

Abstract
A nonheme iron(III) terminal methoxide complex, [FeIII(N3PyO2Ph)(OCH3)]ClO4, was synthesized. Reaction of this complex with the triphenylmethyl radical (Ph3C•) leads to formation of Ph3COCH3 and the one-electron reduced iron(II) center, as seen by UV-vis, EPR, 1H NMR, and Mössbauer spectroscopy. These results indicate that homolytic Fe-O bond cleavage occurs together with C-O bond formation, providing a direct observation of the “radical rebound” process proposed for both biological and synthetic nonheme iron centers.
Reserve for TOC
The radical rebound mechanism is a useful paradigm to explain C-H hydroxylations carried out by both heme and nonheme oxygenases, as well as analogous synthetic complexes.1 The mechanism involves the abstraction of a hydrogen atom from a C-H bond by a high-valent metal-oxo species, followed by recombination of the carbon radical (R•) with the newly formed metal-bound hydroxide ligand. The recombination, or “rebound” step, is presumed to involve homolytic cleavage of the Fe-O bond along with one-electron reduction of the iron center and formation of the new C-O bond (Scheme 1). The C-H cleavage step is typically rate-determining, making the rebound step too fast for direct observation. Efforts to probe this step indirectly have come from a number of experimental and computational studies.2 Alternative pathways to OH rebound are also possible, including radical dissociation in the synthetic systems and trapping by other reactive species (e.g. O2). 2a
Scheme 1. Oxygen and Halogen Radical Rebound Mechanisms.
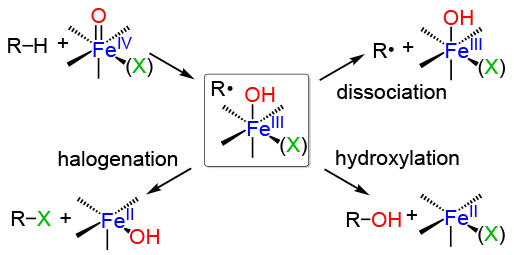
The rebound step is especially important for the nonheme iron halogenases (e.g. SyrB2, CytC3, WelO5).3 These enzymes selectively transfer a halogen ligand over an OH ligand in the putative rebound step, and the question of how these enzymes direct halogenation, versus the more common hydroxylation seen for the hydroxylases, has been the focus of synthetic model studies.4
Synthetic nonheme model systems involving high-valent Fe(O) species have provided evidence for both rebound and non-rebound pathways in C-H oxidations.5 Related computational studies have shown that when radical dissociation rates are competitive with rebound rates, a nonrebound mechanism may prevail, and possible causes for this reactivity were suggested.2a
The factors that control the rebound process, and sometimes result in nonrebound pathways, are critical in determining the final outcome of nonheme iron-mediated oxidations. For example, substrate orientation has been invoked as a key factor influencing rebound in the nonheme iron halogenases.3 However, first-coordination sphere effects, such as the identity of the coligands on Fe, the relative metal-O(X) bond strengths, Fe redox potentials, and the electronic structure of the Fe complex all may have a significant influence over directing rebound versus non-rebound reactivity in both enzymes and synthetic catalysts.
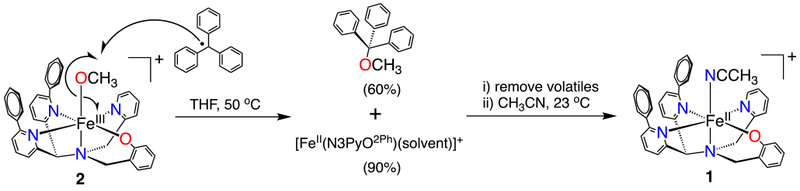
We have initiated efforts to develop complexes in which the rebound step can be directly interrogated. We recently described the first example of such a system for a heme-related iron-hydroxide complex.6 However, to our knowledge there has thus far been no direct observation of a radical rebound reaction with nonheme iron to produce a new C-O bond.7 Herein we report the synthesis and structural characterization of a rare, terminal iron(III)-methoxide complex, and show that it reacts directly with stabilized carbon radicals to give a methoxy ether and a reduced iron(II) complex.
