

Abstract
Streptococcus pneumoniae commonly resides asymptomatically in the nasopharyngeal (NP) cavity of healthy individuals but can cause life-threatening pulmonary and systemic infections, particularly in the elderly. NP colonization results in a robust immune response that protects against invasive infections. However, the duration, mechanism, and cellular component of such responses are poorly understood. In this study, we found that repeated NP exposure of mice to S. pneumoniae TIGR4 strain results in pneumococcal-specific Ab responses that protect against lethal lung challenge. Abs were necessary and sufficient for protection because Ab-deficient μMT mice did not develop postexposure protection, only becoming resistant to lung infection after transfer of immune sera from NP-exposed mice. T cells contributed to immunity at the time of NP exposure, but neither CD4+ nor CD8+ T cells were required. The protective activity was detectable 20 wk after exposure and was maintained in irradiated mice, suggesting involvement of long-lived Ab-secreting cells (ASC), which are radioresistant and secrete Abs for extended periods of time in the absence of T cells or persistent Ag. CD138+ bone marrow cells, likely corresponding to long-lived ASC, were sufficient to confer protection. NP exposure of aged mice failed to protect against subsequent lung infection despite eliciting a robust Ab response. Furthermore, transfer of CD138+ bone marrow cells or sera from NP-exposed old mice failed to protect naive young mice. These findings suggest that NP exposure elicits extended protection against pneumococcal lung infection by generating long-lived CD138+ ASC and that the protective efficacy of these responses declines with age.
Streptococcus pneumoniae (pneumococci) are a group of bacteria consisting of >90 capsular polysaccharides serotypes that cause infections ranging from asymptomatic colonization of the nasopharynx to invasive diseases such as bacteremia, meningitis, and pneumonia (1). Nasopharyngeal (NP) colonization (also known as carriage) occurs in ~40% of adults and as many as 100% of children in developing countries (2). Lung infection is the most common invasive form of pneumococcal disease, resulting in >1 million deaths each year worldwide (3). The elderly (>65 y old) are particularly susceptible to invasive pneumococcal infections and in the United States account for most hospitalizations and deaths due to this disease (4).
Ab-mediated responses against S. pneumoniae, particularly against the surface structures such as phosphorylcholine (PC), the polysaccharide capsule, as well as several proteins, contribute to immunological protection from invasive pneumococcal disease (5–8). Both T cell–dependent and T cell–independent Ab responses have been shown to confer long-lived protection against invasive pneumococcal infections (9–12). Traditionally, T cell-dependent responses, such as those elicited by the protein-conjugated pneumococcal capsular vaccine Prevnar (13), are more closely associated with long-lived responses. However, T cell–independent Ab responses, such as those elicited by the nonconjugated pneumococcal polysaccharide (PPS) vaccine Pneumovax, can also lead to long-term protection (9, 14). Aging leads to defects in both T cell–dependent and T cell–independent Ab production (15, 16), limiting the efficacy of the current vaccines (15, 17) and contributing to the susceptibility of the elderly to invasive pneumococcal infection.
Humoral immunity can be maintained over time by memory B cells that develop into Ab-secreting cells (ASC) upon re-encountering the Ag (18). However, a second memory compartment consisting of long-lived ASC resides in the bone marrow (19). Although it was previously thought that formation of long-lived ASC is T cell–dependent (20), it was recently demonstrated that T cell–independent Ags such as serotype 3 PPS also promote generation of these cells (12, 21). These long-lived ASC express the plasma cell marker CD138 and, unlike naive and memory B lymphocytes, are resistant to radiation. Furthermore, unlike traditional memory B cells, which remain quiescent until Ag reexposure, CD138+ long-lived ASC maintain Ab production independent of the continued stimulation by Ag (22–24). These cells are found in niches such as the bone marrow, and for maintenance require receptors such as CD93 (25), a protein detected on early B cells that is lacking on traditional memory B cells and found to be dispensable for B cell development and traditional germinal center Ab responses (25).
NP colonization elicits both T cell-dependent or T cell–independent Ab responses in mice and humans (5–7, 26–28), resulting in the recognition of both capsular polysaccharides and pneumococcal protein Ags. The precise nature of the response generated may depend on both duration of carriage and serotype of the bacterium (5, 7, 10, 27–31), but the incidence of NP colonization declines in adulthood, suggesting that successive episodes of S. pneumoniae colonization may be a natural means of inducing protective NP immune responses (26, 27, 32). In fact, pneumococcal colonization with S. pneumoniae serotype 6B results in protection against subsequent NP colonization when individuals are rechallenged with the same strain (7). Epidemiological evidence suggests that carriage is also associated with serotype-independent postcolonization immunity (33). Postcolonization protection from subsequent recolonization in mice depends not on Abs but on CD4+ T cells that produce IL-17 (6, 26, 34–38), which accelerates bacterial clearance after rechallenge by boosting neutrophil influx into the nasopharynx as well as their ability to clear pneumococci (39).
NP colonization also provides protection from invasive disease. Prior colonization protects mice from septicemia (6, 26, 27) and lung infection (5, 26) in a manner that requires Ab production (5, 27) and can be replicated by the transfer of immune sera from colonized human donors (7). Antipneumococcal Abs are thought to confer protection in part by increasing Ab-mediated phagocytosis by neutrophils (5, 27). However, the key parameters of these responses are still to be fully defined. For example, the role of T cells in the development of a protective response varies depending on the experimental system (5, 27). In addition, although humans are protected against reinfection up to a year after experimental carriage (7), and epidemiological data suggest that protection could be long-lived (33), very few investigations into the duration of postcolonization immunity have been performed.
In this study, we found that asymptomatic exposure of the nasopharynx of young, inbred mice to S. pneumoniae serotype 4 (TIGR4 strain) elicited robust, pneumococcal-specific Ab responses that were necessary and sufficient to confer significant protection against subsequent lung challenge for extended periods of time. The protective response did not require CD4+ T lymphocytes at the time of NP exposure, or at the time of lung challenge. Rather, radiation-resistant CD138+ bone marrow cells, likely representing long-lived ASC, conferred extended protection. Although exposure resulted in a robust Ab response in aged mice, this response was not sufficient to protect against lung infection, and protection mediated by CD138+ bone marrow cells was lost upon aging.
Materials and Methods
Mice
Young (~2-mo-old) and old (~20- to 22-mo-old) C57BL/6 male mice were purchased from The Jackson Laboratory and the National Institute on Aging colonies, respectively and housed for 3 wk at our facilities prior to performing experiments. CD8–/– mice and BALB/cByJ (BALB/c) wild-type controls were purchased from The Jackson Laboratory. Mice were housed in a specific-pathogen free facility at Tufts University, and all procedures were performed in accordance with Institutional Animal Care and Use Committee guidelines. All other animals were housed in a pathogen-free facility at the University of Massachusetts Medical School (UMMS) and maintained according to the standards and policies of the Institutional Animal Care and Use Committee. Mice were 8–10 wk old at the time of colonization and were sex matched in each experiment. Wild-type BALB/cByJ and C57BL/6 mice were purchased from The Jackson Laboratory and Charles River Laboratories, respectively. TCR β–chain–and TCR-δ-chain–deficient TCR βδ–/–, CD93–/–, μMT–/–, IgA–/–, MHC class II–/–, and Jh–/– mice in a C57BL/6 background were bred and maintained in our colony at UMMS (40). CD4–/– mice in a BALB/c background were courtesy of Dr. S. Swain at UMMS. Ab-deficient μMT–/– mice were originally obtained from Dr. K. Rajewsky at the Immune Disease Institute of Harvard Medical School. IgA–/– mice (41) were backcrossed to C57BL/6 and deposited at the University of Missouri Mutant Mouse Regional Resource Center by Dr. D.W. Metzger at the Albany Medical College. JH–/– mice in a C57BL/6 background were originally obtained from Dr. K. Rajewsky. CD93–/– mice were from Dr. A. Tenner at the University of California Irvine (originally from Dr. M. Botto of the Rheumatology Section at the Imperial College London, London, U.K.). In all experiments, mice were sex matched and in experiments other than those involving aged mice, the mice were also age matched. For survival experiments, at least five mice per group were included unless the specific experimental condition functioned as a control that was a repetition of previously published results.
Bacteria
S. pneumoniae TIGR4 strain (serotype 4) and Hungary strain (ATCC 700673; serotype 19A) were grown at 37°C in 5% CO2 in Todd-Hewitt broth (BD Biosciences) supplemented with 0.5% yeast extract and Oxyrase (Fisher Scientific) until the midexponential phase. Aliquots were then frozen at −80°C in the growth media with 25% (v/v) glycerol. Prior to use, bacterial aliquots were thawed on ice, washed once, and diluted in PBS. The titers were then confirmed by plating on tryptic soy agar plates supplemented with 5% sheep blood agar (Northeast Laboratory Services).
Animal infections
Intranasal inoculation.
Mice were restrained without anesthesia and given 10 μl (5 × 105 CFU) of bacteria equally distributed between the nostrils with a pipette. This procedure was repeated at weekly intervals for 3 wk to establish stable NP carriage. Naive mice received 10 μl of sterile PBS into the nares. One week after the third intranasal (i.n.) inoculation, all mice were treated with i.p. injections with 1 mg of rifampicin (Fitzgerald Industries International) for 2 consecutive days to clear the infection from the colonized mice. As described in Results, nearly all (>96%) mice that were capable of producing Abs were free of apparent illness after this protocol.
Intratracheal infection.
Three weeks following the last i.n. inoculation (2 wk after rifampicin treatment), mice were lightly anesthetized with isoflurane and 50 μl of bacteria was instilled directly to the trachea with the tongue pulled out to facilitate delivery of bacteria directly into the lungs as previously described (17). Mice were monitored closely after intratracheal (i.t.) infection and moribund mice were euthanized.
i.p. infection.
To model systemic spread of the bacteria, 102 CFU were injected into the peritoneum of mice.
i.p. immunization.
S. pneumoniae TIGR4 (107 CFU) was killed by heating at 65°C for 2 h and then suspended in 250 μl of 1 × PBS for i.p. injection into each mouse. Mice were i.p. immunized with two doses of heat-killed S. pneumoniae TIGR4, administered 2 wk apart. i.t. challenges were performed 2 wk after immunization.
Sample collection
Ten microliters of tail vein blood collected in 90 μl of anticoagulant (75 μl of 1 M sodium citrate, 50 μl of 1 M citric acid, and 12.375 ml of PBS pH was used for ELISAs and enumeration of bacterial loads. The lungs were aseptically removed and homogenized in sterile Todd-Hewitt broth using a tissue homogenizer. The bronchoalveolar lavage fluid (BALF) was obtained by washing the lungs with PBS via a needle attached to the end of polyethylene tubing (BD Biosciences) inserted into the trachea. For each sample, serial dilutions were plated on blood agar plates to enumerate CFU.
Adoptive transfer of serum or Ab
Three weeks after NP exposure, mice were sacrificed and blood was collected by cardiac puncture, incubated at 37°C for 1 h, and then spun for 8 min at 0.8 relative centrifugal force in a microfuge. The clear serum portion was carefully collected and pooled. Naive mice were given 250 or 400 μl of pooled sera by i.p. injection and challenged 24 h later. Anti-PC mAb (IgG) clone HPCG11 was a gift from Dr. A. Rothstein (UMMS) (42). Anti-PC mAb (IgA) clone TEPC 15 was purchased from Sigma-Aldrich. Abs were dialyzed against sterile 1× PBS before use, and mice were treated with 50 μl of anti-PC mAb or 50 μg of anti-PC mAb by i.p. injection 24 h prior to i.t. challenge.
