

Abstract
Patients with chronic hepatitis C virus (HCV) infection risk complications of cirrhosis, liver failure, and hepatocellular carcinoma (HCC). Previously, our proteomic examination of hepatocytes carrying a HCV-replicon revealed that deregulation of cytoskeletal dynamics may be a potential mechanism of viral-induced HCC growth. Here, we demonstrate the effect of HCV replication on the microtubule regulator stathmin (STMN1) in HCC cells. We further explore how the altered activity or synthesis of stathmin affects cellular proliferation and sensitivity to apoptosis in control HCC cells (Huh7.5) and experimental HCV-replicon harboring HCC cells (R-Huh7.5). The HCV-replicon harboring HCC cells (R-Huh 7.5) lack viral structural genes/proteins for acute infectivity and thus is the standard model for in vitro chronic infection study. Knockdown of endogenous stathmin reduced sensitivity to apoptosis in replicon cells. Meanwhile, constitutively active stathmin increased sensitivity to apoptosis in replicon cells. In addition, overexpression of constitutively active stathmin reduced cell proliferation in both control and replicon cells. These findings implicate, for the first time, a novel role for stathmin in viral replication–related apoptosis. Stathmin’s potential role in HCV replication and HCC make it a candidate for the future study of viral-induced malignancies.
Keywords: Hepatitis C virus, hepatocellular carcinoma, STMN1, microtubule, apoptosis, phosphorylation
Introduction
Despite recent pharmaceuticals’ milestone achievement in providing highly effective drugs to treat hepatitis C virus (HCV), the high cost of these interferon-free combination regimens continues to limit access of these potentially curative and life-saving treatments to many chronically infected patients. Meanwhile, the global infection rates of the HCV are predicted to increase throughout the next 2 decades, with third-world countries experiencing the heaviest burden of morbidity.1 Although HCV infection can commonly remain asymptomatic, persistent infection can cause serious inflammation and scarring of liver tissue that will eventually lead to cirrhosis.2 Hepatitis C patients with cirrhosis are also at high risk for developing irreversible liver failure and hepatocellular carcinoma (HCC), the sixth most common cancer worldwide.3 The HCV’s role in HCC transformation is a current field of scientific interest, as virus targeting therapeutics also present a unique opportunity for liver cancer prevention in the hepatitis C patient demographic.
Our previously published mass spectrometry study of HCC cells characterized the phospho-proteomic changes induced by the presence of HCV to uncover candidate proteins that potentially alter the intracellular environment for enhanced viral replication.4 Notably, our analysis highlighted the deregulation of cytoskeletal dynamics as a particular mechanism of interest. Cytoskeletal structural elements (microtubules, actin microfilaments, and intermediate filaments) regulate several important cellular functions including maintaining a cell’s shape, facilitating cellular movement, vesicle transport, and ensuring proper stepwise cell division.5 To interrogate the potential role of cytoskeletal changes in relation to HCV replication and its connection to HCC, we focused on the microtubule regulator stathmin/oncoprotein18 (STMN1/Op18).
Stathmin, a microtubule regulator detected by our proteomic experiments, presents a compelling challenge to investigate the molecular pathogenesis of HCV on HCC growth through alterations to the cytoskeleton. Past studies have characterized the oncogenic potential of stathmin by its overexpression in several different cancers including breast,6 ovarian,7 and lung carcinoma.8 Higher stathmin is associated with an increased risk for metastases in esophageal squamous cell carcinoma9 and a worse prognosis in liver cancer.10 Overexpression of stathmin was also previously reported to inhibit macrophage interferon-γ (IFNγ) and lipopolysaccharide innate immune defense against viral infection.11 The HCV has been shown to interact with the protein signal transducer and activator of transcription 3 (STAT3), an upstream transcription factor that antagonizes stathmin’s destabilizing effect on microtubules.12 In its active form, stathmin induces the breakdown of tubulin polymers in a process known colloquially as “microtubule catastrophe.”13 The deactivation of stathmin activity may be important to HCV as a means of controlling the stability of the microtubule cytoskeleton, which serves as a highway of vesicle trafficking.14 Various research widely supports the significance of intracellular microtubule network in mediating interactions between HCV core proteins, hepatocyte lipid droplets, and the viral replication complex.15 During the initial phase of viral replication, the virus uses microtubules and motor proteins for movement within a cell to assemble and distribute viral progeny for eventual release into the extracellular matrix.16 Yet, the specifics of how stathmin expression and activity influence HCV remain unclear. Due to the broad range of cellular and viral processes that could be disrupted by microtubule destabilization, stathmin provides a unique opportunity to elucidate how deregulation of this protein affects the growth and survival of HCC cells in chronic hepatitis C.
Here, we report the examination of HCV’s influence on hepatocyte apoptosis sensitivity and growth through the manipulation of stathmin expression and activity. These in vitro phenotypic responses may display how stathmin-facilitated changes to the microtubule cytoskeleton can create an intracellular environment that promotes HCV replication and HCC proliferation.
Methods
Cell culture
Huh7.5, R-Huh7.5, and 293T cells were cultivated in Dulbecco’s Modified Eagle Medium (DMEM) with 10% fetal bovine serum, 1% glutamine, and 1% penicillin streptomycin at 5% CO2 and 37°C. Huh7.5 was a gift from Charles Rice (The Rockefeller University, New York, NY, USA). Subgenomic replicon HCV cells (R-Huh7.5) were created as previously described17 and incubated with 500 µg/mL of Geneticin for selection and maintenance. 293T cells were purchased from ATCC (CRL-11268).