Previous work from our lab described a pentadentate ligand, N3PyS, that produced a mononuclear iron(II) complex which reacted readily with O2 and NO.8 It was shown that incorporation of the single thiolato donor facilitated O2 reactivity.9 Reaction of [FeII(N3PyS)](BF4) with O2 led to S-oxygenation.8
For the current study, we replaced the phenylthiolate arm with a phenoxo donor and added phenyl substituents to give the new ligand N3PyOH2Ph (Scheme 2), with the objective of maintaining the electron-rich nature of the FeII center for O2 reactivity while avoiding ligand oxygenation. The bulky phenyl groups were incorporated to discourage µ-oxo dimer formation. The unsubstituted analog N3PyOH gives an oxo-bridged diferric complex from FeII and O2.10 A similar diphenyl-substituted N4Py system has led to our recent isolation and structural characterization of a metastable FeIV(O) complex.11
Scheme 2. Synthesis of [FeII(N3PyO2Ph)(CH3CN)] (ClO4) (1) and [FeIII(N3PyO2Ph)(OCH3)](ClO4) (2).
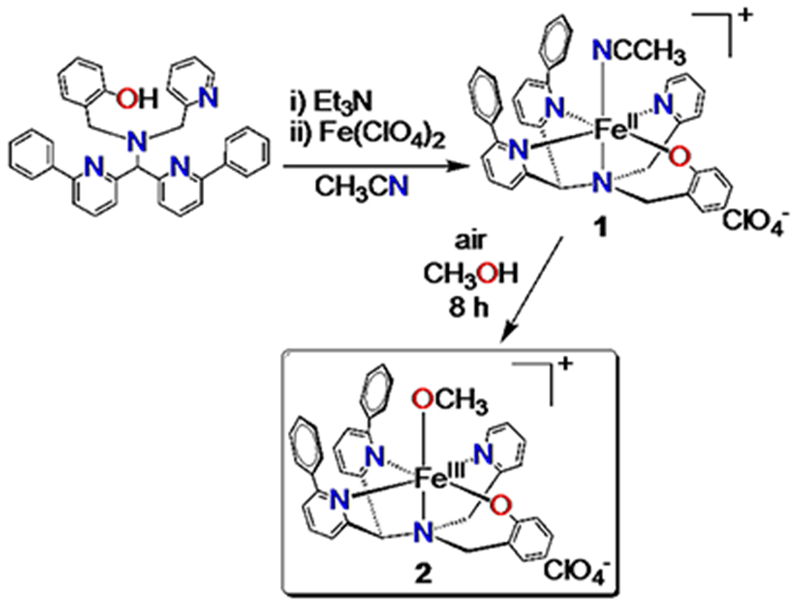
Reaction of N3PyOH2Ph with FeII(ClO4)2 and base in acetonitrile led to the isolation of a high-spin mononuclear FeII complex, [FeII(N3PyO2Ph)(CH3CN)](ClO4) (1) (Scheme 2). Dark yellow crystals were grown from MeOH/Et2O, resulting in the crystal structure shown in Figure 1. The ligand binds pentadentate to FeII, while the sixth site is occupied by acetonitrile. The Fe-N distances in 1 are similar to the Fe-N distances found in high-spin FeII complexes of both N3PyS and N4Py2Ph derivatives, with the exception of the phenyl-substituted pyridine donor trans to the phenolate group, which exhibits an elongated distance of 2.473(3) Å. A similarly elongated distance is seen for one of the phenyl-pyridyl donors (Fe-N = 2.378(13) Å) in [FeII(N4Py2Ph)(NCCH3)](BF4)2.11a The Fe-O distance in 1 is in line with other FeII-OAr complexes.12
Figure 1.
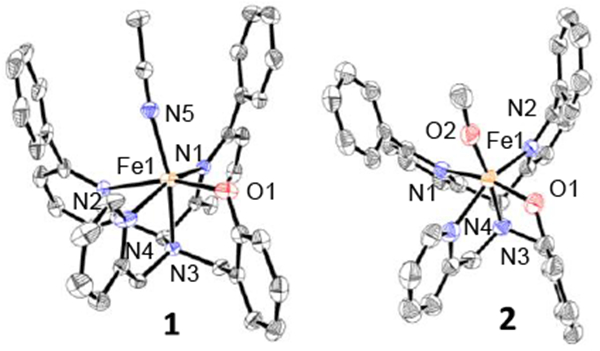
Displacement ellipsoid plots (50% probability) of the cations of 1 and 2 at 110(2) K. H atoms are not shown for clarity.
