

Abstract
Spectroscopic methods, which are used to establish RNA structure-function relationships, require strategies for post-synthetic, site-specific incorporation of chemical probes into target RNAs. For RNAs >50 nt, the enzymatic incorporation of a nucleoside or nucleotide monophosphate guanosine analog (G-analog) at their 5'-end is routinely achieved by in vitro transcription (IVT) using T7 RNA polymerase (T7RNAP) and a DNA template with a GTP-initiating class III Φ6.5 promoter. However, when high G-analog:GTP ratios are used to bias G-analog incorporation at the 5'-end, RNA yield is compromised. Here, we show that the use of a T7RNAP P266L mutant in an IVT with 10:1 thienoguanosine (thG):GTP increased the percent incorporation and yield of 5'-thG-initiated precursor tRNA for a net ~3-fold gain compared to an IVT with T7RNAP. We also demonstrate that a one-pot multi-enzyme approach, which consists of transcription by T7RNAP P266L and a post-transcriptional clean-up by polyphosphatase and an exonuclease, led to essentially near-homogeneous 5'-thG-modified transcripts. This approach should be of broad utility in preparing 5'-modified RNAs.
Keywords: RNA, Fluorescent probes, T7 RNA polymerase, In vitro transcription, Thienoguanosine
Table of Contents
Premature termination of RNAs initiated with guanosine analogs has rendered difficult the introduction of 5'-guanosine modifications into RNAs generated by in vitro transcription with T7 RNA polymerase (T7RNAP). Here, we provide a solution using a mutant T7RNAP with a decreased propensity for abortive transcription.
RNAs with site-specific chemical modifications are powerful tools in biochemical studies.[1] Although chemical modifications can be site-specifically incorporated into RNAs using solid-phase synthesis, this method entails the non-trivial synthesis of a phosphoramidite-bearing NTP-analog and is limited to RNAs < 50 nt.[2] In contrast, the enzymatic method for site-specifically introducing chemically-modified nucleoside/nucleotide monophosphate analogs at the 5'-ends of RNAs during in vitro transcription (IVT) is particularly attractive due to its low cost and applicability to RNAs of any length. Bacteriophage-derived RNA polymerases (e.g., SP6, T3, T7) have long been used to synthesize RNAs, with T7 RNA polymerase (T7RNAP) being a well-established and preferred choice.[3] A desired RNA is generated by placing the sequence of interest immediately downstream of the 17-bp T7RNAP promoter and by performing an IVT with recombinant, single-polypeptide T7RNAP.
While there are many T7RNAP promoter variants, the commonly employed class III Φ6.5 promoter allows high transcriptional yields when the RNA is initiated with two guanosines (Gs).[3] Several investigators[2,4] have exploited this attribute to bias T7RNAP to initiate with G-analogs, which can support initiation but not elongation since they lack a triphosphate. Despite the remarkable tolerance of T7RNAP to utilize non-native purine mimics as initiators, an intrinsic limitation of this approach is the low yield of RNA resulting from: (i) the use of high G-analog:GTP ratios (up to 100:1) in the IVTs, and (ii) a large fraction of the RNAs initiated with G-analogs finishing up as aborted transcripts. Here, we sought to address these limitations.
In this report, we show that the use of a T7RNAP mutant with a decreased propensity for abortive transcription enhances yield and incorporation of the G-analog. Moreover, we used moderate G-analog:GTP ratios (10:1) during IVTs to help enhance the yield of RNAs initiated with GTP and the G-analog, but then employed a post-transcriptional (one-pot) enzymatic clean-up to selectively eliminate 5'-GTP-initiated RNAs. Overall, this simple approach solves a long-standing problem to prepare 5'-modified RNAs with high yield and purity.