Depletion of CD4+ and CD8+ T cells
At days −2, −1 and +1 relative to i.t. challenge, mice were treated with mAbs against CD4 clone GK1.5 (Bio × Cell) or CD8 clone 2.43 (Bio × Cell). Depletion of cells was confirmed by FACS analysis of peripheral blood lymphocytes. B220-FITC (clone RA3–6B2), CD4-PE (clone RM4–5), and CD8-PE (clone 53–5.8) Abs were purchased from BD Biosciences and used for staining and analysis at the UMMS and Tufts FACS core facilities. The data were analyzed using FlowJo software.
Sublethal irradiation
For testing the role of radiation-resistant cells, mice received total, whole-body irradiation at a sublethal dose of 6.0 Gy (600 rad, gamma ray source) 3 wk following NP exposure. Mice were administered a mixture of antibiotics (0.5 mg/ml each of neomycin sulfate and polymyxin sulfate) in the drinking water starting 2 d prior to irradiation, for 2 wk until 3 d before i.t. challenge. Challenges were performed 2 wk after irradiation.
Adoptive transfer of CD138+-enriched cells
Three weeks after NP exposure, mice were sacrificed and the bone marrows harvested. The cells were labeled with a biotinylated anti-CD138 Ab (clone 281–2), followed by incubation with streptavidin-conjugated microbeads (BD Biosciences), and the CD138+ fractions were positively selected by magnetic separation (BD IMagnet). Both CD138+-enriched and -depleted fractions were collected, resuspended in PBS, and 2 × 107 cells were transferred to mice via i.p. injections. Enrichment of cells was confirmed by FACS analysis of the enriched, depleted, and unfractionated bone marrow cells. CD138-allophycocyanin (clone 281–2) Ab and streptavidinallophycocyanin were used for staining and analysis at the UMMS and Tufts FACS core facilities. The data were analyzed using FlowJo software, and dead cells were excluded based on forward and side scatter. For bone marrow transplantation, C57BL/6 mice were treated once with 5-fluorouracil at 150 mg/kg body weight delivered i.p. 2 d before transfer. JH–/– mice receiving bone marrow cells were subjected to a total, whole-body dose of 5.0 Gy (500 rad, gamma ray source). Mice were challenged i.t. 2 wk following transfer.
ELISA
Nunc MaxiSorp flat-bottom 96-well plates were coated overnight at 4°C with 106 CFU per well of heat-killed whole TIGR4 pneumococci, 1 μg per well of PC-BSA (Biosearch Technologies), or PPS serotype 4 (PPS4; American Type Culture Collection) in 100 μl of coating buffer (0.05 M Na2CO3 [pH 9.0]). The next day, plates were washed three times with wash buffer (0.05% PBS-Tween) and blocked for 2 h in blocking buffer (0.05% PBS-Tween with 1% BSA). Samples (serum, lung homogenates, or BALF) were then added to the well for a 3-h incubation at room temperature. After washing, pneumococcal-specific Abs were detected by incubating with HRP-conjugated goat anti-mouse IgM, IgG, or IgA Abs (1:10,000 dilution) for 1 h at room temperature. After washing out the secondary Abs, 100 μl of SureBlue Reserve tetramethylbenzidine two-component microwell peroxidase substrate system (Kirkegaard and Perry Laboratories) was added to each well and plates were read at OD650 using a Spectramax 250 plate reader. Kinetic ELISAs were performed with readings taken every 30 s for 12 or 15 min. To confirm that the Ab assays were specifically measuring anti-PPS4 Abs, in pilot experiments and in experiments comparing Ab responses between young and old mice, the sera were preabsorbed for 30 min with a pneumococcal cell wall polysacharride mixture (CWPS-multi from Cedarlane) to neutralize Abs not specifically recognizing capsule prior to addition of the samples to the ELISA plate. We found no significant differences in levels of anti-PPS4 Abs in untreated versus preabsorbed sera (Supplemental Fig. 4). All Ab units were calculated as percentages of a control hyperimmune serum included in each ELISA plate. In the initial experiment to characterize the humoral immune response to NP exposure (Fig. 1), the Ab responses to immunization with heat-killed S. pneumoniae were included as a positive control. Naive unimmunized control values were subtracted from all data points presented. Hyperimmune sera were prepared from wild-type C57BL/6 mice that were i.n. inoculated with S. pneumoniae TIGR4 at weeks 0 and 1, i.p. immunized with heat-killed TIGR4 at week 3, and i.t. challenged with the same strain at week 5. Sera from these mice were collected, pooled, and frozen in aliquots at − 80°C.
FIGURE 1.
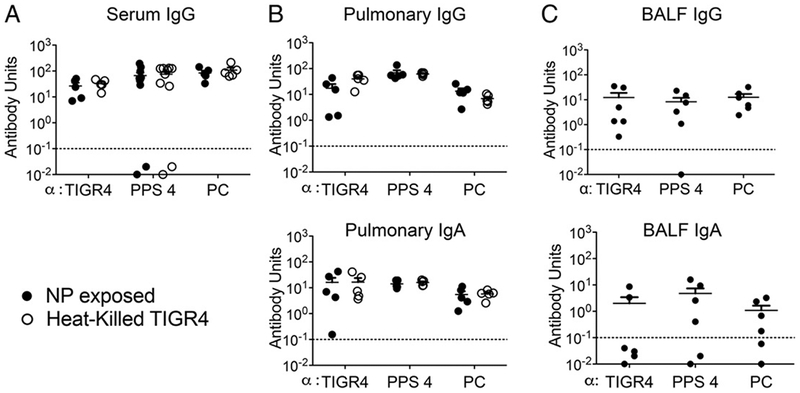
NP exposure elicits serum and pulmonary antipneumococcal Abs. Three weeks after a series of three i.n. inoculations of 8- to 10-wk-old C57BL/6 mice with S. pneumoniae TIGR4, IgG (top panels) or IgA (bottom panels) responses to heat-killed whole bacteria (TIGR4), purified PPS4 and PC from serum (A), lung homogenates (pulmonary) (B), and bronchoalveolar lavages (BALF) (C) were analyzed by ELISA (•). Ab units were calculated based on a hyperimmune standard included in each ELISA plate, and the background titers, determined by ELISA of serum or homogenates of uninfected control mice, were subtracted from the data (see Materials and Methods). The titers elicited by i.p. immunization with heat-killed pneumococci (heat-killed TIGR4; O) were included for comparison. Each point represents an individual mouse; dotted lines represent limit of detection, and bars represent the means. Data were pooled from n =10 (A), n = 5 (B), or n = 6 (C) mice per group.
Data analysis
Statistical significance between two groups was determined using the unpaired Student t test, and survival analyses were performed using the log-rank (Mantel–Cox) test in GraphPad Prism.
Results
B cells promote survival following i.n. inoculation
NP colonization with wild-type pneumococci elicits robust protective Ab responses (5, 7). To explore the immunological basis of this protection against pneumonia, we established in inbred C57BL/6 mice, a strain for which many mutant strains have been generated, a NP colonization model with serotype 4 S. pneumoniae TIGR4 strain that induces the development of a protective immune response against serious lung infection. The serotype 4 S. pneumoniae TIGR4 strain is a human bacteremia isolate that is highly invasive in mice (43) and can establish NP colonization after i.n. inoculation of wild-type C57BL/6 mice (34). We inoculated 5 × 105 CFU in 10 μl equally distributed into each of the two nares of C57BL/6 mice. We were able to recover an average of ~105 pneumococci from nasal washes or nasal tissue homogenates immediately after inoculation (Supplemental Fig. 1), suggesting that a significant portion of the inoculum is lost either due to host responses or drainage of a large portion of the inoculum to other sites (44). On average, approximately the same number of pneumococci was recovered 10 d after inoculation (Supplemental Fig. 1), but with greater mouse-to-mouse variation (Supplemental Fig. 1). These results suggest that i.n. inoculation with serotype 4 S. pneumoniae TIGR4 results in stable high-level colonization of the nasopharynx in most mice.
Although low-level aspiration of the bacteria into the lungs has been documented following NP inoculation (45), the lungs and blood of i.n. inoculated mice remained free of bacteria (Supplemental Fig. 1). Most wild-type mice survived following NP colonization, with only 10% dying during the course of 2 wk (Table I).
Table I.
B cells promote survival following i.n. inoculation
| Strain | Deficiency | Survivors/Total (% Survival) |
|---|---|---|
| Wild-type | None | 18/20 (90%) |
| rag1−/− | B and T cells | 6/16a (37%) |
| TCRβδ−/− | T cells | 15/15 (100%) |
| μMT−/− | Mature B cells | 7/11a (63%) |
| JH−/− | Mature B cells | 12/20a (60%) |
Survival was significantly different from wild-type mice as measured by the log-rank (Mantle–Cox) test.
To investigate the immune mechanisms required to restrict colonization to the nasopharynx, we monitored the survival of i.n. inoculated mice lacking specific immune cells. B and/or T lymphocytes were required to fully limit colonization to the nasopharynx, because only 37% of rag1–/– mice, which lack both cell types, survived S. pneumoniae i.n. inoculation (Table I). T cell-deficient TCR ßδ–/– mice uniformly survived i.n. inoculation. In contrast, the survival rates of μMT –/– and JH–/– mice (60–63%), which lack B cells, were significantly lower than for wild-type mice (90%), suggesting that mature B cells contributed to resistance. Nevertheless, μMT –/– and JH–/– mice survived at a significantly higher rate than mice deficient in both B and T cells, suggesting that in the absence of B cells, T cells may play a role in limiting colonization to the nasopharynx (Table I).
NP exposure elicits serum and pulmonary antipneumococcal Abs
Owing to the considerable mouse-to-mouse variation in colonization 10 d postinoculation (Supplemental Fig. 1), and owing to evidence that multiple rounds of i.n. inoculation can enhance protective responses (26, 46), we established NP carriage by inoculating mice i.n. three times at weekly intervals with low volume (10 μl) of S. pneumoniae as previously described (35). In the cumulative experiments described below, >96% of mice subjected to this inoculation protocol are free of discernable illness. To eliminate persistent Ag stimulation of immune cells by live pneumococci beyond this 3-wk interval (35, 46), 1 wk after the third i.n. inoculum mice were treated with rifampicin to terminate S. pneumoniae colonization. We termed this procedure “NP exposure.”
Given that B cells play an important role in limiting infection after i.n. inoculation, 2 wk after rifampicin treatment we measured pneumococcal-specific Abs in sera, lung homogenates, and BALF samples that should reflect both local and systemic Ab responses. Ag-specifc IgG and IgA ELISA (see Materials and Methods) were performed using either Ag heat-killed TIGR4 bacteria, purified PPS (the basis of a current antipneumococcal [Pneumovax] vaccine that elicits a protective T cell–independent Ab response) (47), or PC, a component of the pneumococcal cell wall (1). For serum and lung homogenate titers, sera from mice immunized i.p. with heat-killed bacteria, which are known to protect against lethal systemic infection (48), were included for comparison.
IgG Abs against all tested Ags were present in the sera and lung homogenates of NP-exposed mice at levels comparable to the robust responses elicited by i.p. immunization (Fig. 1A, 1B, top panel, compare filled versus open circles). Consistent with infection of the NP mucosal surface, we also found significant IgA titers in lung homogenates of i.n. inoculated mice (Fig. 1B, bottom panel). Analysis of BALF (Fig. 1C) confirmed high titers of IgG (top panel) and IgA (bottom panel) against the three pneumococcal Ag preparations, indicating that antipneumococcal Abs were also present in the airways of NP-exposed mice.