Reaction of 1 with air in methanol leads to a slow color change from dark yellow 1 to a new dark purple species. UV-vis spectroscopy shows the conversion of 1 to a new spectrum with a broad band at 550 nm (ε = 1290 M−1 cm−1), which is consistent with a phenolate-to-iron(III) charge-transfer transition.13 Crystallization of the purple product from MeOH/Et2O led to the crystal structure shown in Figure 1. The structure reveals an iron(III) complex with a terminal methoxide ligand in the open site, with formula [FeIII(N3PyO2Ph)(OCH3)](ClO4) (2) (Scheme 2). Crystals of 2 were examined by EPR spectroscopy, and revealed a relatively sharp feature at g = 4.26 (13 K, THF). This spectrum is consistent with a high-spin FeIII (S = 5/2) ground state. An Evans method measurement14 of 2 in THF-d8 gave μeff = 5.3 μB, close to the spin-only value (5.91 μB) for a mononuclear high-spin FeIII ion. The distance of the Fe-N bond for the phenyl-substituted pyridine donor trans to the phenolate donor of 2 is shorter than the same bond in 1 (2.341(2) Å vs. 2.473(3) Å), as expected for the increase in oxidation state. Terminal iron-alkoxide complexes are relatively rare. The closest analog to 2 is FeIII(N4Py)(OCH3)]2+.15 The Fe-OCH3 bond length for 2 of 1.784(2) Å) is similar when compared to the same bond in the closest analog, FeIII(N4Py)(OCH3)]2+ (1.772(3) Å), and is on the short end of the range seen for other FeIII-OCH3 complexes (FeIII-O: 1.77–1.90 Å).15–16
Given that 2 is a close analog of the FeIII(OH) rebound intermediate in Scheme 1, we hypothesized that 2 could be used for the direct examination of a radical rebound process. A suitable carbon radical (R•) might react with 2 to give a methoxy ether (ROCH3) product and the one-electron reduced iron(II) complex. Trityl radical (Ph3C•) is stable in organic solvents, and readily prepared in the solid state as a closed-shell dimer (Ph3C)2 (Gomberg’s dimer), which dissociates to give a known amount of the radical (~2% at 25 °C) in solution.17 Reaction of (Ph3C)2 with 2 in THF at 23 °C led to the slow decay (~48 h) of the FeIII-OCH3 complex as seen by UV-vis spectroscopy. At higher temperatures, Gomberg’s dimer dissociates to a greater extent,17 and heating the former reaction to 50 °C led to ~80% decay of the 570 nm band for 2 within 1 h (Figure S7). Monitoring the same reaction by EPR spectroscopy showed that the singlet at g = 4.26 assigned to 2 decreases upon reaction with Ph3C•, with an 80% loss of 2 after 1 h at 50 °C (Figure S9), in good agreement with the UV-vis data. The final product is EPR-silent, consistent with integer-spin FeII, the anticipated product.
Thin-layer chromatography (TLC) indicated that the rebound product Ph3COCH3 was formed after 1 h in reaction mixtures of 2 and (Ph3C)2 in THF at 50 °C. Analysis by 1H NMR spectroscopy in THF-d8 revealed the formation of a peak at δ 3.04 ppm after 60 min (Figure S13) which can be assigned to Ph3COCH3. Quantitation of this peak gave a yield of the methoxy ether of 60% (based on total 2). This result was corroborated by GC-FID, which revealed production of Ph3COCH3 in 58% yield. The GC and NMR yields are in good agreement with each other. Further improvements in yield could not be obtained by increasing reaction times up to 24 h. The yield for this reaction is likely limited by the concomitant formation of a side product of the trityl radical, which was identified by 1H NMR as a tautomeric form of Gomberg’s dimer that does not dissociate to release trityl radical,18 as well as slow background decomposition of 2 under the reaction conditions (Figure S8).