To obtain a high yield of full-length RNAs containing a G-analog at the 5'-end, we chose a test G-analog and examined a set of modified IVT conditions. Three reasons inspired our choice of thienoguanosine (thG; Figure 1A), a fluorescent guanosine surrogate (Figure S1 in the Supporting Information),[5] as the pilot analog. First, thG permits facile fluorescence detection[5a] post-synthesis, thus allowing us to rapidly examine different IVT conditions and fine-tune variables as needed. Second, optimizing 5'-incorporation is expected to have payoffs for site-specific (internal) modification because 5'-thG-modified RNAs can be 5'-phosphorylated and ligated to another RNA to yield the desired longer RNA with an internal thG.[5b] Finally, given the value of emissive nucleoside analogs as bona fide isomorphs, methods to enhance incorporation of these isomorphs into RNAs will greatly aid mechanistic and spectroscopic studies, as was demonstrated elegantly in a recent study of the hammerhead ribozyme.[5b] As a model RNA for our IVTs, we used a 155-nt precursor-tRNACys (pre-tRNACys; Figure 1B).
Figure 1.
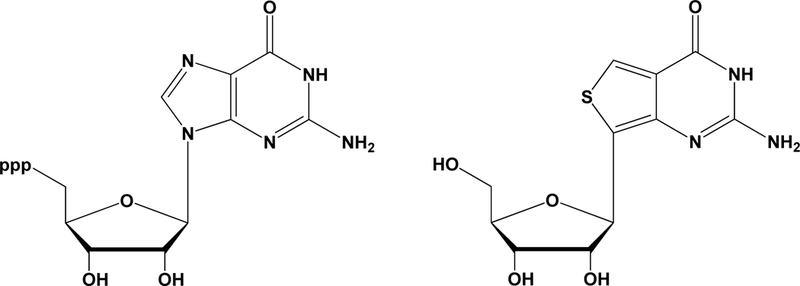
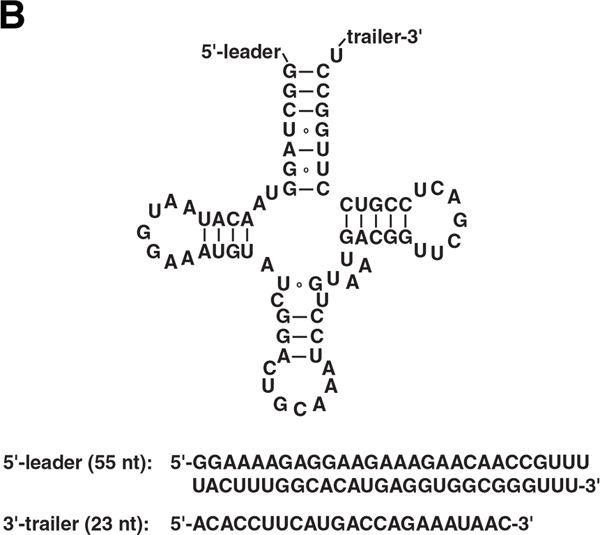
(A) Structure of guanosine triphosphate and thienoguanosine (thG). (B) Secondary structure of pre-tRNACys, the model RNA used in this study. The sequences of the 5'-leader and 3'-trailer are provided below the secondary structure.