Infectious virus production
pNRLFC was transcribed in vitro, and the purified RNA was electroporated into Huh7.5 cells to generate infectious viral supernatant as previously described.18
Tissue microarray immunohistochemistry
Staining was performed on patient core liver samples on tissue microarray slides prepared as previously described.19 A total of 95 cases (3 cores for each case) were used for staining and subsequent analysis (36 uninfected patients and 59 hepatitis C patients, patients with hepatitis B infection were excluded). Slides were deparaffinized and steamed in sodium citrate buffer (10 mM sodium citrate, 0.05% Tween 20, pH 6.0) for 20 minutes. Slides were cooled in running tap water for 10 minutes and then washed twice in distilled water for 5 minutes. Blocking was performed using phosphate-buffered saline (PBS) with 0.01% Tween (PBS-T) with 0.5% bovine serum albumin for 1 hour and then primary total stathmin antibody (Cell Signaling Technology, Danvers, MA, USA) was incubated at room temperature for 1 hour. Slides were washed with PBS-T. Secondary HRP antibody and DAB staining performed using DAKO LSAB System-HRP (Carpinteria, CA, USA) per manufacturer’s protocol. The staining procedure was performed using the Cell Signaling Technology Signal Stain Boost per manufacturer’s protocol. Slides were digitized on a ScanScope AT (Leica Biosystems, Inc., Vista, CA, USA) and morphometric analysis performed with Definiens’ Tissue Studio (Definiens, Inc., Parsippany, NJ, USA) to determine the percentage of stathmin-positive cells in a non-biased method. Briefly, stain-specific algorithms were created using the predefined cytoplasmic detection module and classification tool. Positive, and negative stained cells within each core were identified. Thresholds were set to classify hematoxylin stain for nuclei and DAB stain for positive cytoplasmic staining. The data were exported to Excel for further statistical analysis.
Western blot
Lysates were prepared with Triton X-100 lysis buffer (0.5% Triton X-100, 10 mM Tris, 150 mM NaCl, 5 mM EDTA, 1 mM Sodium Orthovanadate, 40 mM Sodium Fluoride, 50 mM Beta-Glycopyrophosphate, 5% Sigma Aldrich Protease Inhibitor Cocktail, and 10% Glycerol). Protein samples were separated using sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred overnight onto polyvinylidene difluoride membrane (EMD Millipore, Darmstadt, Germany). Western blots were incubated with primary antibodies (overnight, 4°C) and appropriate secondary antibodies (1 hour, room temperature). Primary antibodies for stathmin, β-actin, and green fluorescent protein (GFP) were purchased from Cell Signaling Technology. Primary antibody for tubulin was purchased from the Developmental Studies Hybridoma Bank at the University of Iowa (Iowa City, IA, USA). Secondary antibodies were purchased from Jackson ImmunoResearch (West Grove, PA, USA). Western blots were visualized with ECL Prime Western Blot Detection Reagent (GE Healthcare, Buckinghamshire, UK) and Odyssey Fc (Li-Cor, Lincoln, Nebraska, USA). All Western blot analyses were performed using biological duplicates.
Small-interfering RNA and transfection materials
The small-interfering RNA (siRNA) and controls were obtained from Santa Cruz Biotechnology (Dallas, TX, USA). Transfection (20 nM siRNA) was performed using Lipofectamine 2000 obtained from Invitrogen (Carlsbad, CA, USA) as per the manufacturer’s protocol.
Caspase 3 apoptosis assay
Cells were transfected with siRNA for 24 hours. About 1 µM staurosporine was added for 24 hours to induce apoptosis and then lysed with Triton X-100 lysis buffer. About 40 µL of each cell lysate was incubated with 50 µL HEPES protease assay buffer (20 mM HEPES, 2 mM DTT, 10% glycerol in distilled water) and 5 µL caspase 3 substrate from BD Pharmingen (Franklin Lakes, NJ, USA) for 2 hours. Caspase 3 activity was recorded using fluorescence at EX 365 and EM 410-460. Fluorescence was quantified using a Modulus Microplate Reader from Turner Biosystems (Sunnyvale, CA, USA). All caspase 3 assay analyses were done using biological triplicates.
Plasmid cloning
RNA was extracted from Huh7.5 cells using QIAGEN RNeasy Mini Kit (Hilden, Germany) and complementary DNA (cDNA) for wild-type (WT) STMN1 was created from the extracted RNA using Invitrogen SuperScript III First-Strand Synthesis System for RT-PCR Kit. Using standard PCR and ligation protocols, WT STMN1 cDNA was cloned into lentivirus vector pRRL-sin-cPPT-MCS-IRES-emdGFP (pRRL) which was obtained from the UCLA Vector Core.
Site-directed mutagenesis
Stathmin phosphorylation site mutants (Ser16, Ser25, Ser38, Ser63, and Quad) were created using the Q5 Site-Directed Mutagenesis Kit (New England BioLabs, Ipswich, MA, USA). The following primers were used: Ser16 Forward 5′GAAGCGTGCCGCAGGCC AGGCTT3′ and Ser16 Reverse 5′TCCAGTTCTTTCACCTGGATATCAGAAGAAGC3′, Ser25 Forward 5′CTTTTGAGCTGATTCTCGCCCCTCGGTCAAAAG3′ and Ser25 Reverse 5′TCAAAAGCCTGGCCTGAG3′, Ser38 Forward 5′GTTCCAGAATTCCCCCTTGCCCC TCCAAAGAAGAAGG3′ and Ser38 Reverse 5′TCTGGAACAGATTCTTTTGACC3′, Ser63 Forward 5′GAAGAAAGACGCAAGGCCCATGAAGCTGAGG3′ and Ser63 Reverse 5′TCTTCTGCAGCTTCTAATTTCTTC3′. STMN1 mutagenic primers changed serine phosphorylation sites to alanine. All mutagenesis was performed according to manufacturer’s protocol. DNA was extracted from transformed colonies selected with ampicillin. Sequencing confirmed successful mutagenesis of the serine phosphorylation sites and no additional unintended mutations.