NP exposure results in protection against subsequent systemic challenge by S. pneumoniae
Given that Ab titers following i.n. inoculation were comparable to those resulting from immunization with heat-killed bacteria, which was previously shown to protect against i.p. challenge (48), we tested whether NP-exposed mice acquired immunity to systemic challenge. Mice that had been NP exposed, or naive controls, were challenged i.p. with 100 CFU of S. pneumoniae strain TIGR 4. Whereas naive mice uniformly died within 48 h, NP-exposed mice uniformly survived the challenge (Fig. 2A, left panel). To evaluate protection from a highly lethal pulmonary dose of S. pneumoniae TIGR4, mice were challenged i.t. with 107 CFU, which is a dose ~1000-fold greater than the LD50. Naive mice challenged with this dose uniformly died within 84 h, whereas 80% of the colonized mice survived this challenge (Fig. 2A, right panel). Our results demonstrate that NP exposure affords significant protection against subsequent i.p. or lung challenge.
FIGURE 2.
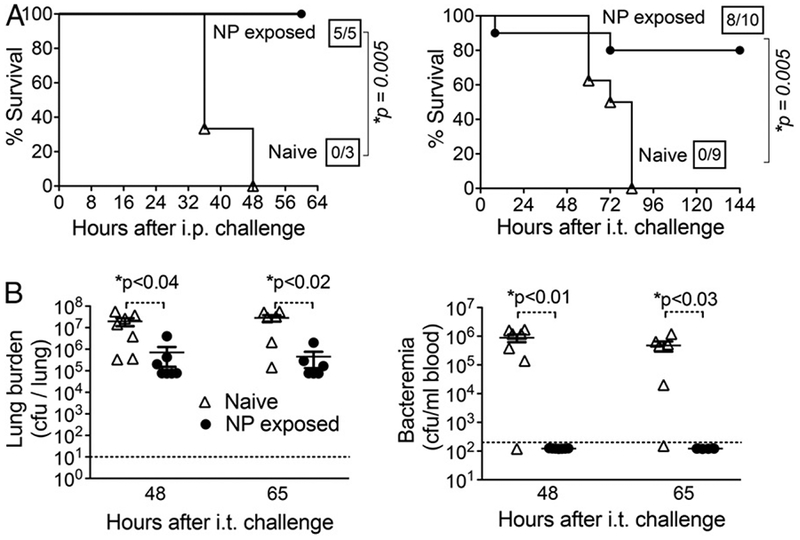
NP exposure results in protection against subsequent systemic challenge by S. pneumoniae. (A) After 3 wk of NP exposure to S. pneumoniae TIGR4, 8- to 10-wk-old C57BL/6 mice were challenged i.p. (left panel) with 100 CFU or i.t. with 1 × 107 CFU (i.t.; right panel) and survival was assessed. Boxed fractions denote survivors over total mice 2 wk after challenge. For i.t challenge, data pooled from two independent experiments are shown. (B) At the indicated times after i.t. challenge with 2 × 105 CFU of S. pneumoniae TIGR4, naive (n = 7) or NP-exposed (n = 6) mice were sacrificed and bacterial burdens in the blood (left) or lung homogenates (right) were determined. Dotted lines represent the limit of detection, and bars represent the mean. Points underneath the dotted lines illustrate that no bacteria were detected. For both panels, asterisks indicate significance calculated by a Student t test
To determine whether NP exposure resulted in lower bacterial numbers in the lungs and bloodstream, naive or NP-exposed mice were challenged i.t. with 2 × 105 CFU of S. pneumoniae TIGR4, a dose that results in >80% survival at 3 d postinfection in naive mice (49). At 48 and 65 h after challenge, mice were sacrificed and pneumococci in lung homogenates or sera were quantitated. Naive mice contained ~107 pneumococci per lung at both time points, whereas the bacterial load in the lungs of NP-exposed mice was ~15-fold lower (Fig. 2B, left panel). Notably, whereas nearly all naive mice suffered bacteremia approaching 106/ml at 48 and 65 h after i.t. challenge, and eventually succumbed to infection, blood of NP-exposed mice remained free of any detectable bacteria (Fig. 2B, right panel). Our data suggest that the significantly higher survival of NP-exposed mice is associated with the ability to both control bacterial burden in the lungs and prevent bacteremia.
A humoral immune response is essential to confer significant protection from pulmonary S. pneumoniae challenge following NP exposure
To determine whether the Ab response induced by NP exposure was required for protection against lethal systemic or lung challenge, wild-type or B cell–deficient μMT–/– or JH –/– mice were subjected to NP exposure. As indicated in Table I, ~60% of the B cell-deficient mice survived i.n. inoculation. We then subjected 6 μMT–/– or 10 JH –/– survivors to subsequent i.t. challenge with 2 × 106 CFU of S. pneumoniae TIGR4. Whereas previously colonized wild-type mice survived at the expected rate of >80%, only 1 of the 6 (17%) μMT mice and none of the 10 JH –/– mice survived lung infection, indicating that B cells are essential for protection from lung challenge after NP exposure (Fig. 3A).
FIGURE 3.
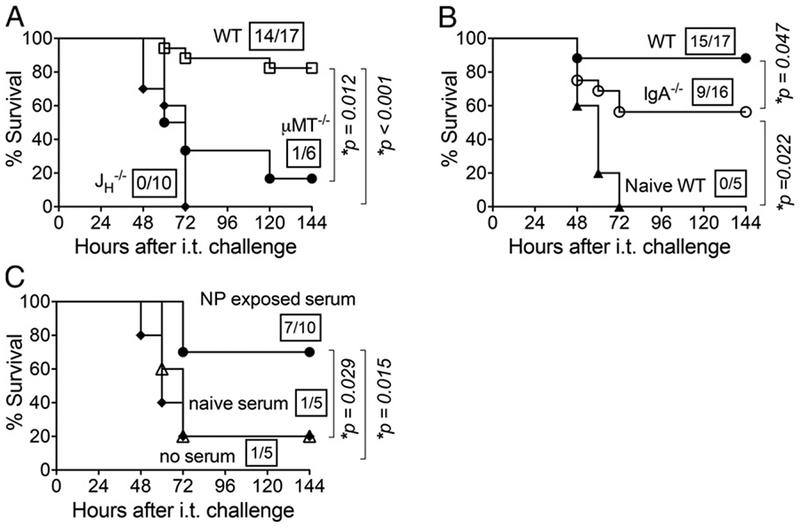
A humoral immune response is essential to confer significant protection from pulmonary S. pneumoniae challenge. (A) Survival of NP-exposed wild-type or B cell–deficient (i.e., JH–/– or μMT–/–) 8- to 10-wk-old C57BL/6 mice was assessed at various time points after i.t. challenge. (B) Survival was assessed at the indicated time points after i.t. challenge of 3-wk NP-exposed wild-type or IgA-deficient mice, or naive wild-type mice. (C) Naive, wild-type 8-wk-old C57BL/6 mice received no serum or 400 μl of pooled serum from indicated donors i.p., and then 24 h later were challenged i.t. with 2 × 106 CFU of S. pneumoniae TIGR4. Survival was assessed at the indicated time points. For all panels, boxed fractions denote survivors over total mice 2 wk after challenge; data pooled from two independent experiments are shown, and statistical significance was calculated by the log-rank (Mantel-Cox) test.
As IgA was present at high titers in the lungs of NP-exposed mice (Fig. 1), to assess the role of an IgA response elicited by NP exposure in subsequent immunity, we inoculated IgA-deficient mice i.n. weekly for 3 consecutive weeks (Fig. 3B). All of the mice survived the i.n. inoculations. When i.t. challenged with 2 × 106 CFU of S. pneumoniae TIGR4, 9 of 16 (56%) IgA-deficient mice survived challenge, a rate significantly (p = 0.047) lower than the 88% survival rate of previously colonized wild-type mice. Nevertheless, the 56% survival rate reflected significant (p = 0.02) protection compared with naive mice, which uniformly suffered a lethal infection. These results indicate that protection is mediated by IgA as well as IgA-independent factors.
To determine whether serum Abs were sufficient to promote protection of naive mice, pooled sera from NP-colonized mice, or as a control, from naive mice, were adoptively transferred by i.p. injection into naive wild-type mice. The recipient mice were then subjected to i.t. challenge with 2 × 106 CFU of S. pneumoniae TIGR4. The survival rate among mice receiving control sera from naive mice was 20%, identical to that of mice that received no serum (Fig. 3C). In contrast, sera from NP-exposed mice conferred survival of 70% of recipients (p = 0.014). These results indicate that Abs induced during NP exposure provide a degree of protection against subsequent pulmonary infection.
NP exposure induces cross-protection
The sera of NP-exposed mice contain high IgG and IgA titers against PC, a structural component of pneumococci irrespective of capsular serotype (Fig. 1) (1). Anti-PC Abs isolated from human sera were found to provide protection from i.p. challenge against S. pneumoniae serotype 6 in mice (8). Therefore, we tested whether NP exposure–induced protection from pulmonary challenge can be achieved when challenged with a heterologous strain. Mice that had been exposed to S. pneumoniae TIGR4 were challenged i.t. with 107 CFU of serotype 19A strain Hungary. By 48 h after challenge, the average bacterial burden in the lungs of NP-exposed mice (filled circles) was ~30-fold (p = 0.009) and 15-fold (p = 0.048) lower than their naive counterparts (open triangles) at 48 and 72 h postinfection, respectively (Fig. 4A).
FIGURE 4.
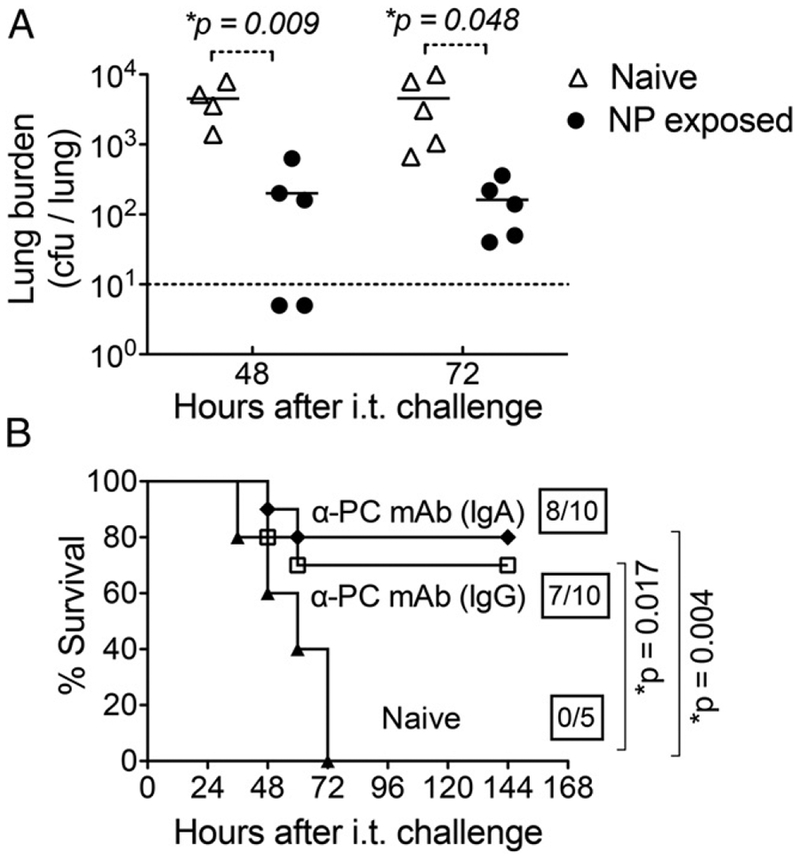
NP exposure induces protection independent of capsular serotype, and Abs to PC confer protection from pulmonary challenge by S. pneumoniae. (A) Eight- to ten-week-old C57BL/6 wild-type mice were NP exposed to S. pneumoniae TIGR4 strain and challenged i.t. with 107 CFU of serotype 19A strain Hungary. S. pneumoniae in lung homogenates prepared at 48 and 72 h after i.t. challenge were enumerated by plating for viable counts. Dotted line represents limit of detection. Statistical significance was determined by a Student t test and asterisks indicate p < 0.05. (B) Naive 8- to 10-wk-old C57BL/6 wild-type mice were given 50 μg of purified Abs against PC (anti-PC IgA, ●; anti-PC IgG, ☐) via i.p. injection. Mice were challenged i.t. with 2 × 106 CFU of S. pneumoniae TIGR4 24 h later. Survival was assessed at the indicated time points after challenge and compared with naive controls (▴). Boxed fractions denote survivors over total mice 2 wk postchallenge, and statistical significance determined by the log-rank (Mantel–Cox) test is indicated.