We considered various modifications to the IVT conditions to increase incorporation of the G-analog. Foremost, based on the premise that poor IVT yields arise from the rapid depletion of limited GTP, which is typically used at low concentrations to favor incorporation of the modified G-analog, we used a phased-addition strategy wherein GTP was supplemented at timed intervals. Although this approach engendered 5'-incorporation gains, the increase was not uniform for different G-analogs (Adib and Gopalan, unpublished data). Second, we included varying concentrations of Mn2+ in the IVT to influence T7RNAP fidelity[5] and thereby favor the G-analog over GTP, but these attempts were unsuccessful. Results from these studies revealed that abortive transcription is a critical roadblock, and inspired us to consider the use of T7RNAP mutants that have been shown to increase incorporation of various 2'-modified NTP-analogs into RNAs synthesized during IVTs.[6] From this panel of mutants, we chose T7RNAP Pro266Leu (P266L) because kinetic and footprinting studies revealed that this mutant exhibits a delayed transition from transcriptional initiation to elongation and generates fewer aborted transcripts.[7] We sought to test the postulate that the T7RNAP P266L mutant would decrease aborted transcripts typically observed when 5'-G-analogs are used with T7RNAP wild-type (WT; Figure 2A). The problem of low G-analog incorporation, however, necessitates a combination of transcriptional and post-transcriptional strategies since a fraction of transcripts will always be initiated with GTP. Therefore, we investigated the utility of an one-pot multi-enzyme (OPME) approach wherein a clean-up step (post-transcription with T7RNAP P266L) with a polyphosphatase and an exonuclease is used in tandem to selectively degrade the 5'-GTP-initiated RNAs and yield near-homogeneous 5'-thG-initiated RNAs (Figure 2B).
Figure 2.
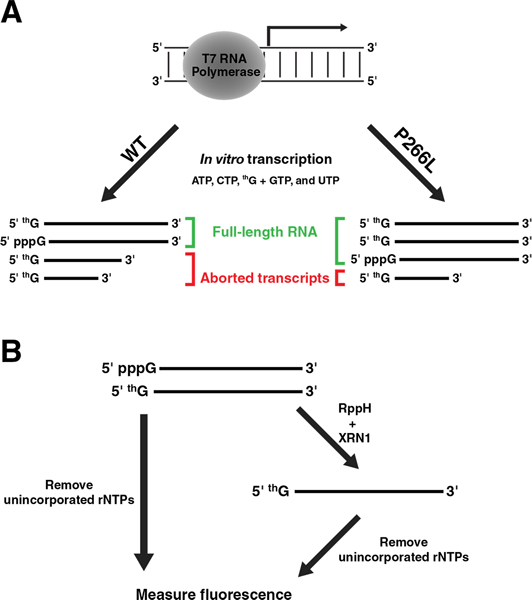
(A) Schematic of the expected outcomes during an IVT that has a fixed ratio of thG:GTP and was peformed using either T7RNAP WT or P266L mutant. This illustration depicts qualitatively the gains with respect to abortive transcription (also, see Figure S2). (B) Comparison of a typical IVT versus the one-pot multi-enzyme approach designed to eliminate 5'-GTP-initiated RNAs.
Prior to comparisons of T7RNAP WT and P2666L mutant, we first confirmed the use of roughly equivalent amounts of these variants in our IVTs (Figure 3A). Because of our long-standing interest in 5'-incorporation of 5'-deoxy-5'-azido-guanosine (azG), we had performed IVTs with this analog and obtained data indicating that percent incorporation was higher when the G-analog:GTP ratio was 10:1 compared to 4:1, regardless of whether we used the WT or the P266L mutant. This conclusion is also consistent with a previous report that used azG.[4a] Additionally, by using an IVT spiked with an α-[32P]-labeled rNTP, we found that the fraction of short aborted transcripts relative to full-length was significantly lower with T7RNAP P266L compared to the WT when azG was the analog (data not shown); we confirmed this trend using an IVT performed with thG as well (Figure S2).
Figure 3.