Lentivirus packaging and transduction
Lentiviruses were created using 293T cells. About 1.9 µg of purified plasmid DNA, 0.5 µg pSL3 (envelope), 3.8 µg pSL4 (packaging), and 1.9 µg pSL5 (Rev) were cotransfected into 293T cells with X-tremeGENEene 9 (Roche, Basel, Switzerland) and DMEM media. The lentivirus media was collected 48 hours later using a 0.45 µM filter unit and stored at 4°C. About 293T media was replaced and collected in the same way 24 hours later. The lentivirus media was concentrated with the Clontech Lenti-X Concentrator according to the manufacturer’s protocol for 24 hours in 4°C. The virus pellet was then resuspended in DMEM media and aliquoted for storage at −80°C. For transduction, lentiviral media was added to cells in a 6-well format (40% confluent) for a total volume of 500 µL. The media was replaced with 1 mL fresh DMEM media after 5 days.
MTT cell viability assay
Cells were transduced with stathmin mutant lentiviruses on 96-well plates. MTT assays were completed using ATCC MTT Cell Proliferation Assay Kit (Manassas, VA, USA) while following the manufacturer’s protocol. The cells were incubated with 10 µL of MTT reagent for 2 hours at 37°C and then lysed with 100 µL of detergent reagent. After overnight incubation at room temperature in the dark, cells were read with absorbance at 600 nm. All MTT assay analyses were done using biological triplicates.
Plasmid transient transfection
Stathmin WT and mutant overexpression vectors were transfected into cells using Lipofectamine 2000 (Invitrogen) as per the manufacturer’s protocol. The media was replaced 8 hours after transfection to prevent cytotoxicity. Transfection was repeated again 24 hours later. On visual confirmation that transfection reached at least 20% to 30% by GFP fluorescence, cells were harvested for further experimentation.
Flow cytometry apoptosis assay
Flow cytometry staining was performed on transiently transfected cells using the BD Pharmingen PE AnnexinV Apoptosis Detection Kit I. Prior to harvesting, cells were treated for 24 hours with 10 µM staurosporine (Cell Signaling Technology) to induce apoptosis. Cells were harvested using 0.25% trypsin (ThermoFisher Scientific, Waltham, MA, USA) and washed with cold PBS. After removing the supernatant, cell pellets were resuspended with 1× annexin-binding buffer and stained with AnnexinV-PE and 7-AAD at room temperature for 10 minutes. Stained samples were taken immediately for analysis and AnnexinV positivity was measured on GFP-gated cells. All flow cytometry analyses were done using biological triplicates.
Statistics
All statistical tests (P values from 2-way Student t test, standard error, standard deviation) were performed using Microsoft Excel. A P value of <.05 was significant.
Results
HCV induces stathmin expression
Endogenous levels of total stathmin were compared between control, replicon-harboring HCC, and NRLFC HCV-infected HCC cells: Huh7.5, R-Huh7.5 (HCV-replicon), V-Huh7.5 (HCC cells infected with a J6/JFH-based reporter virus NRLFC).18 Both replicon and HCV-infected cells exhibited a higher amount of stathmin as detected by Western analysis (Figure 1A). Furthermore, under serum starvation, stathmin expression was significantly reduced in control Huh7.5 but remained upregulated in replicon R-Huh7.5 (Figure 1A). The expected decrease in stathmin expression in growth-arrested cells, which was noted in control Huh7.5 but not evident in replicon R-Huh7.5, suggests the HCV-replicon–induced stathmin upregulation in a fashion that is independent of cell cycle processes. V-Huh7.5 with and without serum starvation is included to show that it has similar effects on stathmin expression as R-Huh7.5 does.
Figure 1.

Stathmin levels are elevated in HCV-infected livers and in replicon-harboring Huh7.5 cells. (A) Western blot with control Huh7.5, replicon R-Huh7.5, and NRLFC HCV-infected V-Huh7.5 to determine total stathmin levels in whole cell lysates. (+) Cells are not serum starved, whereas (−) cells are serum starved. (B) Tissue microarray immunohistochemistry staining for total stathmin in cirrhotic liver tissue from (−HCV) uninfected patients compared with (+HCV) patients infected with hepatitis C. Staining was quantified and analyzed based on percentage of cells showing positive staining. Percent cells positive for stathmin staining was significantly higher in tissue from patients with hepatitis C (P < .001). Error bars reflect standard error. Immunostaining for phospho-stathmin was attempted and no signal could be detected suggesting that phospho-stathmin epitope is lost during formalin fixation and paraffin embedding as is commonly seen with phospho-epitopes (data not shown). (C) Representative immunostaining for stathmin in liver tissue from cirrhotic patients with and without HCV infection. Additional images are shown in supplemental Figure 1.
To determine the effect of HCV infection in vivo, immunohistochemistry staining of nontumor tissue samples from HCC patients with and without HCV was performed. Increased staining for stathmin was observed in infected patients (Figure 1B and 1C, and Supplemental Figure 1) suggesting that HCV infection either directly or indirectly induces stathmin expression. The viral-mediated increase in stathmin staining allowed us to observe the intrinsic effect of HCV noted in cell lines on patient tissue samples. This correlation directed the next experimental steps in altering the messenger RNA (mRNA) expression or protein activity of stathmin to further explore downstream cellular effects.
Stathmin inhibition affects apoptosis
Changes in stathmin expression in replicon cells suggested the possibility that this protein may affect cellular processes such as apoptosis. The siRNA-mediated knockdown studies were performed to interrogate the effect of endogenous stathmin protein levels on hepatocyte cell death. Western blot analysis confirmed stathmin siRNA knockdown through decreased protein detection (Figure 2A). Positive detection of viral nonstructural protein 5A (NS5A) in replicon R-Huh7.5 cells depicted persistent viral replication after stathmin siRNA transfection, suggesting that any differential apoptosis noted in subsequent experiments is not the result of an siRNA effect on HCV replication. After stathmin knockdown, the resulting level of apoptosis varied. Staurosporine was used to induce apoptosis prior to assessing caspase 3 levels. Given that staurosporine is an effective inducer of caspase dependent and independent apoptosis, it provides a useful model to determine the impact of stathmin on apoptosis sensitivity. This methodology has been previously applied to other viral-related apoptosis studies.20-24 In the absence of the virus, there was greater apoptosis with stathmin knockdown as compared with siRNA control. In contrast, stathmin knockdown resulted in decreased apoptosis in replicon cells (Figure 2B).