To test whether anti-PC Abs were sufficient to protect against i.t. challenge, naive mice were treated with 50 μg of monoclonal IgA or IgG anti-PC Abs 24 h prior to i.t. challenge [Fig. 4B, “α-PC-mAb (IgA)” and “α-PC-mAb (IgG)”]. Mice that received IgG or IgA anti-PC by i.p. injection (Fig. 4B, open squares) had a survival rate of 70 and 80%, respectively, significantly (p = 0.017) higher than for untreated mice, which uniformly suffered lethal infection (Fig. 4B, filled triangles). These results suggest that postcolonization protection against lung infection could be mediated by anti-PC Abs that recognize all pneumococci independent of serotype.
T cells play a role in antipneumococcal Ab responses following NP exposure and subsequent protection from i.t. challenge
We next wanted to determine the cellular components required to mount the protective Ab-mediated response after NP exposure. T cell–independent responses are sufficient for protection against systemic infections following immunization with PPS (11, 12). As described in Table I, we found that prevention of lethal infection after i.n. inoculation also did not require T cells. We further confirmed that T cell–deficient mice were completely protected from i.p. challenge after systemic immunization with heat-killed whole S. pneumoniae TIGR4 (Supplemental Fig. 2). In analyzing the immune response to NP exposure, we found that NP-exposed TCR ßxδ–/– mice displayed slightly diminished Ab IgG (Fig. 5A, top) and IgA (Fig. 5A, bottom) responses against PPS4 and PC compared with wild-type mice. IgG and IgA responses against whole bacteria (TIGR4) were also lower in TCR ßxδ–/– mice compared with wild-type mice, although the difference did not reach statistical significance (Fig. 5A). To determine whether this muted IgG and IgA response corresponded to a lower level of protection from pulmonary challenge, we infected these mice i.t. with 2 × 106 CFU of S. pneumoniae TIGR4. Following i.t. challenge, 14 of 17 (83%) NP-exposed wild-type mice and 1 of 10 naive wild-type mice survived (Fig. 5B), as expected from our previous findings. Only 6 of 15 (40%) NP-exposed T cell–deficient mice survived i.t. challenge (Fig. 5B), a rate that was significantly (p = 0.022) lower than that of colonized wild-type mice and statistically indistinguishable from that of naive mice. These results suggest that full protection from pulmonary challenge following NP exposure depends on T lymphocytes.
FIGURE 5.
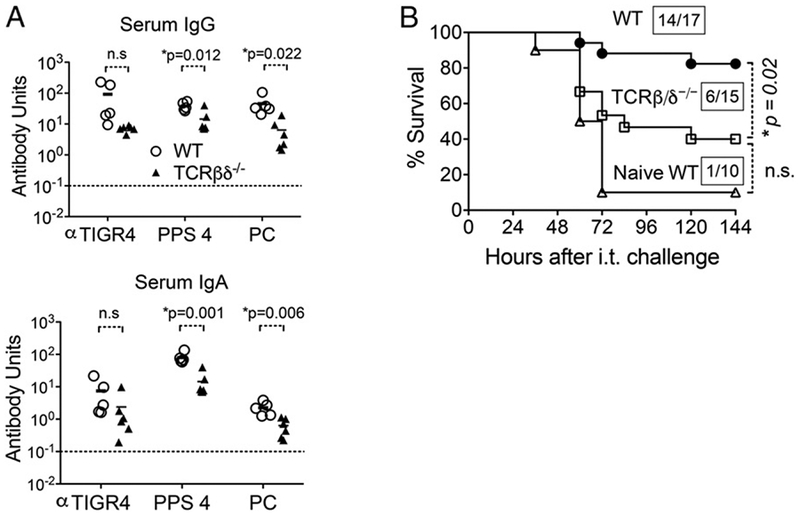
T cells contribute to Ab production following i.n. exposure and protection against subsequent i.t. challenge. (A) Three weeks after NP exposure of 8- to 10-wk-old wild-type or TCRβxδ–/– mice with S. pneumoniae TIGR4, IgG (top) and IgA (bottom) Abs against indicated Ags were titered in the sera of wild-type (○) or T cell–deficient (▴) mice. Ab units were calculated based on a hyperimmune standard included in each ELISA plate, and the background titers, determined by ELISA of serum of naive mice, were subtracted from the data (see Materials and Methods). The indicated p values were determined using a Student t test. Asterisks represent statistical significance (p < 0.05). Dotted lines represent limit of detection. (B) Three weeks after NP exposure, wild-type and T cell–deficient mice (TCRβxδ –/– ) were challenged i.t. with 2 × 106 CFU of S. pneumoniae TIGR4. Survival was assessed at the indicated time points after challenge. Fractions denote survivors 2 wk after challenge over the total number of mice, and asterisks indicate statistical significance (p < 0.05) by the log-rank (Mantel–Cox) test. n.s., not significant.
CD4+ T cells were shown to be critical in preventing lung infection and recolonization of the nasopharynx after NP colonization (5, 34), but whether these cells are absolutely required for the development of immunity at the time of lung challenge is unclear (5, 27). We found that 14 of 15 (93%) mice immunodepleted of 98% of their CD4+ lymphocytes just prior to challenge (data not shown) survived lung infection with 2 × 106 S. pneumoniae TIGR4 (Fig. 6A), indicating that CD4+ cells were not essential effectors at the time of challenge.
FIGURE 6.
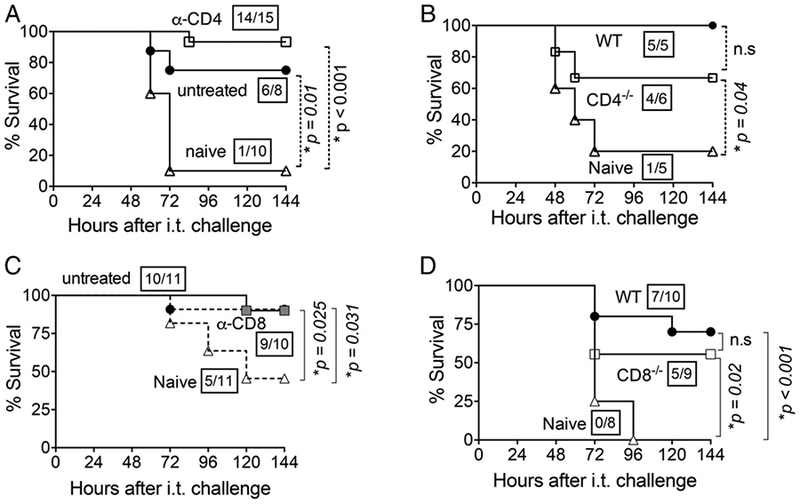
CD4+ and CD8+ T cells are individually dispensable for NP exposure-mediated protection. (A) Three weeks after NP exposure of 8- to 10-wk-old C57BL/6 mice, they were treated with 0.5 μg of GK1.5 anti-CD4 mAb at days −2, −1, and +1 relative to i.t. challenge to deplete 98% of CD4+ T cells (data not shown). Survival of CD4-depleted mice (α-CD4; □), untreated NP-exposed C57BL/6 mice (●), and naive C57BL/6 controls (Δ) was assessed serially after i.t. challenge with 2 × 106 CFU of S. pneumoniae TIGR4. (B) Survival of NP-exposed CD4-deficient BALB/c mice (□), NP-exposed wild-type (WT; ●), or naive wild-type mice (naive; Δ) was assessed after i.t. challenge with 2 × 106 CFU of S. pneumoniae TIGR4. (C) Three weeks after NP exposure, wild-type BALB/c mice were treated with 0.5 μg of clone 2.43, an mAb against CD8, at days −2, −1, and +1 relative to i.t. challenge to deplete 98% of CD8+ T cells (data not shown). Survival of CD8-depleted mice (α-CD8; ■) was assessed at indicated time points after i.t. challenge with 2 × 105 CFU of S. pneumoniae TIGR4 and compared with NP-exposed, nondepleted mice (●) and naive controls (Δ). (D) Three weeks after NP exposure, CD8-deficient mice in BALB/c background (CD8–/–; □) and wild-type BALB/c mice (WT; ●) or naive BALB/c wild-type mice (naive; Δ) were i.t. challenged with 1 × 106 CFU of S. pneumoniae TIGR4. Survival was assessed at the indicated time points after challenge and compared with naive controls (□). For (A)–(D), fractions denote survivors 2 wk after challenge over the total number of mice, and asterisks indicate statistical significance (p < 0.05) by the log-rank (Mantel–Cox) test. Data pooled from two independent experiments are shown. n.s., not significant.
To determine whether these cells were required for the development of postexposure protection, we used CD4–/– mice on a BALB/c background . We first established that, similar to wild-type C57BL/6 mice, NP colonization of wild-type BALB/c mice fully protected mice from subsequent i.t. challenge, whereas only a small fraction (20%) of naive mice survived the challenge (Fig. 6B). Eight of 11 (73%) NP-exposed, CD4-deficient BALB/c mice survived pulmonary challenge, a rate that was significantly (p = 0.04) higher than the (20%) rate for naive mice (Fig. 6B) and not significantly different from the (100%) survival rate observed for wild-type BALB/c mice. We conclude that although CD4 T cells may contribute to the humoral immune response after NP exposure, they are not absolutely required for protection against lung infection, either during initial colonization or subsequent lung infection.
CD8+ T cells potentially promote Ab production against PPS (50). To determine whether CD8+ T cells were required for full protection after NP exposure, we immunodepleted 98% CD8+ T cells of wild-type BALB/c mice (data not shown) prior to i.t. challenge with 2 × 105 CFU of S. pneumoniae TIGR4. CD8+ T cell-depleted mice (Fig. 6C, “α-CD8”) displayed similar survival following i.t. challenge (90% survival; 9 out of 10) as for untreated mice (90.9% survival; 10 out of 11). Survival of these depleted mice was significantly different from those of naive controls, suggesting that CD8+ T cells were not absolutely indispensable for protection following NP exposure at the time of subsequent lung challenge.