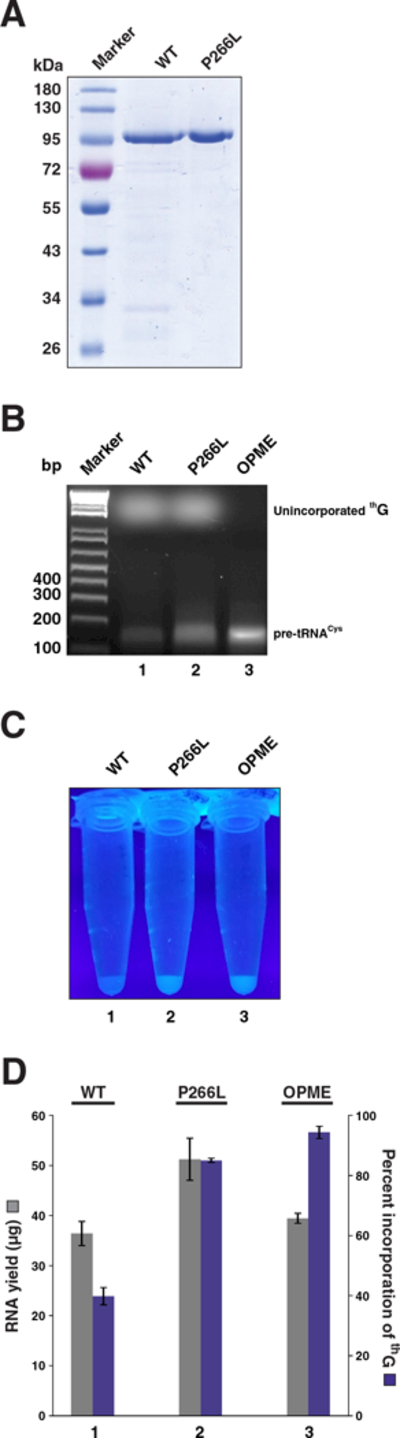
(A) Purity of recombinant T7RNAP used in this study, as demonstrated by SDS-PAGE [10% (w/v) polyacrylamide] analysis. (B) One µl from a 100-µl IVT performed using either T7RNAP WT (lane 1) or P266L (lane 2) was electrophoresed on a 2% (w/v) agarose gel and stained with ethidium bromide; samples examined were from post-DNase I treatment. Lane 3 has 1 µg of the final pre-tRNACys 55–23 RNA that was generated using the OPME (post-IVT RppH and Xrn-1) approach. (C) Fluorescence (λexc, 312 nm) of 5 µM RNA obtained after IVT with T7RNAP WT (1) or P266L (2), or using the OPME method with P266L (3). (D) RNA yields (grey) and % incorporation of thG (purple) with T7RNAP WT (1) or P266L (2), or using the OPME method with P266L (3). Mean and standard deviation values were calculated from three independent measurements.
Using a 10:1 ratio of thG:GTP, we determined that T7RNAP WT initiates 40% of pre-tRNACys transcripts with thG affording a total RNA yield of 37 µg from a 100 µl IVT (Figures 3B, 3C, and 3D). However, when T7RNAP P266L was employed in the IVT, thG percent incorporation increased to 85% with a total RNA yield of 50 µg (Figures 3B and3D). Thus, the net gain in 5'-thG-modified RNA is ~3-fold. While visual inspection post-agarose gel electrophoresis revealed that the IVT yield is higher with the P266L mutant compared to the WT (Figure 3B), the percent incorporation of thG in pre-tRNACys was accurately determined by first eliminating the unincorporated thG and then measuring the fluorescence in the final RNA product. The efficacy of the desalting column in removing the unincorporated thG was evident from examining the washes on a trans-illuminator (λexc, 312 nm); the ability to easily monitor thG is an appealing feature in its use and allows for rapid visual comparison of its incorporation efficiency into RNA across different IVT conditions (Figure 3C; Figure S3). While use of the desalting column was essential for complete removal of unincorporated thG, we consistently recovered only ~70% of the RNA input to the column. Replacing this step with an alternative that results in a lower loss will be desirable.