Figure 2.
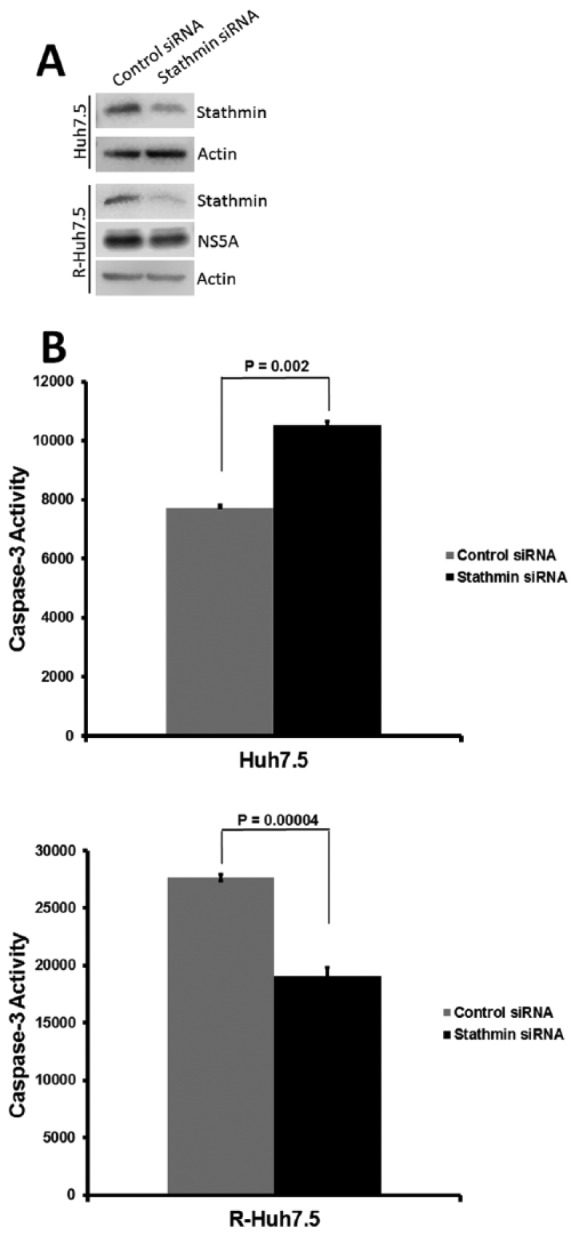
Stathmin knockdown reduces staurosporine induced caspase 3 activity in replicon cells. (A) Western blot for stathmin with and without knockdown. The control Huh7.5 and replicon R-Huh7.5 Western blots were developed with different exposure times. (B) Effect of stathmin knockdown on staurosporine-induced apoptosis as measured by caspase 3 activity in control Huh7.5 and replicon R-Huh7.5 cells.
Stathmin activation affects cellular viability
To further investigate the role of stathmin activity on cell viability, we generated and sequenced GFP-tagged mutant stathmin lentiviruses in which either 1 (S16A, S25A, S38A, S63A) or all 4 (Quad) serine phosphorylation sites are replaced with alanine. These mutated sites cannot be phosphorylated by upstream kinases, potentially allowing stathmin to remain in a constitutively active state. The GFP, WT, and mutant stathmin lentiviruses were transduced into 293T cells to amplify the plasmids, which were then transfected into control Huh7.5 and replicon R-Huh7.5 cells. The GFP transfection efficiency reached around 30% in Huh 7.5 cells and 20% in R-Huh7.5 cells (Supplemental Figure 2) and was similar across all plasmid transfections (GFP, WT, S16A, S25A, S38A, S63A, and Quad) as demonstrated by flow cytometry (data not shown). Overexpression of the lentivirus vectors was observed in both cell lines through detection of stathmin on Western blot (Figure 3A). Expression of the stathmin phospho-site mutants altered cell viability as indicated by MTT assay (Figure 3B and C). For both cell lines, the cell viability of GFP and WT stathmin was not altered when compared with 3 of the phospho-site mutants (S16A, S25A, and S63A). However, S38A stathmin and Quad stathmin exhibited significantly lower amount of cellular viability compared with GFP and WT stathmin for both cell lines. For control Huh7.5, the difference in cellular viability for S38A and Quad as compared with WT stathmin was 36.9% and 20.1% (Figure 3B). For replicon R-Huh7.5, the cellular viability for S38A and Quad as compared with WT stathmin were 25.9% and 20.3% (Figure 3C). Individual phospho-site mutants’ impact on the cellular viability of control cells was compared with the effect of each respective mutant in replicon cells. A statistically significant difference for S38A (P = .0002) and Quad (P = .012) mutants was detected between the 2 cell lines.
Figure 3.