To determine whether CD8+ T cells were required for protection at the time of NP exposure, we NP exposed CD8–/– mice generated on a BALB/c background with S. pneumoniae TIGR4 and subsequently challenged them i.t. with 1 × 106 CFU of the same strain. Five of nine (56%) CD8–/– BALB/c mice survived pulmonary challenge at this dose, a rate that was slightly but not significantly lower than the (70%) survival rate of wild-type BALB/c mice (Fig. 6D), and was significantly (p = 0.02) higher than the (0%) rate for naive mice (Fig. 6D). Taken together, these findings suggest that T cells are required for full protection, but neither CD4+ nor CD8+ T cells could be demonstrated to be absolutely essential for significant protection following NP exposure.
NP exposure induces extended protection from lethal lung challenge
In a human carriage model, NP colonization induced protection from recolonization for up to a year (7). We found that the levels of pneumococcal-specific IgG (Fig. 7A, left panel) and IgA (right panel) Abs were maintained at comparable levels for 10 and 18 wk after NP exposure of wild-type C57BL/6 mice. To test whether the presence of an extended Ab response offered protection, naive or NP-exposed mice were challenged i.t. with 2 × 106 CFU of S. pneumoniae TIGR4 at 10 or 20 wk after colonization. First, all naive mice suffered lethal infection, but those challenged at 20 wk after exposure succumbed significantly (p = 0.0025) earlier than did naive mice challenged at 10 wk (Fig. 7C versus Fig. 7B), indicating that this 10-wk difference in challenge timing is associated with diminished resistance to pneumococcal infection. Second, 70 and 33% of NP-colonized mice survived i.t. pulmonary challenge at 10 and 20 wk after exposure, respectively (Fig. 7B, 7C), rates significantly higher than the (0%) survival rate of naive mice, suggesting that the elevated Ab titers provide at least partial protection at 10 and 20 wk after exposure.
FIGURE 7.
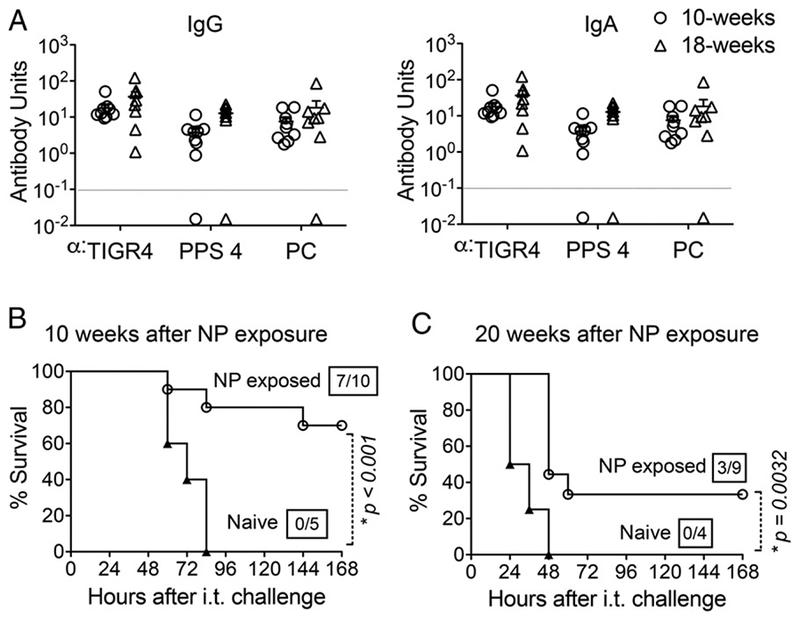
Abs elicited by i.n. exposure confer extended protection against i.t. challenge. (A) At10 and 18 wk after NP exposure of 8-wk-old C57BL/6 mice to S. pneumoniae TIGR4, IgG (left panel) and IgA (right panel) Abs against indicated Ags were measured in sera. Each point represents an individual mouse, and dotted lines indicate the limit of detection. (B) Ten weeks and (C) 20 wk after NP exposure, NP-exposed (○) or naive mice (a) were i.t. challenged with 2 × 106 CFU of S. pneumoniae TIGR4 and survival was assessed at indicated times after i.t. challenge. Fractions denote survivors 2 wk after challenge over the total number of mice. The log-rank (Mantel–Cox) test was used to calculate p values. Asterisks indicate p < 0.05.
Long-lived CD138+ Ab-producing bone marrow cells play a significant role in exposure-induced protection against i.t. challenge
The sustained Abs detected after eradication of NP exposure by rifampicin treatment (Fig. 7) (46) indicates that the long-lived Ab response described above is unlikely to be the result of continuous Ag restimulation of memory B cells. To assess the role of quiescent memory B cells in long-lived protection, we subjected naive or NP-exposed mice to sublethal whole-body irradiation, which results in fewer bone marrow T and B cells and compromises the ability of quiescent memory B cells to differentiate into Ab-producing plasma cells (12). Upon i.t. challenge with 2 × 106 bacteria 2 wk after irradiation, only one of five (20%) of naive mice survived (Fig. 8A). In contrast, 11 out of 18 (62%) previously NP-exposed and irradiated mice survived i.t. challenge, a frequency that was statistically (p = 0.02) different from that of naive mice and indistinguishable from the survival rate (78%) of control nonirradiated NP-exposed mice (Fig. 8A). This suggests that quiescent memory cells are not required for protection and instead implicate long-lived radioresistant cells in this process.
FIGURE 8.
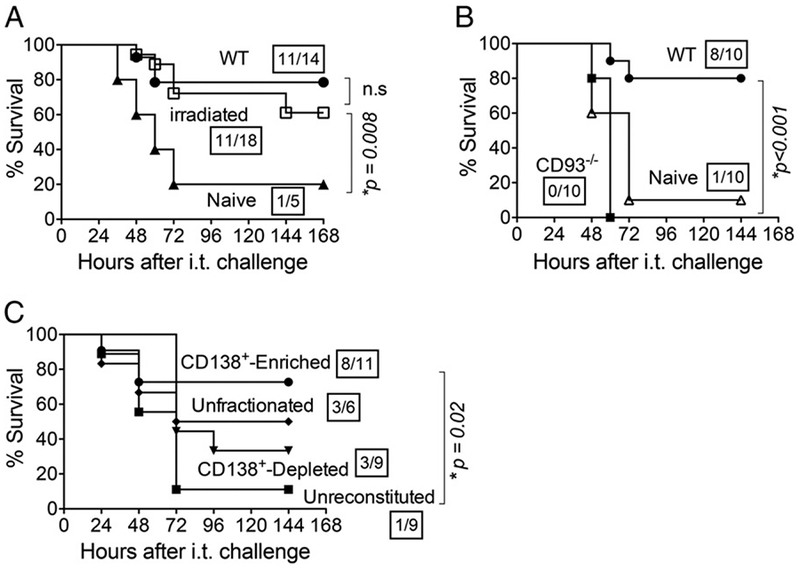
Long-lived CD138+ ASC mediate protection against pulmonary challenge. (A) Three weeks after NP exposure 8- to 10-wk-old C56BL/6 mice to S. pneumoniae TIGR4, mice were exposed to nonlethal whole-body radiation (600 rad). Two weeks after irradiation, mice were challenged i.t. with 2 × 106 CFU of S. pneumoniae TIGR4. Survival was assessed at the indicated time points after i.t. challenge. (B) Three weeks after NP exposure, CD93-deficient mice (CD93–/–; ■) and wild-type mice (●) were i.t. challenged with 2 × 106 CFU of S. pneumoniae TIGR4. Survival was assessed at the indicated time points after challenge and compared with naive controls (Δ). (C) Three weeks after NP exposure, unfractionated bone marrow cells or those enriched or depleted for CD138+ cells were transferred into JH–/– mice exposed to nonlethal whole-body radiation (500 rad). Mice were then challenged i.t. with a lethal dose (2 × 106 CFU) of S. pneumoniae TIGR4. Survival was assessed at the indicated time points after challenge. For (A)–(C), fractions denote survivors 2 wk after challenge over the total number of mice. Asterisks indicate a statistical significance (p < 0.05) by the log-rank (Mantel–Cox) test. Data were pooled from two independent experiments.
Long-lived CD138+ bone marrow ASC are radioresistant due to the fact that they, unlike quiescent memory cells, are not obligated to proliferate to confer effector function (22–24). Notably, CD93 is expressed at high levels on these cells, but not on germinal center or memory B cells, and it is required for the maintenance of long-lived CD138+ ASC (25). NP-exposed CD93–/– mice uniformly survived, indicating that CD93 is not required to prevent the development of lethal systemic infection from NP inoculation (data not shown). To investigate the potential role of these cells in postexposure immunity to S. pneumoniae, these NP-exposed CD93 mice were infected i.t. with 2 × 10 S. pneumoniae TIGR4. Strikingly, none of the 10 NP-exposed CD93–/– mice survived beyond 60 h after lung challenge (Fig. 8B), demonstrating that protection induced by NP colonization is CD93-dependent.
To more directly test the suggestion that CD138+ ASC provide protection, B cell–deficient JH–/– mice were exposed to sublethal whole-body irradiation and reconstituted with ~10 cells of unfractionated bone marrow, or bone marrow fractions either enriched for or depleted of CD138+ cells. Two weeks after reconstitution, mice were challenged i.t. with 2 × 106 CFU of S. pneumoniae TIGR4. Irradiated mice reconstituted with unfractionated bone marrow cells displayed more than twice the survival rate of unreconstituted controls (i.e., 50% versus 11%), although this increase did not reach statistical significance (Fig. 8C). Eight out of 11 (73%) mice reconstituted with bone marrow cells enriched for CD138+ cells survived i.t. challenge, a rate significantly (p < 0.02) higher than the 11% rate of unreconstituted controls (Fig. 8C). In contrast, only three of nine (33%) mice reconstituted with CD138-depleted bone marrow cells survived, a rate statistically indistinguishable from that of unreconstituted controls (Fig. 8C). These data suggest an important role for radioresistant CD138+ bone marrow cells in exposure-induced protection against pneumococcal lung challenge.
NP exposure of aged mice does not confer significant protection from lung challenge
The elderly are at an increased risk of invasive pneumococcal infections, suggesting that the protection afforded by NP exposure declines with age (4, 51–53). To investigate this, the Ab responses of young (2-mo-old) or old (22- to 24-mo-old) C57BL/6 male mice were measured after NP exposure to S. pneumoniae TIGR4 promoted by the three-round inoculation protocol. NP-exposed old mice generated IgG Ab titers against whole S. pneumoniae, capsule, or PC that were indistinguishable from those of young controls (Fig. 9A), and none of the old mice succumbed to disease following NP exposure (data not shown), indicating that the immune mechanisms required to restrict lethal disease following colonization of the nasopharynx are intact in aged hosts.
FIGURE 9.