To synthesize 5'-NAD-capped RNA, Jäschke and coworkers[8] employed an elegant multi-pot, chemo-enzymatic approach. Post-IVT, the RNA product was subjected to a polyphosphatase treatment to remove the pyrophosphate and yield 5'-GMP transcripts, which were then reacted with nicotinamide mononucleotide-phospho-rimidzolide. Any unreacted 5'-GMP transcripts were then removed by Xrn-1. We have now extended this approach to enhance the fraction of 5'-thG-pre-tRNACys albeit with an OPME set-up. Post-IVT, RppH was utilized to convert 5'-GTP-pre-tRNACys into 5'-GMP-pre-tRNACys that was then degraded using commercially available Xrn-1. This exonuclease was not expected to act on 5'-thG-pre-tRNACys given the absence of a 5'-monophosphate. An advantage of the OPME approach is that these enzymes are added post-IVT with no intervening processing steps. Our OPME method resulted in 40 µg of near-homogenous (~95%) 5'-thG-pre-tRNACys from a 100-µl IVT (Figure 3D). That a small fraction of the 5'-GTP initiated pre-tRNACys was not removed indicates that the activities of RppH and Xrn-1 may not be optimal under our OPME reaction conditions, but these can be tweaked through customized optimization trials.
Owing to the poor solubility of guanosine in water, the stock of thG was prepared in DMSO. Inclusion of 20% DMSO in IVTs was reported[9] to afford an increase in RNA yield and lower 3'-heterogeneity. To test the idea that inclusion of DMSO in the IVT might similarly influence the bias of T7RNAP to initiate RNAs with thG, we performed IVTs of pre-tRNACys as described above except in the presence or absence of 5% DMSO. However, we did not observe any significant effect on RNA yield or incorporation of thG upon inclusion of DMSO (Figure S4).
Because the total RNA yield (50 µg/100 µl IVT) of pre-tRNACys from IVTs containing thG was ~2.5-fold lower than IVTs containing only 3 mM rNTPs, we sought two different approaches to bridge this gap. First, we explored the use of a mutant T7RNAP containing 10 mutations (termed ‘RGVG-M6’), including P266L, which exhibited a 25-fold increase in RNA yield of fully 2'-modified RNA over other commonly used mutant T7RNAPs.[7d] When we purified this mutant and tested the recombinant version in IVTs, we did not observe an increase in the total yield of 5'-thG-initiated RNAs. Second, we examined the effects of retaining the [thG]:[GTP] ratio but increasing their absolute concentrations on T7RNAP P266L’s activity. Indeed, changing [thG]:[GTP] from 4.8 mM:0.48 mM to 10 mM:1 mM led to doubling of the yield of total pre-tRNACys, although percent incorporation of thG decreased by one-fourth (duplicate experiments, data not shown). A modest compromise in the incorporation of thG is offset by the yield payoff especially since the OPME approach helps selectively eliminate the GTP-initiated RNA. Due to the template-dependent variability in IVT yields, pilot tests of different rNTP concentrations in IVTs as well as different [thG]:[GTP] ratios will be gainful for any new thG-initiated RNA.
Our results show that the T7RNAP P266L mutant enhances the total RNA yield and percent of 5'-thG-initiated RNAs, with additional gains resulting from the post-IVT use of RppH and Xrn-1 to selectively remove the 5'-GTP-initiated RNAs. The findings with thG reaffirm the increased tolerance of T7RNAP P266L mutant to initiate RNA transcripts with modified nucleosides, an attribute noted before while investigating use of modified 2'-NTPs.[7c] Given the value of thG as a probe of RNA folding/dynamics and RNA function,[5b] the facile synthesis of 5'-thG-initiated RNAs should increase the use of this guanosine isomorph. Moreover, we expect that the OPME approach described here will improve percent incorporation and yields of other G-analogs at the 5'-end of RNAs of any length and sequence. Our work should also motivate an examination of the ability of different T7RNAP mutants to initiate RNA transcripts with adenosine analogs from a class II Φ2.5 promoter.[10]
Experimental Section
Purification of T7RNAPs
Both the T7RNAP WT and P266L were overexpressed in Escherichia coli and purified to near-homogeneity using immobilized metal-affinity chromatography. Details are provided in the Supporting Information.