Mössbauer spectroscopy provided further insights regarding the rebound reaction of 2 with trityl radical. Isotope-enriched 2 (57Fe, 95.93%) was reacted with Gomberg’s dimer in THF at 50 °C for 70 min, and then frozen at 77 K for Mössbauer analysis (Figure 2). The spectrum of 2 before reaction, measured at 5.2 K, shows a 6-line pattern typical of a magnetically split iron(III) complex, overlaid with a quadrupole doublet. These signals are consistent with a mixture of FeIII in both slow- and fast-relaxation regimes, respectively. This analysis was confirmed when the sample was measured at 100 K (Figure 2), showing that the sextet collapsed into a single quadrupole doublet with parameters consistent with high-spin FeIII (δ = 0.50, ΔEQ = 1.29 mm s−1). The Mössbauer spectrum of a solid state sample of 2 at 100 K is nearly identical (Figure S17), further corroborating this analysis. Upon reaction with Ph3C•, the FeIII starting material disappears, and a new, sharp quadrupole doublet with parameters δ = 1.38 and ΔEQ = 3.35 mm s−1 is observed. The parameters of this new doublet are distinctive of high-spin iron(II), and quantification of the signal indicates that the final FeII species accounts for 90% of the total iron content. This result indicates that the slow background decay of 2 gives the same one-electron reduced FeII product.
Figure 2.
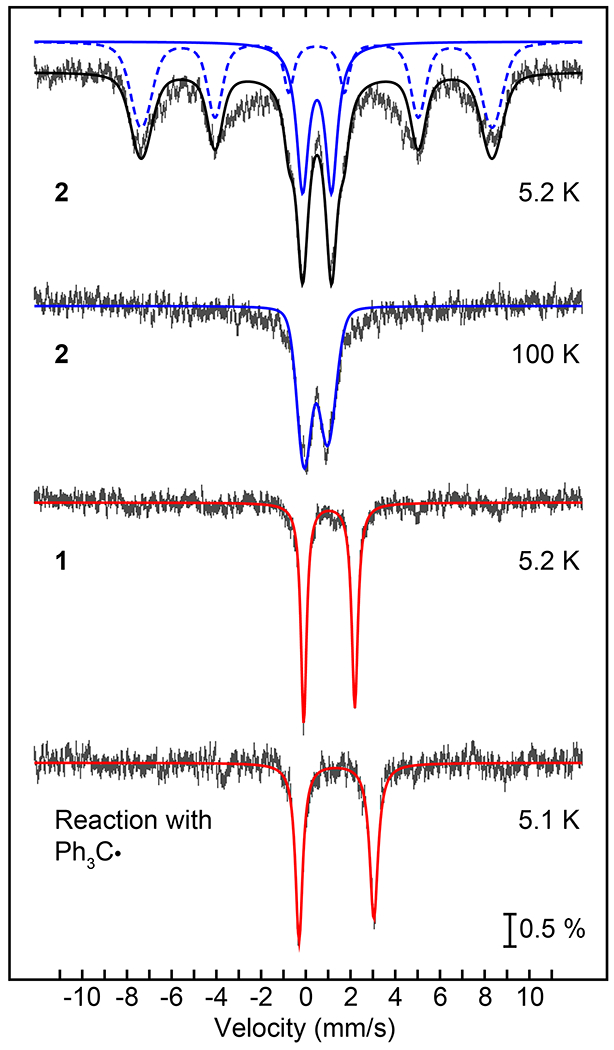
57Fe Mössbauer spectra (hatched lines) for: complex 2 in THF at 5.2 K together with the best fits for hs-FeIII in both the slow-(blue dashed line) and fast-relaxing (blue solid line) regimes (top); same sample as in (top) at 100 K and best fit (blue line) for a fast-relaxing quadrupole doublet as the major component (top middle); complex 1 in THF at 5.2 K and best fit (red line) for a hs-FeII quadrupole doublet (bottom middle); the reaction mixture of 2 and Gomberg’s dimer in THF at 50 °C after 70 min and best fit (red line) for a hs-FeII quadrupole doublet (bottom).
A Mössbauer spectrum of a recrystallized sample of 1 in THF shows a high-spin FeII quadrupole doublet (δ = 1.05, ΔEQ = 2.29 mm s−1), albeit with different parameters than the final reaction mixture described above. The differences observed between this spectrum and the final reaction mixture are most likely due to different solvent ligands occupying the sixth site of [FeII(N3PyO2Ph)]+. A THF solvent molecule likely occupies the open site in the reaction with Ph3C•, as compared to the coordinated CH3CN seen in 1. To gain further insight, DFT calculations were performed on optimized geometries of 1, 1-THF, and a 5-coordinate analog of 1. The calculated isomer shift of a 5-coordinate complex is lower than both the experimental data and calculated 6-coordinate complexes (Table S3). DFT calculations also show the isomer shift of 1-THF is higher than that for 1, as seen experimentally. Thus, DFT calculations support that a 6-coordinate product is formed. This analysis is further supported by paramagnetic 1H NMR, which revealed that the initial spectrum for 2, with peaks between +100 and –40 ppm, disappears upon reaction with trityl radical, leaving poorly-resolved features. However, removal of the THF and dissolution of the remaining brown residue in CD3CN gives an NMR spectrum that can be assigned to the acetonitrile-bound FeII complex (Figure S14).