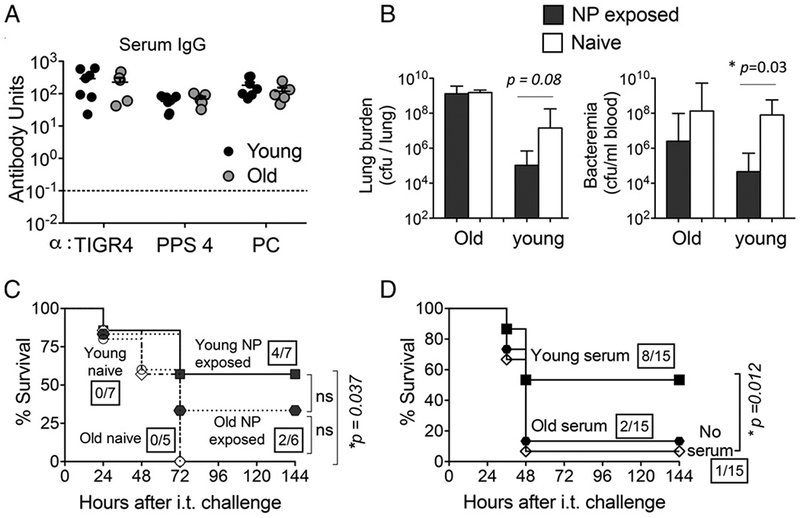
I.n. exposure is not protective against subsequent S. pneumoniae challenge in old mice. Young (2-mo-old) and old (20- to 22-mo-old) C57BL/6 male mice were NP exposed to S. pneumoniae TIGR4, and (A) 3 wk later sera IgG levels against whole bacteria (TIGR4), PC, and purified PPS4 were assessed. Ab units were calculated based on a hyperimmune standard included in each ELISA plate, and the background titers, determined by ELISA of serum of naive mice within each group, were subtracted from the data (see Materials and Methods). Dotted lines represent limit of detection. Data were pooled from n = 7 young (black circles) and n = 5 old (gray circles) mice per group. (B) Mice were then challenged i.t. with 1 × 106 CFU of S. pneumoniae TIGR4 and bacterial numbers in the blood and lungs of NP-exposed (filled bars) and naive (open bars) young and old mice were determined 48 h postchallenge. Bars represent mean ± SEM, and significant differences, determined by Student t test, are indicated by asterisks. Representative data from one of two independent experiments with n = 4 mice per group are shown. (C) Survival over time of young naive (O), young NP-exposed (■), old naive (◇)), and old NP-exposed (●) mice following i.t. challenge was also determined. (D) Naive, wild-type male mice received no serum or 250 μl of pooled serum from NP colonized young or old donors i.p., prior to challenge with S. pneumoniae TIGR4. Data were pooled from three independent experiments with a total of 15 mice per group. Boxed fractions denote survivors over total mice 2 wk after challenge and statistical significance (*p < 0.05) was calculated by the log-rank (Mantel–Cox) test. n.s., not significant.
To determine whether the Ab responses mounted in an aged host protect from pulmonary infection, the NP-exposed or naive (male) mice were challenged i.t. with 1 × 106 CFU of S. pneumoniae TIGR4. As observed with young female mice (Fig. 2), prior exposure boosted the resistance of young male mice against invasive infection (Fig. 9). Whereas the bacterial burden of naive young male mice was ~107 CFU of S. pneumoniae per lung and 108 CFU per milliliter of blood, the burden of NP-exposed mice was ~ 100-fold and ~1000-fold lower, respectively. Notably, whereas all young naive mice succumbed to the infection by day 3 following i. t. inoculation, four out of seven (57%) NP-exposed young mice survived this challenge (p = 0.037; Fig. 9C).
In contrast, prior NP exposure did not reduce the burden of infection in old mice after i.t challenge with 1 × 106 CFU of S. pneumoniae TIGR4 (Fig. 9B). Aged naive mice and aged NP-exposed mice had equal numbers of pneumococci per lung (Fig. 9B, left panel), and although aged NP-exposed mice had 12-fold lower levels of bacteremia (Fig. 9B, right panel) and a higher rate of survival (33% versus 0%) compared with aged naïve mice, these differences were not statistically significant. Thus, although it is possible that NP exposure confers some degree of resistance of lung challenge in aged mice, significant protection was not demonstrated.
Although the amount of Abs produced by aged mice following NP exposure was indistinguishable from the amount produced by young mice (Fig. 9A), anti-PC Abs from aged BALB/c mice were shown to be inherently less protective than those from young mice (54). To assess the protective activity of the Abs generated in our aged C57BL/6 mice following NP exposure with pneumococci, naive young mice were injected i.p. with pooled sera from NP-exposed young or old mice and challenge i.t. with S. pneumoniae TIGR4. Most (8 of 15) mice receiving sera from young NP-exposed mice survived challenge, reflecting significant (p = 0.012) protection compared with the 7% (1 of 15) survival rate for control mice that received no serum. In contrast, only 13% (2 of 15) of mice receiving sera from old mice survived infection, indicating that Abs induced during NP exposure of old mice are not sufficient to provide protection against subsequent pulmonary infection (Fig. 9D).
CD138+ bone marrow cells of NP-exposed aged mice are not sufficient to confer significant protection against lung challenge
To explore whether defects in the CD138+ cell compartment contribute to the observed age-associated decline in protective immunity after NP exposure, we compared the percentage of CD138+ bone marrow cells in NP-exposed young and old male mice. We found that the percentage of CD138-expressing cells in the bone marrow of old mice was significantly lower than in the bone marrow of young mice (19.2% versus 42.45%; p = 0.023; Fig. 10A). To test whether bone marrow CD138+ cells of old mice generated after NP exposure confer protective immunity, young naive C57BL/6 male mice were exposed to 5-fluorouracil to deplete endogenous cells and were reconstituted with ~107 CD138+-enriched bone marrow cells from either old or young NP-exposed mice. We determined the percentage of CD138+ cells in each enriched fraction and found that although CD138+ cells comprised a slightly lower fraction of cells in old versus young mice (i.e., 61.5% versus 69.3%), the difference was not statistically significant (Supplemental Fig. 3B).
FIGURE 10.
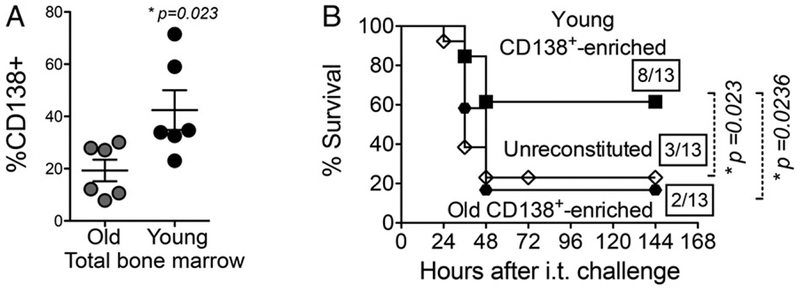
CD138+ cells from NP-exposed aged hosts are not sufficient for protection of naive mice against pulmonary challenge. (A) Young (2-mo-old) and old (20- to 22-mo-old) C57BL/6 male mice were NP exposed to S. pneumoniae TIGR4, the bone marrow was harvested, and the percentage od CD138+ cells was measured by flow cytometry. Data shown are pooled from two experiments with a total of six mice per group. Asterisks indicate p < 0.05. (B) Three weeks after NP exposure, bone marrow cells from young and old mice enriched for CD138 were transferred into mice pretreated with 5-fluorouracil. Mice were then challenged i.t. with 2.5 × 10 CFU of S. pneumoniae TIGR4. Survival was assessed at the indicated time points, and fractions denote survivors over the total number of mice. Asterisks indicate a statistical significance (p < 0.05) by the log-rank (Mantel–Cox) test. Data were pooled from three independent experiments.
In pilot infection experiments, we found that 5-fluorouracil-treated mice were exquisitely susceptible to pneumococcal challenge, reflected by 100-fold higher bacteremia compared with naive mice and uniform lethality within 36 h following challenge with 2 × 105 CFU of S. pneumoniae TIGR4 (data not shown). To evaluate potential protective activity of CD138+-enriched fractions, mice were challenged i.t. with 8-fold fewer (2.5 × 104 CFU) pneumococci. Using this challenge dose, mice reconstituted with CD138+-enriched fractions from young NP-exposed male mice displayed more than twice the survival rate of unreconstituted controls (61% versus 23%; p = 0.023; Fig. 10B), consistent with our finding using irradiated mice. Furthermore, transfer of CD138+-enriched bone marrow fractions from young naive mice failed to enhance survival (Supplemental Fig. 3A), indicating that NP exposure with pneumococcus triggers a specific protective CD138+ memory response. Only 4 of 13 (30%) mice reconstituted with CD138+-depleted bone marrow cells survived, a rate statistically indistinguishable from that of unreconstituted controls (Supplemental Fig. 3A). Importantly, survival of mice reconstituted with CD138+-enriched fractions from old NP-exposed male mice was not different from unreconstituted controls, displaying only 15% survival, a rate significantly (p = 0.0236) lower than those receiving CD138+-enriched cells from young mice (Fig. 10B). These data suggest that the ability of CD138+ bone marrow cells to induce exposure-driven protection against pneumococcal lung challenge wanes with age.
Discussion
Experimental pneumococcal carriage elicits immune responses, including a strong Ab response, that protect against both recolonization of the nasopharynx by S. pneumoniae (6, 26, 34) and more invasive infections such as pneumonia and bacteremia (5, 6, 26, 27). Furthermore, passive transfer of sera from humans NP colonized with S. pneumoniae confers upon mice protection from pulmonary challenge by a different serotype (7). In this study, we show in a murine model that repeated NP exposure to S. pneumoniae triggers a long-lived Ab response, mediating extended protection against lethal lung challenge.
NP colonization of mice resulted in the production of IgA and IgG Abs that, in addition to recognizing whole bacteria and capsular polysaccharide, are also directed against PC, a conserved cell wall component and potential cross-protective Ag (55). Anti-PC Abs are known to be protective against invasive pneumococcal infections (56, 57). In this study, we also found that anti-PC mAb provided passive immunity to lung challenge by S. pneumoniae TIGR4, and antisera from mice NP exposed to strain TIGR4 provided protection against a heterologous serotype 19A S. pneumoniae strain. Our findings are thus consistent with previous results suggesting that S. pneumoniae carriage in mice can induce Ab responses protective against pneumococcal strains of different capsular serotypes (26).
In contrast to protection induced by immunization with whole killed bacteria, we found that protection induced by NP exposure was partially dependent on T lymphocytes. In some murine NP-colonized models, protection from recolonization at the same site depended on CD4+ T cells producing IL-17 that boosted neutrophil influx to the site of infection, accelerating bacterial clearance at rechallenge (34, 37, 39). In models of NP-induced protection against lung infection, CD4+ T cells were shown to be dispensable at the time of lung challenge for protection from serotype 2 S. pneumoniae D39 strain (27), but they were required for postcolonization lung protection from S. pneumoniae serotype 19F (5). Furthermore, NP colonization with 19F elicits Abs directed against protein Ags but not capsular polysaccharides, and the requirement for CD4+ T cells for postcolonization protection is consistent with the presumed T cell-dependent nature of that Ab response (5). Our studies demonstrated that protection elicited by NP exposure did not require CD4+ T cells. We also found no statistically significant evidence for a specific role for CD8+ T cells in protection at either initial colonization or subsequent challenge. Note that this study used a protocol involving three rounds of i.n. inoculation (35), whereas many other studies investigating immunity induced by i.n. inoculation used only a single inoculation event (5, 27). Thus, the degree to which specific T cell subsets are required for protection may vary not only with the specific S. pneumoniae strain but also with the particular experimental system. Future work will be needed to better understand the specific contribution of T cells to NP-induced protection from lung infection.