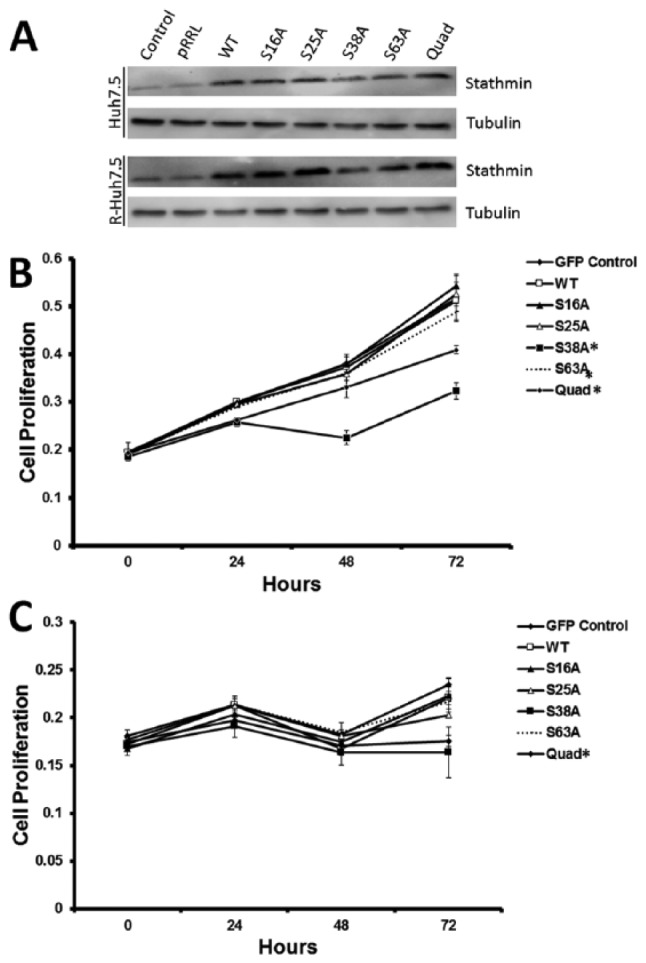
Activation of stathmin reduces cell viability. (A) Western blot of control Huh7.5 and replicon R-Huh7.5 cells with lentiviruses carrying empty GFP vector (pRRL), stathmin wild-type (WT), and phospho-stathmin mutants (S16A, S25A, S38A, S63A, and Quad). MTT cell proliferation assays with a 72-hour time course on (B) control Huh7.5 and (C) replicon R-Huh7.5 cells transduced with lentiviruses carrying empty GFP vector (pRRL), stathmin wild-type (WT), and phospho-stathmin mutants (S16A, S25A, S38A, S63A, and Quad). (* denotes P < .05 compared with WT). GFP indicates green fluorescent protein.
Stathmin activation sensitizes cells to apoptosis in the setting of viral replication
Because the S38A and Quad stathmin mutants negatively affected cell proliferation, we further investigated the impact of the GFP-tagged phospho-site mutant vectors on cellular sensitivity or resistance to the induction of apoptosis. AnnexinV-PE positivity was measured by flow cytometry, gating on GFP-positive cells to measure apoptosis. In the control cell line, there was no alteration in apoptosis (P > .05) when compared with WT stathmin (Figure 4A). However, in the replicon cell line, there was increased sensitivity to apoptosis (P < .05) in cells expressing Quad stathmin when compared with WT stathmin (Figure 4B).
Figure 4.
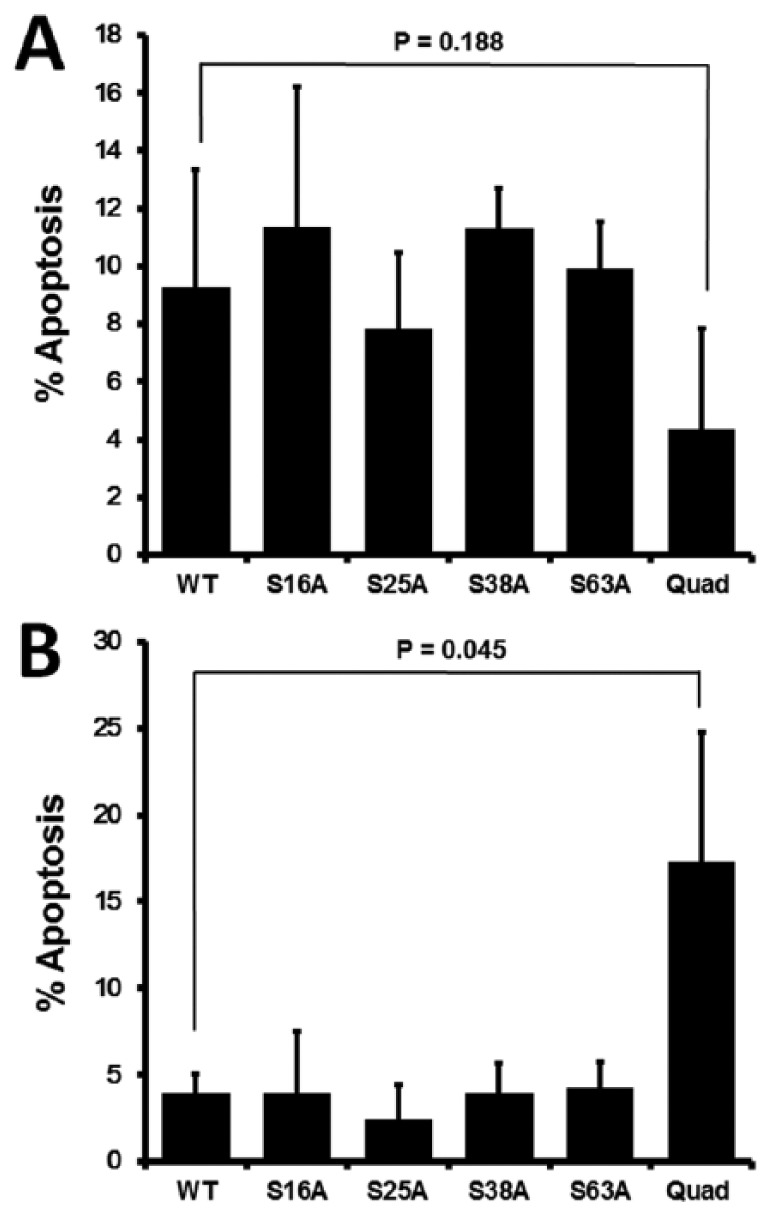
Quad phospho-site mutant stathmin increases sensitivity to apoptosis only in the context of HCV replication. Staurosporine-induced apoptosis as measured by AnnexinV staining in cells transfected with the stathmin wild-type (WT) or phospho-site mutant (S16A, S25A, S38A, S63A, and Quad) vectors. AnnexinV positivity was measured in GFP positive cells and displayed as percent apoptosis for (A) control Huh7.5 and (B) replicon R-Huh7.5. GFP indicates green fluorescent protein.