Taken together, the UV-vis, EPR, NMR and Mössbauer data confirm that 2 reacts with Ph3C• to give the one-electron-reduced, radical rebound product [FeII(N3PyO2Ph)]+ and a new C-O bond. The rebound reaction is summarized in Scheme 3. In our work on Fe(OH) rebound with trityl radical, we have shown that a concerted rebound mechanism is the most likely scenario.6 We suggest a similar mechanism here, as shown in Scheme 3.
Scheme 3. Radical Rebound Reaction.
The synthesis and structural characterization of a rare, mononuclear terminal FeIII-methoxide complex allowed for the first direct observation of radical rebound with a nonheme iron complex to give a new C-O bond. The FeIII-OCH3 complex reacts efficiently with trityl radical in a rebound process that involves homolytic cleavage of the Fe-O bond and formation of Ph3COCH3 and the one-electron-reduced iron(II) complex. The current work provides support for the feasibility of the rebound pathway in C-H activation processes by nonheme iron-oxo species. However the probability of rebound versus cage escape of the carbon radical is a key question that remains challenging to assess. The direct examination of the radical rebound reaction described here foreshadows further experimental approaches to address these fundamental mechanistic issues.
Supplementary Material
ACKNOWLEDGMENT
The NIH (GM119374 to D.P.G. and G.N.L.J.) is gratefully acknowledged for financial support. G.N.L.J. further thanks the University of Otago and the Faculty of Science, the University of Melbourne for financial support.
Footnotes
ASSOCIATED CONTENT
Supporting Information. Syntheses, spectroscopy, X-ray crystallographic files (CIF) are available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Huang X; Groves JT, J. Biol. Inorg. Chem 2017, 22, 185–207. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ortiz de Montellano PR, Chem. Rev 2010, 110, 932–948. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kovaleva EG; Lipscomb JD, Nat. Chem. Biol 2008, 4, 186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).(a) Cho KB; Hirao H; Shaik S; Nam W, Chem. Soc. Rev 2016, 45, 1197–210. [DOI] [PubMed] [Google Scholar]; (b) Shaik S; Kumar D; de Visser SP; Altun A; Thiel W, Chem. Rev 2005, 105, 2279–2328. [DOI] [PubMed] [Google Scholar]
- (3).(a) Mitchell AJ; Zhu Q; Maggiolo AO; Anath NR; Hillwig ML; Liu X; Boal AK, Nat. Chem. Biol 2016, 12, 636–640. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Matthews ML; Neumann CS; Miles LA; Grove TL; Booker SJ; Krebs C; Walsh CT; Bollinger JM Jr., Proc. Natl. Acad. Sci 2009, 106, 17723–17728. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Martinie RJ; Livada J; Chang W.-c.; Green MT; Krebs C; Bollinger JM Jr.; Silakov A, J. Am. Chem. Soc 2015, 137, 6912–6919. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Wong C; Fujimori DG; Walsh CT; Drennan CL, J. Am. Chem. Soc 2009, 131, 4872–4879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) Planas O; Clemancey M; Latour J-M; Company A; Costas M, Chem. Commun 2014, 50, 10887–10890. [DOI] [PubMed] [Google Scholar]; (b) Puri M; Biswas AN; Fan R; Guo Y; Que L Jr., J. Am. Chem. Soc 2016, 138, 2484–2487. [DOI] [PubMed] [Google Scholar]
- (5).(a) Cho K-B; Wu X; Lee Y-M; Kwon YH; Shaik S; Nam W, J. Am. Chem. Soc 2012, 134, 20222–20225. [DOI] [PubMed] [Google Scholar]; (b) Bae SH; Seo MS; Lee YM; Cho KB; Kim WS; Nam W, Angew. Chem. Int. Ed. Engl 2016, 55, 8027–8031. [DOI] [PubMed] [Google Scholar]; (c) Rana S; Dey A; Maiti D, Chem. Commun 2015, 51, 14469–14472. [DOI] [PubMed] [Google Scholar]