In vitro transcription reactions
A PCR DNA template containing a class III Φ6.5 promoter and gene encoding Arabidopsis thaliana mitochondrial precursor tRNACys was prepared as previously described.[11] This pre-tRNA has a 55-nt 5'-leader and a 23-nt 3'-trailer (Figure 1B), and was chosen due to its availability and use in our laboratory for studies of RNase P. One hundred-µl IVTs were performed at 37°C for 5 h in 80 mM HEPES (pH 7.5), 1 mM spermidine, 33 mM MgCl2, 5% (v/v) DMSO, 3 mM ATP, 3 mM CTP, 3 mM UTP, 0.2 U of thermostable inorganic pyrophosphatase [New England Biolabs (NEB), Ipswich, MA], 400 ng of DNA template and approximately 2 µg of either T7RNAP WT or P266L. IVTs containing thG were performed using 4.8 mM thG and 0.48 mM GTP (10:1 ratio). Post-transcription, the DNA template was degraded by addition of 10 U of DNase I (Roche, Basel, Switzerland) and incubation at 37°C for 30 min.
For the OPME approach, 5 U of the RppH polyphosphatase (NEB) and NaCl (to a final concentration of 50 mM) was added to the DNase I-treated IVT reaction and incubated for 1.5 h at 37°C. Subsequently, 1 U of the Xrn-1 exonuclease (NEB) and NaCl (to a final concentration of 100 mM) was added to the reaction and incubated for 1.5 h at 37°C. To ensure higher Xrn-1 activity, we increased the temperature to 45°C for 5 s every 5 min; these reactions were performed using a thermal cycler.
Post-transcription and RNA processing, all reactions were subjected to a phenol-chloroform extraction and subsequently to a Zymo Clean and Concentrator™-25 (Zymo Research, Irvine, CA) to remove unincorporated nucleotides and short, abortive transcripts <18 nt in length (Figure S3). Total RNA yield was determined using Abs260 measurements on a NanoDrop 2000c spectrophotometer and the extinction coefficient of pre-tRNACys.
Fluorescence measurements
To generate a standard curve, increasing concentrations of thG in 20 mM Tris-HCl (pH 7.5) were aliquoted into a 384-well black microplate (Nunc) and fluorescence intensity was recorded using a Tecan M1000 microplate reader (λexc, 380 nm; λem, 452 nm). Relative fluorescence units were determined by subtracting the background fluorescence of blanks lacking thG, and RFU was then plotted against concentration of thG to generate a linear curve-fit (Excel). This standard curve was subsequently used to determine the concentration of thG in pre-tRNACys (Figure S1). To investigate if there was any quenching associated with thG present at the 5' terminus of pre-tRNACys, we compared the fluorescence of thG-initiated pre-tRNACys before and after alkaline hydrolysis, and found that the fluorescence values were nearly indistinguishable (data not shown). This finding lends confidence to the estimates of % 5'-modified RNAs that were determined from the thG-based standard curve.
Supplementary Material
Acknowledgements
We are grateful to Drs. Yuhong Zuo and Thomas Steitz (Yale University) for providing the T7RNAP P266L expression vector as well as a detailed purification protocol, and to Dr. Yitzhak Tor (University of California, San Diego) for generously providing thG used in this study and for his valuable feedback. We deeply appreciate the insightful comments and suggestions of Drs. Mikhail Kashlev (NCI), Craig Martin (UMass) and Irina Artsimovitch (OSU). We thank Prof. E. J. Behrman (OSU) for encouragement and support, and members of the Gopalan laboratory for their helpful suggestions. We thank the Ohio State University (Arts & Sciences Research Scholarships), NSF REU grant DBI-1062144, the Pelotonia Undergraduate Fellowship Program, and NIH R01GM120582 for supporting this work. Research reported in this publication was supported by the Office of Director, NIH (under award S10OD023582).