Discussion
Overview
Previous studies have analyzed the effect of pseudophosphorylated stathmin on microtubule polymerization25 and the influence of stathmin phosphorylation mutants on Xenopus embryo development.26 However, this is the first study that uses phospho-site stathmin mutants to interrogate the impact of constitutively active stathmin on HCC proliferation and sensitivity to apoptosis. Through modulation of stathmin expression or activity this study examines stathmin’s role in apoptosis and proliferation in the setting of HCV replication. Replicon cells (R-Huh7.5) were used as a model to examine effects of chronic HCV replication and thus does not apply to acute HCV infection.
In summary, stathmin was manipulated through 2 approaches: siRNA inhibition of stathmin mRNA and mutation of stathmin regulatory phosphorylation sites. Decreased stathmin levels resulted in decreased sensitivity to apoptosis only in the presence of viral replication. In addition, constitutive activation of stathmin increased sensitivity to apoptosis. These results suggest that constitutive stathmin activation affects cell viability in the setting of viral replication, potentially through a combination of cell cycle inhibition and apoptosis.27 Active and inactive forms of stathmin are required at different points within the cell cycle to regulate cell divison.28 These observations raise the possibility that HCV modulates the activity of stathmin, which could lead to a deregulated ratio of active to inactive stathmin that promotes a permissive intracellular environment.
Hypothesized mechanism
Part of the host response to counter some viruses includes upregulation of immune mechanisms that can enhance apoptosis. Many chronic viral infections, subsequently, persist partly due to how the virus can successfully alter its host response to inhibit apoptosis.29 By suppressing this host defense mechanism, the virus hypothetically would have a longer time to replicate, therefore ensuring its higher chances of survival. This immune viral-escape molecular mechanism is supported by previously examined viruses that are known to resist apoptosis during chronic active infection, such as adenovirus,30 herpesvirus, and Epstein-Barr virus.29,31,32
Upregulation of stathmin may reflect an initial protective host cell defense by promoting apoptosis. However, the virus may manipulate this response to create an intracellular environment that is proviral (Figure 5). As knockdown of endogenous stathmin allowed replicon cells to have greater resistance against apoptosis, a lower stathmin abundance may be advantageous to HCV replication. Therefore, a potential viral mechanism to circumvent any upregulation of stathmin via host immune response would be inactivation of stathmin through phosphorylation to simulate low abundance. As the stathmin mutant responsible for the highest apoptosis (Quad) had the combination of all 4 serine residues altered to alanine, it is conceivable that when all phospho-sites are disrupted, the virus loses the ability to inactivate stathmin leading to greater apoptosis sensitivity. Therefore, modulation of upstream kinases and their signal transduction pathways may be important means through which HCV could influence stathmin function.
Figure 5.
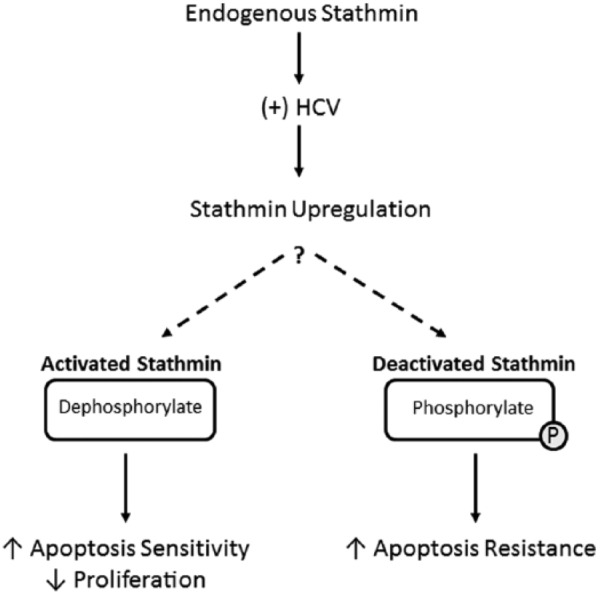
Hypothesized mechanisms of stathmin activity. Replication of HCV may affect stathmin expression in HCC cells. This could lead to viral-mediated deactivation of stathmin through serine phosphorylation, possibly through increase upstream kinase activity. Deactivation of stathmin may increase cellular resistance to apoptosis. HCC indicates hepatocellular carcinoma; HCV, hepatitis C virus.
Targets for future stathmin investigation
This investigation of stathmin in the setting of chronic HCV replication led us toward identifying potential candidates for further therapeutic development. Only 2 stathmin mutants negatively affect cell proliferation. These 2 vectors, S38A and Quad, both contain the phosphorylation site at the Ser38 residue altered to alanine. Previous studies have identified phosphorylated Ser38-stathmin as a characteristic biomarker of aggressive endometrial cancer.33 Moreover, the Ser38 residue is regulated by several kinases including cyclin-dependent kinase 1 (CDK1), mitogen-activated protein kinase 1 (MAPK1), mitogen-activated protein kinase 3 (MAPK3), and c-Jun N-terminal kinase (JNK).34 The HCV could affect stathmin activity through altering Ser38 itself or by indirectly modulating these upstream kinases. Thus, future anti-HCC and anti-HCV studies could focus on examining 2 potential candidates: (1) manipulating the Ser38 phosphorylation site to repress HCC proliferation and (2) modulating the intracellular environment of chronic HCV infection to emulate the expression of Quad stathmin to sensitize cells to apoptosis.