- (6).Zaragoza JPT; Yosca TH; Siegler MA; Moënne-Loccoz P; Green MT; Goldberg DP, J. Am. Chem. Soc 2017, 139, 13640–13643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) The observation of metal-bound C-O products following a related rebound process for a nonheme RuIV(O) complex was described. Kojima T; Nakayama K; Ikemura K; Ogura T; Fukuzumi S, J. Am. Chem. Soc 2011, 133, 11692–11700. [DOI] [PubMed] [Google Scholar]; (b) Smith JM; Mayberry DE; Margarit CG; Sutter J; Wang H; Meyer K; Bontchev RP, J. Am. Chem. Soc 2012, 134, 6516–6519. [DOI] [PubMed] [Google Scholar]; (c) Jang ES; McMullin CL; Kass M; Meyer K; Cundari TR; Warren TH, J. Am. Chem. Soc 2014, 136, 10930–10940. [DOI] [PubMed] [Google Scholar]; (d) Iovan DA; Betley TA, J. Am. Chem. Soc 2016, 138, 1983–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).McQuilken AC; Jiang Y; Siegler MA; Goldberg DP, J. Am. Chem. Soc 2012, 134, 8759–8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Draksharapu A; Li Q; Roelfes G; Browne WR, Dalton Trans 2012, 41, 13180–13190. [DOI] [PubMed] [Google Scholar]
- (10).(a) Ligtenbarg AGJ; Oosting P; Roelfes G; La Crois RM; Lutz M; Hage R; Spek AL; Feringa BL, Chem. Commun 2001, 385–386. [Google Scholar]; (b) Unjaroen D; Swart M; Browne WR, Inorg. Chem 2017, 56, 470–479. [DOI] [PubMed] [Google Scholar]
- (11).(a) Sahu S; Widger LR; Quesne MG; de Visser SP; Matsumura H; Moënne-Loccoz P; Siegler MA; Goldberg DP, J. Am. Chem. Soc 2013, 135, 10590–10593. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sahu S; Zhang B; Pollock CJ; Durr M; Davies CG; Confer AM; Ivanović-Burmazović I; Siegler MA; Jameson GNL; Krebs C; Goldberg DP, J. Am. Chem. Soc 2016, 138, 12791–12802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Boyle TJ; Ottley LAM; Apblett CA; Stewart CA; Hoppe SM; Hawthorne KL; Rodriguez MA, Inorg. Chem 2011, 50, 6174–6182. [DOI] [PubMed] [Google Scholar]
- (13).Shakya R; Allard MM; Johann M; Heeg MJ; Rentschler E; Shearer JM; McGarvey B; Verani CN, Inorg. Chem 2011, 50, 8356–8366. [DOI] [PubMed] [Google Scholar]
- (14).(a) Evans DF; Jakubovik DA, J. Chem. Soc. Dalton Trans 1988, 2927–2933. [Google Scholar]; (b) Confer AM; Mcquilken AC; Matsumura H; Möenne-Loccoz P; Goldberg DP, J. Am. Chem. Soc 2017, 139, 10621–10624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Roelfes G; Lubben M; Chen K; Ho RYN; Meetsma A; Genseberger S; Hermant RM; Hage R; Mandal SK; Young VG Jr.; Zang Y; Kooijman H; Spek AL; Que L Jr.; Feringa BL, Inorg. Chem 1999, 38, 1929–1936. [DOI] [PubMed] [Google Scholar]
- (16).(a) Jonas RT; Stack TDP, J. Am. Chem. Soc 1997, 119, 8566–8567. [Google Scholar]; (b) Chang K-C; Huang C-J; Chang Y-H; Wu Z-H; Kuo T-S; Hsu H-F, Inorg. Chem 2016, 55, 566–572. [DOI] [PubMed] [Google Scholar]; (c) Moon D; Lah MS, Inorg. Chem 2002, 41, 4708–4714. [DOI] [PubMed] [Google Scholar]; (d) Kerber WD; Perez KA; Ren C; Siegler MA, Inorg. Chem 2014, 53, 11507–11516. [DOI] [PubMed] [Google Scholar]
- (17).(a) Gomberg M, J. Am. Chem. Soc 1914, 36, 1144–1170. [Google Scholar]; (b) Ihde AJ, Pure Appl. Chem 1967, 15, 1–14. [Google Scholar]
- (18).Chen C; Lee H; Jordan RF, Organometallics 2010, 29, 5373–5381. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.