References
- [1].Alexander S, Devaraj N, Biochemistry 2017, 56, 5185–5193. [DOI] [PubMed] [Google Scholar]
- [2].Paredes E, Evans M, Das SR, Methods 2011, 54, 251–259. [DOI] [PubMed] [Google Scholar]
- [3].a) Masquida B, Beckert B, Jossinet F, N. Biotechnol. 2010, 27, 170–183; [DOI] [PubMed] [Google Scholar]; b) Milligan JF, Uhlenbeck OC, Methods Enzymol. 1989, 180, 51–62; [DOI] [PubMed] [Google Scholar]; c) Milligan JF, Groebe DR, Witherell GW, Uhlenbeck OC, Nucleic Acids Res. 1987, 15, 8783–8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Lee GH, Lim HK, Jung W, Hah SS, Bull. Korean Chem. Soc 2012, 33, 3861–3863; [Google Scholar]; b) Skipsey M, Hack G, Hoope TA, Shankey MC, Conway LP, Schroder M, Hodgson DRW, Nucleos. Nucleot. Nucl 2013, 32, 670–681; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhang L, Sun L, Cui Z, Gottlieb RL, Zhang B, Bioconjug. Chem. 2001, 12, 939–948; [DOI] [PubMed] [Google Scholar]; d) Williamson D, Cann MJ, Hodgson DRW, Chem Commun. 2007, 47, 5096–5098. [DOI] [PubMed] [Google Scholar]
- [5].a) Shin D, Sinkeldam RW, Tor Y, J. Am. Chem. Soc 2011, 133, 14912–14915; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li Y, Fin A, McCoy L, Tor Y, Angew. Chem. Int. Ed 2017, 56, 1303–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Tabor S, Richardson CC, Proc. Natl. Acad. Sci. USA 1986, 86, 4076–4080; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Walmacq C, Kireeva ML, Irvin J, Nedialkov Y, Lubkowska L, Malagon F, Strathern JN, Kashlev M, J. Biol. Chem 2009, 284, 19601–19612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Kotkowiak W, Pasternak A, Kierzek R, PLOS One 2016, 11, e0148282; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Padilla R, Sousa R, Nucleic Acids Res. 2002, 30, e138; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sochor F, Silvers R, Muller D, Richter C, Furtig B, Schwalbe H, J. Biomol. NMR 2016, 64, 63–74; [DOI] [PubMed] [Google Scholar]; d) Meyer AJ, Garry DJ, Hall B, Byrom MM, McDonald HG, Yang X, Yin YW, Ellington AD, Nucleic Acids Res. 2015, 43, 7480–7488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Durniak KJ, Bailey S, Steitz TA, Science 2008, 322, 553–557; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Guillerez J, Lopez PJ, Proux F, Launay H, Dreyfus M, Proc. Natl. Acad. Sci. USA 2005, 102, 5958–5963; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ramirez-Tapia LE, Martin CT, J. Biol. Chem. 2012, 287, 37352–37361; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Tang GQ, Nandakumar D, Bandwar RP, Lee KS, Roy R, Ha T, Patel SS, PLOS One 2014, 9, e91859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hofer K, Abele F, Schlotthauer J, Jaschke A, Bioconjug. Chem. 2016, 27, 874–877. [DOI] [PubMed] [Google Scholar]
- [10].Helmling C, Keyhani S, Sochor F, Furtig B, Hengesbach M, Schwalbe H, J. Biomol. NMR 2015, 63, 67–76. [DOI] [PubMed] [Google Scholar]
- [11].a) Huang F, He J, Zhang Y, Guo Y, Nat. Protoc. 2008, 3, 1848–1861; [DOI] [PubMed] [Google Scholar]; b) Huang F, Shi Y, Bioorg. Med. Chem. Lett 2010, 20, 6254–6257. [DOI] [PubMed] [Google Scholar]; c) Li N, Yu C, Huang F, Nucleic Acids Res. 2005, 33, e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Chen TH, Tanimoto A, Shkriabai N, Kvaratskhelia M, Wysocki V, Gopalan V, Nucleic Acids Res. 2016, 44, 5344–5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.