Microtubule deregulation as dual target for antiviral/anticancer research
Both assembly and disassembly of the microtubule spindle affect mitotic cellular division. Interference of this highly controlled process can trigger apoptosis.35 Given that deregulation of apoptosis and proliferation are features of carcinoma,36-39 viral disruption of these pathways may facilitate hepatocarcinogenesis. If HCV manipulates microtubules through proteins such as stathmin to create an ideal environment for viral replication, the resulting cytoskeletal deregulation and inhibition of apoptosis may lead to carcinoma development. Therefore, microtubule dynamics may serve as a dual target for antiviral drug development and liver cancer prevention to treat infected hepatocytes that are primed for malignant transformation.
As standard HCV treatment uses direct-acting antivirals (DAAs) that target viral proteins such as the RNA polymerase nonstructural protein 5B (NS5B),40 looking at host intracellular environment provides an alternative approach to identify targets for therapeutic development. A broader methodology that takes advantage of a more inclusive view of host-virus interactions may help to address hepatitis treatment from a different angle. Therapy that seeks to induce intolerant cellular conditions for viral reproduction offers an additional strategy that can complement current Food and Drug Administration–approved options to address future areas of concern, such as viral relapse.41 Previous studies of HCV in HCC patients have shown that the genetic variance of the virus is higher in cancerous liver tissue compared with benign liver tissue.42 These studies imply that within virulent cells there may be a potential risk of mutation that could lead to resistance against standard DAAs. By maintaining an antiviral host cell environment after DAA treatment, a synergistic compound may help minimize relapse and hepatocarcinogenesis. The ability to target both the virus and the risk of HCC increases the potential utility of intracellular therapy in patients with chronic hepatitis C.
Supplementary Material
Acknowledgments
The authors thank UCLA’s Janis V. Giorgi Flow Cytometry Core Facility and Jeffrey Calimlim for their services. In addition, they thank the David Dawson lab for providing us with lentivirus packaging vectors and Chau Tran for the helpful discussion and mutagenesis kit recommendation.
Footnotes
Funding:The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Tower Cancer Research Foundation, Landon Foundation-American Association for Cancer Research Innovator Award for Cancer Prevention Research, and the NIH National Institutes of Diabetes, Digestive and Kidney Disease (grant number R01DK090794-SWF).
Declaration of conflicting interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: NTL, DP, and SWF conceived of the presented idea. NML, DP, JQV, and LL designed and carried out the experiments and analyzed the data. CYK, PC, NP, EG, and RK helped with the experiments. CEM created TMA and performed the IHC. VA and AD engineered the HCV replicon. NTL and NML wrote the manuscript. All authors reviewed and approved the final manuscript.
References
- 1. Messina P, Humphreys I, Flaxman A, et al. Global distribution and prevalence of hepatitis C virus genotypes. Hepatology. 2015;61:77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kanwal F, Kramer JR, Ilyas J, Duan Z, El-Serag HB. HCV genotype 3 is associated with an increased risk of cirrhosis and hepatocellular cancer in a national sample of U.S. Veterans with HCV. Hepatology. 2014;60:98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ananthakrishnan A, Gogineni V, Saeian K. Epidemiology of primary and secondary liver cancers. Semin Intervent Radiol. 2006;23:47–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lu NT, Liu NM, Vu JQ, et al. Phospho-network analysis identifies and quantifies hepatitis C virus (HCV)-induced hepatocellular carcinoma (HCC) proteins regulating viral-mediated tumor growth. Cancer Genomics Proteomics. 2016;13:339–358. [PMC free article] [PubMed] [Google Scholar]
- 5. Etienne-Manneville S. Actin and microtubules in cell motility: which one is in control? Traffic. 2004;5:470–477. [DOI] [PubMed] [Google Scholar]
- 6. Baquero MT, Hanna JA, Neumeister V, et al. Stathmin expression and its relationship to microtubule-associated protein tau and outcome in breast cancer. Cancer. 2012;118:4660–4669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sonego M, Schiappacassi M, Lovisa S, et al. Stathmin regulates mutant P53 stability and transcriptional activity in ovarian cancer. EMBO Mol Med. 2013;5:707–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nie W, Xu D, Gan L, et al. Overexpression of stathmin 1 is a poor prognostic biomarker in non-small cell lung cancer. Lab Invest. 2015;95:56–64. [DOI] [PubMed] [Google Scholar]
- 9. Akhtar J, Wang Z, Yu C, Zhang ZP, Bi MM. STMN-1 gene: a predictor of survival in stage IIA esophageal squamous cell carcinoma after Ivor-Lewis esophagectomy? Ann Surg Oncol. 2013;21:315–321. [DOI] [PubMed] [Google Scholar]
- 10. Hsieh SY1, Huang SF, Yu MC, et al. Stathmin1 overexpression associated with polyploidy, tumor-cell invasion, early recurrence, and poor prognosis in human hepatoma. Mol Carcinog. 2010;49:476–487. [DOI] [PubMed] [Google Scholar]
- 11. Xu K, Harrison RE. Down-regulation of stathmin is required for the phenotypic changes and classical activation of macrophages. J Biol Chem. 2015;290:19245–19260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McCartney EM, Helbig KJ, Narayana SK, Eyre NS, Aloia AL, Beard MR. Signal transducer and activator of transcription 3 is a proviral host factor for hepatitis C virus. Hepatology. 2013;58:1558–1568. [DOI] [PubMed] [Google Scholar]
- 13. Nouar R, Breuzard G, Bastonero S, et al. Direct evidence for the interaction of stathmin along the length and the plus end of microtubules in cells. FASEB J. 2016;30:3202–3215. [DOI] [PubMed] [Google Scholar]