Asymptomatic NP carriage in infants and young children is associated with serotype-independent immunity to pneumococcal carriage, but the length and mechanism of the protective effect of carriage remains relatively unexplored (33). In this study, we showed that in mice, antipneumococcal Ab titers remained high at 18 wk postexposure and retained detectable protection from lung challenge at 20 wk. In addition to traditional (radiation-sensitive) memory B cells that develop, with CD4+ T cell help, into Ab-secreting plasma cells upon re-encountering Ag, immunological memory can be provided by long-lived, radiation-resistant CD138+ ASC, which reside in the bone marrow and secrete Abs in the absence of circulating Ags (19, 23, 24). Long-lived ASC do not require reactivation for Ab production and thus function independently of CD4+ T cells at the time of secondary challenge (20). Exposure to T cell-independent Ags, such as serotype 3 PPS and haptenated LPS, can give rise to these cells, which provide protection from systemic S. pneumoniae challenge in mice (12, 21). We found that CD4+ T cells were not required for protection at the time of initial NP exposure or at the time of pulmonary challenge, and the production of Abs against the capsular polysaccharide, a T cell–independent Ag, was long-lived. Furthermore, we found that CD93, which is required for maintaining CD138+ ASC but not traditional memory B cells (25), was absolutely required for protection. CD93 is expressed on many cell types, so this exquisite susceptibility of CD93-deficient mice may be multifactorial. Nevertheless, adoptive transfer of bone marrow enriched for CD138+ cells, isolated from NP-exposed but not naive mice, to naive mice conferred protection from i.t. lung challenge, suggesting that these cells make a major contribution to postexposure immunity.
B1 B cells contribute T cell–independent protection from systemic infection by S. pneumoniae (11). The B1a subset generates natural Abs that provide a measure of innate immunity to systemic challenge, and our observation that B cell–deficient mice did not survive as well as control mice during initial NP exposure (Table I) is consistent with a role for these cells in protection from challenge at that site. Additionally, the B1b subset generates anti-PPS Abs that contribute to adaptive immunity to systemic infection by S. pneumoniae (11, 55). Some experimental systems indicate that B1 cells can secrete Abs without adopting the canonical ASC phenotype (59), and if this transition to secreting cells does not require cell division, it might be expected to be relatively radioresistant. Hence, we also cannot rule out the possibility that a population of radioresistant B1b cells contributes to a postexposure memory response. In preliminary studies, adoptively transferred B1-enriched peritoneal cells failed to protect rag1–/– mice from i.t. challenge after NP exposure (data not shown), suggesting either that B1 cells might not be sufficient or that transferred peritoneal cells do not transit to NP-proximal lymphoid tissue in this experimental system. Further studies are required to assess the role of B1 cells in immunity following NP exposure to S. pneumoniae.
The elderly are at increased risk of life-threatening invasive pneumococcal infections, and previous work has demonstrated age-associated susceptibility to S. pneumoniae infection in mice (17, 60). Although aged (20- to 22-mo-old) mice were able to survive NP exposure similarly to young controls and generated comparable levels of antipneumococcal Abs, they did not acquire significant postcolonization protection against pulmonary challenge. In fact, despite similar levels of Abs against PC and PPS4, sera from colonized old mice did not demonstrate the protective activity of sera from young mice. A possible explanation is that the functionality of antipneumococcal Abs against PC and/or capsular polysaccharides declines with age. The opsonic capacity of Abs elicited by administration of the PPS vaccine is lower in elderly compared with young volunteers (61), and an anti-PC mAb mixture isolated from old mice, in contrast to a mixture of mAb isolated from young mice, fails to protect naive recipients against systemic infection with serotype 3 S. pneumoniae (54). In colonization models, although S. pneumoniae establishes a comparable level of NP colonization in young and old mice in the first week after i.n. inoculation, only young mice begin clearing the bacteria soon thereafter (62). Future work will be required to determine whether diminished opsonic activity of Ab contributes to ineffective postexposure immune response in aged mice.
Finally, we found that defects in the CD138+ compartment may contribute to the blunted protection following NP exposure associated with aging. Following colonization, old mice had a lower percentage of CD138-expressing cells in the bone marrow than did young mice, and when adoptively transferred, these cells failed to protect naive young mice from lung infection. Thus, the long-lived CD138+ bone marrow cells that promote postexposure protection from lethal pulmonary infection in young mice appeared to lose function upon aging. In humans, both the absolute counts and percentages of circulating plasma cells (CD138+ and CD1382) decline with aging (63), suggesting that the protective capacity of CD138+ cells revealed in this murine system may have a functional counterpart in humans, and that their decline may contribute to the age-associated susceptibility of the elderly to pneumococcal lung infection. Further analysis of the generation and protective capacities of these long-lived ASC may provide new avenues to develop preventative strategies against invasive pneumococcal disease.
Supplementary Material
Acknowledgments
We thank Mark Lipsitch for help in establishing the NP colonization model, Rick Malley for helpful advice, Pilar Alcaide and Ann Rothstein for reagents, Susan Swain, Klaus Rajewsky, Dennis W. Metzger, Andrea Tenner, and Marina Botto for mice, Hardy Kornfeld for i.t. infection protocols, as well as Ognjen Sekulovic and Marcia Osburne for critical reading and discussion of the manuscript.
This work was supported in part by National Institute on Aging Pathway to Independence Award K99 AG051784–02 (to E.N.B.G.).
Abbreviations used in this article:
- ASC
Ab-secreting cell
- BALF
bronchoalveolar lavage fluid
- i.n.
intranasal(ly)
- i.t.
intratracheal(ly)
- NP
nasopharyngeal
- PC
phosphorylcholine
- PPS
pneumococcal polysaccharide
- PPS4
PPS serotype 4
- UMMS
University of Massachusetts Medical School
Footnotes
E.N.B.G. and N.H.T.M. designed research, conducted research, analyzed data, and wrote the paper; A.E.G., N.S., and S.C. conducted research; A.C. and E.J.M.-E. provided essential reagents and expertise; R.M.G. and J.M.L. designed research and wrote the paper; J.M.L. had primary responsibility for final content; and all authors read and approved the final manuscript.
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Kadioglu A, Weiser JN, Paton JC, and Andrew PW. 2008. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat. Rev. Microbiol 6: 288–301. [DOI] [PubMed] [Google Scholar]
- 2.Obaro S, and Adegbola R. 2002. The pneumococcus: carriage, disease and conjugate vaccines. J. Med. Microbiol 51: 98–104. [DOI] [PubMed] [Google Scholar]
- 3.Song JY, Choi JY, Lee JS, Bae IG, Kim YK, Sohn JW, Jo YM, Choi WS, Lee J, Park KH, et al. 2013. Clinical and economic burden of invasive pneumococcal disease in adults: a multicenter hospital-based study. BMC Infect. Dis 13: 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chong CP, and Street PR. 2008. Pneumonia in the elderly: a review of the epidemiology, pathogenesis, microbiology, and clinical features. South. Med. J 101: 1141–1145; quiz 1132, 1179. 10.1097/SMJ.0b013e318181d5b5 [DOI] [PubMed] [Google Scholar]
- 5.Wilson R, Cohen JM, Jose RJ, de Vogel C, Baxendale H, and Brown JS. 2015. Protection against Streptococcus pneumoniae lung infection after nasopharyngeal colonization requires both humoral and cellular immune responses. Mucosal Immunol 8: 627–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roche AM, King SJ, and Weiser JN. 2007. Live attenuated Streptococcus pneumoniae strains induce serotype-independent mucosal and systemic protection in mice. Infect. Immun 75: 2469–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferreira DM, Neill DR, Bangert M, Gritzfeld JF, Green N, Wright AK, Pennington SH, Bricio-Moreno L, Moreno AT, Miyaji EN, et al. 2013. Controlled human infection and rechallenge with Streptococcus pneumoniae reveals the protective efficacy of carriage in healthy adults. [Published erratum appears in 2013 Am. J. Respir. Crit. Care Med. 187: 1153.] Am. J. Respir. Crit. Care Med 187: 855–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goldenberg HB, McCool TL, and Weiser JN. 2004. Cross-reactivity of human immunoglobulin G2 recognizing phosphorylcholine and evidence for protection against major bacterial pathogens of the human respiratory tract. J. Infect. Dis 190: 1254–1263. [DOI] [PubMed] [Google Scholar]
- 9.Adderson EE 2001. Antibody repertoires in infants and adults: effects of T-independent and T-dependent immunizations. Springer Semin. Immunopathol 23: 387–403. [DOI] [PubMed] [Google Scholar]
- 10.Basset A, Thompson CM, Hollingshead SK, Briles DE, Ades EW, Lipsitch M, and Malley R. 2007. Antibody-independent, CD4+ T-cell-dependent protection against pneumococcal colonization elicited by intranasal immunization with purified pneumococcal proteins. Infect. Immun 75: 5460–5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haas KM, Poe JC, Steeber DA, and Tedder TF. 2005. B-1a and B-1b cells exhibit distinct developmental requirements and have unique functional roles in innate and adaptive immunity to S. pneumoniae. Immunity 23: 7–18. [DOI] [PubMed] [Google Scholar]
- 12.Taillardet M, Haffar G, Mondiere P, Asensio MJ, Gheit H, Burdin N, Defrance T, and Genestier L. 2009. The thymus-independent immunity conferred by a pneumococcal polysaccharide is mediated by long-lived plasma cells. Blood 114: 4432–440. [DOI] [PubMed] [Google Scholar]
- 13.Paradiso PR 2012. Pneumococcal conjugate vaccine for adults: a new paradigm. Clin. Infect. Dis 55: 259–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alugupalli KR, Leong JM, Woodland RT, Muramatsu M, Honjo T, and Gerstein RM. 2004. B1b lymphocytes confer T cell–independent long-lasting immunity. Immunity 21: 379–390. [DOI] [PubMed] [Google Scholar]
- 15.Ridda I, Macintyre CR, Lindley R, Gao Z, Sullivan JS, Yuan FF, and McIntyre PB. 2009. Immunological responses to pneumococcal vaccine in frail older people. Vaccine 27: 1628–1636. [DOI] [PubMed] [Google Scholar]
- 16.Buffa S, Bulati M, Pellicano M, Dunn-Walters DK, Wu YC, Candore G, Vitello S, Caruso C, and Colonna-Romano G. 2011. B cell immunosenescence: different features of naive and memory B cells in elderly. Biogerontology 12: 473–483. [DOI] [PubMed] [Google Scholar]
- 17.Bou Ghanem EN, Clark S, Du X, Wu D, Camilli A, Leong JM, and Meydani SN. 2015. The a-tocopherol form of vitamin E reverses age-associated susceptibility to streptococcus pneumoniae lung infection by modulating pulmonary neutrophil recruitment. J. Immunol 194: 1090–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Phan TG, and Tangye SG. 2017. Memory B cells: total recall. Curr. Opin. Immunol 45: 132–140. [DOI] [PubMed] [Google Scholar]
