gilbert K. F.; McIntyre C. J.; Nguyen K. T.; Nizi E.; Carroll S. S.; Ludmerer S. W.; Burlein C.; DiMuzio J. M.; Graham D. J.; McHale C. M.; Stahlhut M. W.; Olsen D. B.; Monteagudo E.; Cianetti S.; Giuliano C.; Pucci V.; Trainor N.; Fandozzi C. M.; Rowley M.; Coleman P. J.; Vacca J. P.; Summa V.; Liverton N. J. Discovery of MK-5172, a macrocyclic hepatitis C virus NS3/4a protease inhibitor. ACS Med. Chem. Lett. 2012, 3, 332–336. 10.1021/ml300017p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawitz E.; Yang J. C.; Stamm L. M.; Taylor J. G.; Cheng G.; Brainard D. M.; Miller M. D.; Mo H.; Dvory-Sobol H. Characterization of HCV resistance from a 3-day monotherapy study of voxilaprevir, a novel pangenotypic NS3/4A protease inhibitor. Antiviral Ther. 2017, 10.3851/IMP3202. [DOI] [PubMed] [Google Scholar]
- Soumana D. I.; Kurt Yilmaz N.; Prachanronarong K. L.; Aydin C.; Ali A.; Schiffer C. A. Structural and thermodynamic effects of macrocyclization in HCV NS3/4A inhibitor MK-5172. ACS Chem. Biol. 2016, 11, 900–909. 10.1021/acschembio.5b00647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthew A. N.; Zephyr J.; Hill C. J.; Jahangir M.; Newton A.; Petropoulos C. J.; Huang W.; Kurt-Yilmaz N.; Schiffer C. A.; Ali A. Hepatitis C virus NS3/4A protease inhibitors incorporating flexible P2 quinoxalines target drug resistant viral variants. J. Med. Chem. 2017, 60, 5699–5716. 10.1021/acs.jmedchem.7b00426. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.