- 14. Granger E, McNee G, Allan V, Woodman P. The role of the cytoskeleton and molecular motors in endosomal dynamics. Semin Cell Dev Biol. 2014;31:20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wong MT, Chen SS. Emerging roles of interferon-stimulated genes in the innate immune response to hepatitis C virus infection. Cell Mol Immunol. 2016;13:11–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Greber UF, Way M. A superhighway to virus infection. Cell. 2006;124:741–754. [DOI] [PubMed] [Google Scholar]
- 17. Blight KJ, McKeating JA, Rice CM. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol. 2002;76:13001–13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Arumugaswami V, Remenyi R, Kanagavel V, et al. High-resolution functional profiling of hepatitis C virus genome. PLoS Pathogens. 2008;4:e1000182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Uthaya Kumar DB, Chen CL, Liu JC, et al. TLR4 signaling via NANOG cooperates with STAT3 to activate Twist1 and promote formation of tumor-initiating stem-like cells in livers of mice. Gastroenterology. 2016;150:707–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Halder UC, Bagchi P, Chattopadhyay S, Dutta D, Chawla-Sarkar M. Cell death regulation during influenza A virus infection by matrix (M1) protein: a model of viral control over the cellular survival pathway. Cell Death Dis. 2011;2:e197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Salako MA, Carter MJ, Kass GE. Coxsackievirus protein 2BC blocks host cell apoptosis by inhibiting caspase-3. J Biol Chem. 2006;281:16296–16304. [DOI] [PubMed] [Google Scholar]
- 22. Wasilenko ST, Stewart TL, Meyers AF, Barry M. Vaccinia virus encodes a previously uncharacterized mitochondrial-associated inhibitor of apoptosis. Proc Natl Acad Sci U S A. 2003;100:14345–14350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gubser C, Bergamaschi D, Hollinshead M, Lu X, van Kuppeveld FJ, Smith GL. A new inhibitor of apoptosis from vaccinia virus and eukaryotes. PLoS Pathog. 2007;3:e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu X, Li Q, Dowdell K, Fischer ER, Cohen JI. Varicella-Zoster virus ORF12 protein triggers phosphorylation of ERK1/2 and inhibits apoptosis. J Virol. 2012;86:3143–3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Amayed P, Pantaloni D, Carlier MF. The effect of stathmin phosphorylation on microtubule assembly depends on tubulin critical concentration. J Biol Chem. 2002;277:22718–22724. [DOI] [PubMed] [Google Scholar]
- 26. Kuntziger T, Gavet O, Sobel A, Bornens M. Differential effect of two stathmin/Op18 phosphorylation mutants on Xenopus embryo development. J Biol Chem. 2001;276:22979–22984. [DOI] [PubMed] [Google Scholar]
- 27. Mistry SJ, Atweh GF. Role of stathmin in the regulation of the mitotic spindle: potential applications in cancer therapy. Mt Sinai J Med. 2002;69:299–304. [PubMed] [Google Scholar]
- 28. Rubin CI, Atweh GF. The role of stathmin in the regulation of the cell cycle. J Cell Biochem. 2004;93:242–250. [DOI] [PubMed] [Google Scholar]
- 29. Benedict CA, Norris PS, Ware CF. To kill or be killed: viral evasion of apoptosis. Nat Immunol. 2002;3:1013–1018. [DOI] [PubMed] [Google Scholar]
- 30. Hay S, Kannourakis G. A time to kill: viral manipulation of the cell death program. J Gen Virol. 2002;83:1547–1564. [DOI] [PubMed] [Google Scholar]
- 31. Baik SY, Yun HS, Lee HJ, et al. Identification of stathmin 1 expression induced by Epstein-Barr virus in human B lymphocytes. Cell Prolif. 2007;40:268–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen PW, Lin SJ, Tsai SC, et al. Regulation of microtubule dynamics through phosphorylation on stathmin by Epstein-Barr virus kinase BGLF4. J Biol Chem. 2010;285:10053–10063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wik E, Birkeland E, Trovik J, et al. High phospho-Stathmin(Serine38) expression identifies aggressive endometrial cancer and suggests an association with PI3K inhibition. Clin Cancer Res. 2013;19:2331–2341. [DOI] [PubMed] [Google Scholar]
- 34. Ng DC, Zhao TT, Yeap YY, Ngoei KR, Bogoyevitch MA. c-Jun N-terminal kinase phosphorylation of stathmin confers protection against cellular stress J Biol Chem. 2010;285:29001–29013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mollinedo F, Gajate C. Microtubules, microtubule-interfering agents and apoptosis. Apoptosis. 2003;8:413–450. [DOI] [PubMed] [Google Scholar]
- 36. Pierce RH, Vail ME, Ralph L, Campbell JS, Fausto N. Bcl-2 expression inhibits liver carcinogenesis and delays the development of proliferating foci. Am J Pathol. 2002;160:1555–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wong RS. Apoptosis in cancer: from pathogenesis to treatment. J Exp Clin Cancer Res. 2011;30:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hughes R, Parry J, Beynon J, Jenkins G. Molecular changes consistent with increased proliferation and invasion are common in rectal cancer. Clin Transl Oncol. 2011;13:753–759. [DOI] [PubMed] [Google Scholar]
- 39. Femia AP, Salvianti F, Luceri C, et al. Sustained proliferation and resistance to apoptosis after a cytotoxic insult are early alterations in rat colon carcinogenesis. Int J Cancer. 2012;131:529–536. [DOI] [PubMed] [Google Scholar]
- 40. Stedman C. Sofosbuvir, a NS5B polymerase inhibitor in the treatment of hepatitis C: a review of its clinical potential. Therap Adv Gastroenterol. 2014;7:131–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sarrazin C, Dvory-Sobol H, Svarovskaia ES, et al. Prevalence of resistance-associated substitutions in HCV NS5A, NS5B, or NS3 and outcomes of treatment with ledipasvir and sofosbuvir. Gastroenterology. 2016;151:501.e1–512.e1. [DOI] [PubMed] [Google Scholar]
- 42. Harouaka D, Engle RE, Wollenberg K, et al. Diminished viral replication and compartmentalization of hepatitis C virus in hepatocellular carcinoma tissue. Proc Natl Acad Sci U S A. 2016;113:1375–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.