- 19.Radbruch A, Muehlinghaus G, Luger EO, Inamine A, Smith KG, Dorner T, and Hiepe F. 2006. Competence and competition: the challenge of becoming a long-lived plasma cell. Nat. Rev. Immunol 6: 741–750. [DOI] [PubMed] [Google Scholar]
- 20.Bortnick A, and Allman D. 2013. What is and what should always have been: long-lived plasma cells induced by T cell-independent antigens. J. Immunol 190: 5913–5918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bortnick A, Chernova I, Quinn WJ III, Mugnier M, Cancro MP, and Allman D. 2012. Long-lived bone marrow plasma cells are induced early in response to T cell-independent or T cell-dependent antigens. J. Immunol 188: 5389–5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manz RA, Lohning M, Cassese G, Thiel A, and Radbruch A. 1998. Survival of long-lived plasma cells is independent of antigen. Int. Immunol 10: 1703–1711. [DOI] [PubMed] [Google Scholar]
- 23.Slifka MK, and Ahmed R. 1998. Long-lived plasma cells: a mechanism for maintaining persistent antibody production. Curr. Opin. Immunol 10: 252–258. [DOI] [PubMed] [Google Scholar]
- 24.Slifka MK, Antia R, Whitmire JK, and Ahmed R. 1998. Humoral immunity due to long-lived plasma cells. Immunity 8: 363–372. [DOI] [PubMed] [Google Scholar]
- 25.Chevrier S, Genton C, Kallies A, Karnowski A, Otten LA, Malissen B, Malissen M, Botto M, Corcoran LM, Nutt SL, and Acha-Orbea H. 2009. CD93 is required for maintenance of antibody secretion and persistence of plasma cells in the bone marrow niche. Proc. Natl. Acad. Sci. USA 106: 3895–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richards L, Ferreira DM, Miyaji EN, Andrew PW, and Kadioglu A. 2010. The immunising effect of pneumococcal nasopharyngeal colonisation; protection against future colonisation and fatal invasive disease. Immunobiology 215: 251–263. [DOI] [PubMed] [Google Scholar]
- 27.Cohen JM, Khandavilli S, Camberlein E, Hyams C, Baxendale HE, and Brown JS. 2011. Protective contributions against invasive Streptococcus pneumoniae pneumonia of antibody and Th17-cell responses to nasopharyngeal colonisation. PLoS One 6: e25558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCool TL, Cate TR, Moy G, and Weiser JN. 2002. The immune response to pneumococcal proteins during experimental human carriage. J. Exp. Med 195: 359–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goldblatt D, Hussain M, Andrews N, Ashton L, Virta C, Melegaro A, Pebody R, George R, Soininen A, Edmunds J, et al. 2005. Antibody responses to nasopharyngeal carriage of Streptococcus pneumoniae in adults: a longitudinal household study. J. Infect. Dis 192: 387–393. [DOI] [PubMed] [Google Scholar]
- 30.Wilson R, Cohen JM, Reglinski M, Jose RJ, Chan WY, Marshall H, de Vogel C, Gordon S, Goldblatt D, Petersen FC, et al. 2017. Naturally acquired human immunity to pneumococcus is dependent on antibody to protein antigens. [Published erratum appears in 2017 PLoS Pathog. 13: e1006259.] PLoS Pathog 13: e1006137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wright AK, Ferreira DM, Gritzfeld JF, Wright AD, Armitage K, Jambo KC, Bate E, El Batrawy S, Collins A, and Gordon SB. 2012. Human nasal challenge with Streptococcus pneumoniae is immunising in the absence of carriage. PLoS Pathog 8: e1002622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lipsitch M, Whitney CG, Zell E, Kaijalainen T, Dagan R, and Malley R. 2005. Are anticapsular antibodies the primary mechanism of protection against invasive pneumococcal disease? PLoS Med 2: e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Granat SM, Ollgren J, Herva E, Mia Z, Auranen K, and Makela PH. 2009. Epidemiological evidence for serotype-independent acquired immunity to pneumococcal carriage. J. Infect. Dis 200: 99–106. [DOI] [PubMed] [Google Scholar]
- 34.Malley R, Trzcinski K, Srivastava A, Thompson CM, Anderson PW, and Lipsitch M. 2005. CD4+ T cells mediate antibody-independent acquired immunity to pneumococcal colonization. Proc. Natl. Acad. Sci. USA 102: 4848–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trzcinski K, Thompson C, Malley R, and Lipsitch M. 2005. Antibodies to conserved pneumococcal antigens correlate with, but are not required for, protection against pneumococcal colonization induced by prior exposure in a mouse model. Infect. Immun 73: 7043–7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moffitt KL, Gierahn TM, Lu YJ, Gouveia P, Alderson M, Flechtner JB, Higgins DE, and Malley R. 2011. TH17-based vaccine design for prevention of Streptococcus pneumoniae colonization. Cell Host Microbe 9: 158–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Z, Clarke TB, and Weiser JN. 2009. Cellular effectors mediating Th17-dependent clearance of pneumococcal colonization in mice. J. Clin. Invest 119: 1899–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCool TL, and Weiser JN. 2004. Limited role of antibody in clearance of Streptococcus pneumoniae in a murine model of colonization. Infect. Immun 72: 5807–5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu YJ, Gross J, Bogaert D, Finn A, Bagrade L, Zhang Q, Kolls JK, Srivastava A, Lundgren A, Forte S, et al. 2008. Interleukin-17A mediates acquired immunity to pneumococcal colonization. PLoS Pathog 4: e1000159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alugupalli KR, Gerstein RM, Chen J, Szomolanyi-Tsuda E, Woodland RT, and Leong JM. 2003. The resolution of relapsing fever bor-reliosis requires IgM and is concurrent with expansion of B1b lymphocytes. J. Immunol 170: 3819–3827. [DOI] [PubMed] [Google Scholar]
- 41.Harriman GR, Bogue M, Rogers P, Finegold M, Pacheco S, Bradley A, Zhang Y, and Mbawuike IN. 1999. Targeted deletion of the IgA constant region in mice leads to IgA deficiency with alterations in expression of other Ig isotypes. J. Immunol 162: 2521–2529. [PubMed] [Google Scholar]
- 42.Rodwell JD, Gearhart PJ, and Karush F. 1983. Restriction in IgM expression. IV. Affinity analysis of monoclonal anti-phosphorylcholine antibodies. J. Immunol 130: 313–316. [PubMed] [Google Scholar]
- 43.Tettelin H, Nelson KE, Paulsen IT, Eisen JA, Read TD, Peterson S, Heidelberg J, DeBoy RT, Haft DH, Dodson RJ, et al. 2001. Complete genome sequence of a virulent isolate of Streptococcus pneumoniae. Science 293: 498–506. [DOI] [PubMed] [Google Scholar]
- 44.Abel S, Abel zur Wiesch P, Davis BM, and Waldor MK. 2015. Analysis of bottlenecks in experimental models of infection. PLoS Pathog 11: e1004823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Briles DE, Novak L, Hotomi M, van Ginkel FW, and King J. 2005. Nasal colonization with Streptococcus pneumoniae includes subpopulations of surface and invasive pneumococci. Infect. Immun 73: 6945–6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Malley R, Lipsitch M, Stack A, Saladino R, Fleisher G, Pelton S,Thompson C, Briles D, and Anderson P. 2001. Intranasal immunization with killed unencapsulated whole cells prevents colonization and invasive disease by capsulated pneumococci. Infect. Immun 69: 4870–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang L, Li Z, Wan Z, Kilby A, Kilby JM, and Jiang W. 2015. Humoral immune responses to Streptococcus pneumoniae in the setting of HIV-1 infection. Vaccine 33: 4430–4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stillman EG, and Goodner K. 1933. Resistance to pneumococcus infection in rabbits following immunizing injections of heat-killed pneumococcus suspensions. J. Exp. Med 58: 195–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bou Ghanem EN, Clark S, Roggensack SE, McIver SR, Alcaide P, Haydon PG, and Leong JM. 2015. Extracellular adenosine protects against Streptococcus pneumoniae lung infection by regulating pulmonary neutrophil recruitment. PLoS Pathog 11: e1005126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kobrynski LJ, Sousa AO, Nahmias AJ, and Lee FK. 2005. Cutting edge: antibody production to pneumococcal polysaccharides requires CD1 molecules and CD8+ T cells. J. Immunol 174: 1787–1790. [DOI] [PubMed] [Google Scholar]
- 51.Collins AM, Wright AD, Mitsi E, Gritzfeld JF, Hancock CA, Pennington SH, Wang D, Morton B, Ferreira DM, and Gordon SB. 2015. First human challenge testing of a pneumococcal vaccine. Double-blind randomized controlled trial. Am. J. Respir. Crit. Care Med 192: 853–858. [DOI] [PubMed] [Google Scholar]
- 52.van Deursen AM, van den Bergh MR, and Sanders EA, Carriage Pilot Study Group. 2016. Carriage of Streptococcus pneumoniae in asymptomatic, community-dwelling elderly in the Netherlands. Vaccine 34: 4–6. [DOI] [PubMed] [Google Scholar]
- 53.Obaro SK 2000. Confronting the pneumococcus: a target shift or bullet change? Vaccine 19: 1211–1217. [DOI] [PubMed] [Google Scholar]
- 54.Nicoletti C, Yang X, and Cerny J. 1993. Repertoire diversity of antibody response to bacterial antigens in aged mice. III. Phosphorylcholine antibody from young and aged mice differ in structure and protective activity against infection with Streptococcus pneumoniae. J. Immunol 150: 543–549. [PubMed] [Google Scholar]
- 55.Tanaka N, Fukuyama S, Fukuiwa T, Kawabata M, Sagara Y, Ito HO, Miwa Y, Nagatake T, Kiyono H, and Kurono Y. 2007. Intranasal immunization with phosphorylcholine induces antigen specific mucosal and systemic immune responses in mice. Vaccine 25: 2680–2687. [DOI] [PubMed] [Google Scholar]
- 56.Briles DE, Forman C, Hudak S, and Claflin JL. 1984. The effects of subclass on the ability of anti-phosphocholine antibodies to protect mice from fatal infection with Streptococcus pneumoniae. J. Mol. Cell. Immunol 1: 305–309. [PubMed] [Google Scholar]
- 57.Briles DE, Forman C, Hudak S, and Claflin JL. 1984. The effects of idi-otype on the ability of IgG1 anti-phosphorylcholine antibodies to protect mice from fatal infection with Streptococcus pneumoniae. Eur. J. Immunol 14: 1027–1030. [DOI] [PubMed] [Google Scholar]
- 58.Alugupalli KR 2008. A distinct role for B1b lymphocytes in T cell-independent immunity. Curr. Top. Microbiol. Immunol 319: 105–130. [DOI] [PubMed] [Google Scholar]
- 59.Savage HP, Yenson VM, Sawhney SS, Mousseau BJ, Lund FE, and Baumgarth N. 2017. Blimp-1-dependent and -independent natural antibody production by B-1 and B-1-derived plasma cells. J. Exp. Med 214: 2777–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hinojosa E, Boyd AR, and Orihuela CJ. 2009. Age-associated inflammation and Toll-like receptor dysfunction prime the lungs for pneumococcal pneumonia. J. Infect. Dis 200: 546–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Simell B, Vuorela A, Ekstrom N, Palmu A, Reunanen A, Meri S, Kayhty H, and Vakevainen M. 2011. Aging reduces the functionality of anti-pneumococcal antibodies and the killing of Streptococcus pneumoniae by neutrophil phagocytosis. Vaccine 29: 1929–1934. [DOI] [PubMed] [Google Scholar]
- 62.Krone CL, Trzcmski K, Zborowski T, Sanders EA, and Bogaert D. 2013. Impaired innate mucosal immunity in aged mice permits prolonged Streptococcus pneumoniae colonization. Infect. Immun 81: 4615–4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Caraux A, Klein B, Paiva B, Bret C, Schmitz A, Fuhler GM, Bos NA, Johnsen HE, Orfao A, and Perez-Andres M, Myeloma Stem Cell Network. 2010. Circulating human B and plasma cells. Age-associated changes in counts and detailed characterization of circulating normal CD138- and CD138+ plasma cells. Haematologica 95: 1016–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.